Can an ApoE Mutation Halt Alzheimer’s Disease?
Quick Links
In today’s Nature Medicine, a team of researchers led by Yakeel Quiroz, Joseph Arboleda-Velasquez, Francisco Lopera, and Eric Reiman describe the case of a Colombian woman who inherited the autosomal-dominant E280A mutation in presenilin 1 but stayed cognitively well for decades past this mutation’s typical age of disease onset. Called Paisa, this mutation causes Aβ overproduction, triggering neurodegeneration and cognitive decline by a person’s 40s. Yet this woman stayed sharp for three decades after that. Now in her 70s, she struggles with short-term memory but still remains independent. Astoundingly, amyloid PET scanning revealed a massive buildup of amyloid plaques in her brain, far higher than those seen in young mutation carriers who are cognitively impaired. Despite all this amyloid, she has very little tau pathology, mostly confined to the medial temporal lobe, and her brain glucose metabolism is almost normal. It’s as if the chain of pathogenic events is broken after amyloid. What explains her remarkable resistance?
- The ApoE Christchurch mutation seems to block secondary tau pathology.
- This protective effect is similar to, but greater than, that of ApoE2.
- Protection correlates with lost binding to HSPGs, believed to propagate tangles.
The researchers believe the answer is a rare mutation in ApoE3 that disrupts the protein’s ability to bind lipoprotein receptors and other proteoglycans. The Colombian woman inherited two copies of this ApoE variant, known as the Christchurch mutation after the city where it was first identified. In vitro experiments showed that ApoE3ch behaves like the ApoE2 allele, also known to be protective. Both isoforms poorly bind heparan sulfate proteoglycans (HSPGs), the extracellular matrix molecules that have been implicated in the propagation and uptake of toxic forms of tau. In a related study, posted November 2 as a preprint on medRχiv, the same authors report that inheritance of two copies of ApoE2 also confers extra protection against AD, particularly tau pathology. Homozygous ApoE2 carriers develop far fewer tangles than ApoE2/3 carriers, or any other ApoE genotype.
“Together, the two studies make the case for ApoE having a profound effect on neuroprotection,” Reiman, at Banner Alzheimer’s Institute in Phoenix, told Alzforum. “We’re hoping this will galvanize interest in understanding ApoE-related mechanisms and developing treatments that target ApoE.”
Others shared his excitement. “This new study demonstrates, for the first time, that functional ApoE is truly required for full pathological and clinical development of AD,” Yadong Huang and Maxine Nelson at the University of California, San Francisco, and Kelly Zalocusky at the Gladstone Institute of Neurological Disease wrote in an accompanying Nature Medicine editorial. David Holtzman at Washington University, St. Louis, noted in an email, “[The data] suggest several mechanisms to further explore regarding how ApoE may be modulating inflammation, tauopathy progression, and neurodegeneration.”

Plaques but No Tangles. A woman with a familial AD mutation and two ApoE3 Christchurch alleles (left scans) had a very high plaque (top, red), but low tangle burden (middle) and healthy brain metabolism (bottom) compared with a typical mutation carrier (right scans). [Courtesy of Arboleda-Velasquez et al., Nature Medicine.]
Mouse studies from Holtzman’s lab first suggested that ApoE4 can worsen tau pathology independently of its effect on amyloid (Sep 2017 news), and human studies are starting to hint at the same thing, seeing more tangles in the brain and more phosphorylated tau in the cerebrospinal fluid of ApoE4 carriers than noncarriers, particularly women (Aug 2018 conference news). A recent tau imaging study found faster cognitive decline in ApoE4 carriers than noncarriers with equal tau burden, suggesting the allele somehow speeds up neurodegeneration (Nov 2019 news).
Lessons From Studying Resilience
The scientists working with the Colombian kindred did not set out to study ApoE. Led by Lopera at the University of Antioquia, Colombia, they studied people who appeared to escape the otherwise deterministic Paisa mutation in hopes of elucidating the AD pathogenesis pathway and finding new therapeutic approaches. Most E280A carriers are diagnosed with MCI at age 44, and dementia by age 49. However, the woman in this case study did not develop symptoms until three decades later, and what symptoms she has now are limited to problems remembering recent events. Her other neurological tests are normal, and her memory troubles have remained stable over two years of testing.
Quiroz and colleagues at Massachusetts General Hospital brought her to Boston to examine her biomarkers to try to find an explanation. Strikingly, amyloid PET imaging revealed a brain loaded with plaques. Her PiB distribution volume ratio (DVR) was 1.96, much higher than the 1.50 typically seen in Paisa carriers with MCI. “Her brain has one of the highest levels of amyloid pathology I’ve seen in the family,” said Quiroz, who leads PET studies with the Paisa population.
Even so, flortaucipir PET lit up only the medial temporal and occipital lobes, where tangles tend to start early in disease. What’s more, FDG PET revealed that her brain’s glucose metabolism remained robust, better than that in younger carriers with MCI, particularly in the precuneus (see image above). Her hippocampal volume is similar to those of carriers in their 40s, and she has relatively low levels of plasma NfL, a marker of neuronal injury.
Joseph Arboleda-Velasquez from the Schepens Eye Research Institute of Mass. Eye and Ear led genetic analyses. Whole-exome sequencing turned up but one likely explanation for her resilience: the Christchurch R136S mutation. This variant has been previously reported in the cardiovascular literature. It disrupts the LDL receptor binding domain of ApoE, crippling the protein’s ability to traffic fats. The Christchurch mutation was first identified in a man who had an ApoE2/2 genotype and hyperlipoproteinemia Type III (Wardell et al., 1987). This type of dyslipidemia develops in about 10 percent of homozygous ApoE2 carriers. It is marked by high levels of low-density lipoproteins, triglycerides, and cholesterol. Notably, the Colombian woman also has hyperlipoproteinemia Type III, with a blood cholesterol level of 512 mg/dl and triglycerides at 692 mg/dl, three times the normal level. She takes statins and has not experienced any cardiovascular problems to date.
Comparing Isoforms Yields Clues to Mechanism
The dyslipidemia in this woman suggested that ApoE3ch might share some characteristics with ApoE2. To explore this, Arboleda-Velasquez, Michael O’Hare, and colleagues compared ApoE isoforms in vitro. Normal ApoE3 stimulates aggregation of synthetic Aβ42, and ApoE4 even more so. In contrast, the Christchurch variant of ApoE3 behaved like the E2 allele, producing less clumping. Aggregation was lowest in the absence of any ApoE. This hints that the protective effect of the Christchurch mutation was due to a loss of function. Curiously, the finding implied that the Colombian woman might have even more plaques in her 70s without the Christchurch mutation. Even so, her amyloid load was well high enough to cause impairment in most people, hence there had to be another explanation for her cognitive preservation.
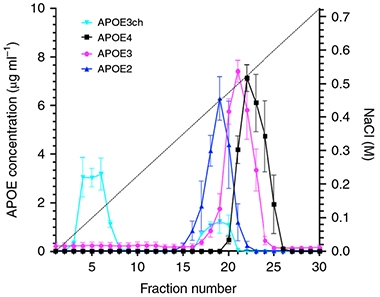
No HSPG for Me. The rare ApoE3 Christchurch allele (aqua) washes rapidly off a heparin column. Among the common alleles, protective ApoE2 (blue) binds most weakly, pathological ApoE4 (black) most strongly. [Courtesy of Arboleda-Velasquez et al., Nature Medicine.]
What about substrate binding? ApoE binds LDL receptors. ApoE4 does it the best, with E3 next and E2 weakest. ApoE3ch has a binding affinity that is intermediate between ApoE3 and ApoE2 (Lalazar et al., 1988). ApoE2 homozygotes with the Paisa mutation fare less well than does the woman with two Christchurch mutations. This made LDLR binding a poor candidate to be the protective mechanism, Reiman said. Somehow, ApoE3ch provides stronger protection than even a double dose of E2.
On a different measure, however, ApoE3ch was in a class by itself. The same domain on ApoE that binds LDLRs also recognizes HSPGs, a large class of polysaccharide molecules found as receptors on the neuronal cell membrane as well as in the extracellular matrix. One such HSPG is heparin, an anticoagulant secreted by mast cells. When the authors ran ApoE isoforms on a commercial heparin column, ApoE4 clung most tightly, while ApoE3 eluted earlier, and ApoE2 even sooner. ApoE3ch, however, barely bound heparin at all, with most of it coming quickly off the column (see image above). The dramatic effect of the Christchurch mutation in this assay means that how well ApoE binds HSPGs could be a factor in triggering downstream neurodegeneration. The HSPG data reinforce the idea that the Christchurch mutation results in a loss of normal ApoE function, Reiman said.
The Christchurch mutation appears to be recessive. Four other Paisa mutation carriers in the Colombian kindred inherited a single copy of it, and all developed cognitive impairment at the normal age. However, Quiroz noted that the sample was small. She has since identified another 20 or so Paisa mutation carriers with a heterozygous Christchurch mutation, and hopes this larger sample will produce a more definitive answer as to whether a single allele confers any protective effect.
“Additional in vitro and in vivo studies are required for unraveling the mechanisms underlying the protective effect,” Danny Michaelson at the University of Tel Aviv wrote to Alzforum (full comment below).
But How Does This Protect the Brain?
HSPGs have been implicated in the buildup of amyloid plaques, as well as in promoting the microglial response to amyloid (Apr 2016 news; Bussini et al., 2005; Zhang et al., 2012). Other studies link HSPGs to tau pathology. Marc Diamond at the University of Texas Southwestern Medical Center in Dallas and others have reported that these sticky receptors allow tau fibrils to glom onto neurons, facilitating uptake and propagation (Mar 2013 conference news; Apr 2015 news; Rauch et al., 2018; May 2018 news). Perhaps ApoE facilitates tau binding to HSPGs, Reiman speculated. In that case, the Christchurch variant’s weak affinity for HSPG would disrupt the spread of toxic tau through the brain. Interventions that weaken ApoE-HSPG binding might have therapeutic potential, he believes.
“This theory warrants experimental testing in cells and animals and, if successful, further clinical studies in humans,” Huang, Nelson, and Zalocusky noted in their editorial. Diamond wrote to Alzforum, “These results imply that we should ask more questions about a direct relationship between ApoE and tau. There are definitely reports of their interaction, so this could be an interesting avenue.”
For his part, Holtzman was most intrigued by the inflammation angle. “One wonders whether the effect of this ApoE variant is somehow decreasing the ability of ApoE to initiate a microglial-mediated inflammatory response to tau pathology,” he wrote (full comment below). His lab recently reported that ApoE4 exerts its toxic effects through microglia in tauopathy mouse models (Oct 2019 news).
Jacob Raber at Oregon Health and Science University in Portland suggested using unbiased omics approaches to explore the mechanisms underlying these isoform effects. “Together with other reports, these data support that tau and ApoE might be especially good therapeutic targets in AD,” he wrote (full comment below).
Potential Interventions
Could ApoE-HSPG binding be targeted therapeutically? The authors raised an antibody, 1343A, against ApoE’s LDLR binding region. When they incubated regular ApoE3 with it prior to adding it to the heparin column, the protein behaved like the ApoE3ch variant, eluting rapidly without apparent contact with this experimental HSPG stand-in. Quiroz said the scientists are further characterizing this antibody to see if it is suitable for clinical development.
A small-molecule approach would be cheaper, and more practical for repeat administration, than an antibody. Researchers led by Carl Frieden at WashU have previously described one such molecule, EZ482, that binds ApoE and alters its shape in such a way as to block HSPG binding (Mondal et al., 2016). Frieden told Alzforum that his group did not study EZ482’s effects in vivo, or develop it further. He is starting a project to look for additional compounds that alter ApoE binding. Meanwhile, researchers at the Gladstone Institute previously identified compounds similar to EZ482 that corrected some of ApoE4’s toxic effects in cell culture (Chen et al., 2012; Wang et al., 2018).
Reiman’s group is also interested in finding small molecules that block ApoE binding to HSPGs. However, he believes silencing ApoE with antisense oligonucleotides or RNA interference might be a simpler approach, and perhaps the first thing to test in clinical trials.
Joachim Herz at the University of Texas Southwestern Medical Center in Dallas noted that weakening the association of ApoE with HSPG receptors at the cell surface would effectively decrease the ApoE concentration in endosomes. Herz believes that ApoE4 wreaks much of its mischief there, because the apolipoprotein unfolds inside these acidic compartments and blocks recycling of neuronal receptors such as AMPA and NMDA. He found that a small molecule that inhibits the endosomal proton leak channel NHE6 sped up receptor recycling, weakening ApoE4 toxicity (Xian et al., 2018). About Arboleda et al., Herz wrote, “[These data] nicely provide circumstantial support to our model that ApoE4 aggregation in the early endosome is a key event in AD pathogenesis that can be effectively reduced or abrogated with small-molecule inhibitors of NHE6.”
“Undoubtedly, this work opens exciting new avenues in AD research and reveals promising possibilities for potential AD therapeutics,” Yadong, Nelson, and Zalocusky noted.
ApoE2 Strengthens Link to Tau Toxicity
The separate ApoE2 homozygote study complements the ApoE3ch findings. Using data from 4,108 autopsy-confirmed AD cases and 989 controls assembled by the Alzheimer’s Disease Genetics Consortium, Reiman and colleagues found that the 24 ApoE2 homozygotes in this sample ran but a third the risk of AD as did ApoE2/3 heterozygotes. In cases where AD did develop, it happened much later in life. Alzforum covered some of these data at AAIC last July in Los Angeles (Aug 2019 conference news).
The manuscript details a new discovery beyond this meeting story. Similar to the Christchurch mutation, ApoE2 in this pathology-confirmed sample seemed to ameliorate tau pathology. ApoE2 homozygotes died with an average Braak tangle stage of 2.3, heterozygotes, 3.2, compared with 4 or higher for the other ApoE genotypes. The results remained significant even after adjusting for plaque burden.
Notably, the protection from ApoE2 appeared unique to Alzheimer’s. There was no difference in the risk for cerebral amyloid angiopathy, Lewy body disease, hippocampal sclerosis, or vascular brain injury in this large sample. This once again suggests a specific effect of ApoE on the secondary tauopathy that develops in Alzheimer’s disease in the presence of amyloid, Reiman said. He added that the results seem specific for secondary tauopathies, because some evidence suggests ApoE2 homozygotes are more susceptible to primary tauopathies such as progressive supranuclear palsy (Zhao et al., 2018). “This result points towards additional mechanisms through which the ApoE2 allele confers its protective effect, beyond decreasing amyloid deposition,” Gemma Salvadó at the Barcelonaβeta Brain Research Center, Spain, wrote to Alzforum (full comment below).—Madolyn Bowman Rogers
References
Mutations Citations
News Citations
- ApoE4 Makes All Things Tau Worse, From Beginning to End
- Do Brain Changes at Menopause Make Women More Prone to Alzheimer’s?
- ApoE4 and Tau in Alzheimer’s: Worse Than We Thought? Especially in Women
- Sticky Matrix Proteins Lead to Amyloid Accumulation, Slow Clearance
- Tau, α-Synuclein Spread: Crazy Stuff—How Might It Work?
- Tau Triple Threat: Do Trimers Make Bad Seeds?
- To Deliver Itself From Cell to Cell, Phospho-Tau Uses UPS
- In Tauopathy, ApoE Destroys Neurons Via Microglia
- Rare Luck: Two Copies of ApoE2 Shield Against Alzheimer’s
Paper Citations
- Wardell MR, Brennan SO, Janus ED, Fraser R, Carrell RW. Apolipoprotein E2-Christchurch (136 Arg----Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest. 1987 Aug;80(2):483-90. PubMed.
- Lalazar A, Weisgraber KH, Rall SC Jr, Giladi H, Innerarity TL, Levanon AZ, Boyles JK, Amit B, Gorecki M, Mahley RW. Site-specific mutagenesis of human apolipoprotein E. Receptor binding activity of variants with single amino acid substitutions. J Biol Chem. 1988 Mar 15;263(8):3542-5. PubMed.
- Bussini S, Meda L, Scarpini E, Clementi E, Conti G, Tiriticco M, Bresolin N, Baron P. Heparan sulfate proteoglycan induces the production of NO and TNF-alpha by murine microglia. Immun Ageing. 2005 Jul 16;2:11. PubMed.
- Zhang X, Wang B, O'Callaghan P, Hjertström E, Jia J, Gong F, Zcharia E, Nilsson LN, Lannfelt L, Vlodavsky I, Lindahl U, Li JP. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-β in murine brain. Acta Neuropathol. 2012 Jun 13; PubMed.
- Rauch JN, Chen JJ, Sorum AW, Miller GM, Sharf T, See SK, Hsieh-Wilson LC, Kampmann M, Kosik KS. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep. 2018 Apr 23;8(1):6382. PubMed.
- Mondal T, Wang H, DeKoster GT, Baban B, Gross ML, Frieden C. ApoE: In Vitro Studies of a Small Molecule Effector. Biochemistry. 2016 May 10;55(18):2613-21. Epub 2016 Apr 27 PubMed.
- Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, Pleiss MA, Ji ZS, Balestra ME, Walker DW, Xu Q, Jeong DE, Budamagunta MS, Voss JC, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012 Feb 17;287(8):5253-66. PubMed.
- Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, Miller BL, Malloy MJ, Huang Y. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018 May;24(5):647-657. Epub 2018 Apr 9 PubMed.
- Xian X, Pohlkamp T, Durakoglugil MS, Wong CH, Beck JK, Lane-Donovan C, Plattner F, Herz J. Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer's disease. Elife. 2018 Oct 30;7 PubMed.
- Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, Attrebi ON, Petrucelli L, Fryer JD, Wszolek ZK, Graff-Radford NR, Caselli RJ, Sanchez-Contreras MY, Rademakers R, Murray ME, Koga S, Dickson DW, Ross OA, Bu G. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018 Oct 22;9(1):4388. PubMed.
External Citations
Further Reading
Primary Papers
- Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
- Zalocusky KA, Nelson MR, Huang Y. An Alzheimer's-disease-protective APOE mutation. Nat Med. 2019 Nov;25(11):1648-1649. PubMed.
- Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, Chen Y, Su Y, Myers AJ, Hardy J, Paul Vonsattel J, Younkin SG, Bennett DA, De Jager PL, Larson EB, Crane PK, Keene CD, Kamboh MI, Kofler JK, Duque L, Gilbert JR, Gwirtsman HE, Buxbaum JD, Dickson DW, Frosch MP, Ghetti BF, Lunetta KL, Wang LS, Hyman BT, Kukull WA, Foroud T, Haines JL, Mayeux RP, Pericak-Vance MA, Schneider JA, Trojanowski JQ, Farrer LA, Schellenberg GD, Beecham GW, Montine TJ, Jun GR, Alzheimer’s Disease Genetics Consortium. Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun. 2020 Feb 3;11(1):667. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Columbia University
This elegant study provides proof-of-principle evidence that the cascading component of the “amyloid cascade hypothesis” can be unhinged. The study shows that a lot of amyloid plaque accumulation does not necessarily lead to tau pathology and neurodegeneration. Namely, amyloid can be the smoke and not the fire.
What is the fire that has been doused by the APOE3ch mutation? The study shows that the mutation prevents uptake by neurons. So, from a neuron’s perspective, ApoE Christchurch is equivalent to an APOE null. Here, three lines of evidence provide an answer. One, the recent Neuron paper by Tessier-Levigne’s group that shows that PS1 mutations cause endosomal traffic jams by intraneuronal accumulation of APP fragments (enlarged endosomes), which typically occurs by blocking endosomal recycling (Kwart et al., 2019). Two, papers by Joachim Herz, which show that APOE nulls accelerates endosomal recycling (e.g. Xian et al., 2019). Three, recent papers showing that blocking endosomal recycling increases tau pathology (Young et al., 2018) and the early manifestations of neurodegeneration (Temkin et al., 2017), independent of APP.
Together, one can plausibly speculate that the APOE3ch mutation effectively normalizes the endosomal recycling defects caused by PS1 mutations, preventing tau pathology, neurodegeneration, and dementia, even with extracellular accumulation of amyloid plaques.
References:
Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, Tessier-Lavigne M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron. 2019 Oct 23;104(2):256-270.e5. Epub 2019 Aug 12 PubMed.
Xian X, Pohlkamp T, Durakoglugil MS, Wong CH, Beck JK, Lane-Donovan C, Plattner F, Herz J. Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer's disease. Elife. 2018 Oct 30;7 PubMed.
Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, Goldstein LS. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer's Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Reports. 2018 Mar 13;10(3):1046-1058. Epub 2018 Mar 1 PubMed.
Temkin P, Morishita W, Goswami D, Arendt K, Chen L, Malenka R. The Retromer Supports AMPA Receptor Trafficking During LTP. Neuron. 2017 Apr 5;94(1):74-82.e5. PubMed.
View all comments by Scott SmallOHSU
This paper is exciting and remarkable for various reasons.
References:
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019 Nov 4;216(11):2546-2561. Epub 2019 Oct 10 PubMed.
Raber J, Wong D, Yu GQ, Buttini M, Mahley RW, Pitas RE, Mucke L. Apolipoprotein E and cognitive performance. Nature. 2000 Mar 23;404(6776):352-4. PubMed.
Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez S, Sullivan PM, Holtzman DM, Sadowski MJ. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE ε2 and ε4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. 2014 Jun 28;2:75. PubMed.
Schultz BG, Patten DK, Berlau DJ. The role of statins in both cognitive impairment and protection against dementia: a tale of two mechanisms. Transl Neurodegener. 2018;7:5. Epub 2018 Feb 27 PubMed.
Villasana LE, Rosenthal RA, Doctrow SR, Pfankuch T, Zuloaga DG, Garfinkel AM, Raber J. Effects of alpha-lipoic acid on associative and spatial memory of sham-irradiated and 56Fe-irradiated C57BL/6J male mice. Pharmacol Biochem Behav. 2013 Jan;103(3):487-93. Epub 2012 Oct 7 PubMed.
View all comments by Jacob RaberWashington University
The finding that this individual with a PS1 E280A mutation did not develop symptoms of cognitive impairment due to AD until her early 70s is very intriguing. The fact that she also has the ApoE3 Christchurch mutation, has very high amyloid deposition, but less tau pathology than one would expect brings up a lot of interesting possibilities.
Since the person has a high amyloid load but less tau pathology, one wonders whether the effect of this ApoE variant is somehow decreasing the ability of ApoE to initiate a microglial-mediated inflammatory response to tau pathology. While this cannot be determined from the information presented in this individual at this point, we have observed in mouse models of tauopathy that ApoE2 results in less neuroinflammation in the brain than with ApoE4 and that ApoE plays a key role in the extent of tau pathology development and tau-mediated neurodegeneration.
The fact that ApoE3 Christchurch does not bind to the LDLR receptor or HSPG very well suggests several mechanisms to further explore regarding how ApoE may be modulating inflammation, tauopathy progression, and neurodegeneration.
View all comments by David HoltzmanTel Aviv University
While much of the genetic study of AD focuses on risk factors and causes of the disease, it is at least as important to identify corresponding protective factors. This study takes this approach by focusing on factors that protect/delay onset of disease by focusing on subjects who are protected from the AD presenilin mutation PSEN1/E280A. This led to the identification of a PSE1/E280A-expressing person who has two copies of the APOE3 Christchurch (R138S) mutation, and did not develop AD three decades after the expected disease onset.
These findings have important AD pathology and ApoE-related therapeutic ramifications. Pathologically, the AD protective effects of R136S are associated with high A plaque deposition but limited tau burden and neurodegeneration, suggesting that tau plays a more important role than Aβ in driving neurodegeneration.
plaque deposition but limited tau burden and neurodegeneration, suggesting that tau plays a more important role than Aβ in driving neurodegeneration.
Therapeutically, these findings suggest that either genetic engineering of ApoE3 by mutating it to R136A and/or corresponding pharmacological approaches could potentially be used to counteract the effects of ApoE4. Such an approach is complimentary to the suggested treatment with the AD protective ApoE2.
However, additional in vitro and in vivo studies are required for unraveling the mechanisms underlying the protective effect of ApoE3 Christchurch, such as why only the homozygous form of this molecule is protective.
View all comments by Daniel MichaelsonMaastricht University; VU University Medical Centre
In general the data confirm previous findings, and highlights that intervening in APOE-related mechanisms is still a useful therapeutic avenue for AD.
The neuropathological confirmation in a subset of 5,000 participants is a strong asset. It is of interest that the APOE e4 allele is not associated with hippocampal sclerosis and vascular brain injury. This may partly explain why the OR of e4 for AD is higher in the pathologically confirmed subgroup, since hippocampal sclerosis and vascular injury may lead to a clinical diagnosis of dementia mimicking clinical AD. The APOE-e4 allele is associated with Lewy body pathology, suggesting that aggregation of amyloid and α-synuclein share a common mechanism.
View all comments by Pieter Jelle VisserLund University
Reiman et al. is a very interesting study, showing that the risk of Alzheimer's disease for APOE-ε2 homozygotes is extremely low even when compared with APOE-ε2 heterozygotes. This result, even not surprising, has an impact on how we should think about the effect of APOE genotype on Alzheimer's disease.
Until now, due to the low frequency of the ε2 allele, few studies had focused their attention on its impact on Alzheimer's disease; and those that have done it have usually studied only the ε2 heterozygote group or ε2 carriers merging heterozygotes and homozygotes. Hence, studies like this, in emphasizing the ε2 homozygosity effect, are most interesting.
One of the strongest points of this paper is the large amount of neuropathological data gathered. This allowed study of the actual APOE effects on Alzheimer's risk without confounding effects of misclassified patients. As shown here, in the non-autopsy cohort, both ε2 and ε2 allele effects are smaller than in the neuropathological cohort. Even if it cannot be confirmed with this data set, this suggests that AD-misclassified cases and preclinical AD cases could account for this difference, reinforcing the significance of autopsy results.
Another interesting point was the differential effect found on tau tangle severity after adjusting by amyloid plaque severity. This result points toward additional mechanisms through which the ε2 allele confers its protective effect beyond decreasing amyloid deposition. We are starting to figure out the different mechanisms through which ε2 is protective. In that regard, we have recently conducted a multicohort study including MRI of an unprecedented number of cognitively unimpaired individuals including APOE-ε2 homozygotes. We analysed the ε2 allele dose-effect on gray-matter volumes, and found bigger volumes in key AD-related areas with increasing number of ε2 alleles. This suggests that ε2 homozygotes may confer additional resilience through distinct brain structural patterns, even before there is pathological protein accumulation.
In summary, this study is a starting point to a more detailed study of the ε2 allele, specifically ε2ε2, regarding the mechanisms behind its protective effects on Alzheimer's disease. As it has been shown on these analyses, even with a limited amount of ε2 homozygotes, the protective effect of this allele is higher than previously reported, and the implications and causes of this should be further studied.
View all comments by Gemma SalvadóInstitute of Neurology, UCL
One should always be aware that this is a single case report. It remains possible there are other reasons for this woman’s protection. However, there are precedents for other APOE variants that change risk of AD (Kamboh et al., 1999; Medway et al., 2014).
A welcome consequence of this finding is that it may help bolster work on the mechanisms of APOE risk.
References:
Kamboh MI, Aston CE, Perez-Tur J, Kokmen E, Ferrell RE, Hardy J, DeKosky ST. A novel mutation in the apolipoprotein E gene (APOE*4 Pittsburgh) is associated with the risk of late-onset Alzheimer's disease. Neurosci Lett. 1999 Mar 26;263(2-3):129-32. PubMed.
Medway CW, Abdul-Hay S, Mims T, Ma L, Bisceglio G, Zou F, Pankratz S, Sando SB, Aasly JO, Barcikowska M, Siuda J, Wszolek ZK, Ross OA, Carrasquillo M, Dickson DW, Graff-Radford N, Petersen RC, Ertekin-Taner N, Morgan K, Bu G, Younkin SG. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer's disease. Mol Neurodegener. 2014 Mar 10;9:11. PubMed.
View all comments by John HardyWashington University in St. Louis
Due to the known nature of their mutations, autosomal-dominant cohorts are powerful models to understand Alzheimer's pathobiology. Any time an ADAD carrier significantly deviates from their expected trajectory, there is the possibility to gain remarkable insight into protective mechanisms.
The work by Arboleda-Velasquez and colleagues describes an ADAD mutation carrier who has maintained relatively preserved cognition beyond the expected age they would decline. This individual has high levels of Aβ measured with PET, but relatively low and atypical presentation of tau pathology.
The association of preserved cognition and altered tau PET presentation with the APOE3ch mutation is fascinating. This result further emphasizes just how strongly tau pathology measured with PET is coupled to cognition. Exploring the impact this unique variant has on APOE functioning and its downstream consequences could open the door to new therapeutic approaches to treat the disease.
View all comments by Brian GordonColumbia University Medical Center
We’d like to pick up on several of the interesting themes raised in these studies as based on our own work in this area.
In a study of 1,557 brains in the NACC v 10 database (which we presented at AAIC, 2019, and which we’ve now submitted) we found that E2 was associated with significantly reduced risk of neuritic plaques, diffuse plaques, and Braak stage. Odds Ratios in E2 v E3/E3 contrasts were around .50 for these three neuropathologies and ORs in E2 v E4 contrasts were around .90, again for the three AD pathologies. In a mediation analysis we found that E2 had direct effects on tau Braak stage, as well as indirect effects via amyloid.
However, there were sharp limits to E2 neuroprotection. When the E2 isoform was in the presence of the e4 isoform, i.e., in the E2/E4 genotype, E2 was not protective (see also Oveisgharan et al., 2018). This genotype “behaved” like other E4 carrier cases in terms of promoting pathology. Second and unexpectedly, E2 demonstrated trends for increases in FTLD related pathologies (including TDP-43 and select tauopathies).
We’ve also conducted microarray transcriptional profiling studies in human postmortem neocortex to identify E2-associated pathways. We found that E2 was linked to increased expression of transcripts in an integrin/extracellular matrix KYOTO pathway (Conejero-Goldberg et al., 2014). The potential importance of this finding (and the HSPG binding related to the APOE mutation) is that matrix neurobiology might play a role in tau propagation as suggested by Lendvai et al., 2012.
References:
Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, Chen S, Davies P, Goldberg TE. APOE2 enhances neuroprotection against Alzheimer's disease through multiple molecular mechanisms. Mol Psychiatry. 2014 Feb 4; PubMed.
Lendvai D, Morawski M, Négyessy L, Gáti G, Jäger C, Baksa G, Glasz T, Attems J, Tanila H, Arendt T, Harkany T, Alpár A. Neurochemical mapping of the human hippocampus reveals perisynaptic matrix around functional synapses in Alzheimer's disease. Acta Neuropathol. 2012 Sep 9; PubMed.
Oveisgharan S, Buchman AS, Yu L, Farfel J, Hachinski V, Gaiteri C, De Jager PL, Schneider JA, Bennett DA. APOE ε2ε4 genotype, incident AD and MCI, cognitive decline, and AD pathology in older adults. Neurology. 2018 Jun 12;90(24):e2127-e2134. Epub 2018 May 11 PubMed.
University of Michigan
This is a very interesting finding, and parallels recent findings from the Kaczorowski lab (in mice). In our recent study, we took the approach to map modifiers that protect/delay onset of cognitive deficits in a genetically diverse population of transgenic mice carrying human familial AD mutations in APP and PSEN1 (Neuner et al., 2019). In this way, we identified genetic variants in the receptor binding domain of mouse Apoe associated with cognitive resilience (Figure 2). Specifically, strains of mice with ADAD mutations that were resilient to cognitive decline despite high amyloid burden had two copies of the C57BL/6J allele at the ApoE locus. Our results are consistent with the present report, that variants in ApoE modify the impact on ADAD mutations on cognition (even absent effects on amyloid).
Perhaps more importantly, since most AD mouse models are maintained on a pure C57BL/6J background, we speculate that preclinical studies that failed to translate to human clinical trials may be due to studies being run in mice that harbor protective variants at ApoE that modify the impact of ADAD mutations.
References:
Neuner SM, Heuer SE, Huentelman MJ, O'Connell KM, Kaczorowski CC. Harnessing Genetic Complexity to Enhance Translatability of Alzheimer's Disease Mouse Models: A Path toward Precision Medicine. Neuron. 2019 Feb 6;101(3):399-411.e5. Epub 2018 Dec 27 PubMed.
Make a Comment
To make a comment you must login or register.