Sticky Matrix Proteins Lead to Amyloid Accumulation, Slow Clearance
Quick Links
In late-onset Alzheimer’s disease, Aβ clears slowly from the brain, leading to the buildup of amyloid plaque. Sticky extracellular proteins known as heparan sulfate proteoglycans (HSPGs) may be partially responsible for this, according to a new paper in the April 1 Science Translational Medicine. Researchers led by Guojun Bu at the Mayo Clinic in Jacksonville, Florida, repressed HSPG production in neurons of mouse models of AD. Without the sticky proteins, amyloid plaque levels plummeted, in line with previous in vitro data suggesting that HSPGs enhance Aβ aggregation. Moreover, soluble Aβ disappeared from interstitial fluid more rapidly in the mice that lacked proteoglycans. Meanwhile, more amyloid accumulated in blood vessels, which still contained HSPGs. Overall, the evidence suggests that proteoglycans trap Aβ, encouraging it to clump and persist in the mouse brain, the authors propose. In Alzheimer’s brains as well, HSPGs crowd amyloid plaques, hinting that the same mechanism could be at work.
Other researchers expressed enthusiasm for the work. “This is an elegant study that demonstrates the importance of HSPGs in Alzheimer’s,” Marcel Verbeek at Radboud University Nijmegen Medical Centre, The Netherlands, told Alzforum. He noted that the study produced novel data on in-vivo clearance, as well as confirming, in a mouse model, previous hypotheses about HSPGs’ role in plaque formation. Lars Nilsson at the University of Oslo, Norway, wrote to Alzforum, “This is an excellent study, and the results are convincing, as large animal cohorts and multiple techniques were used.” Nilsson added that the mouse model developed here will enable further studies of HSPG biology.
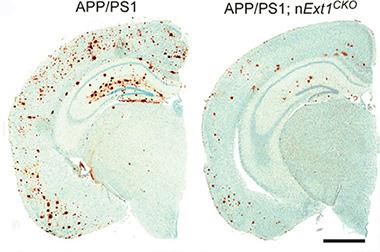
Unsticking Plaques.
In AD mouse brains lacking sticky HSPGs (right), up to two-thirds less amyloid plaque (brown) accumulates than in controls (left). [Courtesy of Liu et al., Science Translational Medicine/AAAS.]
HSPGs consist of sulfated sugar chains attached to a protein backbone. The molecules are named for their protein cores and come in five classes, comprising membrane-bound syndecans and glypicans, as well as secreted agrin, perlecan, and type XVIII collagen. These large gummy molecules carpet cell surfaces and flood the extracellular space, and participate in numerous crucial processes such as molecular adhesion, cell signaling, and development.
Previous data linked these proteoglycans to AD, but it was unclear whether they helped or hindered. Several cellular studies found that HSPGs bound Aβ and promoted its aggregation, suggesting the latter (see Cotman et al., 2000; Watanabe et al., 2004; Cheng et al., 2013). However, Bu’s group and others reported that membrane-bound HSPGs help internalize Aβ, which may allow the peptide to travel to lysosomes for degradation (see Sandwall et al., 2010; Kanekiyo et al., 2011). Recently, researchers led by Nilsson and Jin-Ping Li at Uppsala University, Sweden, overexpressed heparanase, an enzyme that chops up HSPGs, in the brains of mice that carried mutant Swedish APP. Amyloid load dropped roughly in half, hinting that the overall effect of HSPGs in AD might be detrimental (see Jendresen et al., 2015).
Bu wanted to investigate this question using a genetic model that would allow for more spatial and temporal control of HSPG expression. First author Chia-Chen Liu generated mice that lacked Ext1 in adult neurons. This enzyme lengthens heparan sulfate chains. At one year of age, these animals had about half as much heparan sulfate in the cortex and hippocampus as wild-types did, but otherwise they appeared normal. Liu and colleagues then crossed these mice with APPPS1 animals. The offspring accumulated about one-third as much plaque by one year of age as did APPPS1 littermates (see image above). Likewise, levels of insoluble Aβ40 and Aβ42 only reached one-third to one-half those of APPPS1 littermates. Inflammation, which typically accompanies plaques, was also lower. The authors saw one-third to one-half fewer activated astrocytes and microglia, and about half as much of several pro-inflammatory cytokines in the brain.
How might HSPGs promote plaque formation? In part, they seemed to encourage the peptide to clump. Without HSPGs, soluble oligomeric forms of Aβ dropped by more than half in hippocampus and cortex, as seen with an antibody specific for this conformation (see Feb 2009 news). Meanwhile, total soluble Aβ did not change, indicating no effect on production. HSPGs may capture Aβ and increase its local concentration, triggering oligomerization and aggregation, Bu suggested to Alzforum.
Other mechanisms may be at work as well. The authors collaborated with David Holtzman and John Cirrito at Washington University in St. Louis to measure interstitial fluid levels of Aβ via microdialysis in three- to four-month-old APPPS1 and APPS1/Ext1-negative mice, long before plaques form. The authors first infused a γ-secretase inhibitor to block production of the peptide, then recorded how quickly Aβ40 and Aβ42 levels fell. In the conditional Ext1 knockouts, Aβ vanished more rapidly, with a half-life about two-thirds of that in APPPS1 littermates. Curiously, amyloid deposits in blood vessels rose by two-thirds in Ext1-negative mice, suggesting that without the sticky HSPGs around, more of the peptide left the brain by this route and was captured by vascular HSPGs or other vascular structures. In addition, HSPGs may block cleavage sites on Aβ, shielding the peptide from proteases and allowing it to linger in APPPS1 brain, Bu suggested. A previous in-vitro study reported that HSPGs protected fibrillar amyloid from degradation (see Gupta-Bansal, 1995).
How closely do the mouse findings match what happens in human brain? While that is still unknown, Bu and colleagues compared postmortem HSPG levels in the temporal cortices of 20 AD cases to those in 20 age-matched controls. The AD brains contained significantly higher levels of several HSPGs, including syndecan-3, syndecan-4, glypican-3, agrin, and perlecan, particularly in insoluble brain fractions. The findings jibe with previous studies that reported high HSPG levels in amyloid plaques (see Snow et al., 1987; van Horssen et al., 2002; Zhang et al., 2014). “The data give us more confidence that targeting this pathway will be relevant in humans,” Bu said.
However, Verbeek pointed out that while almost all amyloid plaques in AD brain contain HSPGs, only a third or less of the plaques in APPPS1 mice do. This suggests the mouse plaques have a somewhat different, perhaps looser, composition. The difference could mean that plaques in human brain will be harder to bust up, Verbeek suggested. He also noted that any potential therapy would have to target the Aβ-HSPG interaction very specifically, so as not to interfere with the normal functions of these molecules. Bu plans to look for compounds that bind Aβ and keep it from attaching to proteoglycans. Such compounds might be mimics of heparan sulfate. A previous study from Italy reported that the small heparin enoxaparin suppressed plaque formation in AD mice, supporting the idea that this strategy could work (see May 2004 news).
Bu also wants to use his mouse model to investigate other effects of HSPGs. Studies by Marc Diamond at the University of Texas Southwestern Medical Center in Dallas, as well as others, indicate that cell-surface HSPGs internalize pathological tau and α-synuclein, allowing these proteins to propagate from neuron to neuron (see Aug 2013 conference news; Apr 2015 news). Bu will test this by expressing toxic tau and α-synuclein in his conditional knockouts, either by crossing them with appropriate mice, or injecting lysate from brains with tauopathies or synucleinopathies. He will measure how quickly the proteins spread compared to the rate in control mice. —Madolyn Bowman Rogers
References
Research Models Citations
News Citations
- Research Brief: New Methods for Aβ Detection, Production
- Anticoagulants for Alzheimer’s?
- Tales of Traveling Tau: Is Transfer Between Neurons Normal?
- Tau Triple Threat: Do Trimers Make Bad Seeds?
Paper Citations
- Cotman SL, Halfter W, Cole GJ. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Mol Cell Neurosci. 2000 Feb;15(2):183-98. PubMed.
- Watanabe N, Araki W, Chui DH, Makifuchi T, Ihara Y, Tabira T. Glypican-1 as an Abeta binding HSPG in the human brain: its localization in DIG domains and possible roles in the pathogenesis of Alzheimer's disease. FASEB J. 2004 Jun;18(9):1013-5. PubMed.
- Cheng F, Ruscher K, Fransson LÅ, Mani K. Non-toxic amyloid beta formed in the presence of glypican-1 or its deaminatively generated heparan sulfate degradation products. Glycobiology. 2013 Dec;23(12):1510-9. PubMed.
- Sandwall E, O'Callaghan P, Zhang X, Lindahl U, Lannfelt L, Li JP. Heparan sulfate mediates amyloid-beta internalization and cytotoxicity. Glycobiology. 2010 May;20(5):533-41. PubMed.
- Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci. 2011 Feb 2;31(5):1644-51. PubMed.
- Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LN, Li JP. Overexpression of heparanase lowers the amyloid burden in amyloid-β precursor protein transgenic mice. J Biol Chem. 2015 Feb 20;290(8):5053-64. Epub 2014 Dec 29 PubMed.
- Gupta-Bansal R, Frederickson RC, Brunden KR. Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. J Biol Chem. 1995 Aug 4;270(31):18666-71. PubMed.
- Snow AD, Willmer J, Kisilevsky R. Sulfated glycosaminoglycans: a common constituent of all amyloids?. Lab Invest. 1987 Jan;56(1):120-3. PubMed.
- van Horssen J, Kleinnijenhuis J, Maass CN, Rensink AA, Otte-Höller I, David G, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM. Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol Aging. 2002 Jul-Aug;23(4):537-45. PubMed.
- Zhang GL, Zhang X, Wang XM, Li JP. Towards understanding the roles of heparan sulfate proteoglycans in Alzheimer's disease. Biomed Res Int. 2014;2014:516028. Epub 2014 Jul 23 PubMed.
Further Reading
Primary Papers
- Liu CC, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G. Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer's disease. Sci Transl Med. 2016 Mar 30;8(332):332ra44. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Oslo
University of Oslo
This is an excellent study showing that reduced neuronal HSPG biosynthesis in APP/PS1 transgenic mice results in less parenchymal amyloid deposition, but more cerebral amyloid angiopathy (CAA). A conditional knockout of the glycosyltransferase EXT1 in postmitotic neurons in the forebrains of APP/PS1 mice was generated. For the future, a more complete characterization of this bitransgenic as well as our Hpa x APP model might give further insights into the molecular mechanisms which are involved. Interestingly, the findings now reported complement and extend our cross-breeding study, which was done as a joint collaboration with Professor Jin-Ping Li and colleagues at Uppsala University in Sweden. We found decreased plaque burden in the cerebral cortex, as well as a reduced CAA burden when the HSPG-degrading enzyme, heparanase, was overexpressed in a less neuronal-selective manner in APPswe mice (Jendresen et al., 2015). …More
In the animal model now introduced by Liu et al., HSPG in cerebral vasculature was presumably not much affected by the neuron-specific EXT1 knockout as compared with our study, in which heparanase was more ubiquitously overexpressed. In the present report, elegant microdialysis studies were also used to show that the half-life of both Aβ40 and Aβ42 was reduced in the bitransgenic mice. Together the findings point to, as suggested, the importance of the perivascular drainage pathway for Aβ clearance with HSPG affecting Aβ turnover both in the parenchyma and the vasculature and perhaps also across the blood brain barrier (Bakker et al., 2016). The authors showed reduced Aβ oligomer levels in the mouse crosses, arguing that HSPG also increases the aggregation of Aβ. Effects on both Aβ clearance and Aβ aggregation are not surprising. It has also been reported for other amyloid-associated components, e.g., Apolipoprotein E (Nilsson et al., 2004; Hashimoto et al., 2012; Castellano et al., 2011).
These findings suggest multiple pathogenic mechanisms that could be targeted for therapeutics. HSPG biology has been strangely disregarded, in spite of quite strong indirect evidence that it might modify Alzheimer’s disease pathogenesis. Presumably, the neglect relates to the failed clinical trial with tramiprosate, a case of a poorly evaluated drug candidate that entered advanced clinical trials (Karran and Hardy, 2014). Like the old saying goes, “Don’t throw the baby out with the bathwater!”
References:
Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LN, Li JP. Overexpression of heparanase lowers the amyloid burden in amyloid-β precursor protein transgenic mice. J Biol Chem. 2015 Feb 20;290(8):5053-64. Epub 2014 Dec 29 PubMed.
Bakker EN, Bacskai BJ, Arbel-Ornath M, Aldea R, Bedussi B, Morris AW, Weller RO, Carare RO. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell Mol Neurobiol. 2016 Mar;36(2):181-94. Epub 2016 Mar 18 PubMed.
Nilsson LN, Arendash GW, Leighty RE, Costa DA, Low MA, Garcia MF, Cracciolo JR, Rojiani A, Wu X, Bales KR, Paul SM, Potter H. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 2004 Oct;25(9):1153-67. PubMed.
Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, Mitani A, Joyner D, Thyssen DH, Bacskai BJ, Frosch MP, Spires-Jones TL, Finn MB, Holtzman DM, Hyman BT. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J Neurosci. 2012 Oct 24;32(43):15181-92. PubMed.
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011 Jun 29;3(89):89ra57. PubMed.
Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014 Aug;76(2):185-205. Epub 2014 Jul 2 PubMed.
UT Southwestern, Dallas
University of California, San Francisco
University of Texas, Southwestern Medical Center
This is an important study for the field of neurodegeneration as it underlines the relevance of HSPGs in sporadic Alzheimer’s disease (AD). Liu and colleagues clearly demonstrate that the conditional knockout of the Ext1 enzyme in the HSPG synthesis pathway significantly reduces the cortical and hippocampal Aβ plaque burden as well as Aβ in soluble fractions. The paper provides solid evidence for several mechanisms underlying the decrease of Aβ plaque deposition in the knockout mice: The reduction of cerebral heparin sulfate (HS) enhanced the clearance of soluble Aβ from the interstitial fluid (ISF), without affecting Aβ processing or expression. Moreover, consistent with previous findings in the literature (Castillo et al., 1999), the decrease of neuronal HS resulted in a reduction of Aβ aggregation.…More
The authors used microdialysis to assess Aβ metabolism in the ISF. Several possible clearance mechanisms are discussed, including enhanced perivascular drainage, increased degradation by ISF proteases, and decreased cellular Aβ uptake and subsequent lysosomal aggregation and seed formation. Further investigations will show if one or several of the discussed pathways enhanced Aβ clearance in this study. It will be critical to understand in detail the underlying molecular pathways for future therapeutic approaches.
It is remarkable that only some classes of HSPG are increased in AD brains while for other classes no differences could be found between normal and AD brain tissue. This indicates that a certain HSPG pattern within the brain is connected to AD pathology, suggesting that further investigations of potential mechanisms should focus on these HSPG classes in particular. Moreover, AD pathology is known to follow a stereotypical neuroanatomical pattern (Thal et al., 2002) and it is tempting to hypothesize that specific HSPG expression patterns in the brain might contribute to this process. The question about the chronological order of events, i.e., if the AD-related Aβ burden triggers increased HSPG expression or vice versa, remains open and would equally be interesting to address in future studies.
We strongly agree with the authors that HSPG-mediated uptake and transcellular propagation is a relevant mechanism to consider in HSPG-related AD pathology. As previously demonstrated by several groups, HSPG not only mediates cellular Aβ uptake (Kanekiyo et al., 2012) but also the uptake of other amyloidogenic proteins involved in neurodegenerative disorders such as tau, α-synuclein, and prion protein (Holmes et al., 2013; Horonchik et al., 2005). Thus, repeating the current study in other mouse models of neurodegeneration, such as tauopathies and synucleinopathies, will be very informative. The conditional Ext1 knockout seems to be an excellent model for this purpose, since it avoids the absence of HSPG knockout in the early embryological stages, which we would predict to be toxic since HSPG plays a major role in embryological development (Poulain and Yost, 2015). However, because “prion-like” amyloids could potentially play a physiological role in transcellular information trafficking (Sanders et al., 2016), side effects from a complete Ext1 knockout in tau or α-synuclein models need to be monitored carefully.
In summary, the study highlights the relevance of HSPG in AD pathology and underlines the need for future studies to dissect the HSPG pathway for the purpose of finding innovative therapeutic approaches for AD and other neurodegenerative disorders.
References:
Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem. 1999 Apr;72(4):1681-7. PubMed.
Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002 Jun 25;58(12):1791-800. PubMed.
Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer's amyloid-β. J Neurosci. 2012 Nov 14;32(46):16458-65. PubMed.
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013 Aug 13;110(33):E3138-47. Epub 2013 Jul 29 PubMed.
Horonchik L, Tzaban S, Ben-Zaken O, Yedidia Y, Rouvinski A, Papy-Garcia D, Barritault D, Vlodavsky I, Taraboulos A. Heparan sulfate is a cellular receptor for purified infectious prions. J Biol Chem. 2005 Apr 29;280(17):17062-7. Epub 2005 Jan 24 PubMed.
Poulain FE, Yost HJ. Heparan sulfate proteoglycans: a sugar code for vertebrate development?. Development. 2015 Oct 15;142(20):3456-67. PubMed.
Sanders DW, Kaufman SK, Holmes BB, Diamond MI. Prions and Protein Assemblies that Convey Biological Information in Health and Disease. Neuron. 2016 Feb 3;89(3):433-48. PubMed.
Make a Comment
To make a comment you must login or register.