Raw Deal: Lose Ataxin, Gain BACE
Quick Links
CAG trinucleotide repeat expansions in the ataxin-1 gene cause spinocerebellar ataxia type 1, a fatal neurodegenerative disease. Curiously, variants of this same gene, sans trinucleotide repeat expansions, associate with risk for Alzheimer’s. Researchers do not know why. Now, scientists led by Jaehong Suh and Rudolph Tanzi, Massachusetts General Hospital and Harvard Medical School, Boston, and Huda Zoghbi, Baylor College of Medicine, Houston, report in the August 22 Cell that ataxin-1 normally suppresses Aβ production, at least in mice. Knocking out the gene accelerated BACE1 transcription in AD-vulnerable regions of the brain and spurred amyloidogenic processing of the Aβ precursor protein (APP). In APPPS1 mice, loss of ataxin-1 exacerbated Aβ deposition and impaired both hippocampal neurogenesis and olfactory axonal guidance. The results hint that loss-of-function mutations in ataxin-1 raise the risk of AD through an increase in BACE1 expression, though that has yet to be proven.
- Deleting ataxin-1 stimulates plaque growth in mice.
- Loss of ataxin-1 unleashes EVT transcription factors, BACE promoter.
- Ataxin-1 loss of function may partly explain genetic risk for AD.
“This is an impressive story demonstrating convincingly that ataxin-1 suppresses BACE1 transcription,” wrote Stefan Lichtenthaler, German Center for Neurodegenerative Diseases, Munich, who was not involved in the study. “I am amazed by the underlying molecular mechanism, and find it tempting to speculate that additional mechanisms controlling BACE1 transcription exist and remain to be identified.”
Through genome-wide association studies, Tanzi and others previously linked the ataxin-1 gene, ATXN1, to AD risk (Bertram et al., 2008; Bettens et al., 2010). Copy number variations are involved, though the exact genetic variants are unknown (Swaminathan et al., 2011; 2012).
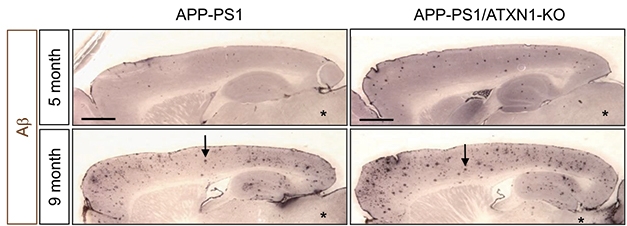
Piling On. APP-PS1 ATXN1 knockout mice (right) deposit more plaques at five and nine months than APP-PS1 mice (left). [Image courtesy of Suh et al., 2019. Cell.]
Suh and colleagues wondered if a loss-of-function mutation, rather than CAG repeat as found in spinocerebellar ataxia, could predispose people to AD. A previous paper from Tanzi’s lab reported that knockdown of ataxin-1 expression in cell culture increased the secretion of sAPPbeta and Aβ, but how this occurred was unclear (Zhang et al., 2010). For answers, Suh turned to ataxin-1 knockout mice (Matilla et al., 1998). At three months, they had 50 percent more BACE1 protein and soluble APPβ (sAPPβ) in the brain than controls. In contrast, sAPPα, cleaved by ADAM10, was down by about 40 percent, likely due to competitive cleavage by BACE1.
Where was BACE1 upregulated? Immunostaining revealed more of the secretase in AD-vulnerable brain regions, including the hippocampus, cortex, and olfactory bulb, but not in the brainstem or cerebellum. Given that BACE1 and ataxin-1 appeared in their normal milieu—presynaptic terminals and neuronal nuclei, respectively—a direct interaction between the two proteins seemed unlikely. So what linked them?
Ataxin-1 interacts with transcription factors, hence the researchers wondered if its absence increased transcription of BACE1. They found more newly synthesized BACE1 mRNA in ataxin knockouts than controls. Next they identified several transcription factors that bind the BACE1 promoter and activate BACE transcription. Expression of three of these, ETV1, ETV4, and ETV5, is normally tempered by a complex of ataxin-1 and a transcriptional repressor called Capicua (CIC); alas, in ataxin-1 knockouts, up to 70 percent more ETV4 and ETV5 appeared in the cortex. Overexpressing either of these two in HEK293 cells activated the BACE1 promoter. Together, the data suggest that loss of ataxin-1 releases the brakes on expression of ETV4/5, which then power BACE1 transcription.
When Suh and colleagues crossed ataxin-1 knockout mice with a BACE1+/- heterozygote that made half the normal amount of BACE1 protein, amyloidogenic processing of APP returned to near-normal levels in the offspring. Hippocampal neurogenesis and axonal guidance in the olfactory bulb, which were off-kilter in the ataxin KOs, were also rescued in BACE1 heterozygotes.
To see if loss of ataxin-1 would exacerbate amyloid plaque pathology, the authors crossed ataxin-1 knockouts with APPPS1 mice. As in the ataxin-1 knockouts, BACE1 expression rose, sAPPα levels dropped, and Aβ plaque load and gliosis doubled (see image above).
To the author’s mind, the findings suggest that BACE1 mediates the association between ATXN1 and AD. The paper reports no data directly connecting AD risk variants to transcriptional regulation of BACE. “If ataxin-1 levels go down in the brain for any reason, this can increase BACE1 transcription in the brain, particularly in AD-vulnerable regions,” Suh said. Despite the failures of clinical trials testing BACE1 inhibitors, he speculated that therapeutically targeting the enzyme in specific brain regions could still be viable, particularly with proper dosage to reduce BACE1 activity by less than previous trials did.
Could elevated BACE1 expression explain cognitive deficits in SCA1? BACE1 mRNA levels were unchanged in mice expressing an ataxin-1 knock-in with a 154 polyglutamine repeat, but the mice did have 30 and 40 percent more BACE1 in the cortex and cerebellum, respectively, than controls. The authors did not report whether this led to more Aβ.
“This work is very interesting,” said Sheng-Han Kuo, Columbia University, New York. “It’s important to understand the normal function of the ataxin-1 gene, and its interplay in different diseases.” Kuo cautioned that the work implies that gene therapy to knock down ataxin-1 in SCA1 could have unintended consequences. “We may need to fine-tune our knockdown strategy to target specific alleles,” he said. It remains to be seen how the transcriptional changes seen in mice play out in people.—Gwyneth Dickey Zakaib
References
Research Models Citations
Paper Citations
- Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008 Nov;83(5):623-32. PubMed.
- Bettens K, Brouwers N, van Miegroet H, Gil A, Engelborghs S, De Deyn PP, Vandenberghe R, Van Broeckhoven C, Sleegers K. Follow-up study of susceptibility Loci for Alzheimer's disease and onset age identified by genome-wide association. J Alzheimers Dis. 2010 Jan;19(4):1169-75. PubMed.
- Swaminathan S, Kim S, Shen L, Risacher SL, Foroud T, Pankratz N, Potkin SG, Huentelman MJ, Craig DW, Weiner MW, Saykin AJ, The Alzheimer's Disease Neuroimaging Initiative Ad. Genomic Copy Number Analysis in Alzheimer's Disease and Mild Cognitive Impairment: An ADNI Study. Int J Alzheimers Dis. 2011;2011:729478. PubMed.
- Swaminathan S, Huentelman MJ, Corneveaux JJ, Myers AJ, Faber KM, Foroud T, Mayeux R, Shen L, Kim S, Turk M, Hardy J, Reiman EM, Saykin AJ, . Analysis of copy number variation in Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. PLoS One. 2012;7(12):e50640. PubMed.
- Zhang C, Browne A, Child D, Divito JR, Stevenson JA, Tanzi RE. Loss of function of ATXN1 increases amyloid beta-protein levels by potentiating beta-secretase processing of beta-amyloid precursor protein. J Biol Chem. 2010 Mar 19;285(12):8515-26. PubMed.
- Matilla A, Roberson ED, Banfi S, Morales J, Armstrong DL, Burright EN, Orr HT, Sweatt JD, Zoghbi HY, Matzuk MM. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci. 1998 Jul 15;18(14):5508-16. PubMed.
Further Reading
Primary Papers
- Suh J, Romano DM, Nitschke L, Herrick SP, DiMarzio BA, Dzhala V, Bae JS, Oram MK, Zheng Y, Hooli B, Mullin K, Gennarino VA, Wasco W, Schmahmann JD, Albers MW, Zoghbi HY, Tanzi RE. Loss of Ataxin-1 Potentiates Alzheimer's Pathogenesis by Elevating Cerebral BACE1 Transcription. Cell. 2019 Aug 22;178(5):1159-1175.e17. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.