CONFERENCE COVERAGE SERIES
Alzheimer's Association International Conference (AAIC) - 2023
Amsterdam, Netherlands and Online
16 – 20 July 2023
CONFERENCE COVERAGE SERIES
Amsterdam, Netherlands and Online
16 – 20 July 2023
Change was in the air at the Alzheimer’s Association International Conference, held July 16-20 in Amsterdam. With the first treatment in 20 years having just earned traditional approval from the U.S. Food and Drug Administration, another with accelerated approval, and a third with positive Phase 3 data, Alzheimer’s researchers marked the end of a long drought, and made plans to build on these gains. Some 7,500 people attended in person, and another 3,500 viewed talks online. A third of attendees were under 35, more than 60 percent were women, and almost a fifth were from low- and middle-income countries, noted the association’s Maria Carrillo.
“I’ve never experienced such a positive vibe at AAIC,” said Philip Scheltens, who has been part of this conference for the last 35 years. “We’ve waited so long [to have new treatments], and now we’re there.” Scheltens, newly retired from leading the Alzheimer’s Center at VU University Medical Center in the meeting’s host city, received the Alzheimer’s Association’s Bengt Winblad Lifetime Achievement Award at AAIC. Many subsequent sessions were introduced by his successor, Wiesje van der Flier, who shares directorship of the Alzheimer’s Center with Yolande Pijnenburg.
In a further sign of how drug approvals are changing the field, the U.S. Centers for Medicare and Medicaid Services announced its plan to end restrictions on covering amyloid PET scans July 18 during AAIC. In effect since 2013, the previous policy had limited beneficiaries to a single scan per lifetime, and only for those participating in a clinical trial (Jan 2013 news; Oct 2013 news). In its proposed decision memo, the agency noted that this once-per-lifetime restriction was no longer appropriate, given the development of anti-amyloid treatments.
Against this backdrop, sessions in Amsterdam featured numerous talks on amyloid immunotherapy. Other notable topics included Down’s syndrome—a genetic form of Alzheimer's that could potentially be treated with such antibodies but has not been studied for that purpose—and anti-tau antibodies and their potential for concurrent trials. A symposium on proposed revisions to the NIA-AA criteria for diagnosing AD drew keen interest. So did sessions on reviving BACE inhibitors, inflammation and vascular contributions to dementia, ADRD biomarkers beyond Aβ and p-tau, the LEAD cohort of early onset AD, APOE research, and many other topics (see upcoming conference stories).
This story summarizes the first in-depth look at Phase 3 data from donanemab, which were discussed July 17 in Amsterdam, and published in JAMA the same day. In Amsterdam, Eli Lilly’s Mark Mintun said the company has applied for traditional approval from the FDA, and will file in other countries this year. Lilly has begun a new safety and efficacy trial, Trailblazer-Alz5, focused on countries outside the U.S.

Diverging Curves? In the primary analysis population, donanemab (green) slowed cognitive decline compared to the placebo group (gray) on the iADRS (left) and CDR-SB (right), with curves continuing to diverge over 18 months. [Courtesy of Eli Lilly.]
Donanemab Benefit Robust, Grows Over Time
Lilly had released Phase 3 top-line results from Trailblazer-Alz2 two months ago, reporting that donanemab slowed decline on the primary and all secondary clinical endpoints by about a third in the primary analysis population, who had low-to-intermediate tau tangle loads (May 2023 news). In Amsterdam, Lilly’s John Sims filled in details. At 18 months, the absolute difference between treatment and control groups was 3.25 points on the iADRS, and 0.67 on the CDR-SB. The latter was slightly larger than the 0.45 points seen with lecanemab, and the 0.53 points notched in the one positive aducanumab trial. Those trials did not use the iADRS.
The Trailblazer-Alz2 cohort was slightly further along in the disease compared to those in other trials, Mintun noted. Participants were about two years older, at an average age of 74. Their baseline MMSE was 23, their plaque load 102 centiloids. These numbers were similar to participants in the negative gantenerumab Phase 3 trials, but more advanced than the population in the positive lecanemab Phase 3 trial, which had an average MMSE of 26 and amyloid load of 76 centiloids (Dec 2022 conference news).
The difference between the donanemab and control groups became statistically significant as early as three months for the iADRS, and at six months for the CDR-SB. The curves continued to separate over 18 months, as expected for a disease-modifying therapy, Mintun said. At 12 months, the numerical difference between treatment and placebo groups was 2.62 for the iADRS and 0.59 for the CDR-SB, both less than the difference at 18 months. The researchers are continuing to follow participants in a further 18-month open-label extension, which is still blinded to the original treatment groups. This will generate data on how the treatment effect evolves over three years.
In Amsterdam, several scientists continued their argument for measuring delay in progression in units of time, rather than numerical differences or percent slowing of decline (Apr 2023 conference news). On this “time saved” scale, donanemab delayed progression on the iADRS by 4.5 months, and on the CDR-SB by 7.5 months, Liana Apostolova of Indiana University School of Medicine in Indianapolis reported in Amsterdam.
The findings were reported to be robust to sensitivity analyses. Efficacy results did not much change when the analyses were adjusted to account for dropouts, when excluding participants who had ARIA, or when using different statistical methods. In all scenarios, donanemab slowed progression between 33 to 40 percent, Sims said.

Slice and Dice by Tau. The Trailblazer-Alz2 trial focused on people with low to intermediate tangle load (middle), the only statistically powered group. Gains were smaller in a high-tau group (right). People with very little tau PET signal (left) were not enrolled, nor followed observationally. [Courtesy of Eli Lilly.]
Treatment Effect Apparent in Early Disease, at Intermediate Tau Loads
In prespecified analyses of the primary outcome data, donanemab appeared to work best at a narrowly defined disease stage. The clinical benefit was largest in the subgroup with mild cognitive impairment. Their decline slowed by 60 percent on the iADRS and 46 percent on the CDR-SB. Grouping participants by age revealed slightly more benefit in those under 75, with decline slowed by 48 percent on the iADRS and 45 percent on the CDR-SB.
Other factors did not seem to matter. The researchers found no difference between men and woman, unlike in the Phase 3 gantenerumab trials, which had a greater effect in men (Apr 2023 conference news). In those trials, women at baseline had worse tau tangles than men, whereas Trailblazer-Alz2 selected participants based on tangle load, resulting in more uniformity on this measure.
Racial and ethnic subgroups were too small to draw conclusions. Lilly has added an ongoing safety addendum to the trial and is using this to try to boost diversity, Mintun noted. There was no difference between APOE4 heterozygotes and noncarriers. The treatment benefit appeared to be slightly less in APOE4 homozygotes, but this subgroup was small and underpowered.
Tangle load did matter, however. The RCT trial population, comprising 1,182 people and used for all the above analyses, had baseline tau PET scans between 1.10 and 1.46 SUVr. Trailblazer-Alz2 enrolled an additional 554 people with scans above 1.46. This high-tau subgroup notched barely 20 percent slowing of decline on the CDR-SB, and below 10 percent on the iADRS. The former was statistically significant, the latter not, though Sims noted that this subgroup was not powered to show significance. Baseline amyloid loads were the same regardless of tangle burden.
This curation of a treatment group by tau PET drew both praise and questions. Christopher van Dyck of Yale School of Medicine in New Haven, Connecticut, called these staging data valuable but noted that as many as a quarter of people with early AD have tau PET loads below 1.10 SUVr. Should this group be treated with donanemab? This registration trial gives no data on this question, as it excluded them.
Sims noted that this population, which Lilly considers to be basically “no tau,” is being evaluated in Trailblazer-Alz3, which enrolls people at the preclinical stage of disease. That trial is not expected to read out until the end of 2027 (Jul 2021 news). This answer did not fully satisfy everyone. In hallway talk at AAIC, several clinician-researchers said they'd prefer to also have data on how donanemab performs in people with mild symptoms and very low tau, many of whom will seek treatment at memory clinics.
Others asked whether tau PET scans will be necessary before treatment with donanemab, so that treating physicians can target the same population as those who benefitted in the trial. Sims argued that this is not needed, given that the combined intermediate- and high-tau groups still showed a statistically significant treatment benefit. Oskar Hansson of Lund University, Sweden, who analyzed biomarker data for Lilly, noted that because people with high tangle loads have a different risk-benefit ratio for treatment, it would be helpful to test for tau burden before prescribing anti-amyloid antibodies. Ideally, this would be done with a blood test rather than tau PET, due to the high cost and limited availability of the latter. Such plasma tests are in development, but not yet broadly available.
Most Biomarkers Moved Toward Normal, But Tangles Stayed Put
The biomarker data from this trial shown at AAIC largely tracked with the clinical findings. Results were similar in the low-to-intermediate tau group, and in the combined analysis with the high-tau group included. As expected, donanemab rapidly removed an average of 88 centiloids of plaque over 18 months. The drop was fastest in the first six months, with a third of participants becoming amyloid-negative then. By the end of the trial, 80 percent had crossed this threshold.
This is faster than with aducanumab. In Amsterdam, Lilly’s Andrew Pain reported 12-month data from the head-to-head comparison of these two drugs in the open-label Trailblazer-Alz4 trial. Six-month data had shown faster clearance with donanemab, in part due to its quicker titration to the effective dose (Dec 2022 conference news). That pattern continued, with donanemab removing 80 centiloids compared to aducanumab's 56, by one year. In this study, 70 percent of people taking donanemab were amyloid-negative by one year, compared with 22 percent on aducanumab.
In the Phase 3 study, plasma tau biomarkers followed plaque. Plasma p-tau181 dropped almost 20 percent on drug, p-tau217, 40 percent. In keeping with the subgroup analyses showing more clinical benefit at earlier stages, people who were in the lowest p-tau217 tertile at baseline did best, declining on the CDR-SB by 46 percent less than placebo, compared with 27 percent less for those with higher baseline p-tau217. This post hoc analysis used the combined tau groups.
Plasma GFAP, a marker of inflammation, fell 21 percent on drug. Plasma NfL, thought to denote neurodegeneration, was mystifying. It initially rose in the donanemab group, before dropping back to slightly below baseline levels by 18 months. Sims noted that this marker varies greatly from one person to another, but did not show spaghetti plots.
As in other amyloid immunotherapy trials, brain volume decreased on donanemab, though hippocampal volume loss resembled that on placebo. The field is grappling with what this accelerated shrinkage means (Apr 2023 news). At AAIC, too, the discussion reprised current arguments that global volume loss perhaps reflects cortical “pseudo-atrophy” as amyloid and attendant inflammation decrease, whereas the hippocampus has little amyloid to begin with.
The most surprising and, to some, concerning finding was that donanemab did not affect tangle growth as measured by tau PET. In Phase 2, the antibody had appeared to slow, though not stop, tangle accumulation in the frontal cortex, with a trend toward slowing in the parietal and temporal lobes (Mar 2021 conference news). In the Phase 3 trial, the tau PET curves of placebo and treatment groups were superimposed.
Hansson noted that ongoing regional analyses may yet surface subtle effects. A post hoc analysis of these Phase 3 data suggested that participants who became amyloid-negative during the trial had less tangle accumulation than those who remained amyloid-positive, Hansson added. Here, too, 18 months of additional study may be instructive.

Staying Power. In participants who stopped donanemab after having dipped below the amyloid threshold by one year, trial outcomes continued to diverge from those who had always been on placebo (arrows). [Courtesy of Eli Lilly.]
What Happens When Treatment Stops?
A unique aspect of donanemab is that treatment stops once plaque load drops below the positivity threshold. In Trailblazer-Alz2, this happened in two-thirds of participants across both tau groups. What did this do to their subsequent clinical progression?
To answer this, Lilly researchers analyzed nearly 300 people who became amyloid-negative at an average of 47 weeks. Their clinical progression continued to diverge from the placebo group, reaching a difference of 3.52 points on the iADRS and 0.75 on the CDR-SB by 18 months, slightly larger than the overall trial benefit.
In Amsterdam, Lilly’s Sergey Shcherbinin presented data on plaque re-accumulation in participants of the earlier Phase 2 study. Analyzing 47 people who became amyloid-negative, with an average final plaque load of 4.7 centiloids, Shcherbinin found that they added about 2 centiloids over the following year. This is similar to the rate of plaque accumulation in the ADNI observational study, even though this trial population has higher tangle loads on average than the ADNI cohort, Shcherbinin noted. This rate was the same regardless of how much amyloid people had cleared, or how low on the centiloid scale they got after a course of donanemab.
Adding data from the Phase 3 trial upped the re-accumulation estimate to 2.8 centiloids per year. At this clip, it would take about five years for a person to become amyloid-positive again after clearance, Shcherbinin said.
ARIA: Still a Fly in the Ointment
The risk of ARIA remains a big concern. In its trials thus far, donanemab had ARIA rates in between the 12 percent on lecanemab and 33 percent on aducanumab, with about one-quarter of people on drug developing it. Six percent of people in Trailblazer-Alz2 developed symptoms from ARIA. In almost 2 percent of participants, nearly all of them APOE4 carriers, these symptoms were considered serious, noted Stephen Salloway of Butler Hospital in Providence, Rhode Island. The risk of ARIA was the same in people taking anticoagulant or anti-platelet medication as in the cohort overall.
As with lecanemab, the risk of macrohemorrhage was higher on drug, doubling from 0.2 to 0.4 percent. Three people on donanemab died from brain bleeds during the trial. Two of them were heterozygous APOE4 carriers and developed severe ARIA-E, Salloway said. The third was not a carrier, but had superficial siderosis at baseline, a sign of previous brain hemorrhages. None were on anticoagulants.
Numerous talks in Amsterdam grappled with how to predict the occurrence of ARIA, with a growing number of scientists tying it to cerebral amyloid angiopathy and its attendant inflammation (see upcoming AAIC story).—Madolyn Bowman Rogers
Scientists once hoped that blocking β-secretase would slow or prevent Alzheimer’s disease. Then came the rude awakening. The inhibitors caused the very thing they were supposed to prevent—cognitive decline. Enthusiasm tanked. Pharmaceutical companies canned their BACE inhibitor programs. But a few lone voices kept calling for another try, and by AAIC 2023, held July 16-20 in Amsterdam, the vibe had shifted. A small but vocal group of basic scientists had always maintained that the field gave up too quickly on these compounds, and now some clinicians have come around.
“BACE inhibition in primary prevention holds great potential,” said Paul Aisen, University of Southern California, San Diego, during a scientific session on the future of BACE inhibition. Even Reisa Sperling, a self-professed skeptic of BACE inhibition, said she could envision a small trial. Sperling, from Brigham and Women’s Hospital, Boston, co-chaired the session with Robert Vassar from Chicago's Northwestern University, who, with Martin Citron, had cloned BACE 24 years ago (Vassar et al., 1999).
What’s changed? For BACE inhibition itself, not much. Secretase experts had always advised low doses to avoid adverse events. That is because BACE not only catalyzes the first step in Aβ production—the cleavage of amyloid precursor protein—but also snips dozens of other substrates. Many are found in neurons; some support axon guidance and synaptogenesis.
What has changed is that scientists now have a way to identify and monitor at least some of those cleavages. In Amsterdam, Stefan Lichtenthaler, Technical University of Munich, reported analysis of cerebrospinal fluid samples taken at baseline and after treatment with three different BACE inhibitors, Merck’s verubecestat, Novartis’s umibecestat, and Shionogi’s atabecestat. The Merck samples came from a Phase 3 trial, while the others came from Phase 2s.
Stephan Muller and Pieter Giesbertz in Lichtenthaler’s lab ran mass-spectrometry-based proteomic analyses of the samples, looking for peptides snipped from cell-surface BACE substrates. “All three BACE inhibitors produced qualitatively and quantitatively similar proteome changes,” said Lichtenthaler. The three inhibitors lowered CSF concentrations of peptides cleaved from the same dozen or so substrates, including Sez6, IL6ST, CACHD1, CHL1, Sez6L, and L1CAM.

BACE Proteomes. These volcano plots show down- (blue) and upregulated (red) protein fragments in the CSF of people who had been treated in BACE inhibitor trials. The profiles for umibecestat (left) and verubecestat (right) were very similar. [Courtesy of Stefan Lichtenthaler, Technical University of Munich.]
These reductions were dose-dependent, and, in the case of the learning and memory-linked protein Sez6, the soluble fragments were almost fully suppressed at the highest inhibitor doses given in those trials. Indeed, reduction of soluble Sez6 directly correlated with reduction of Aβ40 in the CSF.
For some other substrates, however, BACE inhibition, even at the highest doses, only partially reduced their soluble fragments, suggesting other proteases help process them. This was the case for the neural cell adhesion molecules L1CAM and CHL1, for example. Whether that absolves these substrates from any part in the cognitive decline caused by BACE inhibition in the trials remains to be seen.
Nobody knows for certain which of the BACE substrates are needed for cognition. Jochen Herms, Ludwig-Maximilians University, Munich, believes Sez6 is important. At AAIC, he reported that in Sez6 knockout mice, spine density is a bit below that of control mice, but that verubecestat and other BACE inhibitors did not reduce it further. The data suggest that spines supported by this neuronal signaling molecule are vulnerable to BACE inhibition.
Importantly, Lichtenthaler noted that reductions of all these soluble fragments in the CSF reached a maximum at even the lowest doses of umibecestat and verubecestat used in the clinical trials—15mg and 12mg, respectively. Partial suppression occurred in the case of 5 mg atabecestat, which, incidentally, caused no significant cognitive decline. “Cognitive worsening only occurs at drug doses where BACE1 substrate cleavage is nearly maximally inhibited,” Lichtenthaler concluded.
He believes that low doses of BACE inhibitors would allow sufficient substrate cleavage to still occur such that cognitive decline would be avoided, while also sufficiently slowing amyloid accumulation. He said trials to test this hypothesis should collect enough CSF to correlate cleavage of these BACE substrates with any cognitive changes.
What could such a trial look like? Julie Stone, a pharmacokineticist at Merck, has been crunching the numbers on such questions for years (e.g., May 2015 webinar). Stone modelled two scenarios discussed in Amsterdam—primary prevention among people who have tested positive for low levels of amyloid, and a post-immunotherapy “maintenance dose” to keep plaques from regrowing once they had been cleared. In both cases, Stone estimated how BACE inhibition would affect plaque load.
Regarding prevention, Stone concluded that a high dose of verubecestat, or around 27 mg daily, would be needed to halt amyloid growth at a stage where baseline levels are around 10 centiloids. Such a high dose would be needed because, in kinetic terms, plaque growth is a zero-order process, proceeding fastest early in pathogenesis. Alas, that 27 mg dose would suppress Aβ production by 80 percent, a level associated with cognitive side effects in prior trials.
What about when baseline amyloid levels are higher? Perhaps counterintuitively, Stone's model predicted that lower doses of inhibitor would suffice in that case. At a baseline of 50 or 60 centiloids, daily doses of 2.9 mg or 1.7 mg verubecestat, respectively, would prevent further plaque growth. Whether this would slow progression of the disease, including tau pathology and cognitive decline, is unknown, Stone said. The 1.7 mg dose would inhibit BACE by a third, likely low enough to avoid cognitive side effects.
Rather than stopping plaque growth, what about slowing it down? Stone modeled what 35 percent BACE inhibition at various baseline amyloid loads would achieve. At 10, 25, or even 50 centiloids, this would limit plaque growth to reach 50 to 60 centiloids after 20 years, instead of the 80 to 90 centiloids that would be reached in untreated controls.
“The big open question is, is that clinically meaningful?” Stone asked at AAIC. Many AD-related pathologies and symptoms don’t show up until plaque loads surpass 50 centiloids, suggesting this slowing could be beneficial. “Instinctively, we expect that slowing of amyloid would slow progression of the disease,” she said.

Slow Those Plaques Down. Kinetic modeling suggests that 1.7 mg verubecestat, given when amyloid burden had reached 10 (blue line), 25 (red line), or 50 centiloids (yellow line), would slow plaque growth relative to untreated controls (dashed lines). [Courtesy of Julie Stone, Merck.]
Stone predicted a similar scenario if BACE inhibitors come in after immunotherapy has removed plaques. Around 12 mg verubecestat would be able to maintain a 20 centiloid plaque load. That is in the range known to cause cognitive deficits. A dose of 1.7 mg would slow regrowth, again plateauing at about 50-60 centiloids after 20 years.
What about real-life experiments? Preclinical work makes low-dose inhibition look attractive. Elyse Watkins, a postdoc in Vassar’s lab, treated 8-month-old PDAPP mice with 109, 33, or 11 mg/Kg of MBi-10, a BACE inhibitor from Merck. Six months later, mice on the lowest dose had 40 percent less amyloid than untreated controls. The human equivalent dose would be around 0.9 mg/Kg, likely sparing cognition.
Watkins’ data suggests that this dose might even help clear plaques. She had injected the 8-month-old animals with Methoxy O4 to label existing plaques. At 14 months, she measured less of the label in treated mice than in controls, suggesting that some of the plaque present at baseline had cleared.

Going Low. At a low dose of 11 mg/Kg, the BACE inhibitor MBi-10 reduced soluble Aβ42 and plaque load in the cortices and hippocampi of PADPP mice. It even cleared some of the Methoxy O4 injected into the brain six months prior, hinting that it may help clear existing plaques. [Courtesy of Elyse Watkins, Northwestern University.]
Are trialists ready to try? “We have to be very careful,” Sperling told Alzforum. She is concerned about suppressing soluble Aβ. Like BACE inhibitors, solanezumab, a monoclonal antibody that binds monomeric Aβ, also tended to cause cognitive decline in the A4 secondary prevention trial, though without affecting BACE substrates (Mar 2023 news). Dave Morgan, Michigan State University, Grand Rapids, echoed that sentiment. Like others in the field, he thinks Aβ has some important, still-unknown function at synapses.
Even so, Sperling has warmed to the idea of testing these inhibitors once more. “I would be open to a small trial where we test for cognitive changes over a very short time, even as little as a few weeks,” she said.
Aisen is more optimistic. He pointed out that solanezumab did have a small cognitive/clinical benefit in the pooled analyses of two Phase 3 trials in LOAD, and that the solanezumab dose was subsequently increased in A4 (Oct 2012 news; Jun 2017 news). Aisen suspects A4 may have simply lowered soluble Aβ too much for synapses to function properly.
“I think if we embed sufficient cognitive monitoring, we would be comfortable in planning a significant [BACE inhibitor] trial,” he told Alzforum. “Clearly there are cognitive effects, but these are dose-related, reversible, and we can monitor them,” he said.
Aisen believes that in preclinical AD, the A4 trial validated the PACC and RBANS as being sensitive enough to quantify cognitive decline, and would pick up decline caused by BACE inhibition, as well (Mar 2023 news). “I believe we can develop BACE inhibitor trials at lower than 50 percent inhibition, and do it now,” he claimed.
Others welcomed the idea. Ralph Nixon, New York University, noted that BACE inhibition would not only reduce Aβ, but also APP's β-C-terminal fragments. Nixon and others have reported that β-CTFs scupper the endolysosomal system, disrupting proteostasis and exacerbating amyloid pathology (Jun 2010 news; Jun 2022 news; Jul 2023 news).
Lichtenthaler agreed that tempering β-CTF could be an important benefit—as could an increase in sAPPα, a fragment generated by α-secretase, which completes with BACE to cleave APP (Jan 2019 news). “We could have a triple-positive effect of BACE inhibition,” he suggested. Low-dose BACE inhibition might even correct lysosomal dysfunction in AD, he predicted (see Lichtenthaler’s recent comment).
Injecting a cautionary note, Bart De Strooper, University College, London, said that in addition to suppressing cleavage of BACE's other substrates, inhibitors would also lead to more of the shorter, more hydrophobic Aβ peptides generated by α-cleavage of APP between amino acids 16 and 17 of the Aβ peptide sequence. Also called p3 fragments, these Aβ17-x peptides tend to be neglected, but can insinuate themselves into plaques (Iwatsubo et al., 1996). “These are difficult to detect, but I would not overlook them,” said De Strooper.
De Strooper wants to see γ-secretase modulators (GSMs) reconsidered. “Mechanism-based side effects that are difficult to circumvent are always a struggle with BACE inhibitors,” he said. “With GSMs, you aim to lower the toxic Aβ fragments instead of lowering total Aβ. This is a better rationale, because you keep all other biology intact.”
Lichtenthaler countered that the protective Icelandic A673T mutation in APP, which slows its cleavage by BACE and reduces Aβ production by a third, supports the rationale for BACE inhibition, as do mice having only one copy of the BACE1 gene (Maloney et al., 2014). The latter lack the neurological dysfunction of the full knockouts.
BACE inhibitors have been tested up to Phase 3 and could be retested without delay. “I’m pretty convinced low-dose BACE inhibition will work,” said Lichtenthaler.
What would such a trial look like? Aisen is enthusiastic about both types of trial Stone modeled, one for primary prevention and one to augment immunotherapy. Whether pharmaceutical companies would be willing to restart their BACE programs remains to be seen.
“I’m not confident we’ll get that kind of support, but am optimistic that pharma will be willing to supply the drugs,” Aisen said. One pharma scientist told Alzforum her company would likely sell its BACE inhibitor for $1 to a group able to fund and run new trials.
One possibility is an A2 trial akin to the A3 and A45 trials of lecanemab targeting people with amyloid levels between 20 and 40 centiloids, and above 40 centiloids, respectively. Aisen and Sperling are investigators on both. Indeed, the A3 trial was initially slated to test a BACE inhibitor until the cognitive side effects put the kibosh on that. “It seemed plausible then, and it seems plausible now, that BACE inhibition will have a clinically meaningful effect,” said Aisen.
As for A2, Aisen said the goal was always to go lower, to 20 or 10 centiloids. He believes it may be possible to treat people before they test positive for amyloid, using plasma tests for various p-taus or Aβ42/40 as indicators of imminent amyloid deposition. “We are discussing A2,” noted Aisen. “Our intention is to continue to move earlier, toward primary prevention, but we are not ready to pin down a trial design,” he told Alzforum.
“Our goal is to devise a path forward for new clinical trials to test low-level BACE inhibition,” said Vassar, “because we are going to need an oral disease-modifying therapy for Alzheimer’s disease.” De Strooper agreed. "It is extremely important to emphasize that anti-amyloid antibodies cannot be the primary prevention we are all looking for.” —Tom Fagan
More than a decade before memory loss, fragments of phospho-tau start to rise in biofluids. These biomarkers, particularly p-tau217, have proven to be exquisite detectors of amyloid, but they plateau once symptoms surface and don’t track tightly with tau tangles. Here’s where MTBR-tau-243 comes in. According to a paper published July 13 in Nature Medicine, this particular fragment from tau’s microtubule-binding domain rises in step with tau-PET, and continues to climb even in the symptomatic stages of AD. Led by Randall Bateman of Washington University in St. Louis and Oskar Hansson of Lund University in Sweden, the study found that, more than any other tau biomarker tested so far, MTBR-tau-243 also correlates with cognitive decline, offering clinicians an important tool for both diagnosis and prognosis. What’s more, the putative tangle detector comes at a time when it’s sorely needed for tracking treatment responses in trials aimed at tau.
Case in point, at the Alzheimer’s Association International Conference, held July 16-20 in Amsterdam, first data from early phase trials of Eisai’s E2814—an antibody that binds tau’s midsection—indicate that the drug safely engaged its target in people with familial AD. Importantly, it also cut CSF MTBR-tau-243 in half. Together, these findings hint at the possibility that finally, a tau antibody may be putting a dent tau tangles. Of course, the field still needs to find out if this target engagement stems cognitive decline.
In the AD brain, tau tangles comprise fibrils with a C-shaped protofilament core, which itself includes the third and fourth microtubule binding domains near the C-terminus of the protein (Jul 2017 news). Yet it is N-terminal fragments of phosphorylated tau, including p-tau181 and p-tau217, that are secreted by neurons in response to amyloid accumulation, making them sensitive fluid biomarkers for amyloid, not tau tangles (Mar 2018 news).
In search of chunks of tau in fluids that would signal the presence of tangles without the need for costly tau-PET scans, the WashU team previously identified MTBR-tau-243. Among a small number of participants in the Dominantly Inherited Alzheimer’s Network (DIAN), the researchers found that the mid-section fragment tracks with tau-PET and disease progression (Dec 2020 news).
The new study tests MTBR-tau-243 in two sporadic AD cohorts: Swedish BioFINDER-2, and the Knight AD Research Center. First author Kanta Horie, an Eisai-sponsored professor at WashU, and colleagues used mass spectrometry to measure MTBR-tau-243, as well as several phospho-tau markers, in the CSF of 448 BioFINDER-2 and 219 Knight ADRC participants. Both cohorts included people across the spectrum of AD, but BioFINDER-2 has a higher proportion of cognitively impaired people. Horie et al. asked which CSF markers tracked most closely with amyloid- and tau-PET. All phospho-tau markers were measured as a ratio of phosphorylated to unphosphorylated tau. When it came to tracking with amyloid-PET, p-tau217 emerged as the clear winner. For tau-PET, MTBR-tau-243 did the best. More than any of the p-tau species, MTBR-tau-243 tracked with tau-PET signal across all Braak regions.
Viewed from a different angle, the researchers found that amyloid-PET explained most of the variation in CSF p-tau217, while tau-PET best explained variability in MTBR-tau-243. In contrast, p-tau205 variability was equally accounted for by amyloid- and tau-PET.

Pin the Tail on the Tangle. Percentage of variation in each CSF biomarker explained by amyloid-PET (blue) versus tau-PET (green). Most of the variation in p-tau231, 181, 217 was explained by amyloid; p-tau205 was equally explained by both. Tau-PET accounted for most of the variance in MTBR-tau-243 (right). [Courtesy of Horie et al., Nature Medicine, 2023.]
How would MTBR-tau-243 change as AD progressed? Among 220 participants in BioFINDER-2 who had serial CSF measurements, the scientists found that p-tau217, p-tau181, and p-tau231 increased dramatically when people became amyloid-positive, then plateaued when they became tau-positive. In contrast, MTBR-tau-243 hardly changed in response to amyloid positivity, but shot up once tau tangles had inundated the brain. The finding suggests that once a person becomes tau-PET positive, MTBR-tau-243 is the marker that best reflects their further disease progression. In line with this, MTBR-tau-243 tracked more closely than any other CSF biomarker with sinking scores on the mini-mental state exam (MMSE).
Finally, the researchers used an algorithm to pick out which combinations of fluid biomarkers best predicted amyloid accumulation, tau tangles, and cognitive decline. For amyloid, p-tau217 was the best single predictor; a combination of p-tau217, p-tau205, and Aβ42/40 did even better. For both tau-PET and MMSE, MTBR-tau-243 was the best single predictor. However, combining it with p-tau205 improved predictions for both measures. In fact, a combination of p-tau205 and MTBR-tau-243 rivaled tau-PET in predicting cognitive decline.
Hansson thinks the marker has both diagnostic and prognostic potential, particularly when used as part of a panel of fluid biomarkers for Aβ and tau. Because biomarkers such as p-tau217 rise many years before cognitive symptoms surface, and then plateau, a positive result leaves open the possibility that a person’s current cognitive impairment is caused by something other than AD, Hansson said. If MTBR-tau-243 was also elevated, this would give clinicians more confidence in calling AD the culprit. The marker will be most useful if detected in plasma, Hansson said, and researchers are working on that now.
Bateman agreed about the biomarker’s potential in the clinic. He also thinks it could help clinicians predict how well a given patient might respond to a therapy such as lecanemab, which worked better among people with low tau accumulation, as measured by PET, than among those with a high tau-PET signal. The same was true for donanemab, as reported at AAIC (see Part 1 of this series)
Gil Rabinovici of the University of California, San Francisco, noted that while convincing at the group level, the correlations between CSF MTBR-243 and tau PET are dogged by significant variability among individuals. “I think the utility of this biomarker for disease staging at the individual patient level is still to be determined,” he commented, adding that the impact of the new biomarker will depend on whether it can be detected in plasma. “With these caveats, MTBR-tau243 seems to be an interesting new biomarker, and it is especially exciting to see it utilized to measure target engagement in early phase clinical trials of MTBR-targeting tau antibodies,” Rabinovici wrote (comment below).
CSF MTBR-tau243 made its trial debut at AAIC, when Jin Zhou of Eisai presented findings from early-phase studies of E2814, a monoclonal IgG1 antibody that binds to the second and fourth microtubule-binding domains of tau. The researchers propose that the antibody intercepts seeding-competent, MTBR-containing fragments of tau released from cells, thwarting propagation of tau pathology.
Zhou showed findings from a multiple-ascending-dose study in 40 healthy participants, as well as results from a separate study conducted in people with dominantly inherited AD (DIAD). The latter included seven participants with mild to moderate dementia, who were slated to receive monthly infusions of E2814 over 18 months. Over that time, doses increased from 750 mg to 4,500 mg. The DIAD study is ongoing, and the remaining participants have reached the highest dose.
At AAIC, Zhou reported that the drug was safe and well-tolerated at all doses tested, in both healthy volunteers and those with AD. One person had two serious adverse events not attributed to treatment. Three of the seven participants in the DIAD trial dropped out due to cognitive deterioration, an expected problem in studies that include people with moderate dementia, Zhou told Alzforum. Among those with AD, target engagement appeared robust in the CSF, as the researchers detected dose-dependent binding of the antibody to both epitopes within tau’s MTBR.
Among the five participants with AD in which MTBR-tau-243 was measured, its concentration in the CSF plummeted in response to E2814 treatment. For four participants, it dropped between 40 to 70 percent within the first three months, in response to the lowest dose used. For one participant, the biomarker did not drop until dosing was doubled to 1,500mg; then it declined more slowly over the following months (see below).

Taking Down Tangles? CSF-MTBR-tau243 dropped in response to treatment with E2814 in people with dominantly inherited AD. [Courtesy of Jin Zhou, Eisai.]
The biomarker findings suggest that E2814 lowered MTBR-tau-243, but did it knock down tangles? Only post-treatment tau-PET scans will tell. Zhou expects that scan data from the remaining participants within the next few months. “We are anxious and excited about that data,” she said.
Einar Sigurdsson of New York University believes the therapeutic potential of E2184 is well supported by its reduction of MTBR-tau-243. However, he noted that the majority of the antibody’s target is inside of neurons. “With this in mind, it would be interesting if the company examined whether the antibody is taken up into neurons, which would then greatly increase the pool of targetable tau,” he wrote. “If it is not, then it may be efficacious at a lower dose if its neuronal uptake could be enhanced, for example by altering its charge (Congdon et al., 2019).
Tiny single-domain antibodies are also being developed to target intracellular tau (May 2023 conference news). Sigurdsson added that an antibody’s ability to prevent tau seeding versus toxicity may not always go hand in hand. “It will be important to determine if this antibody and related ones against the MTBR region can reduce tau neurotoxicity in vivo, and reduce functional impairments,” he wrote.
E2814 is currently being evaluated in the DIAN-TU Tau NexGen trial, concurrently with lecanemab. The trial is running at 39 locations around the world, and expected to finish in 2027. Zhou said the new tangle-tracking biomarker comes at a perfect time, and the researchers are planning to integrate this measure into the ongoing DIAN trials. “There will be a cross-validation, for both the biomarker and the drug,” Zhou said.
Bateman said he expects the marker to be deployed broadly in clinical studies targeting both Aβ and tau. “If we start using it more across the trials, it will give us more information on this part of the pathology, so we can make better decisions based on what the drugs are doing.” For example, it could be possible that amyloid-targeted therapies that lower p-tau217 but not MTBR-tau-243 are less effective at slowing cognitive decline than drugs that lower both.
E2814 forms part of a field of MTBR-targeted therapies wending their way through preclinical and early clinical development. At AAIC, scientists from Prothena presented findings from the first in-human dosing of PRX-005. This human monoclonal antibody recognizes three epitopes within the first, second, and third microtubule-binding domains of tau. The single-ascending-dose study included 25 healthy participants, who received one of three doses of the antibody, or placebo. The drug appeared safe at all doses tested, and reached a sufficient concentration in CSF, suggesting it successfully crossed the blood-brain barrier. Currently, a multiple-ascending-dose study is ongoing in people with AD.
Prothena also reported preclinical findings on PRX-123, a dual peptide vaccine against Aβ and tau. The vaccine comprises the N-terminus of Aβ and the MTBR region of tau, complexed to an adjuvant that riles T-helper and B cells. At AAIC, Prothena reported that APP/PS1 mice immunized with PRX-123 produced a robust antibody response, and that antibodies in the mice's sera bound to both Aβ plaques and tau neurofibrillary tangles in brain sections from people with AD. The immunization also substantially lowered plaque burden in mice. —Jessica Shugart
No Available Further Reading
When people think of early onset Alzheimer’s disease (EOAD), autosomal-dominant mutations in the APP or presenilin genes come to mind. But these account for fewer than 15 percent of EOAD cases. For the remainder, the Longitudinal Early-Onset Alzheimer’s Disease Study (LEADS), a collaboration led by Liana Apostolova at Indiana University School of Medicine, Indianapolis, Brad Dickerson of Massachusetts General Hospital, Boston, Gil Rabinovici at the University of California, San Francisco, and Maria Carrillo of the Alzheimer’s Association aims to characterize their disease and decipher its root cause. At AAIC, held July 16-20, scientists shared baseline and longitudinal data on genetics, imaging, and fluid biomarkers.
“Our ultimate goal is to launch clinical trials," Apostolova wrote to Alzforum. "We consider EOAD to be the ideal Phase 2 cohort, because these young patients have a ‘pure’ form of AD and few medical comorbidities, yet advance rapidly” (full comment below).
What is Early Onset AD?
Sporadic EOAD strikes before age 65. Because of their young age, people with EOAD have largely been excluded from research. They might make for a suitable clinical trial population, because they have few co-pathologies and their disease progresses rapidly. What causes EOAD is unknown, but scientists believe the answers may lie in undiscovered genetic risk variants.
While most adults with EOAD primarily have memory problems, a greater proportion than is the case in LOAD are diagnosed with rare, non-amnestic subtypes, including primary progressive aphasia (PPA) or posterior cortical atrophy (PCA; Jul 2012 conference news). At AAIC, Angelina Polsinelli of Indiana U described two such cases from LEADS.
A 55-year-old man developed progressive memory and language problems over 18 months, which cost him his job because he mispronounced words, asked people to repeat themselves, and could no longer follow directions. His Mini-Mental State Exam (MMSE) score was 17 out of 30, working memory and language being weakest. He scored 29 out of 60 on the Boston Naming Test, on which controls his age score in the 50s. MRI showed an abnormally small left lateral-temporal lobe, an area that is key to speech and language and is often atrophied in PPA. Since his cerebrospinal fluid Aβ42/40 ratio was low, CSF total tau and phospho-tau181 high, and amyloid and tau PET scans positive, his diagnosis was PPA due to AD.
A 56-year-old woman struggled with depth perception and finding her way. She'd swerve out of her lane while driving and got lost going to the grocery store or doctor’s appointments. Her memory began declining two years later. Her MMSE was 22, with visuospatial perception faring worst. When copying an image, her version appeared “exploded,” a quintessential sign of PCA (McMonagle and Kertesz, 2006). Indeed, MRI showed shrunken temporal, parietal, and occipital cortices. Her low CSF Aβ42/40 ratio, high CSF t-tau and ptau-181, and positive amyloid and tau PET scans prompted a diagnosis of PCA due to AD.

Looks Like PCA. A woman with EOAD rendered a typically disjointed copy of a drawing and was diagnosed with PCA. [Courtesy of Angelina Polsinelli, Indiana University.]
The LEADS Cohort
Most EOAD cases are not so easily defined. PPA and PCA each account for 6 percent of the LEADS cohort, which was created to characterize EOAD cases that lack a clear clinical diagnosis. Designed after the Alzheimer’s Disease Neuroimaging Initiative (ADNI), LEADS will enroll 400 and 100 cognitively normal adults, ages 40 to 64, at 18 U.S. sites. Participants must not carry known mutations for autosomal-dominant AD or frontotemporal dementia. Researchers will follow controls for two years, impaired participants for four, collecting PET and MRI data, cerebrospinal fluid and blood, and administering cognitive and neuropsychologic tests.
LEADS began enrolling in 2018 and expected slow enrollment over three years because EOAD is rare, and COVID slowed things down further, Apostolova explained. Complicating the issue, 25 percent of those who were recruited tested negative for amyloid. These early onset non-AD volunteers came as a surprise to the LEADS investigators. They decided to call them EOnonAD and keep them in the study.
At AAIC, Dustin Hammers of Indiana U presented the baseline characteristics of the cohort (Hammers et al., 2023). As of June 2022, researchers had enrolled 89 controls, 212 people with clinically diagnosed EOAD, and 70 with EOnonAD. Participants were in their mid- to late-50s on average, half were women, 86 percent Caucasian.
At baseline, the EOAD group had worse memory scores than the EOnonAD group, averaging 22 on the MMSE versus 25.5 for the latter. Specifically, they did worse on episodic memory, executive function, and attention. These differences held true after accounting for the severity of global cognitive decline, age, sex, and years of education.
APOE genotype seemed to play a big role. Compared to EOnonAD participants, five times as many people with EOAD were APOE4 homozygous. Half as many had an E2 allele. In the EOAD group, 54 percent carried an APOE4 allele. In EOnonAD and controls, 41 percent did. “We were surprised by the high level of E4 heterozygosity in these two groups,” said Hammers.
What is EOnonAD? The LEAD investigators suspect an FTD-like disease, because these participants had more neuropsychiatric symptoms than their EOAD counterparts. In Amsterdam, Polsinelli reported that they were more apathetic and irritable, had less impulse control, and took more mood stabilizers at baseline (Polsinelli et al., 2023). “Although affective symptoms were common in all early onset AD cases, they were more so in EOnonAD,” she said.
In the EOnonAD group, 62 were not diagnosed with PPA, PCA, or non-amnestic dementia. Some of them had cognitive, fluid marker, and imaging characteristics of FTD. Hee Jin Kim, a visiting professor at Indiana U from South Korea’s Sungkyunkwan University, reported that some scored poorly on executive function, visuospatial, and speed/attention tasks, had high CSF and plasma levels of the neurodegeneration marker neurofilament light, and marked atrophy in their frontoparietal lobes.
What Does Genetics Say?
Genotyping, too, suggested an FTD component to EOnonAD. Kelly Nudelman of Indiana U used whole-exome sequencing in search of known pathogenic mutations. In the EOnonAD sample, two carried pathogenic variants in progranulin (GRN), one in MAPT, and two had expansions in C9ORF72. The same search in the EOAD group netted three participants carrying pathogenic PSEN1 variants.
Genetic screening was not an enrollment criterion in LEADS, hence these eight people's genotype status was determined after they had had baseline data collected. They were subsequently excluded from further data analysis and referred to the ALLFTD Study or the Dominantly Inherited Alzheimer Network. No other FTD or ADAD mutation carriers have turned up since, according to Apostolova.
Some other genetic variants already appear to be involved. One person with EOAD and one with EOnonAD carried a variant in the sphingolipid hydrolase gene SMPD1; variants in this gene cause Niemann-Pick’s disease in homozygous carriers. Fifteen people with EOAD and two with EOnonAD carried a dominant pathogenic mutation in the Parkinson’s risk genes GBA or LRRK2. Another six, four EOAD and two EOnonAD, had a recessive variant in a copy of one of three other PD genes—PRKN, PARK7, or PRKRA.
“In a small subset of LEADS cases, the SMPD1 and PD variants may contribute to overall risk for early onset AD, without necessarily being the sole cause,” Nudelman concluded. Strangely, at baseline, only two PD variant carriers had motor symptoms—tremor or slow gait—though nine had peripheral neuropathy, REM-sleep behavior disorder, or other non-motor signs of PD.
Still, most EOAD and EOnonAD cases could not be explained by known pathogenic variants in genes related to neurodegenerative diseases. Two AD risk variants in TREM2, R47H and R62H, turned up in the cohort, but were as prevalent in controls as in symptomatic participants. “We think EOAD is enriched with novel genetic risk factors that might inform our understanding of disease mechanisms and identify novel pathways and drug targets,” Rabinovici wrote to Alzforum (comment below).
Clues from Imaging
Volumetric brain scans revealed differences between EOAD and EOnonAD. Alexandra Touroutoglou, MGH, spotted a pattern whereby the posterior cingulate cortex, lateral temporal cortex, posterior temporal lobe, inferior parietal lobe, and precuneus were smaller in EOAD than in controls. In contrast, EOnonAD participants had no signs of cortical degeneration. This atrophy signature distinguished EOAD from controls and from EOnonAD more accurately than did scanning for the typical atrophy pattern seen in people with LOAD (Dickerson et al., 2009). “The LOAD signature captured atrophy less well than our new EOAD signature. This supports the use of a measure tailored to EOAD, especially to detect small longitudinal changes, as might be expected from disease-modifying therapies,” Touroutoglou said (image below).

EOAD Signature. Nine cortical regions withered in people with EOAD (top), but not in EOnonAD (bottom). [Courtesy of Alexandra Touroutoglou, Massachusetts General Hospital.]
Greater atrophy of the EOAD signature regions correlated with poorer scores on the Clinical Dementia Rating–Sum of Boxes (CDR-SB) and MMSE. Touroutoglou will follow this cohort and look for different signatures in subtypes, such as PCA and PPA.
Comparing amyloid plaque and neurofibrillary tangle pathology between EOAD and EOnonAD, UCSF’s Renaud La Joie saw that, on baseline PET, all EOnonAD participants were amyloid-negative and most were tau-negative. In contrast, everyone with EOAD was amyloid-positive, and 90 percent were tau-positive (see image below). Plaque and tangle load varied greatly in EOAD, but more amyloid generally tracked with more tangles.

Pretty Separate. EOnonAD participants (orange) were amyloid-negative (x-axis) and most were tau-negative (y-axis). Most people with EOAD (black) were amyloid-positive, and varied greatly in their tangle loads. [Courtesy of Renaud La Joie, University of California, San Francisco.]
What might influence tangle load? Age at onset showed a dramatic link. Compared to people who became symptomatic in their mid-60s, people for whom this happened in their late 40s or early 50s had more cortical tangles at the baseline point of the LEADS study, and for 17 who had a follow-up scan 3.5 years later, they accumulated additional tangles faster if their age at onset was earlier (see image below). Daniel Schonhaut, also at UCSF, predicted that a 55-year-old person with EOAD would amass tangles twice as fast as someone a decade older when all other demographics matched, even if they had a similar tau load on their first scan.
Why? Perhaps tau aggregates more rapidly in younger brains. Previously, scientists correlated seeding activity of tau in brain extracts with AAO, with seeds from younger people being more aggressive (Jun 2020 news). Rabinovici thinks stronger network connectivity in younger people might also play a role. “If tau spreads trans-synaptically, the higher levels of neocortical activity in young patients may provide many more neuronal pathways for tau to spread,” he hypothesized.
Hammers found that 45- to 50-year-olds with EOAD had worse memory, visuospatial function, and attention than their older counterparts, suggesting that earlier onset signals more severe disease. This phenomenon matches prior findings in LOAD. People who developed AD between ages 65 to 74 dropped 2.0 points annually on the MMSE, while those who first showed symptoms over age 75 slipped 1.4 points per year (Stanley et al., 2019). “Age of onset should be considered a continuous variable, as the association between earlier onset and worse disease progression doesn’t start or stop at age 65,” La Joie said.
Age at onset did not influence baseline amyloid burden or amyloid accumulation over time.
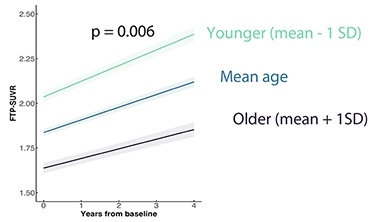
Getting Worse Faster. People whose EOAD began in their late 40s to early 50s (green) had higher tangle load at the LEADS baseline scan and accumulate tau faster than those whose disease began in their early 60s (navy). [Courtesy of Renaud La Joie, University of California, San Francisco.]
Digging deeper into how tau aggregates in EOAD, Schonhaut found that, at baseline, tangles were primarily in the basal-lateral-temporal lobe, parietal association cortex, and mid-frontal gyrus. Over an average of three years, every region except the amygdala accumulated tangles, with the frontal, temporal, lateral, and occipital lobes amassing them quickest. Regions in the first three were part of the EOAD atrophy signature Touroutoglou identified.
Wondering how LEADS baseline characteristics other than age related to tau pathology, Schonhaut looked at sex, amyloid burden, CDR-SB score, and APOE genotype. Only the latter stood out. Over their follow-up, homozygous APOE4 carriers accumulated tangles twice as fast as noncarriers, even after accounting for their baseline amyloid PET load. Scientists are just beginning to appreciate how ApoE4 worsens tau pathology through Aβ-dependent and -independent pathways (Feb 2023 news; Jul 2022 news; Yamazaki et al., 2019).
As for amyloid accumulation, it is strikingly similar in EOAD and LOAD. When UCSF’s Ganna Blazhenets looked at this question, she did not notice this at first, because people with EOAD had more baseline amyloid yet added plaques more slowly than their LOAD counterparts in ADNI. However, Blazhenets realized that EOAD participants were, on average, farther up their amyloid staging curve than ADNI participants.
“Patients with EOAD face a longer path to diagnosis,” Rabinovici said. “Clinicians often don’t have AD on their radar because of the patient’s young age and atypical presentation, often with executive functioning, language, or visuospatial problems rather than memory issues.” Younger people also have high brain reserves and few co-pathologies, enabling them to tolerate a high amyloid burden before showing symptoms, he added. In fact, Blazhenets found that EOAD and LOAD participants with similar baseline amyloid loads accumulated plaques at the same rate (see image below).

Cut From Same Cloth? Amyloid accumulation tends to be detected at different time points in early and late-onset AD (top), since EOAD is often diagnosed at a later stage. When compared at the same baseline amyloid load (bottom left), EOAD and LOAD had the same accumulation rates in a linear mixed-effects model (bottom right). [Courtesy of Ganna Blazhenets, University of California, San Francisco.]
Given that, Blazhenets calculated that the average 75-year-old person in ADNI became amyloid-positive at age 52, while the average 59-year-old in LEADS had turned positive at age 32.
Being amyloid- and tau-negative and unlikely to carry APOE4, people with EOnonAD resemble another mysterious group with suspected non-Alzheimer's pathophysiology. Remember SNAP (Sep 2015 news)? These people have biomarkers of neurodegeneration, yet can be cognitively normal, whereas EOnonAD means dementia. “Technically, some of the EOnonAD individuals in LEADS would likely fall in the SNAP category,” Polsinelli said.
Fluid Biomarkers
If amyloid deposition in EOAD parallels that of LOAD, how about CSF and blood markers? At baseline, they do, too, according to Jeff Dage at Indiana U. Compared to controls, EOAD participants not only had low CSF Aβ42/40 ratios and high CSF p-tau181, t-tau, and NfL, but also abnormal reads of other markers of neuronal health, such as presynaptic protein SNAP25, the postsynaptic protein neurogranin, and the neuronal calcium sensor Vilip1. Only NfL consistently correlated with worse cognition as per CDR-SB, ADAS-Cog1, or MoCa (Dage et al., 2023).
In EOnonAD participants, CSF NfL was slightly up over controls, indicating some neurodegeneration. All other markers were normal, in keeping with a non-AD etiology.
Plasma biomarkers told the same tale. Paige Logan, in Apostolova’s lab, reported that EOAD baseline p-tau231, NfL, and GFAP were high and correlated with more tangles, but not plaques, while an uptick in NfL came with cortical gray-matter thinning. Ralitsa Kostadinova in the same lab found that, in EOAD, these three markers tracked with worse scores on the ADAS-Cog13, MoCA, MMSE, and CDR-SB.
In EOnonAD, only plasma NfL and GFAP were up, and correlated with cognitive decline.
Notably, marker levels differed by sex. Sára Nemes in the Apostolova lab detected more NfL and GFAP in the blood, and more p-tau181, t-tau, neurogranin, and Vilip1 in the CSF of women with EOAD than in blood and CSF from men. Even women with EOnonAD had higher levels of plasma GFAP than men, suggesting that these sex differences occur regardless of amyloid positivity. Nemes thinks this reflects worse pathology or neurodegeneration in women.
“This study provides the first comprehensive analysis of fluid biomarkers in sporadic EOAD. It can inform clinical trial designs that incorporate such markers in patient selection or as endpoints,” Dage said.
LEADing the Way
Apostolova said that as of July 2023, LEADS had enrolled 99 out of 100 cognitively normal controls, 361 people with EOAD, and 112 with EOnonAD. Hammers expects the study to finish enrolling by this fall once it has included 400 EOAD cases. In May 2023, five research papers and a commentary on data generated by LEADS appeared together in a special issue of Alzheimer’s & Dementia. It will contain 11 manuscripts total.
LEADS is set to add five international study sites: University College London, Vrije University Medical Center in Amsterdam, Lund University in Sweden, Sant Pau Memory Center in Barcelona, and Fleni, a nonprofit neurological hospital in Buenos Aires. This is to establish a global network for future cohort studies and, ultimately, clinical trials for EOAD. “The first round of LEADS is observational to optimize the planning of trial outcomes, with the goal of renewing the study to add trial units, like DIAN-TU,” Rabinovici wrote.—Chelsea Weidman Burke
PET tracers that illuminate α-synuclein in the brain have been hard to come by, but scientists may be getting closer. According to a study published August 3 in Cell, an 18F- tracer developed at Emory University, Atlanta, binds to α-synuclein aggregates in human Parkinson’s disease brain samples, in mouse models of synucleinopathy, and in nonhuman primates. Crucially, it does so while shunning Aβ plaques or tau tangles, which commonly co-occur with Lewy bodies. Led by Keqiang Ye, now at the Chinese Academy of Sciences in Shenzhen, the study used cryo-electron microscopy to unveil the structural details of the tracer’s specific liaison with α-synuclein. 18F-F0502B joins a growing field of candidate tracers, including three presented at the Alzheimer’s Association International Conference, held July 16-20 in Amsterdam. One, made by AC Immune, binds to α-synuclein aggregates when they are highly concentrated in the brain, including in people with multiple system atrophy and in a genetic form of PD. The company’s other up-and-coming tracer candidate holds promise in latching onto the smaller inclusions that predominant in the PD brain. Another, made by Merck, bound synuclein in mouse models of PD.
“We are on the cusp of having a selective α-synuclein PET tracer that performs well in Parkinson’s disease,” said Jamie Eberling of the Michael J. Fox Foundation, which contributes funding for several ongoing efforts to develop the tracers, including AC Immune and Merck’s.
A PET ligand for α-synuclein would help immensely with diagnosis and prognosis of synucleinopathies, and with recruitment and monitoring in clinical trials. Yet, suitable tracers have eluded the field for years, due to their subpar affinity for α-synuclein within the brain, as well as off-target binding to other types of aggregates, including Aβ plaques and neurofibrillary tangles. Making matters worse, α-synuclein deposits in the brain can be dwarfed by other protein inclusions. In a landmark for the field last year, AC Immune’s 18F-ACI-12589 detected α-synuclein aggregates in people with multiple system atrophy, but failed to label inclusions in people with other synucleinopathies, including PD (Mar 2022 conference news).
In their hunt, co-first authors Jie Xiang, Youqi Tao, and Yiyuan Xia and colleagues took hints from compounds known to bind and block α-synuclein oligomerization, including molecules with catechol groups, such as dopamine and its derivatives. After screening an initial batch of 23 commercially available compounds for binding to preformed, recombinant α-synuclein fibrils, the researchers ran more extensive screens with derivatives of the top hits. F0502B came out on top. This molecule bound to α-synuclein deposits within brain sections of mice expressing A53T mutant human α-synuclein, but not to Aβ plaques or tau tangles in AD mouse models. F0502B also latched onto α-synuclein in brain sections from people with multiple system atrophy (MSA), dementia with Lewy bodies (DLB), and PD, but far less so to Aβ plaques or tau tangles in AD brain samples. Notably, the molecule, as well as its fluorinated form, 18F-F0502B, bound to α-synuclein from PD or DLB brain sections more tightly than it did to recombinant α-synuclein fibrils.

Path to PET. Chemical candidates, including dopamine derivatives, were screened for specific binding to α-synuclein fibrils (1). Top candidates were further tested in mouse models and in human brain tissue (2). Finally, structure of the α-synuclein/tracer complex was determined, and 18F-F0502B was tested in nonhuman primates (3). [Courtesy of Xiang et al., Cell, 2023.]
To understand how F0502B interacted with α-synuclein fibrils, the researchers turned to cryo-electron microscopy. High-resolution electron density maps revealed the tracer nestled into a deep groove along the surface of recombinant α-synuclein fibrils, which comprised stacked pairs of α-synuclein protofilaments. F0502B filled this binding cavity, stacking along the fibril axis in parallel (image below). While recombinant α-synuclein fibrils are distinct from the “Lewy fold” structures identified in brain samples from people with PD, Parkinson’s disease dementia, and DLB, which are collectively distinct from the fold α-synuclein takes in people with MSA, amino acids forming the F0502B binding pocket are also involved in the Lewy fold (Jul 2022 news). In cryo-EM, this fold is typically occupied by unidentified, nonprotein molecules (Mar 2020 conference news). Further, Ye told Alzforum that only a quarter of the fibrils identified in PD brain samples twisted into the Lewy fold, suggesting substantial structural heterogeneity in fibrils even within a single person. He said his group will discern the structure of tracers in complex with brain derived fibrils.

Finding Fibrils. Cryo-EM image (left) and structural diagram (right) of recombinant α-synuclein fibrils reveal F0502B (orange) binding within a groove formed by stacked pairs of α-synuclein protofibrils. [Courtesy of Xiang et al., Cell, 2023.]
Robert Mach of the University of Pennsylvania in Philadelphia commended the researchers for solving the structure of their tracer in complex with α-synuclein fibrils. He noted that although recombinant fibrils are structurally distinct from those found in the brain, several small-molecule binding sites may be common across fibril isoforms (Hsieh et al., 2018). F0502B binds one of these sites, he said. Still, he thinks one potential concern for F0502B is its dependence on a specific tyrosine residue—Y39—for binding. This residue is a well-known hot spot for phosphorylation and nitration, either of which could influence the structure of the binding pocket.
The scientists put their tracer to the test in rhesus macaques. First, they injected into the striatum preformed, recombinant α-synuclein fibrils, or an adeno-associated virus encoding human A53T-α-synuclein. Eighteen months later, dopamine transporter (DAT) scans of the monkeys revealed substantial nigrostriatal degeneration, akin to that in PD. After infusing the tracer, the researchers found scant retention within the brains of controls, but a significant signal from monkeys burdened with synucleinopathy.
With a standard uptake value ratio of around 0.7, 18F- F0502B may not pass muster as a clinical PET tracer. Rather, Mach views it as a great starting place to make analogs. “We know that very small changes in structure can have a large influence on how these molecules perform in PET imaging studies,” he said. “It is highly possible that a close structural analog could work much better.”
Eberling agreed, noting that the tracer could benefit from further optimization, including improved brain penetrance.
Still, Ye said that although next-generation tracers are under investigation, 18F-F0502B is being tested in people with PD, DLB, and MSA. Braegen Pharmaceuticals, Ltd., a company in Shanghai co-founded by Ye, sponsors development.

Signaling Synucleinopathy. PET scans display 18F-F0502B retention in macaques previously injected with α-synuclein PFFs (bottom left panels) or with AAV-A53T-α-synuclein (right) but not substantially in controls (top left). [Courtesy of Xiang et al., Cell, 2023.]
18F-F0502B joins a handful of other up-and-coming α-synuclein ligands. At AAIC, Francesca Capotosti of AC Immune updated the audience on 18F-ACI-12589, which the company had previously reported to work in people with MSA, but not in other synucleinopathies. Since then, the Capotosti and colleagues, in collaboration with Oskar Hansson at Lund University, Sweden, have scanned more participants, including five with AD, three with PSP, and three with ataxia, as well as 23 with a synucleinopathy. Of the latter group, eight had PD, 13 MSA, and two DLB. For the most part, their initial conclusions held, in that substantial uptake in the brain was seen only in people with MSA. Was the tracer’s penchant for α-synuclein in MSA due to the protein’s conformation, or concentration?
To chip away at this question, the researchers infused the tracer into people with genetic PD or DLB caused by a duplication in the α-synuclein gene. These tend to have a higher burden of α-synuclein pathology relative to people with other forms of PD. Lo and behold, Capotosti did observe ACI-12589 uptake in disease-relevant brain regions, supporting the hypothesis that a high concentration of α-synuclein deposits is needed for them to be detected in the scanner, and that differences in fibril conformation do not explain the tracer’s differential uptake in MSA versus other synucleinopathies. In other words, it’s all about the amount of synuclein, Capotosti said. In support of this idea, 18F-ACI-12589 tracer uptake increased in monkeys after they were inoculated with AAV-A53T-α-synuclein.
Mach has a different interpretation. To his mind, conformational differences in α-synuclein fibrils are still likely to play a strong part in the differential performance of the tracer. Based on screening hundreds of α-synuclein-binding compounds, he said he would be surprised if one worked across all synucleinopathies. “We have radioligands specific for PD, and some for MSA, but not any that bind both,” he said. His group is developing separate tracers for each.
Determined to find a PET agent that can work for PD, AC Immune scientists have developed a new crop of tracers with a higher affinity for α-synuclein aggregates. The top contender, ACI-15916, binds α-synuclein with a fourfold higher affinity than its predecessor. In brain sections from people with PD, the new tracer appeared to detect very small inclusions of α-synuclein, as well as Lewy body neurites, whereas ACI-12589 only latched onto larger inclusions. So far, PET scans in monkeys suggest that the new tracer looks hopeful for clinical studies, Capotosti said.
Idriss Bennacef presented the results of Merck’s quest. He cited low target concentration as the biggest hurdle to success. Merck screened its candidates using brain homogenates from people with PD. Provided by Banner Health, the samples were “clean,” in that they were devoid of Aβ plaques or tau tangles, Bennacef emphasized. Its lead compound—11C-MK-7337—bound tightly and specifically to α-synuclein aggregates in brain sections from people with PD, as well as in A30P-α-synuclein transgenic mice. In PET scans, the tracer lit up regions of the mouse brain that also bound α-synuclein antibodies (image below).
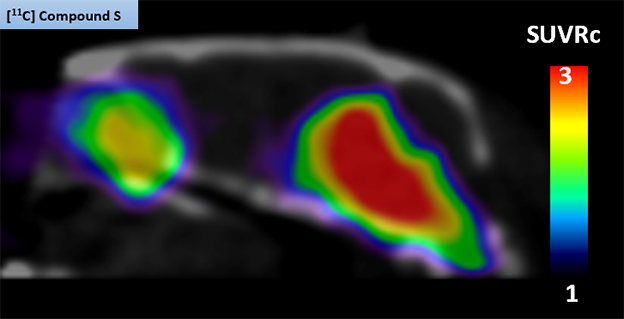
Mouse PET. The 11C-MK-7337 tracer shone brightly in the midbrains of A30P transgenic mice, but not in wild-type mice (not shown). [Courtesy of Idriss Bennacef, Merck.]
Alas, the tracer did display some off-target binding in the cerebella of rhesus macaques, raising uncertainties how it will perform in people, Bennacef said. Ongoing clinical studies aim to find out, he said.—Jessica Shugart
Can scientists who study Alzheimer’s disease find a solution to ARIA? These amyloid-related imaging abnormalities, which reflect brain edema, microbleeds, and occasionally large brain bleeds, have been associated with some deaths in clinical trials. At the Alzheimer’s Association International Conference, held July 16-20 in Amsterdam, scientists discussed the latest data on what causes ARIA.
They homed in on an inflammatory reaction to cerebral amyloid angiopathy. CAA experts noted that ARIA-E resembles CAA-related inflammation, a rare and serious condition caused by auto-antibodies to Aβ. In a plenary talk, Cynthia Lemere of Brigham and Women’s Hospital, Boston, sketched out a possible mechanism, suggesting that anti-amyloid antibodies may bind vascular amyloid, triggering the complement cascade to attack cerebral blood vessels. This would punch small holes, leading to fluid leakage and microhemorrhages.
Lemere and other speakers proposed therapeutic targets that might blunt the severity of ARIA, while highlighting the need for biomarkers of CAA to aid in prognosis and patient management (see Part 7 of this series). Pharma scientists noted that this is an active area of investigation at their companies, as well, with ongoing efforts to develop algorithms and blood tests to predict and better detect ARIA. “I’m hopeful we will be able to find a way around ARIA,” Lemere said in Amsterdam.

Complement As Hatchet Man. According to a new hypothesis, anti-amyloid antibodies (light blue) bind to vascular amyloid and trigger the complement cascade, prompting the membrane attack complex to form (dark blue), which damages blood vessels. [Courtesy of Cynthia Lemere and Maria Tzousi Papavergi.]
Vascular amyloid is common in AD. In Amsterdam, Roxana Carare of the University of Southampton, U.K., described how soluble Aβ in the parenchyma normally gets flushed out of the brain along blood vessels, draining into the basement membrane that packs the vessel wall. Excess Aβ can become trapped there, clogging the plumbing and forming CAA. White matter, which has fewer vessels and drains solutes more slowly than gray, is particularly vulnerable to this phenomenon. In postmortem analysis of brains that had CAA, fluid buildup can be seen in the perivascular space around white-matter vessels, damaging vascular architecture, Carare noted.

White Matter at Risk. Vascular amyloid (yellow) clogs leptomeningeal arteries and penetrating arterioles. Vascular damage (green) tends to be worse in white matter, which has slower blood flow. [Courtesy of Roxana Carare and Roy Weller.]
What happens during immunotherapy? Carare believes antibody therapy worsens the blockage, because antibody-antigen complexes become stuck in the already-obstructed basement membrane. In people immunized with Elan’s active vaccine AN-1792, the first attempt at immunotherapy 20 years ago, postmortem analysis revealed a worsening of CAA, along with damaged white matter (Boche et al., 2008).
Is Complement to Blame?
Lemere focused on how the innate immune system might contribute to ARIA. Praveen Bathini in her group investigated the effects of immunotherapy in 16-month-old APP/PS1 mice expressing human APOE4. These mice develop extensive vascular amyloid. Treating them for 15 weeks with 3D6, the mouse forerunner of the anti-amyloid antibody bapineuzumab, left cerebral vessels coated with the complement proteins C1q and C3. These vessels sprang leaks, leading to many microhemorrhages. In mice treated with a control antibody, vessels remained intact, as well as free of complement activation.

Antibodies Attract Complement. In amyloidosis mice treated with the 3D6 antibody (top), but not in those that receive an isotype control antibody (bottom), vascular amyloid (blue) in cerebellum (left) and meninges (right) becomes coated with complement protein C1q (red). The antibodies also attract activated microglia (green) and macrophages (gold). [Courtesy of Cynthia Lemere and Praveen Bathini.]
Lemere noted that after a patient has received an infusion of therapeutic antibodies, i.e., a big bolus entering their veins, these circulating anti-Aβ antibodies encounter and bind vascular amyloid first. She thinks these antibodies may get tagged by C1q, which recruits C1r and C1s, forming the C1 complex right there at the cerebral vessel wall. This complex would activate the classical complement cascade, producing C3a and C5a, and eventually resulting in formation of the membrane attack complex C5b-9. The complex pokes holes in cells, in this case along the blood-brain barrier, resulting in leakiness and microhemorrhages. She believes this may be the mechanism by which anti-amyloid antibodies give rise to ARIA-E and -H (see image at right).
In addition, complement proteins C3a and C5a likely activate microglia in the area, triggering vascular inflammation, Lemere said. Other talks in Amsterdam hinted at a microglial contribution, as well (see below).
If the complement cascade does trigger ARIA, then interrupting it, for example with antibodies directed against C1s, might halt this calamitous process. However, Lemere cautioned that therapies would have to be precisely directed, and brief, so as not to leave people immunocompromised. For example, while inhibiting C5 would disrupt the membrane attack complex, it would also hobble the body's essential antimicrobial defenses. C1q itself would also be a tricky target, because it promotes essential cellular function, such as differentiation, migration, and survival. Nonetheless, the complement cascade offers myriad targets, and Lemere said her group is exploring several of them.
These data caught the eye of pharma, with Rachelle Doody at Roche calling the findings an exciting lead. Roche is using data from its gantenerumab trials to model possible mechanisms of ARIA (Aldea et al., 2022).

Myriad Targets. The complement cascade is complex, affording many points where therapeutic interventions could be aimed (red stars). [Courtesy of Garred et al., 2021.]
Is ARIA Treatment-Induced CAA-ri?
Others at AAIC elaborated on the links between ARIA and CAA-related inflammation (CAA-ri). Steven Greenberg of Massachusetts General Hospital, an expert on CAA, noted that they look suspiciously similar on MRI scans. Both conditions cause headaches and seizures. Both CAA-ri and ARIA tend to occur in APOE4 carriers, who have more vascular amyloid. Like ARIA, CAA-ri can trigger microbleeds, or occasionally larger hemorrhages. Most tellingly, CAA-ri events are associated with endogenous anti-amyloid antibodies, which peter out as the condition resolves (Kinnecom et al., 2007; Zedde et al., 2023). “ARIA-E is iatrogenic CAA-ri,” Greenberg concluded in Amsterdam.
Fabrizio Piazza of the University of Milano-Bicocca, Italy, linked Alzheimer’s to CAA-ri. He showed case studies of four patients with CAA-ri. Two, who were also positive for AD biomarkers, had more microglial activation, as seen by TSPO PET, in brain areas showing edema. The other two did not. With corticosteroid treatment, this microglial activation waned along with the edema and Aβ autoantibodies (Piazza et al., 2022). The data suggest there may be a heightened inflammatory response to CAA in the context of AD pathology, due to the disassembly of parenchymal plaques that floods clearance pathways with soluble Aβ, Piazza told Alzforum.
Notably, two of these four patients later developed microhemorrhages, seen at their five-month follow-up appointments. This jibes with findings from amyloid immunotherapy trials, where as many as 70 percent of these small bleeds occur after ARIA-E appears, Piazza noted (Salloway et al., 2022). By contrast, the occurrence of isolated ARIA-H, i.e., in the absence of ARIA-E, is the same in treated and control groups in all anti-amyloid trials to date. This suggests that it is the edema that amplifies the risk of microhemorrhages.
Piazza recommended treating ARIA-E with corticosteroids, such as methylprednisolone, as a way to prevent ARIA-H and larger bleeds. This would be a change from the current appropriate-use recommendations for immunotherapy, in which ARIA-E is only treated if serious symptoms develop.
Greenberg agreed, noting that in CAA-ri, several months of immunosuppressive treatment clears up the condition faster, with 94 percent of treated patients improved at the first follow-up appointment compared with half of untreated. In addition, only about a quarter of treated patients had another episode of CAA-ri within the next six years, compared with three-quarters of untreated (Regenhardt et al., 2020). For his part, Piazza found that stopping corticosteroid therapy abruptly, rather than tapering off, hikes the risk of CAA-ri recurrence, again pointing to the importance of careful management (Antolini et al., 2021).
CAA-ri also occurs in familial AD, said Natalie Ryan of the Dementia Research Centre at University College London. In Amsterdam, Ryan discussed the case of a woman with a presenilin 1 mutation, as well as two APOE4 alleles, who developed ARIA-E as seen on brain imaging, and who declined steeply. An autopsy confirmed the presence of CAA-ri (Ryan et al., 2015).
At AAIC, interest in learning how CAA-ri may relate to AD, and how it could inform ARIA management, was widespread. After all, the coming rollout of lecanemab, and perhaps also donanemab, will expose many more people to these antibodies than have been studied in trials.
Piazza runs the Inflammatory CAA and AD Biomarkers International Network. To date, iCAβ has enrolled more than 500 people with CAA-ri. The goal is to gather more data on this condition and use it to improve safety in immunotherapy trials.
Not Just Amyloid Immunotherapy?
One surprise at AAIC was that other AD immunotherapies can cause ARIA, too. Gary Romano, of the biotech Alector, presented data from the Phase 2 trial of Alector’s anti-TREM2 antibody in early AD. AL002 activates the TREM2 receptor, dialing up microglial activation and phagocytosis. To the researchers’ dismay, three participants, all APOE4 homozygotes, developed ARIA-E within the first three months of the trial. The condition resolved after halting dosing and treating the participants with corticosteroids.

E Before H. In the INVOKE-2 clinical trial with Alector’s Trem2 modulating antibody AL002, some participants developed MRI imaging abnormalities indistinguishable from ARIA-E and ARIA-H, just as in amyloid immunotherapy. [Proprietary images and information courtesy Alector.]
Alector has since changed its trial protocol to add MRI monitoring. The company now excludes E4 homozygotes, a more drastic measure that the anti-Aβ antibody programs have not taken. Alas, despite avoiding this highest-risk population, about a quarter of people on AL002 have developed ARIA-E or -H, with about one in eight cases being symptomatic. Romano showed two cases, one in an APOE4 heterozygote and one in a noncarrier, in which ARIA-E resolved within a few months, but was followed by microhemorrhages. This again mimics what happens with amyloid immunotherapy. The ARIA percentages are similar to those seen in those trials, as well.
It is unknown if the mechanism is the same, Romano said. However, the data are thought-provoking in light of Lemere and Piazza’s findings linking microglial activation to ARIA.
In the Clinic: Immunotherapy and ARIA
Initial clinical experiences with anti-amyloid antibodies reinforce the importance of careful management of ARIA. In Amsterdam, Matthew Howe, who works with Stephen Salloway at Butler Hospital in Providence, Rhode Island, presented data from the first 24 patients treated with aducanumab at Butler after its accelerated approval. Six developed ARIA-E, slightly fewer than the one-third who did in the Phase 3 trials. All six were APOE4 carriers, and three were homozygotes. Two of the six had symptoms of ARIA. For them, plus for two others who had ARIA deemed moderate to severe based on MRI, Howe stopped aducanumab permanently. For all six, their ARIA-E cleared up in about three months without corticosteroid treatment.
Despite the immediate stoppage, one initially asymptomatic patient, an E4 homozygote man, developed headaches a month later. Follow-up MRIs showed 18 new microhemorrhages scattered across his brain, well beyond the region of ARIA-E. His headaches and ARIA subsided with time, and he remained enthusiastic about trying new therapies. He has since enrolled in an anti-tau trial, Howe noted. The case illustrates that ARIA-E can have delayed effects. It also highlights the importance of halting dosing quickly for moderate ARIA, even when the person has no symptoms at the time of the MRI. Also at AAIC, Salloway reported that in the aducanumab trials, half of people with asymptomatic ARIA upon imaging went on to develop symptoms if dosing continued.
Lastly, in Amsterdam, researchers debated the link between anticoagulant use and higher risk of large brain bleeds in people on amyloid immunotherapy. In the open-label extension of the Phase 3 lecanemab trial, five people taking anticoagulants, or 3.6 percent of participants, had macro (not micro) hemorrhages. A macrohemorrhage is a bleed larger than 10 mm. Only 0.6 percent of people in the OLE who took lecanemab but no anticoagulants had macrohemorrhages, and only 0.2 percent of people on placebo in the original trial.
Greenberg called this risk substantial, noting that almost half of people who suffer a macrohemorrhage while on anticoagulants die within the next 30 days. Lecanemab’s appropriate-use recommendations warn against prescribing it to people on these medications (Jan 2023 news; Apr 2023 conference news).
Salloway noted that, so far, there is no evidence that donanemab increases the risk of macrohemorrhages for people on anticoagulants. Anticoagulants were allowed in donanemab’s Phase 3 trial, though the numbers were small, with 84 people taking both medications. For aducanumab, which is being given to some people who pay for it out of pocket, the AUR recommends against anticoagulant use. Aducanumab trials excluded the drugs. Every therapeutic antibody will get its own AUR, Salloway added.—Madolyn Bowman Rogers
How can researchers make amyloid immunotherapy safer? At the Alzheimer’s Association International Conference, held July 16-20 in Amsterdam, several speakers linked the amyloid-related imaging abnormalities that are the worst side effects of immunotherapy to cerebrovascular amyloid. ARIA may be an inflammatory reaction to cerebral amyloid angiopathy, they proposed (see Part 6 of this series). If so, screening patients for CAA could help gauge their risk of ARIA and guide treatment decisions. The problem? There are no reliable biomarkers for CAA.
Researchers contacted by Alzforum said there are, as yet, few studies in this area, but that research is heating up. Speakers at AAIC proposed some candidate markers. Meanwhile, pharma scientists are interested in developing fluid biomarkers for ARIA, to help catch the condition earlier and cut down on the expense of MRI scans.
“Biomarkers for CAA and ARIA are critically needed right now,” Suzanne Schindler of Washington University in St. Louis told Alzforum. Mathias Jucker of the University of Tübingen, Germany, agreed this should be a research priority. He noted that early studies of immunotherapy in amyloidosis mouse models had suggested a link with CAA (e.g., Pfeifer et al., 2002; Burbach et al., 2007), but because of the failure of the first such trials, research funding and interest waned. “This is a good example of why basic research is important,” Jucker wrote to Alzforum. “Had we all continued to work on this topic over the last 10 years, we would probably know much more today about the mechanisms of ARIA.”
CAA on MRI. Currently, clinicians diagnose CAA on MRI scans, where excess fluid around blood vessels shows up as bright white areas (top, vessels in cross-section), distinguishing it from intracerebral hemorrhages that occur without CAA (bottom). [Courtesy of David Werring, University College London.]
Detecting CAA in Living Brains
For now, clinicians look for CAA by assessing white-matter damage on MRI scans. However, this is unreliable. “Radiographic imaging markers of CAA have been clearly demonstrated to have insufficient specificity and sensitivity,” Fabrizio Piazza of the University of Milano-Bicocca, Italy, told Alzforum.
Scientists are searching for fluid biomarkers, and have a few leads. In Amsterdam, Cynthia Lemere of Brigham and Women’s Hospital, Boston, proposed certain complement proteins as candidate markers of vascular amyloid. In particular, the concentration of the complement factor C3 in the blood could flag the presence of CAA in the brain, she noted.
Why is this? Cristian Lasagna-Reeves of Indiana University School of Medicine, Indianapolis, found that when mice had cerebrovascular amyloid, their brain endothelial cells triggered astrocytes to become neurotoxic, releasing C3 in the process (Taylor et al., 2022). In a study of 55 people with MCI, elevated serum C3 helped distinguish the 16 with CAA from the 39 without, with every 0.1 unit/mL increase in C3 bumping up their odds of having CAA by 1.4 times (Saito et al. 2022).
In addition, Lemere discussed a potentially more exotic CAA marker, small vesicles known as migrasomes. These membrane blobs are left behind by migrating macrophages. Last month, scientists led by Wei Cai of Sun Yat-Sen University, Guangzhou, China, reported that cultured macrophages exposed to vascular amyloid spit out migrasomes stuffed with the glycoprotein CD5L. CD5L modulates macrophage activity and is part of the body’s defense against infections. However, this protein attaches to vascular amyloid and triggers the complement cascade, activating the C5b-9 membrane attack complex and damaging the blood-brain barrier in the process (Hu et al., 2023). CD5L and C5b-9, as well as migrasomes themselves, may be useful biomarkers of CAA, Lemere suggested.
Jonas Neher of the German Center for Neurodegenerative Diseases in Tübingen previously nominated medin, a protein fragment that aggregates into a vascular amyloid that promotes CAA, as a potential marker (Nov 2022 news). His group has developed highly specific antibodies against medin and is using these to test its association with CAA, Neher told Alzforum. He has also collected CSF from mouse models with and without CAA, and will compare their protein profiles to turn up more candidate markers. Human validation will be challenging, however, because so far there is no large collection of CSF or blood samples from patients with neuropathologically diagnosed CAA, Neher said.
Another recent paper from Marcel Verbeek of Radboud University Medical Center, Nijmegen, The Netherlands, associated a dearth of matrix metalloproteinases MMP-2 and MMP-14 in the CSF with both sporadic CAA and the genetic form caused by the Dutch APP mutation (Vervuurt et al., 2023). CAA was previously linked to heightened MMP activity; it is unclear what the mechanism for lowered MMP might be (Jung et al., 2003; Zhao et al., 2015; Tanskanen et al., 2011).
There are some large-scale efforts to find biomarkers of CAA and vascular damage, and these might provide the needed fluid samples for other studies. Piazza runs the Inflammatory CAA and AD Biomarkers International Network (iCAβ). This study enrolls people with CAA-related inflammation who develop ARIA-like edema, and collects their cerebrospinal fluid and blood. The goal is to identify diagnostic and prognostic markers of CAA and ARIA.
Similarly, the MarkVCID project led by Steven Greenberg at Massachusetts General Hospital, Boston, looks for biomarkers that detect vascular contributions to cognitive impairment and dementia (Mar 2017 news). Though not specifically aimed at CAA, the research may fish out such biomarkers.
Fluid Biomarkers of ARIA-E?
Despite the interest in CAA biomarkers, Piazza cautioned that CAA is only a risk factor for ARIA. Under the right conditions, vascular amyloid can trigger inflammation that leads first to the edema known as ARIA-E, and then to microhemorrhages known as ARIA-H. “The real urgent international research priority is biomarkers for ARIA-E,” Piazza said. Not only would quicker detection of ARIA-E improve safety, but biomarkers could deepen researchers’ understanding of how amyloid immunotherapy works, he added.
Because auto-antibodies to Aβ are associated with CAA-related inflammation, Piazza believes they might identify people at the highest risk for ARIA-E (Piazza et al., 2013). In addition, Piazza suggested that monitoring the levels of such auto-antibodies during immunotherapy, and pausing treatment if they rise, could improve safety.
Kaj Blennow and Henrik Zetterberg of the University of Gothenburg think plasma NfL might be a useful ARIA-E biomarker. They noted that talks in Amsterdam showed that this marker of neurodegeneration first rose and then fell in people treated with donanemab (Jul 2023 conference news). This peak might reflect ARIA, which tends to occur shortly after starting immunotherapy, they speculated. If so, testing plasma NfL levels at baseline and every two weeks could provide an early warning. If NfL spiked, doctors would order an MRI scan. Because plasma NfL can be tested on automated platforms, monitoring for ARIA this way could be more frequent, and much cheaper, than with repeated MRIs, Blennow said.
Ultimately, however, research into ARIA biomarkers will depend on the companies that developed anti-amyloid antibodies, because they have access to the crucial clinical trial samples, Neher noted. In Amsterdam, pharma scientists said this research is a priority for them. Katherine Dawson of Biogen said the company is looking at risk factors that might predict ARIA’s occurrence, for example blood-brain-barrier damage. Roche Diagnostics is trying to develop blood tests for ARIA, investigating molecules such as homocysteine to see if they predict risk. Elevated homocysteine is associated with blood clots and vascular damage. “This is high on the radar screen at Roche,” Rachelle Doody of Roche said.—Madolyn Bowman Rogers
Aliria Rosa Piedrahita de Villegas surprised scientists when she remained sharp 30 years past her expected age of Alzheimer’s disease onset, as dictated by an autosomal-dominant E280A Paisa presenilin mutation. Her resilience was attributed to having two copies of an ApoE3 variant, called Christchurch. At the Alzheimer’s Association International Conference, held July 16-20 in Amsterdam, Yakeel Quiroz of Massachusetts General Hospital, Boston, claimed one copy was sufficient to protect others in Piedrahita de Villegas’ kindred. Among 1,000 Paisa carriers in that extended family who live near Antioquia, Colombia, Quiroz found 12 who also had a copy of Christchurch. They staved off MCI for seven years longer than noncarriers. Though the delay to AD diagnosis was shorter, just four years, this still put their age at onset past the normal range for Paisa carriers.
Like Piedrahita de Villegas, one Christchurch heterozygote had abundant amyloid plaques yet few neurofibrillary tangles. “This is the first evidence that having one copy of Christchurch may give some protection against the Paisa mutation, even if it’s not as strong as in the homozygous case,” Quiroz told Alzforum. However, another study led by Nicholas Cochran, HudsonAlpha Institute for Biotechnology, Huntsville, Alabama, of the same cohort found no delay in cognitive decline in heterozygous Christchurch carriers. The discrepancy comes down to different age-at-onset estimates, the scientists say.
The Christchurch effect might even lead to a therapy. Working with Quiroz, Joseph Arboleda-Velasquez and Claudia Marino of Massachusetts Eye and Ear in Boston have recapitulated the tau-squelching effects using an antibody that makes ApoE behave like the Christchurch variant. When mice expressing human ApoE were injected with the antibody their brains made less hyperphosphorylated tau.
Since 2014, Quiroz and colleagues have studied more than 6,000 members of this extended family through the Colombia-Boston (COLBOS) collaboration. Among this kindred, about 1,200 carry the Paisa presenilin variant. Most turn amyloid positive on PET scans by age 28 and tau positive a decade later. On average, they develop mild cognitive impairment by 44 and dementia by 49 with just a year or two of variation in age at onset (Acosta-Baena et al., 2011).
Despite carrying this pernicious mutation, Piedrahita de Villegas was sharp until age 72. When she died, a month before her 78th birthday, she had but mild dementia symptoms. Subsequent whole-exome sequencing of her DNA revealed she had two copies of the Christchurch mutation. This variant of ApoE3 binds poorly to heparan sulfate proteoglycans (HSPGs), extracellular matrix molecules that help propagate tau seeds (Aug 2013 conference news). Despite an immense burden of amyloid plaques throughout her brain, she had neurofibrillary tangles only in her hippocampus and occipital cortex (Nov 2019 news; Sep 2022 news). In stark contrast, typical Paisa carriers from the Colombian kindred are riddled with tangles, especially in their precuneus, by the time they get MCI in their 40s. They typically die before age 60.
Did ApoE-Christchurch protect others in Piedrahita de Villegas’ extended family? In 2019, Quiroz and colleagues had reported four Paisa carriers with one copy of Christchurch. All became cognitively impaired at the normal age. Last March, Cochran, and co-first authors Juliana Acosta-Uribe at UC Santa Barbara, and Bianca Esposito of the Icahn School of Medicine at Mount Sinai, New York, reported that among 340 Paisa carriers in the Colombian kindred, 11 were heterozygous for APOE-Christchurch. They developed MCI and dementia at the expected ages as well (Cochran et al., 2023).

More Plaques, Fewer Tangles. Amyloid and tau PET scans of a Paisa/Christchurch carrier (right columns) show he had more amyloid yet almost no neurofibrillary tangles when he was diagnosed with MCI at 51 than a typical Paisa carrier diagnosed at age 44 (left columns). [Courtesy of Yakeel Quiroz, Massachusetts General Hospital.]
To cast a wider net, Quiroz and colleagues turned to banked blood samples from 1,000 Paisa carriers and 1,000 noncarriers among the Colombian kindred, sequencing their APOE genes. Among the latter, 100 carried a single Christchurch copy. Twelve of the Paisa carriers did, and 10 of these were the same people analyzed by Cochran.
Quiroz and colleagues comprehensively reviewed their medical records and found that the 12 had delayed AD onset, albeit by less than Piedrahita de Villegas. On average, they were diagnosed with MCI at 51 and dementia at 53, delays of seven and four years, respectively.
Why were the ages at onset later than those estimated by Cochran and colleagues? Quiroz chalks that up to revisions of the ages at onset after more detailed clinical review. In support of that, some have lived until age 60, five years longer than Christchurch noncarriers.
So far, two men with Paisa/Christchurch variants have had PET scans and they support the premise that one copy of the ApoE variant protects. One man was diagnosed with MCI at 51 when his semantic fluency and executive function began to fail. That year, he flew to Boston for structural MRI and amyloid and tau PET scans. Compared to Paisa carriers with a typical MCI age at onset of 44, he had less hippocampal atrophy and almost no tau pathology. Tangles were limited to regions affected during early stage AD, such as the entorhinal cortex, even though he had more amyloid plaques than Christchurch noncarriers of the same age (image above). A follow-up PET scan at age 53 showed only scant spread of tangles to the surrounding medial temporal lobe (image below). By 54, he had progressed to mild dementia.

Slow Spread. In a man carrying Paisa and a copy of ApoE-Christchurch, tau PET scans taken two years apart captured minimal neurofibrillary tangle growth from his entorhinal cortex throughout his medial temporal lobe. [Courtesy of Yakeel Quiroz, Massachusetts General Hospital.]
The other man started having trouble recalling words at age 52, prompting an MCI diagnosis. He developed mild dementia at 57 and progressed to moderate AD by 62. Two years later, he had MRI and FDG PET scans in Colombia. His hippocampus had minimal atrophy and his precuneus metabolism was normal compared to Paisa carriers with MCI in their 40s (image below). Metabolism tanks in the precuneus of Christchurch noncarriers by this age. His preserved metabolism mirrored that seen in Piedrahita de Villegas’s brain.

Preserved Precuneus. Glucose metabolism waned in the precuneus in a Paisa carrier (top), but not in a carrier with one copy of ApoE3-Christchurch (bottom). [Courtesy of Yakeel Quiroz, Massachusetts General Hospital.]
Intriguingly, the benefits of the Christchurch variant may be dependent upon other mutants, such as Paisa. In a few case studies, this APOE variant did not protect against early-onset sporadic AD or cerebrovascular disease in Paisa noncarriers, noted Rik Ossenkoppele and Colin Groot, Amsterdam University Medical Center (Hernandez et al., 2021; Civeira et al., 1996). “These findings point to a complex interplay between APOE and the PSEN1 E280A mutation, which could be exploited to produce new treatment targets for AD,” they wrote (comment below).
Toward a Therapy
In that vein, Quiroz, Arboleda-Velasquez, and Marino are developing an antibody mimicking this isoform.
The Christchurch mutation lies where ApoE binds to HSPGs. These extracellular matrix molecules encourage the dissemination and uptake of tau seeds into neurons, and limiting HSPG-ApoE binding curbs this spread (May 2018 news; Stopschinski et al., 2018). Of all known ApoE isoforms, Christchurch binds most weakly to HSPGs, perhaps explaining the low tangle loads in people with the variant. To block ApoE-HSPG binding, Arboleda-Velasquez and colleagues generated 7C11 an antibody that docks on ApoE’s HSPG-binding site.
To make the antibody, the researchers created an antigen containing a peptide corresponding to the ApoE3 HSPG-binding domain and injected it into mice. One animal produced the antibody, which strongly latched onto ApoE3 and, surprisingly, bound ApoE4 even more tightly. Both isoforms have identical HSPG-binding domains. “Perhaps different conformations or polarities of the ApoE isoforms drive the different affinities,” Marino suggested. 7C11 thwarted most ApoE-HSPG binding, as measured by heparin-affinity chromatography.
Still, targeting the HSPG binding site may have unintended off-target effects. “This antibody might also impede the interaction of ApoE with other proteins required for homeostatic functions, such as the LDL receptor protein, as it binds in the same region,” wrote Victor Montal, Hospital de la Santa Cru, Spain (comment below).
Would the antibody prevent HSPG-induced tau pathology in vivo? The researchers injected 7C11 into the abdomens of 16-month-old APOE4 knock-in mice. By this age, the mice have many phospho-tau aggregates within their neurons, even though they have no tau mutations (Brecht et al., 2004). Compared to controls, mice given 7C11 had about half as many p-tau396-positive cells. This form of p-tau is thought to be an early marker of tau pathology and correlates with denotes toxic tau seeds prone to aggregate (Rosenqvist et al., 2018). The results suggest that the antibody’s ability to mimic Christchurch protects against tau pathology.
Whether 7C11 might work in people remains to be seen. Marino found it bound to postmortem samples of middle temporal gyri from two Colombian Paisa carriers—one APOE3/3, the other APOE4/4.
Groot and Ossenkoppele cautioned that it might not work for everyone. "Given the inconsistent results between protective effects of APOEch in PSEN1 E280A mutation homozygotes, heterozygotes, and noncarriers, and the small sample sizes, there is a reasonable likelihood that APOEch mimicking therapeutics might only benefit specific cases," they wrote.—Chelsea Weidman Burke
Scientists are a step closer to a diagnostic test for Lewy body disease. At AAIC2023, held July 16 to 20 in Amsterdam, Oskar Hansson, Lund University, Sweden, reported that an α-synuclein-based seed amplification assay identified people with LBD among both cognitively impaired and unimpaired volunteers in the Swedish BioFinder cohort. Notably, about half the impaired people who tested positive for Lewy bodies also had amyloid pathology. Having Lewy pathology came with faster cognitive decline, regardless of amyloid.
Within two years, Hansson hopes to use the test to help diagnose LBD in clinical practice. His team collaborated with Piero Parchi’s group at the University of Bologna, Italy. Their findings appeared back-to-back on July 18 in Nature Medicine.
Also at AAIC, Lawren Vandevrede, University of California, San Francisco, reported that a similar CSF assay distinguished autopsy-confirmed LBD from controls with high specificity and sensitivity, but only when the Lewy bodies were predominantly in the cortex and limbic areas of the person's brain. This assay worked less well among people who had Lewy bodies only in the amygdala or brainstem and who typically had milder symptoms.
Still, Vandevrede believes that, overall, seed amplification assays will prove useful for diagnosis. They perform better than relying on clinical symptoms, which are not always present or specific to LBD. Vandevrede and Hansson both emphasized that α-synuclein assays will be particularly important for running and interpreting drug studies. For example, they could explain why some people respond poorly to anti-amyloid removal.
“The quantification of LB pathology in vivo holds dual relevance. One is within a clinical framework, offering improved individualized prognostic insights, and the other in the context of clinical trials for all neurodegenerative disorders where a substantial number of cases are marked by the presence of LB pathology,” wrote Dag Aarsland, King’s College London, and Maria Camila Gonzalez Velez, Stavanger University Hospital, Norway.
Diagnosing DLB
Lewy bodies, those protein inclusions consisting primarily of α-synuclein fibrils, are found in several neurodegenerative diseases, including Parkinson’s, multiple system atrophy, and dementia with Lewy bodies (DLB). As many as half the people diagnosed with Alzheimer’s disease also have these deposits (Gibb et al., 1989; Hamilton, 2000; Jellinger, 2003). Indeed, across age-related dementias, Lewy bodies come second in prevalence only to amyloid plaques and tau tangles in AD.
Unlike plaques and tangles, Lewy bodies have been difficult to detect with fluid markers. Likewise, PET tracers for synucleinopathies are only just being developed (Part 5 of this series). With no good biomarkers, and symptoms that are easily confused with those of related disorders, diagnosing DLB is challenging, and the extent to which Lewy pathology contributes to cognitive decline in mixed pathologies remains undeciphered.
Enter seed amplification assays. For SAAs, scientists take a small sample of cerebrospinal fluid or tissue extract, incubate it with monomers of recombinant α-synuclein, and wait to see if aggregates form. They shake the solution to nudge seeds to nucleate and fibrils to grow, which can take days. In so doing, scientists led by Claudio Soto, University of Texas Medical School at Houston, detected α-synuclein fibrils in the CSF of PD patients seven years ago (Dec 2016 news). Since then, a flurry of SAA studies have reported synuclein seeds in the CSF of people with prodromal or fully fledged PD, culminating in a study of 1,100 people in the Parkinson’s Progression Markers Initiative that appeared last April (Rossi et al., 2020; Orru et al., 2021; Iranzo et al., 2021; April 2023 conference news).
Scientists found synuclein seeds in CSF from people with other LBDs, as well. Last year, Hansson and colleagues reported that, in BioFinder and a neuropathological cohort called the Arizona Study of Aging and Neurodegenerative Disorders, an α-synuclein SAA distinguished people with PD, Parkinson’s disease dementia, or DLB from controls with high sensitivity and specificity (Hall et al., 2022). As for mixed pathology, scientists led by Andrea Pilotto at the University of Brescia, Italy, found α-synuclein seeds in the CSF of 36 out of 80 people with AD (Pilotto et al., 2023). Now, Parchi and Hansson have correlated Lewy body with amyloid and tangle pathology, and estimated how the former changes clinical outcomes over time.
In Amsterdam, Hansson reported that among the 1,182 cognitively unimpaired people in BioFinder, 8 percent tested positive for Lewy body pathology, and its prevalence increased with age. About 60 percent were men, in keeping with their higher likelihood of getting LBD than women. Plaques or tangles were more prevalent, cropping up in 23 and 16 percent, respectively (image below). That said, 13 percent of those with plaques had LBD as well.
“I thought it extremely interesting that Lewy bodies were significantly more common in people who were amyloid-positive,” said Hansson. Co-first authors Sebastian Palmqvist from Lund and Marcello Rossi from the Istituto delle Scienze Neurologiche di Bologna (ISNB) calculated that having amyloid raised a person’s odds of developing Lewy bodies by 70 percent. Tangles had no correlation with Lewy bodies.


Lewy Body Effects. In BioFinder (top), half as many people had Lewy bodies (LB) as had neurofibrillary tangles (T). Having Lewy bodies accelerated disease progression, as seen by changes in global cognition (bottom left), memory (bottom middle), and attention/executive function (bottom right). [Courtesy of Palmqvist et al., 2023.]
Lewy body pathology mattered. People with it did worse on memory tests than controls with no AD or LB pathology. Their scores were on par with those of people who had tangles, a pathology that tracks with cognitive loss in AD. A positive LB test also correlated with loss of smell, a sign of impending LBD. Synuclein pathology seemed to accelerate AD because, over a period of 10 years, people with amyloid, tangles, and Lewy bodies slipped faster on measures of global cognition, memory, and attention/executive function than did people with either AD or LB pathology alone.
The seeding assay was also prognostic for symptomatic LBD. Some who tested positive subsequently developed DLB or PD; nobody who was negative did. “The average follow-up was 4.2 years, so we will need to see what happens over a longer time frame,” said Hansson.
What about people who were already symptomatic? In the second paper, co-first authors Corinne Quadalti at ISNB and Palmqvist reported similar effects. Of 883 BioFinder participants with mild cognitive impairment or dementia, 204, or 23 percent, had LB pathology; half of them also had plaques and tangles.
The prevalence of LB pathology increased with age but, paradoxically, less so than in the cognitively normal group. The authors suspect that people with other pathologies, such as TDP-43 encephalopathy or vascular disease, might dilute the number of LBD cases in a memory clinic setting. As Palmqvist and colleagues had found for the cognitively unimpaired group, Lewy bodies accelerated decline in measures of cognition, attention/executive function, and memory in the symptomatic cohort (image below).


Faster Downhill. Nearly one in four symptomatic BioFinder participants have Lewy body pathology (top, green and purple). They get worse fast, especially if they also have AD (bottom, pink). [Image courtesy Quadalti et al., 2023.]
For the most part, the data confirm prior findings in neuropathological cohorts, namely that people with mixed AD/Lewy pathology got worse faster (Brenowitz et al., 2017; Malek-Ahmadi et al., 2019; Ryman et al., 2021; Gu et al., 2022).
The assay now allows clinicians to sharpen prognosis, said Hansson. “It opens up the possibility to study the clinical effects longitudinally, and that is quite helpful for clinical trials,” Hansson told Alzforum. “It is important to better understand variability in response to trajectories and therapies to see what happens to these individuals.”
Doug Galasko, University of California, San Diego, agreed. “With the approval of amyloid immunotherapy for AD, the effects and benefits of this treatment in patients who have AD plus positive SAA should be formally studied,” he wrote (comment below). “It will be of interest to see whether anti-amyloid antibodies have weaker clinical effects in such patients, or whether indeed their use attenuates synuclein pathology.”
In one case study, Vandevrede reported clinical outcomes for a 60-year-old woman whose memory had been sliding for five years, and who received 10 mg/Kg aducanumab in the Engage trial. Over six months, the treatment cleared some amyloid, 60 to 51 centiloids, yet her MMSE fell from 24 to 19. Eight months later, she was at 14, and dropped out of the trial. During the next three years, she increasingly showed symptoms typical of DLB, including Parkinsonism, paranoia, anxiety, and profound dysautonomia, or loss of regulatory control over autonomic nervous system function, such as heart rate, breathing, and swallowing. When she died at age 64, she was found to have high levels of plaques and tangles and neocortical Lewy body pathology. “I’m not suggesting we don’t include such patients based on this one case, but it does raise the question of what is the response of patients with co-pathology if you treat with amyloid-lowering or -targeting treatments,” said Vandevrede. “Using some of the tools we now have available for CSF will help us answer that question.”
Galasko noted that while SAAs may pave the way for prevention studies, he wants a less-invasive approach than CSF sampling for screening or enriching populations. Scientists in Japan led by Nobutaka Hattori at Juntendo University School of Medicine, Tokyo, recently reported that they detect α-synuclein seeds in serum. Their amplification assay distinguished people with PD and DLB from controls with accuracies of 96 and 90 percent, respectively (Okuzumi et al., 2023). “We don’t yet have a very reliable blood test, but that might just be a matter of time,” said Hansson.
Clinical Diagnosis Versus Biomarkers
Vandevrede’s data suggests that seed amplification assays can identify limbic and neocortical LBD better than clinical diagnosis. He compared the SYNTap α-synuclein seeding amplification test sold by Amprion, San Diego, against autopsy and clinical data from a small cohort of 56 donors at the Memory and Aging Center at UCSF. Of these, nine had predominantly limbic/neocortical LBD, six had brainstem pathology, and 14 had it in their amygdala. Neurologists consider limbic/neocortical LBD to be the full phenotype, since this is typically accompanied by symptoms of DLB and/or PD. What these brainstem- and amygdala-predominant forms mean clinically is unclear, said Vandevrede, though he suspects they represent prodromal stages.
Overall, SYNTap of CSF identified LBD with 96 percent specificity and 59 percent sensitivity. For those who had limbic/neocortical pathology, those numbers were 96 and 100 percent, respectively. For the amygdala and brainstem categories, sensitivities were only 43 and 17 percent, respectively. Hansson and colleagues had found similar sensitivities in the BioFinder cohort. The data suggest that the prodromal forms are releasing far fewer α-synuclein seeds into the CSF. Indeed, people with brainstem/amygdala LBs who did test positive also hallucinated or had REM sleep disorder, suggesting they may be on their way to more severe disease. “SYNTap may be identifying ‘seed-competent’ subgroups with clinical relevance,” said Vendevrede.
Can seed amplification assays help if they miss most cases? “The amygdala-only variant contributes to a substantial proportion of AD cases with α-synuclein pathology in postmortem studies and would thus go largely unnoticed,” noted Hansson and colleagues. “However, the amygdala-predominant variant does not lead to a clinical LBD presentation and is less clinically relevant,” they wrote. The implication is that the seeding assays will identify any LB pathology that is likely to contribute to clinical decline, including among people with AD co-pathology.
As for limbic/neocortical LBD, the assay's near-perfect performance in this small cohort suggests it may outperform clinical diagnosis, said Vandevrede. While everyone in this group was positive on SYNTap, 44 percent had hallucinations, only 22 percent REM sleep disorder, suggesting the assay picks up even mild disease.
On the other side of the coin, SAAs, such as real-time quaking induced conversion, or RT-QuIC, might correct clinical misdiagnoses. Galasko and colleagues tested CSF of about 200 clinically diagnosed DLB cases. Almost 30 percent tested negative in the seeding assay, suggesting that typical DLB symptoms, such as parkinsonism, hallucinations, and REM sleep disorder, occurring in non-DLB disorders might muddy the waters. “The bottom line is that people can overdiagnose DLB based on the clinical criteria, assuming that CSF RT-QuIC is ground truth,” wrote Galasko. Palmqvist was surprised by that interpretation. “Our data show that we often miss LB pathology,” he wrote. Galasko noted that the cohorts and how they are referred to memory centers are different. “When people are being recruited for a DLB study, we need to be careful about mimics—a little Parkinsonism or disrupted sleep that sounds like RBD can drive false positives,” he wrote.—Tom Fagan
Diagnostic criteria for Alzheimer’s disease were revised only five years ago, but the field has learned a lot since then. At the Alzheimer’s Association International Conference, held last month in Amsterdam, Clifford Jack of the Mayo Clinic in Rochester, Minnesota, proposed updates to the current “ATN” system. Like the 2018 criteria, the new ones are the product of a joint National Institute on Aging-Alzheimer’s Association working group Jack leads. The draft criteria take into account recent discoveries about how biomarkers change over time, using them to stage disease. They retain amyloid and tau for diagnosis and staging but relegate the “N” neurodegeneration marker to second-tier status. They also advance the previous research diagnostic criteria toward clinical use by selecting the best-validated biomarkers.
With two disease-modifying AD treatments already in the clinic, and a third expected, accurate diagnosis has become an urgent need.
The criteria debuted to a packed hall at AAIC. Audience members peppered a panel of working group members with questions for nearly an hour, running the session well into dinnertime. While some aspects of the new criteria raised questions, the overall reception was positive.
“This is great progress, absolutely the direction we need to go,” Gil Rabinovici of the University of California, San Francisco, told the panel. Randall Bateman of Washington University in St. Louis was equally enthusiastic. “This [system] matches the biology so well. You’ve nailed it,” he said in Amsterdam. Bateman acted as a reviewer for the working group, but was not part of developing the criteria.

From ATN To ATNIVS. In the proposed new scheme—which is currently a draft meant to solicit input from the ADRD research community—A and T are the core biomarkers for diagnosis and staging. The draft scheme also recognizes an expanded suite of additional markers that detect non-specific disease responses and co-pathologies. [Courtesy of NIA-AA working group.]
Evolving Disease Conceptions
In 2018, the NIA-AA committee established the first biological definition of Alzheimer’s disease, basing diagnosis on the presence of amyloid plaques (A) and tau tangles (T), rather than symptoms. The criteria also made use of biomarkers of neurodegeneration (N) to help stage disease. The system distinguished clinical stages from 1 to 6, with 1 being biomarker change only, 2, subtle cognitive change, 3, mild cognitive impairment, and 4-6, mild, moderate, and severe dementia (Apr 2018 news; Nov 2018 conference news). The criteria were intended to be a work in progress, and to incorporate new data once it was available.
In Amsterdam, researchers said that moment has come. Jack noted several major changes in the field in the last five years. Besides FDA approval of disease-modifying therapies, the advent of plasma biomarkers plus the recognition that fluid and imaging biomarkers are not interchangeable required updating the standards, he said.
To do this, the NIA-AA convened a 22-person working group. In addition to Jack, academic researchers comprised Thomas Beach of Banner Sun Health Research Institute in Sun City, Arizona; Oskar Hansson of Lund University, Sweden; William Jagust of the University of California, Berkeley; Eric McDade of Washington University in St. Louis; Ozioma Okonkwo of the University of Wisconsin, Madison; Luca Pani of the University of Modena and Reggio Emilia, Italy; Michael Rafii of the University of Southern California, Los Angeles; Philip Scheltens, recently retired from VU University, Amsterdam; Reisa Sperling of Brigham and Women’s Hospital, Boston; and Charlotte Teunissen of Amsterdam UMC.
From pharma, members comprised Scott Andrews at Takeda Pharmaceuticals, Ana Graf at Novartis, Carole Ho at Denali Therapeutics, José Luis Molinuevo at Lundbeck, and Eric Siemers at Acumen Pharmaceuticals. The committee included Eliezer Masliah and Laurie Ryan from the NIA, Teresa Buracchio and Billy Dunn from the Food and Drug Administration, and Maria Carrillo and Heather Snyder from the Alzheimer’s Association. Dunn left the FDA in February; he serves on the board of Prothena. The group has been working on the draft criteria for a year, and is now soliciting outside feedback from the field, Jack said.
Diagnosis by Amyloid and Tau Only
What are the new criteria? For starters, the diagnostic scheme ditches the N category, relying only on A and T, which are considered the core biomarkers of the disease. This change was made because amyloid and tau pathology are more specific for AD than are neurodegeneration markers, Jack said. Moreover, the new approach allows clinicians to use either fluid or imaging biomarkers for diagnosis, with equal validity. Researchers hope this will make AD diagnosis feasible even in parts of the world where brain imaging is unavailable.
The committee specified the biomarkers they believe to be sufficiently validated for clinical use (see table above). For A, these are the Aβ42/40 ratio in cerebrospinal fluid or plasma, and amyloid PET imaging. For T, these are p-tau181 or p-tau217 in CSF or plasma, and tau PET imaging. Other biomarkers, such as p-tau231 and p-tau205, may reach this level later, but for now should be restricted to research use.
Bateman questioned the use of p-tau181 and p-tau217 as tau markers, noting they are more closely tied to plaques than tangles (Dec 2019 conference news; Mar 2020 news). P-tau205, MTBR-tau243, or even total tau, would make better markers for tangles, he suggested (Dec 2022 conference news; Part 3 of this series). Hansson explained that the committee conceived of T as denoting a change in tau metabolism, not specifically the presence of tangles, but agreed the inclusion of p-tau181 and p-tau217 in the T category might cause people to misinterpret them as tangle markers.
The draft criteria are still being refined and may change, Jack said. Indeed, part of the purpose in presenting the draft proposals at AAIC was to invite comment from the global community of ADRD clinician-researchers.
Importantly, the draft criteria currently do not specify particular assays or cutoffs to be used for each biomarker. This should be up to clinicians, and the changing state of the field. However, the criteria do recommend that instead of a single cut point, clinicians recognize an “indeterminate zone” for each biomarker, where a person may or may not have the disease. Results in this zone would trigger further testing.
A point of controversy in Amsterdam was that, in the new scheme, AD can be diagnosed by the presence of either A or T alone. This elicited audience pushback. After all, some people with plaques don’t go on to develop tangles. Some argued that amyloid plaques should be considered a risk factor for AD, rather than diagnostic of it.
The panel disagreed. Amyloidosis is the first event in AD, Jack said. In other words, people who have plaques in their brain already have the disease. They are at risk for its symptoms. Sperling concurred. “Not everyone with amyloid will develop symptoms, but we take [its presence] seriously, and we want to treat it,” she said in Amsterdam. McDade noted that the proposed NIA-AA criteria do not advocate screening cognitively healthy people for plaques. Thus, everyone diagnosed as A+ is likely to have come to a memory clinic with cognitive concerns.

Parallel Tracks. The proposed criteria, shown here in draft form dated July 15, allow clinicians to stage disease by either PET or fluid markers; alas, the stages denoted by each are not equivalent, and the modalities cannot be mixed. Late-stage fluid biomarkers are less well-established than the others, with the starred markers meant to be used only in research settings at this time. [Courtesy of NIA-AA working group.]
Disease Staging Via Parallel Tracks—Fluid or Imaging
In addition to diagnosing disease, A and T markers are able stage it, Jack told the audience. This is possible because scientists have recently associated specific A and T markers with distinct disease stages. The new framework specifies four biomarker stages: initial, early, intermediate, and advanced. These are denoted by letters A through D, to distinguish the biomarker stages from the clinical stages carried over from 2018, which are numerical.
The framework allows for two separate diagnostic paths, one using PET imaging and the other fluid biomarkers. However, Jack emphasized that stages determined by fluid markers or imaging are not equivalent to each other. For example, although the early and intermediate fluid biomarkers p-tau205 and MTBR-tau243 are correlated with tau tangles, researchers have not yet determined how they relate to the location and severity of those tangles as seen by PET. Clinicians will need to stick to one modality for a given patient, or when comparing data between patients.
Within the PET staging pathway, initial disease is diagnosed by global amyloid PET positivity, early disease by tau tangles in the temporal cortex, intermediate disease by a low level of tangles in the neocortex, and advanced disease by high neocortical tangles. For tau PET, both the location and magnitude of uptake matters, Jack noted.
How well does this PET staging work? In Amsterdam, Jack showed two different tests of the method. In one, researchers at ADNI and the Australian Imaging, Biomarkers, and Lifestyle study evaluated 1,045 participants staged in the new way. They found that later stages correlated with a greater prevalence of cognitive impairment and dementia, as expected (Villemagne et al., 2023). In the other, researchers led by Hansson and Rik Ossenkoppele at Lund analyzed progression to MCI and dementia over six years among 1,325 people staged by the new method. They found increasingly greater hazard ratios for clinical progression at more advanced PET stages. For example, compared with cognitively healthy people who were A-T-, those who were A+T- were twice as likely to progress to MCI, A+TMTL+ were 15 times as likely, and A+TNEO+ were 19 times as likely (Ossenkoppele et al., 2022).

Better Predict Progression. People at more advanced PET stages of disease (dark blue, green) develop cognitive impairment (left) and dementia (right) faster than do those at early PET stages (red, light blue). [Courtesy of Ossenkoppele et al., 2022.]
In the fluid biomarker pathway, initial disease is diagnosed by an abnormal CSF Aβ42/40 ratio, or elevated p-tau181, p-tau217, or p-tau231. Early disease is marked by elevated p-tau205, intermediate by MTBR-tau-243, and advanced by a new marker, non-phosphorylated tau fragments in the CSF (Montoliu-Gaya et al., 2023). Jack noted that fluid biomarker staging is more tentative than the PET staging scheme, and needs additional validation.
More Markers: Nonspecific Reactions and Co-Pathologies
While amyloid and tangles are the core biomarkers for AD diagnosis, the new draft criteria recognize that nonspecific reactions contribute. Take neurodegeneration (N) and inflammation (I). They are nonspecific because they occur in many neurodegenerative diseases. Validated N biomarkers include NfL, volumetric MRI, and FDG PET; I has only GFAP so far. Many more biomarkers, such as fluid neurogranin and synaptic PET imaging for N, and fluid YKL-40 and sTREM2 for I, are in research use and may become fit for clinical use.
Finally, the criteria specify markers of co-pathology. Chief among these are vascular damage (V) and α-synuclein (S). For the former, markers include MRI of white-matter hyperintensities, expanded perivascular spaces, and infarctions. For the latter, α-synuclein seed amplification assays of CSF samples are up and coming (Apr 2023 conference news; Part 9 of this series). As yet, there are no markers for TDP-43 deposits, another common co-pathology.
In some cases, the full ATNIVS suite of biomarkers will show mismatches, Jack said. He presented an example of a man who had dementia and was positive for A and N biomarkers, but negative for T. Although the man had Alzheimer’s disease, that was not the cause of his dementia, Jack said. Instead, the culprit was advanced limbic predominant age-related TDP-43 encephalopathy (LATE), which often co-occurs with AD pathology and can confound diagnosis (May 2019 news; Mar 2022 conference news).
Several audience members asked how clinicians will know whether a particular pathology in a given patient is driving that person’s clinical syndrome. The answer? Physicians must interpret biomarkers in the context of each patient’s medical history. For example, poor kidney function, obesity, and certain medications can hike a person’s level of AD biomarkers. Head injuries are known to spike p-tau231. All of that will have to be taken into account. “That’s why us clinicians will still have jobs,” Sperling quipped.
The new staging scheme represents the natural progression of disease, in the absence of treatment. An Alzheimer’s patient who becomes amyloid-negative after a course of immunotherapy treatment still has the disease, Jack said, in answer to an audience question. “Disease-modifying treatments may alter the relationships between biomarkers, but the disease does not disappear,” he added.
How Do Clinical Stages Fit In?
For clinical staging, the committee largely kept the method from 2018. They made only one change, adding a stage zero. It is for people who inherit deterministic AD mutations but do not yet have any biomarker or clinical changes.
How to integrate biomarker and clinical staging? The classical progression would be for a person to move from stage 1A (no symptoms, initial biomarkers) to 2B (subtle symptoms, early biomarkers), 3C (MCI, intermediate biomarkers), and finally 4-6D (dementia, advanced biomarkers). However, an individual person's progression might be different, Jack said. Some people will fare worse clinically than would be expected based on their A and T biomarkers. This could indicate the presence of co-pathologies, which can bring on neurodegeneration and cognitive symptoms earlier than otherwise expected. Others will fare better than expected, perhaps due to resilience factors. “Clinical impairment does not scale in lockstep with biomarkers,” Jack said.
Jack acknowledged that the new system is more complicated than the old. This is also true in other diseases such as cancer, where diagnostic and staging schemes become more elaborate as scientists learn more. The greater level of detail will help better tailor diagnosis to individuals, leading to more personalized medicine, he predicted. Jack also believes that the ability to target treatments to particular disease stages will become more sophisticated. “The era of disease-modifying treatments for AD has just begun,” he noted.
The authors invite feedback here. —Madolyn Bowman Rogers
With three positive and three negative Phase 3 trials of second-generation anti-amyloid antibodies to draw upon, Alzheimerologists now have more data to mine for what works and what does not. At last month’s Alzheimer’s Association International Conference in Amsterdam, scientists pored over gantenerumab and lecanemab data, hunting for clues of which parameters might predict success. And clues they found.
Some take-homes: Clinical benefit was linked to higher drug exposure and faster clearance, perhaps because people need to be amyloid-negative for some period of time before cognitive decline slows. Certain patient characteristics, such as age, APOE genotype, and tangle load, affected drug response, as well. Researchers also debated the still-murky relationship between biomarker change and subsequent cognitive response (see Part 12 of this series).
Overall, scientists in Amsterdam were optimistic that these analyses will enable them to improve treatment regimens. “We’re learning exponentially now,” said Jeffrey Cummings of the University of Nevada, Las Vegas.

Late Clearance. Spaghetti plots from the negative Graduate 1 (left) and 2 (right) trials show that plaque load rose on placebo (gray) and fell on drug (blue). Clearance was slow, however, with only a quarter of participants dipping below the threshold for amyloid negativity (red dotted line), and only by the end of the trial. [Courtesy of Roche.]
Gantenerumab’s Near Miss: Need to Get Lower For Longer?
In Amsterdam, researchers chewed over the Phase 3 Graduate results. Roche scientists had previously reported that gantenerumab cleared half as much plaque as they had expected, and that trends toward less decline on cognitive tests fell short of statistical significance (Dec 2022 conference news). In Amsterdam, researchers debated what caused the disappointing result.
First, one factor that did not explain it: Pharmacokinetic analysis of plasma samples showed the same peripheral drug exposure as had been achieved in the earlier open-label extension trials, said Christopher Lane of Roche. Likewise, the binding affinity of the gantenerumab formulation used in Phase 3 was the same as that of earlier formulations, Lane added in response to an audience question. So the problem did not lie in the antibody itself, nor in dosage or administration.
Then what does account for the weak effect? Partly, titration was slow, taking up nine of the 27 months of the trial. This was done to keep ARIA low, but also resulted in delayed plaque clearance. Spaghetti plots revealed that not only did three-quarters of the cohort stay above the threshold of 24 centiloids set for brain-wide amyloid negativity, but those who did cross it did so only by the end of the trial (see image above). Given that cognitive benefits lag behind amyloid removal, there may have been too little time left in the trial to detect a clinical change in most participants, noted Rachelle Doody of Roche. “We got the exposures we were expecting, but it took us longer to get there,” Doody said in Amsterdam.
Compounding the effect of slow clearance, the baseline amyloid load started high in this group, at around 95 centiloids, compared with 76 centiloids in the Phase 3 Clarity trial of lecanemab. This meant fewer people became amyloid-negative, with the group’s average final centiloid value being 43. Why did plaque load start so high? An audience member noted that use of the free and cued selective reminding test (FCSRT) as an inclusion criteria may have selected for people with more advanced disease, and thus higher plaque load. In particular, because women tend to do better than men on this test at the same disease stage, it might explain a previously observed sex difference in this trial, where women started with a higher tangle load than did men (Apr 2023 conference news). Doody called this a great observation. She noted the FCSRT was used to select for people whose disease would progress during the trial.
In panel discussions at AAIC, other researchers agreed that delayed clearance might be important. “Perhaps plaque did not get low enough for long enough,” said Nick Fox of University College London. Kaj Blennow of the University of Gothenburg, Sweden, pointed out that although fluid biomarkers showed improvement, no one knows how soon they changed. CSF biomarkers, which most directly reflect what happens in brain, were measured only at the beginning and end of the trial. Improvement may have come too late. “We are learning for how long we need to touch this pathway to get a clinical benefit,” Doody said.

Negativity is Key. In the gantenerumab trial, the majority of East Asian participants became amyloid-negative (right); they had a larger cognitive benefit than the rest of the cohort (left). [Courtesy of Roche.]
All About Exposure Other data pointed to the importance of high drug exposure. Graduate participants received a fixed dose of gantenerumab, i.e., 510 mg given subcutaneously every two weeks. Other antibody trials have calculated a person’s dose by weight, in mg/kg. This meant that in the Graduate trials, lighter people received a higher effective dose than did heavier people. In Amsterdam, Angeliki Thanasopoulou of Roche showed subgroup analyses that suggested this could have made a difference.
Dividing participants by geographical region, the East Asian subgroup, which comprised 225 people, fared much better than others. For them, cognitive decline on the CDR-SB slowed by 38 percent, similar to the benefit seen in positive antibody trials. This compared with a slowing of 5 percent for other Graduate participants. The East Asian subgroup also cleared more amyloid, with nearly two-thirds of them becoming amyloid-negative, again similar to the benefit in the lecanemab trial.
Demographically, the biggest difference between East Asians and other groups was weight. East Asians averaged 55 kilograms, meaning they received a dose of about 9 mg/kg, compared with 71 kilograms, or 7 mg/kg, for other groups. In addition, East Asians missed fewer doses than other groups (but had no less ARIA), and by the end of the trial had received a 20 percent higher cumulative dose of antibody.
Additional subgroup analyses strengthened the emphasis on exposure. Thanasopoulou showed that for Graduate participants who received 90 percent or more of their scheduled doses, cognitive decline on the CDR-SB slowed by 16 percent, twice as much as the 8 percent in the cohort overall.

Does Speed Matter? In an exploratory posthoc analysis, cognition (right) declined less in people on gantenerumab who cleared amyloid quickly (green, left) than in those who cleared amyloid more slowly (red, blue). [Courtesy of Roche.]
How Fast Does Amyloid Need to Fall?
Clearance speed might matter, as well. In a preliminary posthoc analysis, Roche scientists clustered participants in the amyloid PET subgroup by patterns of amyloid removal. They found three groups (see image above). In one, plaque resisted clearance, falling late; in another, it dropped steadily but slowly. Neither group notched a cognitive benefit compared with placebo. In the third group, however, plaque load dropped rapidly, ending lower than in the other groups, near the negativity threshold. In this group, cognitive decline slowed.
It is unclear if the speed of removal itself mattered, or the fact that these participants spent longer below the negativity threshold. The analysis is exploratory and the three groups were small, at about 25 people each. It is also unclear what caused these different patterns of plaque removal. At baseline, the fast-clearance group tended to be at an earlier disease stage, with three-quarters diagnosed with MCI compared with half of the other groups, Thanasopoulou said.
Data from other antibody programs further informed the speed question. In Amsterdam, Brian Willis of Eisai presented a pharmacokinetic model constructed with data from all lecanemab trials. Its predictions closely matched observed findings, Willis said. The model identified two participant characteristics that influenced the speed of plaque clearance: APOE genotype, and age. APOE4 carriers cleared plaque faster than did other genotypes, but because they started with about 20 centiloids more, they ended up in the same place after 18 months. Likewise, older participants cleared more plaque than younger ones. The difference could be substantial: for example, compared with a 73-year-old, an 83-year-old cleared 30 percent more plaque, and a 57-year-old cleared only half as much. Unlike genotype, age did not affect baseline plaque load.
Willis did not discuss potential explanations for these clearance differences. Previous research has found that the blood-brain barrier becomes leakier with age, and is leakier in E4 carriers, raising the possibility that in these groups, more antibody may enter the brain, resulting in more exposure (Feb 2015 webinar; Jan 2019 news; May 2020 news). Willis noted that in the aducanumab trials, older participants also cleared plaque faster than did younger ones.
Overall, a picture emerged in which small differences in a person’s drug exposure and plaque clearance determine the size of their clinical benefit. For gantenerumab, a high baseline plaque load and slow clearance meant it came up short in Phase 3. “This was a drug that almost worked,” Doody concluded. She noted that Roche is using publicly available anti-amyloid antibody data to model factors such as the amount of amyloid removed, the speed of removal, and the percentage of people who become amyloid negative, to determine which are most important for efficacy.—Madolyn Bowman Rogers
In amyloid immunotherapy trials to date, biomarker change correlates with clinical efficacy, but the relationship is far from perfect. Plaque can fall and fluid biomarkers normalize without a statistically significant clinical benefit, as in the Phase 3 gantenerumab trials. Or, cognitive decline can slow without a change in tau PET signal, as in the Phase 3 donanemab trial. At last month’s AAIC in Amsterdam, researchers wrestled to understand the link between biomarkers and efficacy, emphasizing the need to define these relationships better. Efforts are underway to model the associations between biomarkers and cognition, and to develop a standardized scale for comparing tau PET scans done with different tracers.
“We have to put parameters on biomarker change,” Rachelle Doody of Roche said in Amsterdam.
First, evidence for correlation. Katherine Dawson of Biogen reported that in the positive Emerge trial of aducanumab, a drop in p-tau181 in both cerebrospinal fluid and blood correlated with less decline on cognitive and functional tests. In plasma, the correlations were weak, ranging from 0.11 to 0.21. In CSF, they were stronger, with a high of 0.44 for slowed decline on the ADCS-ADL-MCI functional test. Dawson noted that p-tau181 fell more in people who became amyloid-negative than in those who stayed above this threshold. “Biomarker changes are linked to efficacy,” she concluded.

Inconsistent Sex Difference. In the Phase 3 gantenerumab trials, CSF biomarkers tended to normalize more in women than men, belying the larger clinical benefit in men. [Courtesy of Roche.]
Yet this does not always hold true. In the Graduate Phase 3 trials of gantenerumab, fluid biomarkers normalized in the absence of a statistically significant cognitive effect. More puzzling still, a subgroup analysis by sex found opposing effects for biomarkers and cognition. While only men benefited on cognitive tests, women had more biomarker change in CSF, Tobias Bittner of Roche reported in Amsterdam. The CSF differences were typically small, and biomarkers moved toward normal in both sexes in the treatment group.
For example, CSF p-tau181 fell by about 20 percent in men, and 25 percent in women. Total tau, neurogranin, and GFAP posted similar numbers. Some secondary biomarkers of inflammation and synaptic damage, such as sTREM2, S100B, and α-synuclein, displayed more dramatic differences, improving only in women. A couple of biomarkers bucked the trend—Aβ42 rose by the same amount in both sexes, and NfL fell more in men than women. Overall, however, women seemed to have a stronger biomarker response than men (see image above). It is unclear if the biomarkers/cognition discrepancy might be due to higher drug exposure in women, who weigh less than men on average.
How much do biomarkers need to change for a measurable treatment benefit? This is still unknown. John Sims of Eli Lilly stressed that researchers should examine a panel of biomarkers to gauge efficacy, because individual markers, such as NfL, have at times given puzzling results.
Tau Tangles Are Harder to Budge Than Previously Thought
Tau PET confounded scientists, producing inconsistent results from trial to trial. Still, hopes are high for tau PET as an outcome measure, and in some cases, this marker does seem to track with efficacy. For example, for aducanumab, which reported a treatment benefit in one of its two Phase 3 trials, an admittedly tiny tau PET substudy found a slight drop in tangles in a medial temporal composite region for people on drug, contrasting with a slight increase for those on placebo (Dec 2019 conference news). Meanwhile, in the Graduate trials, the tau PET signal was unaffected by gantenerumab, Bittner said in Amsterdam.
Alas, the correspondence between degree of tangle cleanup and efficacy is far from perfect. In the Phase 3 Clarity trial, lecanemab slowed, but did not stop, tangle accumulation in the medial temporal lobe, and showed a trend toward slowing in other regions (Dec 2022 conference news). This was a lesser effect than reported for aducanumab, which cleared tangles in the MTL, despite a similar reported clinical efficacy in Clarity and Emerge.
In the Phase 2 donanemab trial, Lilly researchers had reported a slight drop in tangles in the medial temporal lobe, and slowing of accumulation in other regions (Mar 2021 conference news). However, in the Phase 3 Trailblazer-Alz2 trial, donanemab had no effect on tangles, despite delivering the same clinical efficacy as in Phase 2 (Jul 2023 conference news).

Stubborn Tangles. The original Q-ATN model (pink) overestimated how much tangle accumulation would slow compared with placebo (green) in the lecanemab (left) and aducanumab (right) Phase 3 trials. The updated model (blue) offers a better fit with actual data (dots). [Courtesy of Norman Mazer and Roche.]
Why this discrepancy between trials? The answer is unclear. One factor could be that amyloid removal touches tangles only indirectly. Norman Mazer, who recently retired from Roche, had previously developed a Quantitative ATN (Q-ATN) model of AD. It used observational and trial data to predict how amyloid removal would affect downstream biomarkers and cognition (Aug 2022 conference news; Mazer et al., 2022). In Amsterdam, Mazer and Frank Boess of Roche presented an update that incorporates data from the Graduate and Clarity trials, but not yet from the Trailblazer-Alz2 results.
The researchers found that the original model, based on sparse tau PET data, had overestimated how strongly plaque clearance would affect tangles. In the updated version, tangle accumulation slows at about half the rate as previously estimated, better matching trial data (see image above). This has a correspondingly smaller effect on CDR-SB as a result. For example, using baseline data from Graduate 1, the original model predicted that decline on the CDR-SB would slow by 0.86 points. The updated model predicted a slowing of 0.39 points, close to the observed value of 0.31. For other antibodies, too, the updated model predictions are more accurate, falling within the 95 percent confidence interval reported for sensitivity analyses of each trial.
The model predicts that tangles in the brain have a half-life of 3.4 years. This implies they barely budge over a typical trial's duration. The model also suggests that, for cognitive decline to slow, antibodies must hit the brakes on MTL tangles. Except for the Trailblazer-Alz2 data, all effective antibodies thus far have reportedly done this. Scientists at AAIC were nonplussed that Trailblazer-Alz2 posted no effect on tangles at all while reporting arguably the biggest clinical benefit thus far.
CenTauR Rides to the Rescue?
One factor in these discrepancies between trials could be differences between tau PET tracers. The aducanumab and lecanemab trials used MK6240; donanemab, flortaucipir; gantenerumab, GTP1. Flortaucipir is generally considered the least sensitive of the bunch, but at the moment, there is no way to directly compare PET data from different tau tracers. Thus, it is unclear if these data are equivalent.
Researchers at the Critical Path Institute are trying to change this. At last year’s AAIC, they convened a public-private partnership to determine how to scale tau PET scans against a common benchmark, with units called CenTauRs, similar to the centiloid scale for amyloid PET. Led by C-Path’s Sudhir Sivakumaran, the Critical Path for Alzheimer’s Disease (CPAD) consortium includes representatives from the companies that make tau PET tracers, as well as data scientists at the University of Southern California’s Laboratory of Neuro Imaging and Australia’s CSIRO. It receives input from regulatory agencies such as the FDA and EMA.

Unified Yardstick. The proposed CenTauR scale achieves a tight correlation between tangles measured with flortaucipir (x axis) and Roche's RO948 tracer (y axis). [Courtesy of Critical Path for Alzheimer’s Disease.]
In Amsterdam, Antoine Leuzy of Lund University, Sweden, presented the consortium’s work to date. First, the researchers tried the same method that had been used to construct the centiloid scale. It requires choosing a reference tracer to measure other tracers against. However, because tau PET does not have a gold-standard reference tracer, as PiB is for amyloid tracers, this method produced more noise and was less accurate than is the centiloid scale.
Instead, the scientists tried a “joint propagation model,” which obviates the need for a reference standard. Instead, it compares all tracers head-to-head while scaling each from 0 to 100. To develop the standard, the researchers mined data from five different trials that included multiple tracers. The BioFinder2 cohort uses both flortaucipir and RO948; a University of Pittsburgh study uses flortaucipir and MK6240. Roche has a study with GTP1 and MK6240, another with GTP1 and PI2620. Finally, the ACE Alzheimer Centre in Barcelona, Spain, uses both RO948 and PI2620. Together, the data encompass the five most commonly used tau tracers, each being directly compared against two others.
The joint propagation model was indeed able to closely align findings from these different tracers, Leuzy showed in Amsterdam. For example, the correlation between flortaucipir and RO948 data as measured in CenTauRs was around 0.98 (see image above). The scale was able to capture longitudinal change in the BioFinder2 cohort, as well. The method appears more precise than the centiloid-like approach, with a root-mean-squares error for alignment strategies being 6 versus 8.
In addition, the CenTauR scale may reduce the number of PET scans needed to detect an effect by anywhere from 13 to 60 percent, depending on the trial population, Leuzy calculated. A unified tau PET scale therefore is achievable and useful, he concluded. However, Leuzy noted that more data are needed from scans with different levels of tangle severity, to ensure the measure performs equally well at very high and low ends of the scale.
Once the CenTauR scale is ready to be deployed, will it better align tangle measurements with clinical efficacy in trials? The answer may help determine how well tau PET would work as an outcome measure.—Madolyn Bowman Rogers
No Available Comments
Is a person born with APOE4 stuck with the lifelong consequences of this risk allele? These days, yes. In the future, perhaps not. APOE4 can be silenced, and the protective APOE2 allele introduced instead, according to Lance Johnson, University of Kentucky, Lexington. Johnson tested this theory by creating APOE4-to-2 “switch” mice. These animals express APOE4 until activation of Cre recombinase shifts expression to APOE2.
“This is a very useful model to address the potential benefits of switching E4 to E2 systemically or in a cell-type-specific manner and at different stages of disease,” wrote Ling Li of the University of Minnesota, Minneapolis.
At the Alzheimer’s Association International Conference, held in Amsterdam from July 16-20, Johnson reported that an astrocyte-specific switch cleared plaques, calmed microglia, and improved memory, even when amyloidosis was in full swing. A switch in microglia evoked no such improvements, suggesting that microglial ApoE is unable to stir a response to plaques.
Bart De Strooper, Dementia Research Institute, London, and Amaia Arranz from the Achucarro Basque Center for Neuroscience, Spain, came to a similar conclusion. They reported that astrocyte-derived ApoE rouses other glia against amyloid, whereas microglial ApoE does not. These findings help researchers understand the consequences of someday changing APOE expression in people through gene therapy or gene editing, both of which are being explored.
“After amyloid and tau, I envision APOE could be the next therapeutic target for AD,” said Julia TCW of Boston University, during a lively Q&A. Johnson summed up the potential impact this way: “If we were to gene edit all homozygous APOE4 alleles to APOE2/2, we would prevent 16 percent of AD cases. Changing all heterozygous APOE4 alleles to APOE2 would prevent 63 percent of cases.”
To weaken APOE4, scientists have silenced the gene with antisense oligonucleotides or knocked down the protein with antibodies. Both reduced plaque load in mice, and the latter improved spatial memory (Huynh et al., 2017; Liao et al., 2014; reviewed by Yang et al., 2021). In neurons derived from human induced pluripotent stem cells, CRISPR editing of APOE4/4 to APOE3/3 decreased production of Aβ and phosphorylated tau (Jun 2018 news).
Recent genetic evidence supports the idea that APOE knockdown might work in people, too. One man who has a loss-of-function variant in his APOE4 allele had no amyloid pathology or signs of dementia in his 90s, while a second in his 70s was similarly protected (Aug 2023 news).
Other groups have used gene therapy to bolster APOE2 rather than knock down E4. This decreased levels of soluble oligomeric Aβ and amyloid plaques in mice (Zhao et al., 2016). Likewise, in a first for the field, cerebrospinal fluid phospho-tau and total tau, markers of AD pathology, dropped in three women who are homozygous for APOE4 after a single intrathecal injection of LX1001, an adeno-associated virus carrying the APOE2 gene (Dec 2022 conference news).
Still, there may be downsides to knocking down APOE4 or boosting APOE2. The former might rob the brain of physiological, lipid-toting duties, while adding ApoE2 still leaves ApoE4 running amok. What if both approaches were combined?
With this in mind, Lesley Golden in Johnson’s lab created the APOE switch mice, dubbed 4s2. In these knock-ins, every cell has its mouse APOE gene replaced with a construct that carries genes for human APOE4, and APOE2, with a stop codon in between. The mouse also has an inducible gene for Cre recombinase, which recognizes sites flanking the E4 gene and the stop codon. How does the switch work? The animals make ApoE4 until they are fed tamoxifen. This induces Cre, which then snips out the APOE4, and the stop, freeing up expression of the APOE2 gene (see image below).

Gene Swap. When APOE4-to-2 (4s2) “switch” mice (left) ingest tamoxifen (right, bottom), Cre recombinase snips out E4, allowing expression of APOE2. 4s2 mice injected with a control (top) kept expressing APOE4. [Courtesy of Lance Johnson, University of Kentucky.]
A month after intraperitoneally injecting 6-month-old 4s2 mice with five daily doses of tamoxifen, Golden detected an almost complete shift in ApoE production. As measured by mass spectrometry, 93 percent and 80 percent of ApoE in the plasma and brain, respectively, was E2.
What happened after the switch? Using single-cell RNA sequencing, the researchers compared gene expression before and after. Switched brain cells resembled cells from APOE2 mice. They expressed many of the same genes unique to E2 cells. For example, 94 percent of the genes expressed by switched astrocytes were APOE2 astrocyte genes. The same was true for 64 percent of microglial genes. “That most of those genes were the same suggests that we can correct most of the differences inherent in APOE4 versus E2 brains with an allele switch in midlife,” Johnson concluded.
Notably, many of these genes support cellular pathways involved in AD, despite these mice having no AD pathology. For example, after the switch, astrocytes and microglia upregulated genes related to lipid metabolism and immune responses, while downregulating genes involved in Aβ precursor protein processing. Some of the downregulated genes had been identified in AD genome-wide association studies, including APOE, microglial-activating cystatin CST3, microglial phagocytosis receptor AXL, and apoptosis-related clusterin.
The switch had a downside, though. Too many triglycerides built up in the blood. A similar disorder, called hyperlipoproteinemia Type 3, occurs in 10 percent of APOE2/2 carriers. It can cause heart attacks or strokes.
To skirt such side effects, Golden used Cre recombinase genes with astrocyte- or microglial-specific promoters to create specific switches. Again, this worked, with 75 percent of astrocytes or microglia expressing ApoE2 after tamoxifen treatment. The scientists have not yet tested if either model developed hyperlipidemia.
Because these astrocyte- and microglia-specific switch mice have no AD pathology, Golden crossed them with 5xFAD mice. The latter begin accumulating plaques and developing gliosis at 2 months of age; spatial memory deficits follow four months later. To see the effect of switching APOE alleles after plaques are established, Golden injected 6-month-old crosses with tamoxifen, then two months later compared their behavior and brains to controls.
The astrocyte-switched mice had half as many amyloid plaques as unswitched mice, but only one-third as many activated astrocytes and microglia (see image below). They did better on memory tests, freezing 40 percent longer when expecting a foot shock. “Astrocyte ApoE influences other cell types involved in AD processes and can drive improvements in pathology and cognition,” Golden concluded.

Potent Astrocyte. Compared to brains of “unswitched” APOE4 5xFAD mice (top left), those whose astrocytes express E2 had fewer amyloid plaques and less gliosis (top middle). Switching APOE in microglia caused a small but non-significant drop in plaques yet increased gliosis (top, right, and inset, bottom). [Courtesy of Lance Johnson, University of Kentucky.]
The microglia switch mice told a more nuanced tale. After tamoxifen, they had no significant change in plaque load, astrogliosis, or memory. Microgliosis worsened. Because microglia produce but a small fraction of the brain's ApoE, switching them barely affects overall ApoE4 production, Johnson suggested. “In the astrocyte-specific switch mice, there is sufficient astrocyte E2 to regulate microglial responses, while in the microglial-specific switches, the astrocyte E4 still outweighs the microglia-derived E2,” he explained.
Similarly, De Strooper and Arranz reported that microglial-derived APOE was unable to rally glia against amyloid, yet APOE from astrocytes did. When Renzo Mancuso was in De Strooper’s lab, he transplanted human microglia into APP NL-G-F mice with or without endogenous APOE. In mice that expressed endogenous ApoE, the human microglia reacted to plaques by shifting from homeostatic to multiple different subtypes. Without murine APOE, the human microglia only mustered the cytokine-producing subtype (Oct 2022 news). “We speculate that the APOE secreted from microglia is a weak modulator of their response,” said De Strooper.
In Amsterdam, Arranz reported that she has now created similar mouse chimeras, but with astrocytes. She transplanted human astrocytes expressing APOE3 or E4 into one brain hemisphere of newborn APP/PS1 mice. Six months later, the side of the brain hosting human E3 cells had fewer amyloid plaques, with fewer microglia surrounding them, than did the contralateral side. In contrast, hemispheres injected with E4 astrocytes had more plaques than the contralateral side, and the plaques swarmed with microglia. “These data support the idea that human astrocytes modify microglial responses to AD pathology and might have a more impactful role than previously thought,” Arranz said.
All told, the three presentations support the idea that astrocyte-APOE potently regulates glial response to AD pathology, suggesting that future therapies should focus on modifying APOE alleles in those cells.
If anyone can pull off such an APOE4-E2 switch in people, then when during the AD continuum would be the time to do it? Before or after plaques have begun to accumulate? Golden is testing if giving tamoxifen early, i.e., at 2 months, will prevent AD pathology in the 5xFAD/4s2 mice. Johnson plans to cross the 4s2 mice with tauopathy models.
Other labs may soon join in. The wild-type 4s2 mouse will be available through Jackson Laboratory (JAX), Bar Harbor, Maine, later this year. Mike Sasner and colleagues at JAX are creating three other switch mice—APOE3-to-2, APOE4-to-3, and APOE3-to-4, so scientists can assess different allele-swapping scenarios. These mice will be available in mid-2024. Scientists in Amsterdam said they are looking forward to using these models.
Could an APOE switch be therapeutic? Amila Zuko and colleagues at UniQure Biopharma in Amsterdam are testing a dual, APOE4 knock-down/gene therapy approach in mice. Into the striata of wild-type animals they injected a virus containing both a microRNA to suppress APOE4 expression and a gene to overexpress a protective form of APOE, which they did not disclose. At AAIC, the scientists reported that a month after injection, the microRNA and the protective ApoE were produced at high levels in the cortex. In mice expressing human APOE4, injecting just the microRNA knocked down the gene by 60 percent. Zuko's next step is to test the dual therapy in APOE4 mice. This approach is not cell-specific. It is targeted to the brain by direct injection, à la LX1001 in the APOE2 gene therapy trial.—Chelsea Weidman Burke
Almost everyone who has Down’s syndrome (DS) is fated to develop Alzheimer’s disease because they have three copies of the amyloid precursor protein (APP) gene. Their cells overproduce Aβ, they become amyloid- and tau-positive on PET by age 40, and they develop dementia by age 55, on average. This would make them candidates for anti-amyloid immunotherapies, yet they have been excluded from most studies on these treatments thus far. Why? Safety is a concern. Many people with DS have severe cerebral amyloid angiopathy (CAA), which scientists suspect contributes to ARIA, an inflammation of the brain that can be provoked by immunotherapy (see Part 6 of this series).
“Focusing on cerebrovascular pathology in DS is critically important for considering clinical trials with the newly approved immunotherapies against Aβ,” Elizabeth Head, University of California, Irvine, wrote to Alzforum.
Figuring out how and when CAA and cerebrovascular disease begin, and how they progress throughout life, are important goals for the Down’s field. At the Alzheimer’s Association International Conference last month in Amsterdam, scientists reported that signs appear in a person’s 30s and worsen with age. Further, people with more markers of cerebrovascular disease had worse amyloid and tangle pathology, suggesting the vascular pathology marches in lockstep with AD.
“To me, this means cerebrovascular disease is not simply a comorbidity—it is an important part of DSAD,” Adam Brickman of New York’s Columbia University told Alzforum.
Cerebrovascular and Alzheimer's Disease Pathology
Brickman and Alexandre Bejanin at the Hospital de Sant Pau, Barcelona, separately found the simultaneous rise in markers of cerebrovascular disease and AD by analyzing brain scans and fluid biomarkers from people enrolled in two cohorts: the NIH-funded Alzheimer Biomarker Consortium‐Down Syndrome (ABC-DS), which has eight U.S. study sites and one at the U.K.’s University of Cambridge, and the Down Alzheimer Barcelona Neuroimaging Initiative (DABNI) cohort based at Hospital de Sant Pau. Brickman assessed around 240 ABC-DS participants with different analyses, while Bejanin studied up to 250 from DABNI.
Patrick Lao from Brickman’s lab reported that people with DS become amyloid-positive, as judged by PET, at around age 35, and tangle-positive at age 39. Signs of vascular injury begin to appear on MRI scans around then too. Spaces around blood vessels enlarged. Infarcts, a marker of large-vessel disease, emerged in the early 30s. Microbleeds, signs of CAA, and white-matter hyperintensities (WMHs), signs of small-vessel disease, ticked up around age 36 (see image below).

Terrible 30s. People with DS begin accumulating amyloid plaques around age 35 and neurofibrillary tangles at 39 (not shown). Perivascular swelling (top left) and infarcts (top right) appear around age 31. Microbleeds (bottom left) and white-matter hyperintensities (bottom right) start to worsen at 36 and 37, respectively. [Courtesy of Adam Brickman and Patrick Lao, Columbia University.]
In a similar vein, Sára Zsadányi reported a rise in microbleed prevalence and severity around age 40, while Alejandra Morcillo-Nieto found WMHs flare up around the same time. Both work in Bejanin’s lab. In volunteers with microbleeds, hippocampi had begun to shrink while CSFAβ42/40 ratios began to fall and phospho-tau181 to climb, both signs of accumulating plaques. CSF neurofilament light had ticked up also, indicating neuronal damage.
Natalie Edwards in Brickman’s lab saw similar fluid marker changes in plasma. People with many WMHs had high p-tau217, NfL, and plasma glial fibrillary acidic protein (GFAP) compared to controls. These data indicate that vascular injuries worsen as AD pathology progresses.
By the time people with DS develop AD dementia in their 50s, their cerebrovascular disease has progressed so much that their blood-brain barrier became compromised, said Lisi Flores Aguilar from Head’s lab at UC Irvine. She reported that DS occipital cortex tissue was flooded with the blood protein fibrinogen, which barely entered the brain parenchyma of age-matched controls. Fibrinogen only leaks into the brain when vessels are damaged, and its presence in parenchyma indicates BBB breakdown. All told, the evidence supports Brickman’s contention that cerebrovascular disease is a core feature of DSAD and worsens in step with neuropathology.
Cardiovascular Risk in DS
Is cerebrovascular disease in Down’s primarily driven by cardiovascular disease, a major risk factor for late-onset AD? No. People with DS generally do not have heart disease or high blood pressure, according to Sarah Pape of King’s College London. Pape analyzed primary care records from 6,500 people with DS ages 18 to 75 and 23,000 age-matched controls. Half as many DS adults had heart disease or high cholesterol as did controls, and only one-quarter as many, or fewer than 1 percent of the DS population, developed high blood pressure. While 99 percent of people with DS will never have high blood pressure, Brickman and Bejanin’s work show that many will develop microbleeds and WMHs. “Vascular risk factors may exacerbate cerebrovascular disease, but they don’t seem necessary for it to emerge in DS,” Brickman concluded.
Those who did have hypertension were twice as likely to develop dementia by age 55. “People with DS who have cardiovascular disease are at increased risk of AD, just like people who do not have DS,” Andre Strydom, King’s College London, who chaired the DS session, told Alzforum.
When Plaques Meet the Cerebrovasculature
The poor correlation between cardiovascular and cerebrovascular disease in DS supports the idea that CAA causes the latter. Researchers know that severe buildup of Aβ in blood vessel walls weakens them enough to burst. Indeed, microbleeds, a sign of this damage, correlate with CAA in postmortem cortical tissue from DS cases beginning in the mid-30s (Helman et al., 2019). This mirrors the rise in amyloid plaques seen on brain scans starting around that age.

CAA in Dup-APP. Compared to arteries from a person who had Down’s (top), those from a person with the Dup-APP mutation (bottom) had even more amyloid (brown). [Courtesy of Marie-Claude Potier, Paris Brain Institute.]
But is amyloid alone sufficient to cause CAA in DS? To gain a more nuanced understanding, Marie-Claude Potier of the Paris Brain Institute compared postmortem cortical tissue from people who had had DSAD and those who had a rare duplication of APP but no DS. Dup-APP causes early onset AD and CAA (Rovelet-Lecrux et al., 2006). Potier found that while DSAD arteries and vessels had lots of CAA, those from people with the duplication had even more. CAA in the Dup-APP cases was so severe, even their brain capillaries had it, a rarity in other forms of AD (see image at right).
What explains the difference? While more Aβ was found in Dup-APP brains than in DS brains, the isoforms also differed. Shorter Aβ peptides—including Aβ34, Aβ38, Aβ40, and their N-truncated varieties—were more abundant in Dup-APP tissue, though the amount of Aβ42 was similar. In collaboration with Jörg Hanrieder at the University of Gothenburg, Potier used imaging mass spec to find that the short and N-truncated Aβ peptides are exclusively embedded in blood vessel walls of Dup-APP cases, while Aβ42 preferentially aggregated in the parenchyma (see image below). These data suggest that the overabundance of short Aβ peptides in people with the duplication, and in DS to a lesser extent, causes CAA. This mirrors where isoforms home to in CAA among the general population (Alonzo et al., 1998).
Short Peptides Flock to Vessels. While Aβ42 (green) aggregated in the parenchyma of Dup-APP tissue, Aβ40 (red) preferentially settled in the blood vessels. So did Aβ39, Aβ38, and their N-truncated forms (not pictured). [Courtesy of Marie-Claude Potier, Paris Brain Institute.]
Implications for Immunotherapy
As a strong risk factor for ARIA, a side effect of anti-amyloid antibodies, CAA might pose an added hurdle for using immunotherapy to help people with DS fend off Alzheimer's dementia. “It raises a cautionary note, given that … people with DS are more vulnerable to cerebrovascular pathology than are people with late-onset AD,” wrote Head. At the same time, demand for treatment in this population will rise as these drugs become more widely available.
Knowing that CAA and cerebrovascular disease both worsen in the mid-30s will help researchers decide when to start people with DS on these anti-amyloid drugs to avoid ARIA as much as possible. “We may want to start a very low dose or start young, when cerebrovascular problems are not fully developed,” Head added. In a consensus statement on amyloid immunotherapy in DS, Head and colleagues recommended including people with Down’s syndrome in clinical trials, and developing protocols to help monitor such drug usage.
No plans to evaluate any anti-amyloid antibody in DS have been announced. “There are ongoing discussions as we consider how best to safely bring these amyloid-lowering immunotherapies to the DS population,” Michael Rafii, University of Southern California, told Alzforum. The one immunotherapy that is being tested in people with DS is the anti-amyloid vaccine ACI-24 (May 2021 news).
To gear up for trials, researchers are looking to find better markers of pathology and better cognitive tests for DS. This has proven challenging given the broad range of intellectual ability accompanying DS. Head, Brickman, and Donna Wilcock of Indiana University in Indianapolis are collaborating to study longitudinal changes in cerebrovascular pathology in DS in search of fluid markers of cerebrovascular disease. This may help screen people at increased risk of CAA and, potentially, ARIA. A similar search for markers of CAA is ongoing in sporadic AD (Part 7 of this series).
Jason Hassenstab at WashU plans to develop a remote cognitive assessment designed for DS—the first of its kind. His will be a smartphone app consisting of short, gamified tasks that measure memory, attention, and speed. People will “play” them multiple times a day for a week until they’ve completed all the tests. Hassenstab plans to launch the app in 2025.—Chelsea Weidman Burke
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.