What BACE Hits: New Substrates Create New Headaches
Quick Links
While the Alzheimer’s field absorbs full Phase 3 data from elenbecestat, the last of the terminated batch of BACE inhibitors (see Part 11 of this series), scientists are also digging deeper into the myriad biological effects of BACE1 itself. They are hoping to unearth clues as to how BACE inhibitors affect the body, and which substrates might be responsible for the cognitive and other adverse effects seen in the trials conducted thus far.
- Meet Gp130, BACE substrate that may affect synapses via IL-6 signaling.
- CSF Gp130 and SEZ6 could serve as biomarkers of BACE inhibition.
- BACE1 protects the brain by degrading Aβ42; will this complicate dosing?
At the 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases in Munich implicated the cytokine receptor glycoprotein 130 (Gp130) as a new BACE1 substrate that might mediate the synaptic effects seen after inhibition. Meanwhile, Gerhard Multhaup of McGill University, Montreal, made a case for a protective effect of BACE1 in degrading Aβ42 to the harmless intermediate Aβ34. This effect might complicate inhibitor dosing, since in Multhaup’s studies low doses hampered BACE1’s protective effects more than its harmful ones. Together, the data underscore the complexity of BACE inhibition as a therapeutic strategy, suggesting researchers might have to thread a needle to achieve the desired effect.
Another Substrate Muddies the Water
Lichtenthaler teamed up with Matthew Kennedy at Merck to identify BACE1 substrates in CSF. Merck provided CSF data taken from three rhesus monkeys that had received a single 100 mg/kg dose of the experimental BACE inhibitor MBI-4 24 hours earlier. An analysis of more than 600 proteins found a handful that dropped with BACE inhibition. Some of these were known BACE1 substrates, such as APP and CHL1. Most changes were small, but two proteins stood out. CSF concentrations fell by more than half for the known BACE1 substrate SEZ6—and a new one, Gp130.

Cytokine Signaling. Gp130 (yellow ovals) blanket an IL-6 complex (blue and green) to activate transcription through a JAK-STAT pathway. New evidence suggests BACE1 can clip Gp130, preventing IL-6 signaling. [Courtesy of Gottschalk et al., Frontiers in Immunology.]
Lichtenthaler confirmed these findings in human CSF samples from Merck’s verubecestat trials. Thirty hours after participants took that drug, soluble SEZ6 had plummeted by three-fourths, and Gp130 by two-thirds. Lichtenthaler noted that this rapid response to verubecestat is likely due to both proteins’ short half-life in CSF, meaning they fluctuate rapidly in response to processing changes. He believes the proteins might make useful target engagement biomarkers to track BACE1 inhibition.
Gp130 normally binds to membranes, and its soluble version was previously believed to arise through alternative splicing, Lichtenthaler said. The new data instead suggest that it is a product of BACE cleavage. Lichtenthaler confirmed this in cell-free assays and primary neuron cultures, where suppressing BACE1 lowered soluble Gp130. He noted that some previous cell culture studies reported an increase in sGp130 upon BACE1 overexpression, strengthening the case for this relationship (Hemming et al., 2009; Stützer et al., 2013).
In its membrane-bound form, Gp130 participates in cytokine signaling. Extracellular IL6 forms a complex with the soluble IL6 receptor, and this complex binds a Gp130 dimer, activating intracellular signaling and gene transcription through the JAK-STAT pathway (see image above). Lichtenthaler noted that Gp130 is expressed in neurons (Watanabe et al., 1996). Its signaling is believed to be pro-inflammatory, with some evidence that it affects synaptic transmission.
For example, chronic IL-6 signaling amped up excitatory neuronal transmission in the mouse cerebellum, but suppressed excitation in cerebral cortex (Nelson et al., 1999; D’Arcangelo et al., 2000). Another study concluded that IL-6 shifts the balance between synaptic excitation and inhibition in rat cortex to favor the former (Garcia-Oscos et al., 2012). IL-6 signaling also has been linked to autism (Wei et al., 2016).
BACE1 cleavage of Gp130 could lower this signaling, while BACE1 inhibition would increase it. Supporting this, inhibiting BACE1 in cultured cells doubled the amount of activated STAT, Lichtenthaler found. It is not yet clear if this causes any harmful effects. Some previous research links alternative APP processing with heightened IL-6 signaling (Dos Santos Guilherme et al., 2019). IL-6 has been found to suppress neurogenesis, hinting at negative effects on cognition (Dec 2003 news).
In future studies, Lichtenthaler will examine whether BACE1 activity affects Gp130 signaling in vivo. He will also analyze whether the amount of sGp130, or any other BACE1 substrate, in the CSF of people taking verubecestat correlates with the cognitive deficits that develop on the drug. Such a relationship could provide clues to the mechanism behind this worsening.
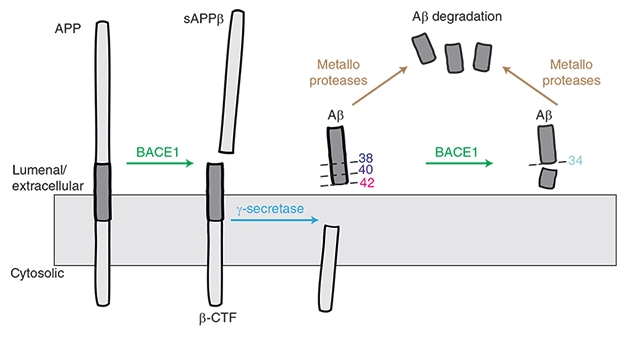
BACE1 Makes (and Breaks?) Aβ. BACE1 starts the cascade that generates Aβ species including 40 and Aβ42, but it may also help to degrade these peptides into harmless Aβ34. [Courtesy of Liebsch et al., Nature Communications.]
Too Much BACE1 Inhibition, or Too Little?
Lest the solution be simply dialing down BACE1 inhibitor levels, Multhaup introduced another layer of complication. Previously, while working in Christian Haass’ lab at Ludwig-Maximilians University in Munich, Multhaup, who now chairs the department of pharmacology and therapeutics at McGill, found that BACE1 not only snips APP to produce the β-CTF fragment that is later cleaved into Aβ40 and Aβ42, but also chops off the end of these peptides to generate Aβ34 (Fluhrer et al., 2003; see image above).
Aβ34 is not toxic. It does not aggregate. Biochemistry and cell-based experiments flagged it as a marker for Aβ degradation and clearance, Multhaup noted (Hernandez-Guillamon et al., 2015). Human data from Randall Bateman and colleagues at Washington University in St. Louis confirmed that higher amounts of CSF Aβ34 correlate with increased turnover of longer Aβ species, as measured by stable isotope labeling kinetics (SILK), in people who have plaque buildup (Patterson et al., 2015; Liebsch et al., 2019).
How would changes in BACE1 affect Aβ34 production? Multhaup found that BACE1 knockout mice produce almost none of the peptide. Heterozygote mice, with half the normal amount of BACE1, have half the normal amount of Aβ34. In fact, Aβ34 tracked BACE1 levels more closely than did Aβ40 or Aβ42. In cultured cells overexpressing β-CTF, rising amounts of BACE1 caused Aβ40 and Aβ42 to drop, while Aβ34 rose. Conversely, inhibiting BACE1 in cultured cells made Aβ34 fall, and Aβ40 and Aβ42 rise slightly. Multhaup believes that this relationship helps explain a paradox in the literature, where overexpressing BACE1 in mice or human cell culture lowered Aβ40, rather than raising it as expected (Lee et al., 2005; Chiocco et al., 2004; Scholz et al., 2018).
What about in people? Multhaup found that Aβ34 tracked BACE1 activity in human brain as well, with both proteins being twice as high in AD as in control brain samples. The amount of Aβ34 varies with disease stage, however. Its levels peak at Braak stages I to III and then fall, perhaps hinting at a failure of clearance mechanisms later in disease. Multhaup had previously reported a similar pattern in CSF, where Aβ34 is high in people with mild cognitive impairment, and a high ratio of Aβ34/Aβ42 serves as a biomarker of preclinical AD (Liebsch et al., 2019).
Does BACE1 inhibition slow Aβ clearance in people? Possibly, Multhaup believes, as there have been independent reports of a drop in CSF Aβ34 after BACE1 inhibition (Mattsson et al., 2012).
To Multhaup’s mind, this hints at trouble for developing a safe and effective BACE1 inhibitor therapy. Multhaup’s group tested two such inhibitors in the same cultured cell line that overexpress β-CTF, and found that Aβ40 tended to rise at low inhibitor concentrations. This suggests that, in this cell line, inhibitors impair BACE’s amyloidolytic activity at lower concentrations than they impair BACE’s amyloidogenic activity, Multhaup said. The exact range of concentrations where this happened depended on the particular inhibitor’s affinity for BACE1.
These findings could complicate attempts to titrate the dosing of BACE1 inhibitors at a low level to avoid deleterious side effects, Multhaup noted. Too low, and the inhibitors might interfere with the clearance of toxic Aβ peptides. Too high—and we have already seen what happens. “Our data imply a partial BACE inhibition might not provide therapeutic benefits,” Multhaup said at AD/PD.
Others are not convinced. Vassar thought this was unlikely to be a problem for inhibitor therapy. “In my view, the main effect of a BACE1 inhibitor is to reduce the production of Aβ in the first place, which would trump any negative effect stemming from secondary inhibition of Aβ42 degradation to Aβ34,” he told Alzforum. Lichtenthaler agreed, pointing out that in clinical trials, low doses of the BACE inhibitor umibecestat did not raise Aβ40 or Aβ42 in CSF (Neumann et al., 2018). “I cannot rule out the possibility that Gerd [Multhaup’s] effect is seen upon single dosing or a short time after dosing, but it is not an issue upon chronic dosing,” he wrote to Alzforum.—Madolyn Bowman Rogers
References
News Citations
- Drop of Hope? No Cognitive Worsening on BACE Inhibitor
- Antiinflammatory Drugs Protect Hippocampal Neurogenesis
Therapeutics Citations
Paper Citations
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS One. 2009;4(12):e8477. PubMed.
- Stützer I, Selevsek N, Esterházy D, Schmidt A, Aebersold R, Stoffel M. Systematic Proteomic Analysis Identifies β-Site Amyloid Precursor Protein Cleaving Enzyme 2 and 1 (BACE2 and BACE1) Substrates in Pancreatic β-Cells. J Biol Chem. 2013 Apr 12;288(15):10536-47. PubMed.
- Watanabe D, Yoshimura R, Khalil M, Yoshida K, Kishimoto T, Taga T, Kiyama H. Characteristic localization of gp130 (the signal-transducing receptor component used in common for IL-6/IL-11/CNTF/LIF/OSM) in the rat brain. Eur J Neurosci. 1996 Aug;8(8):1630-40. PubMed.
- Nelson TE, Campbell IL, Gruol DL. Altered physiology of Purkinje neurons in cerebellar slices from transgenic mice with chronic central nervous system expression of interleukin-6. Neuroscience. 1999 Mar;89(1):127-36. PubMed.
- D'Arcangelo G, Tancredi V, Onofri F, D'Antuono M, Giovedì S, Benfenati F. Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur J Neurosci. 2000 Apr;12(4):1241-52. PubMed.
- Garcia-Oscos F, Salgado H, Hall S, Thomas F, Farmer GE, Bermeo J, Galindo LC, Ramirez RD, D'Mello S, Rose-John S, Atzori M. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol Psychiatry. 2012 Apr 1;71(7):574-82. Epub 2011 Dec 22 PubMed.
- Wei H, Ma Y, Liu J, Ding C, Jin G, Wang Y, Hu F, Yu L. Inhibition of IL-6 trans-signaling in the brain increases sociability in the BTBR mouse model of autism. Biochim Biophys Acta. 2016 Oct;1862(10):1918-25. Epub 2016 Jul 25 PubMed.
- Dos Santos Guilherme M, Stoye NM, Rose-John S, Garbers C, Fellgiebel A, Endres K. The Synthetic Retinoid Acitretin Increases IL-6 in the Central Nervous System of Alzheimer Disease Model Mice and Human Patients. Front Aging Neurosci. 2019;11:182. Epub 2019 Jul 23 PubMed.
- Fluhrer R, Multhaup G, Schlicksupp A, Okochi M, Takeda M, Lammich S, Willem M, Westmeyer G, Bode W, Walter J, Haass C. Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation. J Biol Chem. 2003 Feb 21;278(8):5531-8. PubMed.
- Hernandez-Guillamon M, Mawhirt S, Blais S, Montaner J, Neubert TA, Rostagno A, Ghiso J. Sequential Amyloid-β Degradation by the Matrix Metalloproteases MMP-2 and MMP-9. J Biol Chem. 2015 Jun 12;290(24):15078-91. Epub 2015 Apr 20 PubMed.
- Patterson BW, Elbert DL, Mawuenyega KG, Kasten T, Ovod V, Ma S, Xiong C, Chott R, Yarasheski K, Sigurdson W, Zhang L, Goate A, Benzinger T, Morris JC, Holtzman D, Bateman RJ. Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann Neurol. 2015 Sep;78(3):439-53. Epub 2015 Jul 20 PubMed.
- Liebsch F, Kulic L, Teunissen C, Shobo A, Ulku I, Engelschalt V, Hancock MA, van der Flier WM, Kunach P, Rosa-Neto P, Scheltens P, Poirier J, Saftig P, Bateman RJ, Breitner J, Hock C, Multhaup G. Aβ34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer's disease progression. Nat Commun. 2019 May 20;10(1):2240. PubMed.
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005 Jan 17;168(2):291-302. PubMed.
- Chiocco MJ, Kulnane LS, Younkin L, Younkin S, Evin G, Lamb BT. Altered amyloid-beta metabolism and deposition in genomic-based beta-secretase transgenic mice. J Biol Chem. 2004 Dec 10;279(50):52535-42. PubMed.
- Scholz D, Chernyshova Y, Ückert AK, Leist M. Reduced Aβ secretion by human neurons under conditions of strongly increased BACE activity. J Neurochem. 2018 Oct;147(2):256-274. Epub 2018 Sep 26 PubMed.
- Mattsson N, Rajendran L, Zetterberg H, Gustavsson M, Andreasson U, Olsson M, Brinkmalm G, Lundkvist J, Jacobson LH, Perrot L, Neumann U, Borghys H, Mercken M, Dhuyvetter D, Jeppsson F, Blennow K, Portelius E. BACE1 inhibition induces a specific cerebrospinal fluid β-amyloid pattern that identifies drug effects in the central nervous system. PLoS One. 2012;7(2):e31084. PubMed.
- Neumann U, Ufer M, Jacobson LH, Rouzade-Dominguez ML, Huledal G, Kolly C, Lüönd RM, Machauer R, Veenstra SJ, Hurth K, Rueeger H, Tintelnot-Blomley M, Staufenbiel M, Shimshek DR, Perrot L, Frieauff W, Dubost V, Schiller H, Vogg B, Beltz K, Avrameas A, Kretz S, Pezous N, Rondeau JM, Beckmann N, Hartmann A, Vormfelde S, David OJ, Galli B, Ramos R, Graf A, Lopez Lopez C. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer's disease. EMBO Mol Med. 2018 Nov;10(11) PubMed.
Further Reading
News
- Paper Alert: Trafficking Variants Cause BACE Buildup in Axons
- Raw Deal: Lose Ataxin, Gain BACE
- BACE and γ-Secretase Form Mega-Complex that Processes APP
- In Conditional BACE1 Knockouts, Hippocampal Axons Compromised
- BACE1 Conditional Knockouts Model Adult BACE Inhibition
- Does BACE Drive Neurites into Dystrophy, Shorting Circuits?
Annotate
To make an annotation you must Login or Register.
Comments
Northwestern University Feinberg School of Medicine
Stefan Lichtenthaler gave an outstanding talk at AD/PD 2021. He presented very compelling mass spectrometry data that GP130 is a robust BACE1 substrate. It will be interesting to see what role this new BACE1 substrate may have regarding the cognitive worsening observed in the BACE1 inhibitor drug trials.
Make a Comment
To make a comment you must login or register.