Sans Microglia, Mice Develop CAA and Die Young
Quick Links
Evidence suggests that microglia compact Aβ in brain parenchyma into dense-core plaques, but their effect on vascular deposits of Aβ, namely cerebral amyloid angiopathy (CAA), has been unclear. Now, researchers led by Mathew Blurton-Jones, University of California, Irvine, report that CAA runs amok in a mouse model of amyloidosis that lacks microglia from birth. In the June 14 Cell Reports, they reported that these animals had mostly diffuse, not dense-core, plaques and that these accumulated in and around the tiny blood vessels of the brain.
Without microglia, endothelial integrity faltered, calcium crystals accumulated in the thalamus, and the blood vessels burst. Most animals died by 6 months old. Injecting microglia into the brains of 2-month-old mice repopulated the whole brain within a few months, and this attenuated the pathology. “This fascinating study advances our understanding of the role of microglia in cerebral amyloidosis,” Mathias Jucker, University of Tübingen, Germany, wrote to Alzforum.
- Mice without microglia have diffuse plaques surrounding blood vessels.
- In the brain, endothelial genes are downregulated, and calcium forms deposits.
- Brain tissue from people with CAA also contained calcium crystals.
Researchers have studied microglial effects on amyloid pathology by asking what happens when the cells are ablated. They wiped out microglia by feeding adult mice inhibitors of colony-stimulating factor 1 receptor, which these cells need to survive and proliferate. In some cases this reduced plaques, slowed neurodegeneration, and improved cognition (Mar 2018 news; Apr 2014 news). Others saw plaques mass at blood vessels and neurite dystrophy worsen (Sept 2019 news). Oddly enough, blocking CSF1R had no effect on lifespan, perhaps because the inhibitors did not completely suppress microglia or because they were given once the mice were adults. CSF1R inhibitors may also deplete macrophages, making the findings tricky to interpret.
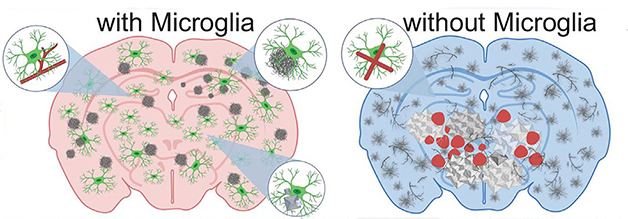
With or Without. In the brains of 5xFAD mice (left), microglia (green) surround blood vessels (red lines) and diffuse amyloid deposits to mop up Aβ and package it into dense-core plaques (gray). When the brain has no microglia (right), it accumulates diffuse amyloid plaques (gray squiggles), cerebral amyloid angiopathy (gray branches), cerebral bleeds (red blotches), and calcium deposits (white). [Courtesy of Kiani Shabestari et al., Cell Reports, 2022.]
To specifically and completely eliminate microglia, first author Sepideh Kiani Shabestari and colleagues turned to mice that lack the fms intronic regulatory element, a major driver of CSF1R expression. Without FIRE, mice make no microglia, though they still have meningeal and perivascular macrophages. The animals seem to develop normally, and live as long as do wild-type mice (Rojo et al., 2019). Kiani Shabestari found they carry a normal complement of peripheral immune cells in their cervical lymph nodes, spleen, and bone marrow. “This mouse model allows researchers to study microglial function more cleanly and in a much more sophisticated way than before,” Oleg Butovsky, Brigham and Women's Hospital, Boston, told Alzforum.
How did lack of FIRE affect plaque pathology? The scientists crossed FIRE mice with 5xFAD transgenic animals, then analyzed cortical, hippocampal, and thalamic tissue from 5-month-old offspring. They measured plaque load using confocal microscopy and insoluble Aβ levels with by ELISA after extraction.
The 5xFAD-FIRE mice had as many plaques in the parenchyma as did their 5xFAD counterparts, but the former were diffuse while the latter had dense cores (see image below). The crosses had one-quarter of the insoluble Aβ40 and Aβ42 and five times as many plaques surrounding endothelial cells as did 5xFAD mice. The authors interpreted this as amyloid accumulating around the vasculature rather than being packed into plaques by microglia.

FIRE and Mice. Microglia (green) tile hippocampal tissue in 5xFAD mice (left) but are absent from 5xFADs lacking FIRE (middle). While both had a similar number of amyloid deposits (white), 5xFAD mice accumulated dense-core plaques (top right), while in 5xFAD-FIRE mice they were diffuse (bottom right). [Courtesy of Kiani Shabestari et al., Cell Reports, 2022.]
Unlike 5xFAD mice, 5xFAD-FIRE animals and FIRE mice with no amyloid pathology had extensive blood vessel damage, most notably in the thalamus where small and large hemorrhages erupted. Single-nucleus RNA sequencing of vascular cells, glia, and neurons from 5xFAD-FIRE mice identified many transcriptional changes in genes associated with endothelial cells and blood-brain barrier integrity. The expression of two key growth factors, PDGF-β and TGF-β, dropped. Produced by microglia, these proteins support pericyte and endothelial cell growth and maintenance. The authors believe that, without microglia, deficiency of these growth factors leads to breakdown of the BBB.
Interestingly, mutations in PDGF-β cause primary familial brain calcification, a parkinsonian-like neurodegenerative disorder where calcium crystals accumulate in none other than the thalamus (Tadic et al., 2015). In a mouse model of this disease, CSF1R inhibitors exacerbated the calcification of blood vessels in the brain (Zarb et al., 2021). To see if something similar might be going on in 5xFAD-FIRE mice, Kiani Shabestari labelled brain slices with alizarin red, a dye that precipitates free calcium, and fluorescent risedronate, an osteoporosis drug that binds calcium crystals. Both molecules revealed calcium deposits mostly in the thalamus of 5xFAD-FIRE but not 5xFAD mice.
The most striking phenotype was early death. More than 70 percent of the 5xFAD-FIRE mice died by 6 months, while 5xFAD mice usually lived until they were at least 15 months old. “The combination of microbleed tendency of the FIRE mice along with CAA seems to be killing the crossed animals,” Charles Glabe of UCI told Alzforum. Kim Green, also at UCI, told Alzforum that he sees similar premature death after pharmacologically knocking down microglia in 5xFAD mice that had been crossed with animals expressing mutant human tau. This emphasizes the importance of having functional microglia when both Aβ and tau pathologies are in the picture.
Why wasn’t this early demise previously seen in mice given CSF1R inhibitors? Some think it is because this treatment does not deplete all the microglia. Blurton-Jones agreed. “The difference could be that FIRE mice never had microglia, whereas CSF1R-treated mice had them until early adulthood,” he said. Butovsky thought that, without any microglia to support other brain cells, their development and function may suffer in FIRE mice.
Others thought the mice’s young death may came down to the lack of CSF1R, which is also expressed by neurons and supports their survival (Nandi et al., 2012). “Genetic CSF1R and microglial ablation in the FIRE mice may ... predispose them to malfunctions … upon a pathological challenge, such as amyloid plaques, that cannot be properly resolved in the absence of microglia,” wrote Dave Holtzman and Yang Shi, Washington University, St. Louis (full comment below). However, Kiani Shabestari found no CSF1R mRNA in neurons from FIRE mice. Blurton-Jones added that FIRE is only expressed in microglia, hence the specific knockout of just those cells.
Could restoring microglia in 5xFAD-FIRE mice prevent pathology and increase survival? Kiani Shabestari injected the hippocampi and cortices of 2-month-old mice with microglia from wild-type animals. At 2 months, 5xFAD-FIRE mice show no signs of brain hemorrhaging or calcification. After 3 months, microglia had repopulated the brain to levels seen in 5xFAD animals, though the transplanted cells had fewer, stubbier branches than their 5xFAD counterparts.
At 5 months old, animals that received the transplants had less CAA, more dense-core plaques, and no cerebral bleeds or brain calcification. The repopulated microglia expressed TGF-β and PDGF-β and gathered near the vasculature, likely flocking to the CAA. “It is quite remarkable that CAA, brain calcification, and intracerebral hemorrhage were all resolved by microglia transplantation,” wrote Marco Colonna and Zhangying Cai, also at WashU (comment below). Glabe wondered about the therapeutic potential of infusing people with microglial precursors derived from their own stem cells in order to prevent downstream calcification and BBB instability.
Indeed, Kiani Shabestari found calcification in prefrontal cortex tissue from five AD cases with CAA, but none in five CAA-negative cases, suggesting that this phenomenon might occur in the AD brain also. Cortical tissue was more readily available than thalamic tissue from the UCI AD Research Center. People with CAA have more microhemorrhages, worse memory, and die sooner than people who have AD (Bell and Zlokovic, 2009; Arvanitakis et al., 2011).—Chelsea Weidman Burke
References
News Citations
- Wiping Out Microglia Prevents Neuritic Plaques
- Microglial Magic: Drug Wipes Them Out, New Set Appears
- Are Microglia Plaque Factories?
Research Models Citations
Paper Citations
- Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, Bradford B, Caruso M, Gazova I, Sánchez A, Lisowski ZM, Alves J, Molina-Gonzalez I, Davtyan H, Lodge RJ, Glover JD, Wallace R, Munro DA, David E, Amit I, Miron VE, Priller J, Jenkins SJ, Hardingham GE, Blurton-Jones M, Mabbott NA, Summers KM, Hohenstein P, Hume DA, Pridans C. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. 2019 Jul 19;10(1):3215. PubMed.
- Tadic V, Westenberger A, Domingo A, Alvarez-Fischer D, Klein C, Kasten M. Primary familial brain calcification with known gene mutations: a systematic review and challenges of phenotypic characterization. JAMA Neurol. 2015 Apr;72(4):460-7. PubMed.
- Zarb Y, Sridhar S, Nassiri S, Utz SG, Schaffenrath J, Maheshwari U, Rushing EJ, Nilsson KP, Delorenzi M, Colonna M, Greter M, Keller A. Microglia control small vessel calcification via TREM2. Sci Adv. 2021 Feb;7(9) Print 2021 Feb PubMed.
- Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, Mehler MF, Stanley ER. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol. 2012 Jul 15;367(2):100-13. Epub 2012 Apr 19 PubMed.
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009 Jul;118(1):103-13. PubMed.
- Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011 Feb;69(2):320-7. PubMed.
Further Reading
Primary Papers
- Kiani Shabestari S, Morabito S, Danhash EP, McQuade A, Sanchez JR, Miyoshi E, Chadarevian JP, Claes C, Coburn MA, Hasselmann J, Hidalgo J, Tran KN, Martini AC, Chang Rothermich W, Pascual J, Head E, Hume DA, Pridans C, Davtyan H, Swarup V, Blurton-Jones M. Absence of microglia promotes diverse pathologies and early lethality in Alzheimer's disease mice. Cell Rep. 2022 Jun 14;39(11):110961. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
Chinese Institute for Brain Research
Many studies have used CSF1R antagonists to deplete myeloid cells, including microglia in amyloid or tauopathy mouse models, and many have observed protective effects against pathology and neurodegeneration. This is a very interesting and well-done study that shows that genetic ablation of microglia via deletion of the CSF1R enhancer FIRE in amyloid mice induces a shift from parenchymal amyloid plaques to CAA, which is accompanied by detrimental effects of brain calcification and hemorrhages. These effects can be rescued by introducing microglia back into the brain. Very surprisingly, genetic ablation of microglia causes premature lethality in 5xFAD mice that is not seen in any of the CSF1R antagonist-treated mice.…More
How are these detrimental effects logically connected? And why does genetic microglial ablation cause early death of mice that develop amyloid?
It’s likely that pathological events such as amyloid plaques induce abnormal cell signaling, disrupting calcium and phosphate homeostasis, leading to calcium crystal formation; and microglial depletion impairs calcium crystal phagocytosis, causing brain calcification, particularly in blood vessels. Meanwhile, loss of PDGFb-PDGFBR signaling between microglia and pericytes due to microglial depletion may also affect pericyte recruitment and impair blood-brain barrier (BBB) functions. Both aspects can contribute to CAA formation and hemorrhages in 5xFIRE mice. It’s worth noting that although CAA is generally associated with hemorrhages in mouse models, CAA in these mice doesn’t seem to be the cause of hemorrhage, since WT-FIRE mice don’t have CAA, yet have similar amount of hemorrhages as 5xFIRE mice.
The formation of CAA and brain hemorrhage upon genetic microglial depletion recapitulates the findings of an earlier study using PLX5622 to deplete microglia in 5xFAD mice (Spangenberg et al., 2019). However, premature lethality does not occur in PLX-treated mice, either in amyloid (Spangenberg et al., 2019) or tauopathy mouse models (Shi et al., 2019) with long-term treatment. What exactly is causing the early death of 5xFIRE mice is unclear. The authors suggest that hemorrhages are the main cause of mortality, but since WT-FIRE mice develop comparable amount of hemorrhages as 5xFIRE mice, but show a normal lifespan; and that PLX-treated mice with hemorrhages don’t show increased mortality, this hypothesis is less likely to be true.
Because microglia play a critical role in regulating neuronal development, genetic ablation of microglia during the developmental stage inevitably affects neuronal functions, whether the effects can be readily detected or not. In fact, embryonic or neonatal microglial ablation was reported to induce persistent behavior changes in mice in the adult stage. In addition, CSF1R was also shown to be expressed by neurons, and CSF1R signaling on neurons is essential for neuronal survival and differentiation during development (Nandi et al., 2012). Therefore, genetic CSF1R and microglial ablation in the FIRE mice may impact neuronal development, and predispose them to malfunctions linked to lethality later in life upon a pathological challenge (such as amyloid plaques) that cannot be properly resolved in the absence of microglia. The fact that young WT-FIRE and 5x-FIRE mice don’t show cerebral hemorrhages or brain calcification doesn’t exclude the possibility of developmental impacts, because certain developmental effects may need time to accumulate in order to manifest. Detailed gene-expression analysis on neurons and other cell types should help address this question.
On the other hand, one key difference is that the FIRE mice get complete microglial depletion, whereas pharmacological methods can only achieve partial microglial depletion. Therefore, the detrimental effects are less severe in drug-treated mice, which may stay below the threshold for triggering lethality.
Lastly, although FIRE deletion doesn’t affect certain populations of peripheral macrophages, it does impact macrophage development in specific peripheral tissues (Rojo, 2019). PLX treatment also affects some aspects of the peripheral immune system, but the effects are not exactly the same. Whether the lethality comes from peripheral effects upon pathology challenge should also be taken into consideration.
References:
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, Habets G, Rymar A, Tsang G, Walters J, Nespi M, Singh P, Broome S, Ibrahim P, Zhang C, Bollag G, West BL, Green KN. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019 Aug 21;10(1):3758. PubMed.
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019 Nov 4;216(11):2546-2561. Epub 2019 Oct 10 PubMed.
Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, Mehler MF, Stanley ER. The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol. 2012 Jul 15;367(2):100-13. Epub 2012 Apr 19 PubMed.
Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, Bradford B, Caruso M, Gazova I, Sánchez A, Lisowski ZM, Alves J, Molina-Gonzalez I, Davtyan H, Lodge RJ, Glover JD, Wallace R, Munro DA, David E, Amit I, Miron VE, Priller J, Jenkins SJ, Hardingham GE, Blurton-Jones M, Mabbott NA, Summers KM, Hohenstein P, Hume DA, Pridans C. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. 2019 Jul 19;10(1):3215. PubMed.
Washington University School of Medicine
Washington University in St. Louis
The paper by Blurton-Jones and colleagues nicely demonstrates that the genetic depletion of microglia alters amyloid pathology in a unique way, different from the outcomes of pharmacological depletion of microglia.
The authors utilized FIRE mice (Rojo et al., 2019)—in which the fms intronic regulatory element (FIRE), a super enhancer of the Csf1r locus, was deleted—as a tool to deplete microglia. These mice were shown to have a complete loss of parenchymal microglia, yet their border-associated macrophages remained intact. The authors crossed FIRE mice with 5xFAD (generating 5x-FIRE) and characterized their pathological features at 5-6 months of age. Strikingly, 5x-FIRE mice had much shorter lifespans compared to 5xFAD controls, which was very likely due to the increased chances of hemorrhage in the thalamus. Compared to the Aβ pathology in 5xFAD, that in the parenchyma of 5x-FIRE mice was dampened (with relatively comparable plaque numbers but much lower staining intensities of dense core plaques), but they had more plaque deposition along the vascular structure in different brain regions, suggesting the development of cerebral amyloid angiopathy (CAA). Singe nuclei RNA-seq and weighted gene co-expression network analysis (WGCNA) revealed a defect of endothelial cells in 5x-FIRE mice, which might be caused by the lack of microglial TGFβ and PDGFβ.…More
The authors also observed the accumulation of hydroxyapatite calcium crystals in the thalamus of 5x-FIRE mice; meanwhile, they characterized a correlation between brain calcification and vascular pathology in human AD patients, which corresponded to the phenotype observed in mice. Importantly, the authors were able to rescue the pathological defects in 5x-FIRE mice by intracranial injection of WT microglia. Although 160k microglia per mouse seems to be a huge number, it’s still quite remarkable that CAA, brain calcification, and intracerebral hemorrhage were all resolved by microglia transplantation.
Overall, this paper advances our understanding of the role of microglia in amyloidogenesis and neurodegeneration. Previously, several studies investigated the outcome of microglia depletion in AD mouse models by administrating CSF1R inhibitors (Dagher et al., 2015; Olmos-Alonso et al., 2016; Spangenberg et al., 2016; Spangenberg et al., 2019). Though these studies used different dosages of CSF1R inhibitors, with different mouse models, most of them (Dagher et al., 2015; Olmos-Alonso et al., 2016; Spangenberg et al., 2016) reached the conclusion that partial microglia depletion can rescue learning and memory deficits, although the amyloid plaque load remains similar. Only one of these studies (Spangenberg et al., 2019) showed a phenotype similar to that of 5x-FIRE mice—impaired plaque formation and development of CAA—but they did not observe shortened lifespan or increased brain calcification.
It is not surprising that genetic and pharmacological ablation of microglia give rise to different phenotypes. In addition to discrepancy in time windows and sterile inflammation triggered by microglia apoptosis, mice treated with CSF1R inhibitors usually do not have 100 percent depletion of microglia. It has been shown that after 10 weeks of treatment causing a depletion of 99 percent microglia, there were still cells in the thalamus (Spangenberg et al., 2019) which might be the reason CSF1R inhibitor-treated mice have no calcium crystal accumulation in the thalamus, whereas FIRE mice do.
Finally, this paper by Blurton-Jones and colleagues provides useful information for researchers who intend to use FIRE mice for microglia replacement experiments. The premature lethality, intracerebral hemorrhage, and alterations of endothelial cells and pericytes are all important features to be taken into consideration when using 5x-FIRE mice as potential AD mouse models.
References:
Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, Bradford B, Caruso M, Gazova I, Sánchez A, Lisowski ZM, Alves J, Molina-Gonzalez I, Davtyan H, Lodge RJ, Glover JD, Wallace R, Munro DA, David E, Amit I, Miron VE, Priller J, Jenkins SJ, Hardingham GE, Blurton-Jones M, Mabbott NA, Summers KM, Hohenstein P, Hume DA, Pridans C. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. 2019 Jul 19;10(1):3215. PubMed.
Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL, Green KN. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015 Aug 1;12:139. PubMed.
Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M, Holscher C, Perry VH, Gomez-Nicola D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's-like pathology. Brain. 2016 Mar;139(Pt 3):891-907. Epub 2016 Jan 8 PubMed.
Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016 Apr;139(Pt 4):1265-81. Epub 2016 Feb 26 PubMed.
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, Habets G, Rymar A, Tsang G, Walters J, Nespi M, Singh P, Broome S, Ibrahim P, Zhang C, Bollag G, West BL, Green KN. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019 Aug 21;10(1):3758. PubMed.
Bennett RE, Bryant A, Hu M, Robbins AB, Hopp SC, Hyman BT. Partial reduction of microglia does not affect tau pathology in aged mice. J Neuroinflammation. 2018 Nov 9;15(1):311. PubMed.
Harvard Medical School
Harvard Medical School
This paper raises two interesting points about progress in microglial transplantation and the problems of using the 5xFAD mouse model of Alzheimer’s disease.
First, microglial transplantation has been proposed as a therapeutic approach to restore function in several neurological conditions, especially in combination with gene therapy to restore biochemistry lost to genetic mutations (Shibuya et al., 2022; Capotondo et al., 2017). However, preclinical transplantation studies use toxic conditioning protocols to enable the microglia-like cells to efficiently engraft in the brain, which may confound interpretations of efficacy and safety.
Here Shabestari et al. use mice carrying homozygous deletions of a Csf1r regulatory element to eliminate microglia in the adult brain (Rojo et al., 2019). They then restored the microglial population by bilateral intracortical and hippocampal transplantation of 160,000 cryopreserved, wild-type primary mouse microglia from a commercial source. The migration of the transplanted microglia over 24 days is striking and reminds us of our work reporting the response of progenitor cells to intrastriatal infusions of TGF-α protein (Cooper and Isacson, 2004; de Chevigny et al., 2008; Fallon et al., 2000). Surprisingly, the transplanted microglia still do not show a morphology consistent with a homeostatic, efficacious state (similar morphologies of microglia-like cells have been reported after bone marrow transplantation (Shibuya et al., 2022; Capotondo et al., 2017)). While five months should be sufficient time for the engrafted mouse microglia to fully integrate into the brain, it remains unclear why the engrafted cells do not grow fine, elaborate processes as other cell transplanted cell types can accomplish, e.g., mouse neurons.…More
From the perspective of human microglial transplantation, it will be interesting to learn if immunocompromised Csf1r hypomorphic mice may enable the efficient migration and integration of human iPSC-derived microglia in vivo for functional analyses.
Even with these remarkable transplantation findings, we still have the same clinical translation problem. The field of cell and gene therapy for non-oncology indications is moving toward clinical transplantation protocols that are free of chemotherapy (Omer-Javed et al., 2022). However, it appears that microglial clearance is required for efficient microglial transplantation. So how do we prepare a patient with neurodegenerative disease to receive new microglia when the current conditioning methods may worsen the individual’s health? New research is needed to solve this problem.
Second, the 5xFAD mouse is a well-characterized model of amyloid accumulation with considerable limitations for drug development (Oakley et al., 2006). The five genetic mutations introduced into the mouse accelerate AD-related pathology and deliver on the false promise of cheaper drug discovery. The model is limited by this aggressive drive toward pathological endpoints, as clinically relevant mechanisms of action cannot be tested. Compounding the problems with the 5xFAD model, Shabestari et al. introduce a sixth genetic mutation into the mouse and, rather unsurprisingly, demonstrate an exacerbated phenotype that drifts further away from representing human disease.
As with efficacy studies in other rodent models that constitutively express high levels of transgenes to generate pathology, the translational value of the reported efficacy of microglial transplantation in the 5xFAD/Csf1r hypomorphic mouse is difficult to interpret. The transplantation clearly looks to have removed the vasculature phenotype of the “6x” transgenic mouse and spared the parenchymal plaques that characterize the 5xFAD mouse. Since it’s a bit of a stretch to think that the “6x” mouse phenotype represents AD, we conclude that the transplantation of wild-type microglia does not protect against AD pathology. But we readily admit that this may not represent the disease process in patients, since the 5xFAD mouse model is so accelerated.
We are left with a negative interpretation of the transplantation data that is difficult to translate to patients, and the wrong model system.
References:
Shibuya Y, Kumar KK, Mader MM, Yoo Y, Ayala LA, Zhou M, Mohr MA, Neumayer G, Kumar I, Yamamoto R, Marcoux P, Liou B, Bennett FC, Nakauchi H, Sun Y, Chen X, Heppner FL, Wyss-Coray T, Südhof TC, Wernig M. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci Transl Med. 2022 Mar 16;14(636):eabl9945. PubMed.
Capotondo A, Milazzo R, Garcia-Manteiga JM, Cavalca E, Montepeloso A, Garrison BS, Peviani M, Rossi DJ, Biffi A. Intracerebroventricular delivery of hematopoietic progenitors results in rapid and robust engraftment of microglia-like cells. Sci Adv. 2017 Dec;3(12):e1701211. Epub 2017 Dec 6 PubMed.
Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, Bradford B, Caruso M, Gazova I, Sánchez A, Lisowski ZM, Alves J, Molina-Gonzalez I, Davtyan H, Lodge RJ, Glover JD, Wallace R, Munro DA, David E, Amit I, Miron VE, Priller J, Jenkins SJ, Hardingham GE, Blurton-Jones M, Mabbott NA, Summers KM, Hohenstein P, Hume DA, Pridans C. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. 2019 Jul 19;10(1):3215. PubMed.
Cooper O, Isacson O. Intrastriatal transforming growth factor alpha delivery to a model of Parkinson's disease induces proliferation and migration of endogenous adult neural progenitor cells without differentiation into dopaminergic neurons. J Neurosci. 2004 Oct 13;24(41):8924-31. PubMed.
de Chevigny A, Cooper O, Vinuela A, Reske-Nielsen C, Lagace DC, Eisch AJ, Isacson O. Fate mapping and lineage analyses demonstrate the production of a large number of striatal neuroblasts after transforming growth factor alpha and noggin striatal infusions into the dopamine-depleted striatum. Stem Cells. 2008 Sep;26(9):2349-60. Epub 2008 Jun 12 PubMed.
Fallon J, Reid S, Kinyamu R, Opole I, Opole R, Baratta J, Korc M, Endo TL, Duong A, Nguyen G, Karkehabadhi M, Twardzik D, Patel S, Loughlin S. In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci U S A. 2000 Dec 19;97(26):14686-91. PubMed.
Omer-Javed A, Pedrazzani G, Albano L, Ghaus S, Latroche C, Manzi M, Ferrari S, Fiumara M, Jacob A, Vavassori V, Nonis A, Canarutto D, Naldini L. Mobilization-based chemotherapy-free engraftment of gene-edited human hematopoietic stem cells. Cell. 2022 May 18; PubMed.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006 Oct 4;26(40):10129-40. PubMed.
Salk Institute
This informative study assesses the functional role of microglia in AD pathogenesis by using “FIRE” mice that deplete microglia selectively without affecting border macrophages or peripheral immune cells.
It shows that 5xFAD mice lacking microglia have many fewer dense-core Aβ plaques but many more diffuse plaques than 5xFAD mice alone, and at the same time display markedly increased vascular cerebral amyloid angiopathy-like pathology. This agrees with previous studies that assessed microglial functions by pharmacologically killing almost all microglia at an early age with CSF1R inhibitors, or by genetically inactivating the key microglial phagocytic genes Mertk and Trem2.
It strongly supports the hypothesis that microglia construct dense-core Aβ plaques via phagocytosis. The authors also characterized, in FIRE mice, hemorrhages and brain calcification, which are potentiated in AD. Interestingly, both were reversed by transplanting 5xFAD/FIRE mice with WT microglia in young adulthood.…More
What is striking but left unexplained in the paper is the dramatic increase in premature death in the 5xFAD/FIRE mice compared to 5xFAD alone; half of the former population dies before significant plaque deposition. This phenotype was not reported previously for 5xFAD mice treated with CSF1R inhibitors as early as 1.5 months. The authors did not speculate as to a causal relationship between the premature mortality and observed deficits. Since the hemorrhages and calcification deficits are not AD-specific (they were observed in WT-FIRE mice as well) and appear at 5-6 months when the majority of 5xFAD/FIRE mice have died, it is unlikely these are the direct cause of death.
A key difference between the current study and previous pharmacological inhibition of microglia is the timing. Microglia play an important role during CNS development by performing routine phagocytosis, synaptic pruning, and the secretion of neurotrophic factors and cytokines. Interestingly, the authors showed altered gene expression in excitatory neurons that “included glutamatergic synapse ontology terms and network hub genes” when comparing between WT FIRE mice and 5xFAD/FIRE mice. To understand the mechanism of premature lethality in AD mice lacking microglia, it may be of interest to investigate deficits in synaptic connectivity and network activity due to microglia depletion, and to see whether these are reversible by adult transplantation of WT microglia in adulthood.
Third Rock Ventures
Kiani Shabestari and colleagues took an innovative genetic approach to understanding the role of microglia in deposition of APP-derived amyloid in the murine brain. They used mice in which an evolutionarily conserved macrophage enhancer element—Fms Intronic Regulatory Element/FIRE, which is required for CSF1R expression—was removed by gene targeting. Deletion of FIRE in the germline abrogates development of tissue macrophages in selected organs (including kidney, skin) as well as absence of brain microglia, leaving monocytes and other macrophage populations unaffected. Upon crossing to 5xFAD mice, the resulting 5x-FIRE mice showed a phenotype of early death, amyloid deposition in vessels at the expense of brain parenchyma resembling CAA, and consequent brain hemorrhages.…More
At 2 months of age, as amyloid deposition began to be detected, stereotactic injections of commercially sourced microglia substantially rescued the phenotype, suggesting that absence of microglia during the onset of amyloid pathology, rather than a developmental effect, accounted for the phenotype. FIRE mice lack parenchymal microglia but not “barrier” macrophages such as perivascular brain macrophages, so that selective deficiency of the microglial population was responsible.
There are several attractive aspects to this report:
1. CAA is an important element of Alzheimer's disease pathology, and this genetic model may provide a means to study aspects of CAA.
2. Both male and female mice were studied—a distinct strength.
3. The authors took extra care to exclude that CSF1R expression by neurons contributed to the phenotype. This is useful, as it provides another line of evidence that CSF1R is not expressed by neurons.
4. The data are well-presented and -discussed, and of high quality.
There are also concerns, as viewed from the standpoint of a translation-minded observer. Previously an academic neurologist-neuroscientist, I now work at Third Rock Ventures, where part of my responsibility is to develop ideas for new biotech companies that address unmet need in neurological disease:
1. What's the next step to exploit this model? Both FIRE mice and 5x-FAD mice are extremely artificial and don't replicate anything seen in human biology. Is the plan to transplant genetically modified microglia (for example, TREM2-null) to begin dissecting which microglial functions are involved in the deposition of amyloid in the parenchyma rather than the vasculature? How tractable will the model be to establishing the molecular basis of the phenotypes?
2. The connection to human disease is underdeveloped, although the investigators did conduct limited preliminary experiments toward establishing this relationship. It is unlikely that in vitro studies with iPSC-derived microglia-like cells in culture will be productive in this regard. Are there similarities between any of the cell clusters emerging from snRNA-Seq in the present studies and those provided by examining human AD brain?
Minor concerns:
What is the mechanism of early mortality for the 5x-FIRE mice? The observation is very striking (70 percent death by 6 months) but unexplained. Humans with CAA can die from extensive cerebral hemorrhage—was this process involved?
2. "DAM microglia" nomenclature, as applied here, is not helpful for understanding the research. The microglial transcriptome can be characterized without jargon terminology, just as is done for other cell types. Further, this approach encourages use of local lab jargon for each RNA-Seq experiment as recently noted by a consensus group (Paolicelli et al., 2022). Senior author Blurton-Jones is a co-author on the consensus report.
Make a Comment
To make a comment you must login or register.