Isotope Labeling Links Tau Production to Aβ Burden
Quick Links
Tau rises in the cerebrospinal fluid as Alzheimer’s sets in, but where does this tau come from, and what does it tell us about the disease? To answer these questions, researchers led by Randall Bateman and Celeste Karch at Washington University, St. Louis, used stable isotope labeling to track tau kinetics in the human central nervous system and in cultured neurons. They reported in the March 21 Neuron that the cultured neurons released N-terminal fragments of the protein about three days after they were made. Tracking total tau in human CSF, they also found that tau production, but not clearance, correlated with Aβ plaque load. Together, the findings suggest that Aβ stokes the production and release of truncated forms of tau from neurons.
- SILK labeling tracks tau kinetics in cultured neurons and the brain.
- N-terminal fragments are actively released from neurons three days after synthesis.
- Full-length tau found outside of cells may come from dying neurons.
- Tau production, but not turnover, correlated with Aβ plaque burden.
“This paper makes a fundamental contribution to our understanding of human tau biology,” said Gil Rabinovici of the University of California, San Francisco. “It also opens the door for more precise integration of tau biomarkers into therapy development.”
Joint first authors Chihiro Sato and Nicolas Barthélemy and colleagues set out to measure tau kinetics in people for the first time. They started by taking a snapshot of the steady-state levels of different forms of tau inside and outside neurons. They used eight antibodies recognizing different epitopes that span the protein to immunoprecipitate then quantify proteoforms by mass spectrometry. To assess intracellular forms of tau, they tested soluble fractions of brain tissue taken postmortem from one person who had no evidence of neuropathology. Three-quarters of the soluble tau was full-length, while the remainder was missing the C-terminal end. In contrast, 99.9 percent of extracellular tau—detected in CSF samples from two cognitively normal volunteers—lacked the C-terminus, even the microtubule binding domains at the end of the protein.
To better understand where these tau fragments originate, Sato and Karch cultured neurons induced from pluripotent stem cells (iPSCs) derived from a healthy 47-year-old man. As for brain tau, most intracellular forms in the cultured cells were full-length, while tau released into the extracellular medium was predominantly truncated before the C-terminus. A small amount of full-length tau also appeared in the culture medium. The researchers speculated this leaked out when neurons died. The cultured neurons also had 100-fold less four-repeat (4R) tau than the human brain.
Armed with these details of steady-state tau levels, Sato and colleagues set out to track tau kinetics using stable isotope labeling kinetics (SILK). The researchers added 13C-leucine to the neuron cultures for two weeks, then replaced the medium with one containing 12C-leucine. They used immunoprecipitation followed by mass spectrometry to measure the concentration of labeled tau peptides inside and outside of the cells over the following three weeks.
At day 14, almost all tau inside and outside of the cells comprised 50 percent 13C. Three days after labeling stopped, the 13C:12C ratio of intracellular tau had fallen. However, it rose in the extracellular medium, suggesting a three-day delay in release of newly synthesized tau into the medium. At day six and subsequent days after labeling stopped, labeled tau both inside and outside the neurons fell as the cells began to replace the 13C-leucine.
Of 14 different labeled tau peptides analyzed in the cell medium, only release of C-terminally truncated versions was delayed. Though much less abundant in the medium, 13C full-length tau had already fallen by day three. The researchers proposed that truncated forms of tau are subject to some sort of regulated processing prior to release, while full-length forms might be released by dead or dying cells.
Overall, tau turned over slowly, with an average half-life of nearly seven days, based on the kinetics of the commonly shared TPSL peptide. Full-length tau had a shorter half-life than truncated versions. However, half-lives were the same for each form whether they were inside or outside of cells. Interestingly, 4R tau had shorter half-lives than 3R species, and phosphorylated forms vanished quicker than unphosphorylated ones. Four-R and phosphorylated tau are more prone to aggregation than 3R and unphosphorylated.
Rabinovici was intrigued by this result. “That 4R isoforms and certain phosphorylated tau species showed more rapid kinetics than other forms of tau suggests that tau species prone to aggregation undergo unique processing in the cell.”
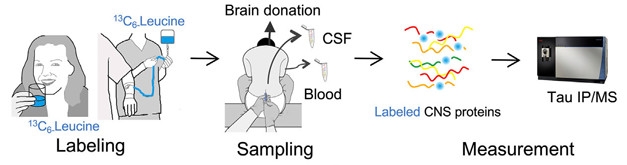
Tracking Tau. Depiction of the SILK method in people. [Courtesy of Sato et al., Neuron, 2018.]
The researchers next tracked tau in people. In their first round of experiments, they used six CSF samples from a recent SILK study they had performed that measured superoxide dismutase 1 kinetics in healthy individuals (Crisp et al., 2015). The participants swallowed one gram of 13C-leucine per day for 10 days, and then CSF was collected approximately 14, 28, 42, and 75 days after labeling started. In a separate protocol developed for the current study, the researchers delivered the stable isotope via continuous intravenous infusion over eight, 16, or 24 hours to four healthy participants in their 50s. Sato and Barthélemy then measured CSF tau approximately one, seven, 14, 28, and 60 days later. The researchers only tracked the common mid-domain TPSL peptide in the human studies, as others did not offer abundant or reproducible signals.
Though there were differences in the rate and extent of tau labeling between the two delivery protocols, the researchers found the protein turned over at about the same rate in both. About 26 pg of tau per mL/day ended up in the CSF and its half-life there averaged just over three weeks. Notably, in people who received the IV leucine, no 13C-tau appeared in the CSF at the first collection time point, which was one or two days after labeling stopped. The researchers detected it at the next time point, at seven days, and it peaked at 14 days. The findings suggested a lengthy delay between tau synthesis and its appearance in the CSF.
The researchers also measured tau in postmortem brain tissue from one other volunteer who died of unrelated causes eight days after receiving the 13C-leucine. Analyzing multiple regions throughout the brain, they calculated that an average of 0.13 percent of tau was labeled. A similar proportion of tau was labeled in the CSF. Together, these findings suggested that not only was the tau label distributed in a roughly even fashion throughout the brain, but the labeled tau in CSF reflected tau production in the brain, Sato told Alzforum.
Would tau kinetics change in the presence of Aβ plaques? To find out, the researchers injected 13C leucine into 11 people with cognitive impairment indicative of very mild AD, and 13 normal controls. Tau half-life was similar between the two groups. Using immunoprecipitation and mass spectrometry to take stock of different forms of unlabeled tau in the CSF, the researchers also found that the two groups had similar tau truncation profiles. However, people with elevated Aβ on florbetapir-PET scans had 1.3 times more tau in their CSF than did those with normal PET scans. Notably, the rate of tau production correlated with the extent of plaque burden, reaching about 40 pg/mL/day in those who bound most PiB.

Delayed Release. Truncated forms of tau show up extracellularly three days after they are synthesized, while full-length tau appears to be passively released from cells. [Image courtesy of Sato et al., Neuron, 2018.]
The findings suggest that the Aβ pathology enhances the production of tau protein. This argues against the idea that elevated CSF tau in people with AD comes primarily from dead and dying neurons, Bateman said, since they would not be making new tau.
Furthermore, dying neurons would release tau from inside the cell—namely, full-length tau. That this form was barely detected in the CSF of healthy controls and people with AD in the steady-state experiments also argues against Aβ-induced tau being from dead cells. This also makes sense in light of CSF tau not being universally elevated in other neurodegenerative diseases, he said. How Aβ pathology or tau’s own aggregation might affect processing and release of tau remains to be investigated, Bateman said
The data also suggest that proteolytic processing and secretion of tau are mechanistically linked. This idea comes from the authors' initial steady-state measurements, showing that tau in the CSF was overwhelmingly truncated at the C-terminus. That SILK uncovered a delay in release of truncated tau from cultured neurons supports that claim. Bateman speculated that cleavage and release might only occur after a certain time bound to microtubules. He speculated that a rising tau in the extracellular space could contribute to the seeding and aggregation of insoluble forms of tau that arise later in disease. Indeed, the researchers also found a correlation between tau production rate and tau accumulation as measured by tau PET.
In an editorial accompanying the paper in Neuron, Henrik Zetterberg of the University of Gothenburg in Sweden wrote that the findings are immediately relevant to biomarker research. Truncated forms of tau in the CSF could reflect newly synthesized proteins elevated in response to Aβ, while full-length tau could indicate ongoing neurodegeneration, he wrote. Current biomarker tests are not designed to distinguish between these different forms, but that could prove useful in the future. Tracking tau production rates could also prove useful as a readout in clinical trials, he added.
“The tau SILK method is likely to increase our understanding of when and how tau pathology starts to accumulate in neurodegenerative diseases, and should also provide information that increases the interpretability of the currently available static CSF and PET tau markers,” wrote Zetterberg.—Jessica Shugart
References
Paper Citations
- Crisp MJ, Mawuenyega KG, Patterson BW, Reddy NC, Chott R, Self WK, Weihl CC, Jockel-Balsarotti J, Varadhachary AS, Bucelli RC, Yarasheski KE, Bateman RJ, Miller TM. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest. 2015 Jul 1;125(7):2772-80. Epub 2015 Jun 15 PubMed.
Further Reading
Papers
- Han P, Serrano G, Beach TG, Caselli RJ, Yin J, Zhuang N, Shi J. A Quantitative Analysis of Brain Soluble Tau and the Tau Secretion Factor. J Neuropathol Exp Neurol. 2017 Jan 1;76(1):44-51. PubMed.
Primary Papers
- Sato C, Barthélemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, Kanaan NM, Yarasheski KE, Baker-Nigh A, Benzinger TL, Miller TM, Karch CM, Bateman RJ. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron. 2018 Mar 21;97(6):1284-1298.e7. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Massachusetts General Hospital, Harvard Medical School
Sato et al. shed new light on the mechanisms of tau proteins in the human CNS. Interestingly, the authors presented strong evidence for tau production as an active process in Alzheimer’s disease. This work brings us from static data points to dynamic measures that may have better predictive potential as we establish mechanistic links between amyloid and tau pathology. These findings will also help elucidate the underlying mechanisms of different tauopathies. They will also inform the design of future clinical trials, especially those testing tau-based therapies.
Harvard Medical School-MGH
This really exciting and groundbreaking study from the Bateman laboratory and colleagues introduces a powerful new set of methodologies highly relevant to efforts to understand human tau biology both in the clinical setting and with ex vivo stem cell models.
By defining the steady-state turnover kinetics of tau using their SILK methodology, this study lays the foundation for future studies seeking to test experimental therapeutic agents that may enhance the rate of tau clearance or its production, as well as to compare the kinetic and metabolic properties of tau across different tauopathies beyond Alzheimer’s disease. That the half-lives of different tau isoforms and post-translationally modified forms are variable is fascinating, begging the question of what the underlying molecular mechanisms are that control the kinetics and stability of different tau species in the CNS.
A critical observation for the field’s efforts working on patient-derived stem cell models was the recognition that the intracellular profile of iPSC-derived cortical neurons was strikingly similar to that observed in human brain despite the relative immature nature of six-week differentiated neurons.
However, the half-life of tau in control human iPSC-derived neurons was ~3.5-fold shorter that in humans (~6.7 days versus 23 days), pointing to differences in the metabolism of tau in ex vivo and in vivo systems.
One area for future investigation will be the impact the detection of peptides by IP-LC-MS methodology will have on our understanding of post-translational modifications of tau. Given the plethora of different tau modifications, these may impact the sensitivity of the assay to detect different peptides. Here, the ability to readily probe these modifications using human iPSC technology will afford a detailed exploration of these variables.
Overall, establishing if and how these tau profiles vary as a function of specific causal mutations for disease will be an exciting future direction for the application of this SILK technology. Moreover, as the mechanisms controlling tau proteolysis remain poorly understood, yet are at the heart of many ongoing therapeutic efforts, the quantitative and scalable nature of these assays provides an invaluable new tool to facilitate more detailed mechanistic studies.
Hertie Institute for Clinical Brain Research, University of Tübingen, and DZNE Tübingen
These very interesting human results are in line with and predicted from the CSF analyses of APP transgenic mouse models (Maia et al., 2013; Schelle et al., 2016).
References:
Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, Jucker M. Changes in amyloid-β and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med. 2013 Jul 17;5(194):194re2. PubMed.
Schelle J, Häsler LM, Göpfert JC, Joos TO, Vanderstichele H, Stoops E, Mandelkow EM, Neumann U, Shimshek DR, Staufenbiel M, Jucker M, Kaeser SA. Prevention of tau increase in cerebrospinal fluid of APP transgenic mice suggests downstream effect of BACE1 inhibition. Alzheimers Dement. 2016 Oct 14; PubMed.
View all comments by Mathias JuckerMake a Comment
To make a comment you must login or register.