Scientists discussed anti-Aβ therapeutic antibodies, confirming their ability to remove brain amyloid and working to boost their brain exposure. They continued their postmortem of BACE inhibitors in hopes of reviving these drugs at lower doses. Efforts at targeting tau are shifting toward tau’s microtubule-binding section, and α-synuclein trials are grappling with rates of disease progression. Emerging neurophysiology-based treatment approached presented promising early stage data, and clinicians shared experiences dealing with a year of COVID disrupting their trials and worsening their patients across neurologic care. Plus: cool news on astrocytes, tau blood tests, better mouse models and more. Check it all out here.
Donanemab Confirms: Clearing Plaques Slows Decline—By a Bit
It has been clear for a while that anti-amyloid antibodies can sweep plaque from the brain, but until now the question of whether this slows cognitive decline has remained hotly contended. Despite some positive signals from four such antibodies, the data have been messy and hard to interpret. At the 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, Mark Mintun of Eli Lilly & Company presented the cleanest data yet on this question. In a Phase 2 trial, the company’s anti-amyloid antibody donanemab met its primary endpoint. Participants did not get better. Even so, donanemab slowed their decline by an average of 32 percent on a combined cognitive and functional measure.
Donanemab banished plaque from the brain in a majority of participants, while nudging down the rate of neurofibrillary tangle accumulation in the frontal cortex and other regions. The trial included several innovative elements, such as screening participants by tangle burden, using tau PET as a secondary outcome measure, and stopping dosing once amyloid was gone. The AD/PD presentation fleshed out previously announced topline data (Jan 2021 news). Full results were published March 13 in the New England Journal of Medicine (Mintun et al., 2021).
Most Alzheimer’s researchers welcomed the findings. “This was the first [disease-modifying] AD drug to meet a clinical endpoint in a Phase 2 trial,” noted Ron Petersen of the Mayo Clinic in Rochester, Minnesota. Michael Weiner of the University of California, San Francisco, found the broader implications encouraging. “In my view, together with data from other trials, this study strongly confirms the ‘amyloid hypothesis’ and demonstrates that treatments aimed at amyloid can slow cognitive decline and modify the progression of AD,” he wrote (full comments below).
At the same time, researchers emphasized that, as with other anti-amyloid immunotherapies, the cognitive benefit was small. “The donanemab story is the most encouraging news on the amyloid front, ever, but whether the effect size is clinically meaningful is questionable,” David Knopman at the Rochester Mayo clinic wrote to Alzforum (full comment below).
Screen By Path. People with few tangles (left) have too little cognitive decline to measure, while those with a heavy tangle burden (right) may have worsened beyond the reach of an anti-amyloid drug. Selecting for the just-right tangle load (middle) may have helped the donanemab trial succeed. [Courtesy of Eli Lilly.]
Plaque-Busting Power
Donanemab is unique among AD immunotherapies in that it targets a modified version of Aβ that has a pyroglutamate attached to the N terminus. This pathological form of Aβ is highly prone to aggregate, depositing in the core of all amyloid plaques, but is found nowhere else in the brain (Dec 2009 conference news; Nov 2010 conference news; Dec 2012 news). In Phase 1 trials, donanemab busted up plaques fast, in many cases clearing all deposits within six months (Aug 2018 conference news; Dec 2019 conference news).
However, even dramatic amyloid clearance has not translated into a clear cognitive benefit in past Phase 2 and 3 immunotherapy trials. In a company call with investors, Lilly’s chief scientific officer, Dan Skovronsky, said part of the problem in obtaining definitive cognitive results may arise from the heterogeneity of AD trial populations, with participants worsening clinically at different rates. To limit this variability, the researchers screened participants in their 18-month Phase 2 Trailblazer study using tau PET. They believed this might work because previous studies had shown that a person’s baseline tau PET signal predicted his or her speed of subsequent cognitive decline.
People with flortaucipir SUVRs below 1.1 were excluded from this trial, since studies have shown almost no cognitive decline in this group within the time span of this trial. Those with SUVRs above 1.46 were also excluded, as the researchers hypothesized that tangle pathology in their brains would be too advanced for an amyloid therapy to do them any good (see image above). Mintun estimates that 30 to 45 percent of people with early symptomatic AD fall into the intermediate tau range where anti-amyloid therapy might be effective.
The researchers ended up enrolling 257 people with this intermediate tangle burden at 56 sites across the United States and Canada. Participants were predominantly white, with an average age of 75, and about three-quarters carried an APOE4 allele. All had early symptomatic AD and were amyloid-positive by florbetapir PET scan, with a mean MMSE of 23.6. Mintun noted that this cognitive average is lower than for many other trials in early AD, where the cutoff for inclusion is often 24, and the average score higher. Selecting participants based on tangle pathology rather than clinical criteria may have allowed for a wider range in clinical status, he suggested. Because of cognitive reserve, people with similar levels of brain pathology often differ in the degree to which their function is preserved (Aug 2017 conference news). “Some patients who would be considered too impaired for inclusion in an early AD trial might still be at an early stage of pathology and respond to treatment,” Mintun noted.
Half the participants, 131 people, received donanemab, the other 126 placebo. Doses were titrated up rapidly for quick plaque clearance, with participants receiving 700 mg for the first three monthly infusions and 1,400 mg per month thereafter. Participants underwent florbetapir scans at weeks 24 and 52 to assess their progress. If their amyloid burden fell below 25 centiloids—the level in healthy young controls—their donanemab dose was lowered to 700. If it fell below 11 centiloids, or below 25 for two consecutive scans, they were switched to placebo.
Why stop dosing? Pyroglutamate-Aβ only occurs in plaques, so once the target is gone, there is no need for further treatment, Mintun said. Commentators applauded this limited course of treatment, given the expense and side effects of antibodies. “That patients could be withdrawn from the treatment is a remarkable prospect for broader use,” Knopman wrote. Petersen suggested it would make AD easier to manage chronically. “We may be able to lower amyloid levels, monitor the patients, and if the levels rise, re-dose. This would be akin to giving a booster immunization,” Petersen said.
In Trailblazer, the baseline amyloid burden was 108 centiloids in the active group and 101 in the placebo group. It stayed stable in the placebo group over the course of the study. In the active group, it dropped by an average of 85 centiloids. The bulk of the clearance came early, with an average drop of 68 centiloids by week 24. At that time, 40 percent of the treatment group were switched to placebo. This rose to 60 percent by 52 weeks and 68 percent by 76 weeks. In other words, two-thirds of participants were amyloid-negative by the end of the trial. Mintun noted that plaque clearance at 18 months was about twice that seen with aducanumab, in agreement with earlier trial results suggesting that donanemab clears plaque more aggressively than do other investigational antibodies.
In an ongoing open-label extension trial, participants who still have plaque, as well as previous placebo patients, will remain on donanemab treatment until they, too, become amyloid-negative. Researchers will follow all patients to assess how they fare over time.
Detecting a Cognitive Signal
Did this clearance translate into a better-functioning brain, though? To answer this, the researchers chose as their primary outcome measure the integrated Alzheimer’s Disease Rating Scale. Lilly had developed the iADRS by combining the ADAS-Cog13 with the ADCS-instrumental Activities of Daily Living scales (Wessels et al., 2015). In the Phase 3 solanezumab Expedition studies, this combined cognitive and functional scale yielded more consistent results than did the CDR-SB, Mintun said. Likewise, in the placebo arm of Trailblazer, the iADRS scores reflected a constant rate of decline, whereas the CDR-SB posted variability from timepoint to timepoint. Mintun said the CDR-SB has proven noisy and unreliable in other large AD studies, as well, for example giving one positive and one negative result in the aducanumab Phase 3 program. “We believe the iADRS is a more consistent and sensitive measure to detect treatment differences than other AD scales,” Mintun said.
In Trailblazer, participants started out with an average iADRS score of 106, with the placebo group declining 10.06 points by the end of the trial, and the active group by 6.86. The treatment groups started to diverge at 24 weeks, around the time plaque clearance became dramatic. The difference between groups reached statistical significance at 36 weeks and maintained that for every timepoint thereafter, with a final p value of 0.04. The one-third slowing of decline is modest. It would translate to a six-month delay in disease progression over the course of the 18-month trial, Mintun noted.
All secondary clinical measures trended in favor of donanemab, but only the ADAS-Cog13 reached significance at p=0.04, with an average slowing of 39 percent. On the CDR-SB, decline slowed only by 23 percent, on MMSE, 21 percent, and on ADL, 23 percent. As with the iADRS, active groups first diverged from placebo at 24 or 36 weeks. Mintun noted that the 23 percent slowing on CDR-SB is no smaller an effect size than has been seen to date in AD trials.
Among individual participants, the pattern of plaque clearance varied, with some getting a large initial drop and others a steady decline. This made no difference to the cognitive benefit, Mintun said.
Given that donanemab completely cleared plaque, the researchers acknowledged that a 32 percent slowing may represent the most it can achieve in people at this stage of AD. “This is probably the ceiling for an amyloid-lowering drug,” Skovronsky said. To do more for patients, researchers likely will have to treat earlier in a prevention paradigm, or combine anti-amyloid treatment with an anti-tau drug, he suggested.
Less Tangles, More Cognition. Trial participants with the lowest tangle burden (left) reaped the biggest benefit, while those with the most tau pathology (right) had none. The findings may help refine selection for future trials. [Courtesy of Eli Lilly.]
When Plaques Vanish, Tangle Formation Slows
About that tau … unlike the loose association amyloid has with cognition, tau tangles are closely linked to cognitive decline. Did donanemab affect them? On a measure of global tau PET, the answer was no. Tau tracer uptake climbed in both groups throughout the study, with the active treatment group gaining only 10 percent less than the placebo group, a nonsignificant difference.
When the researchers looked at regional tracer uptake, they saw something different. Tangle accumulation slowed by 59 percent in the frontal lobe, by 45 percent in the parietal lobe, and 32 percent in the lateral temporal lobe. These differences were statistically significant, the first two at p=0.002. In mesial temporal lobe, the placebo group had no change in tangles, but the donanemab group saw a slight but significant drop. The groups were no different in the occipital lobe.
Plaque clearance was linked to the slowing of tau pathology, Mintun noted. Participants who reached amyloid-negative status during the trial had more slowing on tau PET than those who didn’t. It is unclear mechanistically how plaques affect tangles. Mintun suggested that some toxic aspect of amyloid may be responsible for accelerating tauopathy, such that removing it puts on the brakes. A recent study implicated microglial inflammation as the culprit linking the two pathologies (Nov 2019 news).
Regional tangles, particularly in the frontal cortex, also correlated with the cognitive outcome, with a higher baseline frontal tau signal predicting faster decline on the iADRS. “We’ve provided data to validate regional tau spread as an important surrogate for disease progression and drug effect,” Skovronsky said.
What about the idea that baseline tangle load influences donanemab’s effect? To study this, the researchers stratified the active group into terciles based on their baseline tau PET. The lowest tercile, below 1.14 SUVR, drove most of the cognitive benefit from donanemab; this group’s decline slowed by almost half. The intermediate tercile showed little treatment benefit, and the highest, above 1.27 SUVR, none (see image above). These subgroups were too small for statistical significance, and the analysis is exploratory, Mintun said.
Nonetheless, Lilly researchers believe the data may help explain why this trial succeeded. “Excluding patients with high tau could be a key factor in the efficacy of donanemab. We believe it’s important that all future Alzheimer’s trials and therapies be based on the pathological stage of the patient, as is done in oncology,” Mintun said.
Others wondered whether the tau range should be narrowed further for Phase 3, since people with an SUVR between 1.27 and 1.46 did not benefit. Skovronsky said Lilly will keep the range the same for Phase 3. The idea is to replicate the Phase 2 findings, and Phase 3 will have more power to detect treatment effects.
Gil Rabinovici of UCSF was intrigued by these data. “This suggests that the primary role of amyloid-lowering therapies may be in patients in whom tau is not yet widespread, most of whom will be in the preclinical or very earliest clinical stage. Progress in blood-based biomarkers should greatly facilitate the detection of earliest-stage AD in an accessible, equitable and cost-effective manner,” he wrote (full comment below).
ARIA Still An Issue
Safety data in the Trailblazer trial was similar to previous donanemab studies and to other antibodies in this class. A quarter of the active group, 35 people, developed the brain edema known as ARIA-E. In eight people, about 6 percent of those on donanemab, it was symptomatic. People taking the antibody also developed more superficial siderosis, iron deposits that form on subpial surfaces due to small brain bleeds, than controls, at 14 percent versus 3. They had more ARIA-H, or microhemorrhages, at 8 percent versus 3, and more nausea, 11 percent versus 3. Donanemab administration caused infusion reactions in 10 people, though most were mild or moderate, did not require intervention, and did not reoccur. There were no differences in serious adverse events or deaths between groups.
Erik Musiek of Washington University, St. Louis, noted that the ARIA-E incidence was similar to that in people taking low-dose aducanumab, while the ARIA-H incidence was lower than the 17 percent reported for aducanumab (comment below). Rabinovici believes the overall safety profile of donanemab would be acceptable to most AD patients.
More people on donanemab than placebo, 40 versus nine, discontinued treatment, most due to ARIA-H or superficial siderosis. Most who stopped treatment remained in the study, and their data were included in the final outcome measures. Skovronsky said that those who stopped treatment due to ARIA had already achieved a high degree of plaque clearance, and attained the same drug benefit as those who remained on therapy.
Russell Swerdlow of University of Kansas Medical Center, Kansas City, noted that the occurrence of symptomatic ARIA can confound trial results by inadvertently unblinding participants. Because APOE4 carriers are more likely to develop ARIA and stop treatment, the removal of such fast progressors from the treatment arm also could skew results. “Hopefully the planned Phase 3 studies will implement measures to take into account these potential confounders,” Swerdlow wrote (full comment below).
Lilly researchers contend that ARIA barely affected their Phase 2 results, since an analysis of donanemab subgroups with and without ARIA-E found no difference in their respective rates of decline, and people who stopped treatment were included in the final analysis.
As in the earlier donanemab trial, 90 percent of participants developed anti-drug antibodies. These did not appear to affect treatment efficacy, but Skovronsky acknowledged that it would be better to have a treatment that does not produce them. Lilly is testing such a version of donanemab, dubbed N3pG4, in clinical trials, but Skovronsky said Lilly intends to bring donanemab to market.
Phase 3: Stick With What Worked
Based on the Trailblazer data, Lilly researchers have made several changes to Trailblazer 2, which has been enrolling since last summer. Instead of a Phase 2 trial with 500 participants, it will become a Phase 3 with 1,500. Scientists hope the larger sample will boost the chance of success on secondary outcomes, increase power to see subgroup effects including in the tau tercile groups, and generate a larger safety database, Mintun noted. Trial sites have been enrolling people with both intermediate and high tau scans, but will now limit the primary efficacy analysis to 1,000 participants with intermediate tangle pathology. Data from 500 people with a higher tangle load, above 1.46 SUVR, will help inform future treatment guidelines, Mintun said.
Lilly had previously considered using the CDR-SB as an endpoint for the new trial, but will stick with iADRS instead. In answer to an investor question, Skovronsky said this was discussed with regulators at the Food and Drug Administration. In one change from Phase 2, however, Lilly will analyze efficacy using the Disease Progression Model (DPM), which generates a probability of disease progression based on data from every timepoint across the trial, rather than looking only at the last timepoint, as the standard model does. Mintun said the final timepoint in AD trials is often the noisiest, so he believes a DPM analysis will be more reliable. In the Phase 2 trial, a DPM analysis gave similar results to the standard method, with every endpoint favoring donanemab.
The Global Alzheimer’s Platform will help speed recruitment for Trailblazer 2. GAP co-founder John Dwyer, who leads the Washington, D.C.-based organization, noted that the original Trailblazer was the first trial GAP worked on. His goal is for Trailblazer 2 to enlist 130 sites worldwide, including many in the GAP-North American network. “We expect to have an immediate impact on TRAILBLAZER-ALZ 2, and one of our priorities will be reaching potential participants from diverse communities,” Dwyer wrote to Alzforum.
Skovronsky expects Trailblazer 2 to complete enrollment in the second half of 2021 and read out in the first half of 2023. Lilly will explore its chances for accelerated regulatory approval, but Trailblazer 2 will complete regardless. “Replication will answer important questions for the field, such as confirming subgroups that show no benefit,” Skovronsky said. Still, because the first Trailblazer was designed as a registration trial, he expects a second positive trial could be sufficient for approval.
If donanemab only helps people up to a certain tangle load, how large is the estimated patient group? Skovronsky noted that 4.5 million people have early symptomatic AD in the United States, 5 million in Europe, and 4 million in Japan. If 30 to 45 percent meet the criteria, that would be 1–2 million people in each place.
The FDA is considering a licensing application from Biogen for its anti-amyloid antibody aducanumab (Feb 2021 news). Will the donanemab data influence the agency? Rabinovici, at least, thinks it should not. “While these results are encouraging for the overall drug class, the FDA needs to consider the aducanumab EMERGE and ENGAGE data on their own merits. There are significant differences between aducanumab and donanemab in the antibody-targeted epitopes, study design and patient populations, and one cannot generalize results from one trial to the other,” he wrote.
Others think eventual approval of an anti-amyloid therapeutic is now inevitable. “The donanemab results provide powerful support for the amyloid therapeutic hypothesis; this strategy will bring the first disease-modifying drugs for AD into clinical use,” Paul Aisen of the University of Southern California, San Diego, wrote to Alzforum (full comment below).—Madolyn Bowman Rogers
When stuck with lemons, make lemonade. The “lemon” for Nobuyuki Okamura and colleagues at Tohoku University, Sendai, Japan, was THK-5351, a tracer they had developed to detect neurofibrillary tangles. Alas, the compound bound monoamine oxidase B, as well, souring its usefulness for tau PET. Now for the sweet pivot. Astrocytes predominantly produce MAO-B, and they squeeze out more of this enzyme when stressed, as during the astrogliosis that occurs in response to amyloid and tau pathology. What if a few tweaks to THK-5351 might turn it from a bad tau tracer into a good astrocytosis tracer? The field sure is thirsty for a good astrocytosis tracer.
At this year’s ADPD meeting, which began online March 9, Victor Villemagne, who has moved to the University of Pittsburgh, Pennsylvania, reported that 18F-SMBT-1—just such a derivative—bound MAO-B in the human brain reversibly and with high specificity. In PET scans, people who tested positive for amyloid bound much more of the new ligand in their brains than did amyloid-negative controls. The upshot: Researchers may soon have a better PET tracer for astrogliosis to accompany those that already detect amyloid, neurofibrillary tangles, changes in glucose metabolism, and, perhaps, synapse number.
Care for a chaser with that lemonade? A blood test for astrogliosis may soon come your way as well. At ADPD, Andrea Benedet, University of Gothenburg, Sweden, reported that the concentration in plasma of glial fibrillary acidic protein—which is produced by astrocytes in the central nervous system—climbs twice as high in people who have AD than it does in age-matched controls. Plasma GFAP correlated with brain amyloid and tangles, though the latter association was primarily driven by amyloid, Benedet reported. Curiously, plasma GFAP tracked with amyloid more tightly than did its level in cerebrospinal fluid (CSF). “This is the first time we see a plasma biomarker performing better than a CSF biomarker in predicting amyloid,” said Benedet.
“It is surprising that GFAP seems more specific in plasma than in CSF, but we, too, see better correlation with plasma GFAP and Aβ in our cohorts,” Oskar Hansson, Lund University, Sweden, told Alzforum.
Recent studies led by Ralph Martins at Edith Cowan University, Joondalup, Western Australia, and Charlotte Teunissen at Vrije Universiteit, Amsterdam, support Benedet’s findings. They found that amyloid-positive volunteers had more GFAP in their plasma than did amyloid-negative controls, and that, when combined with the plasma Aβ2/40 ratio, GFAP improved predictions of amyloid positivity.
Astrogliosis PET
Okamura and colleagues used THK-5351 as a starting point to finesse PET ligands for MAO-B (see image below). As reported in the February Journal of Nuclear Medicine, first author Ryuichi Harada and colleagues removed an amino group that was essential for the compound to bind tangles (Harada et al., 2021). This simple tweak dramatically increased the compound’s affinity and specificity for the oxidase. In vitro, the dissociation constant for binding MAO-B was 3.7 nM for the new compound, dubbed SMBT-1. Binding to MAO-A was almost 200-fold weaker, with a Kd of 713 nM. Affinities for Aβ and tau aggregates from human brains were weaker still, with Kd’s of more than 1,000 nM, indicating that SMBT-1 specifically binds MAO-B.
Lose that Amine. Clipping off an N-H group (circled) turned the experimental tau PET tracer THK-5351 into SMBT-1, a reversible and specific ligand for MAO-B. SMBT-1 has much better tracer properties that other MAO-B ligands (top row). [Courtesy of Harada et al., Journal of Nuclear Medicine, 2021.]
But would it work in human tissue? Using autoradiography, Harada and colleagues found that more SMBT-1 bound to sections from AD brains than control brains.
How about for imaging? After injection into the blood streams of mice, the compound rapidly flooded the brain and then quickly washed out—just what the tracer developer ordered.
This contrasts with deprenyl, the most widely used tracer for MAO-B (Feb 2012 news). Deprenyl ends up binding irreversibly to the oxidase when it forms a covalent bond with a flavin cofactor in the enzyme’s catalytic site. Even deuterating deprenyl to slow this chemical dalliance does not fully eliminate the covalent binding.
Voilá, Astrocytosis. Axial, coronal, and sagittal images showing more widespread retention of SMBT-1 in a person with AD (bottom) compared to an age-matched healthy control (top). [Courtesy of Victor Villemagne.]
Toxicity studies indicated that the teeny amounts of SMBT-1 needed for PET would be safe. Villemagne, Harada, and colleagues tested SMBT-1 in 82 participants in Australia’s AIBL longitudinal study. Ten were healthy young controls, 55 healthy old controls, 12 were old with mild cognitive impairment, and five had AD. All were also scanned for plaques and tangles using NAV4694, aka AZD4694, and MK-6240, respectively.
In his AD/PD presentation, Villemagne showed that 18F-SMBT-1 rapidly enters the brain and most of it quickly clears, resulting in high-contrast images of retained compound (see image above). The tracer reached steady-state levels in the brain within about 50 minutes, as judged by standard uptake value ratios using white matter as the reference region. Its regional distribution in the brain coincided with known expression levels of MAO-B, i.e., the thalamus and caudate retained the greatest amount, and cerebellum and white matter the least. Older, healthy, amyloid-negative volunteers retained twice as much SMBT-1 as did young controls, in keeping with increasing expression of MAO-B in the brain with age.
Now You See it, Now You Don’t. Retention of SMBT-1 in the brain of a person with AD and in an age-matched control (left) is all but wiped out by selegiline, an irreversible MAO-B inhibitor (right). The drug quenched up to 85 percent of the signal. [Courtesy of Victor Villemagne.]
Selegeline, the L-enantiomer of deprenyl, cut down the 18F-SMBT-1 signal by 80 to 85 percent, depending on brain region. Because selegeline competes with 18F-SMBT-1 for binding to MAO-B, this reduction is another indication that 18F-SMBT-1’s binding to MAO-B is quite specific. This displacement also suggests that SMBT-1 binds to, or near, the MAO-B catalytic site since deprenyl binds there. Selegeline is approved for treatment of major depression and for people with Parkinson’s disease, in whom it can reduce the need for L-dopa.
What does SMBT-1 reveal about astrogliosis? In this small AIBL cohort, people with AD retained more of this tracer in the posterior cingulate, the supramarginal gyrus, lateral occipital cortex, primary visual cortex, and the angular gyrus, and less in regions such as the hippocampus, parahippocampus, caudate, globus pallidus, and pons. “We found that the close relationship between Aβ deposition and astrogliosis only occurs in some regions of the brain, but not in other regions characterized by high Aβ deposition, such as the ventral frontal cortex and anterior cingulate gyrus,” said Villemagne. “This suggests a more complex and regional relationship than just a 'global ’ [astrocyte] response.” It might be that SMBT-1 PET reflects the progression of disease pathology. Some of the areas that had highest tracer retention, such as the supramarginal gyrus and posterior cingulate, accumulate amyloid early in the disease, Villemagne said.
Next, Villemagne compared SMBT-1 retention in amyloid-positive versus -negative volunteers. The same pattern held; however, additional regions showed higher retention in the amyloid-positive people, including regions of the temporal cortex and the dorsolateral prefrontal cortex. “The most important finding is that those [cognitively normal] controls with higher amyloid in the brain have significantly higher SMBT-1 retention in regions such as the orbitofrontal, posterior cingulate, supramarginal gyrus, temporal occipital regions, and in lateral temporal cortex,” said Villemagne. This suggests that, like deprenyl, the new tracer picks up astrogliosis early in the disease process, albeit with better performance. SMBT-1 has the added advantage of having the 18F isotope for tracking rather than the 11C that labels deprenyl. The short half-life of 11C, circa 20 minutes, limits its use to imaging centers that can create it on-site.
Researchers led by Agneta Nordberg at Stockholm’s Karolinska Institute had previously reported that in autosomal-dominant AD, astrocytosis flares up at the same time as plaques, but not necessarily in the same place (Schöll et al., 2015). This led the scientists to suggest that astrocytosis might even precede plaques. To Villemagne’s mind, astrogliosis does not start a pathological process, but responds to some process going on in the brain. Whether that might be oligomers of Aβ, or some other inflammatory molecule, remains to be seen. "Although I tend to agree with Professor Nordberg, one problem is that whenever the field does not really know what is going on, the default position is to blame ‘the oligomers,’” Villemagne quipped.
SMBT-1 correlated strongly with amyloid, only weakly with tau, and not at all with cognition. “This tells us that Aβ is much better at setting off astrogliosis than is tau,” Villemagne said. Even so, he plans to use the new tracer to study astrogliosis in other diseases, such as ARTAG and LATE, which some believe to be driven primarily by non-Aβ pathologies, such as tau tangles and aggregates of TDP-43, respectively (Kovacs et al., 2016; May 2019 news).
Plasma Portends Astrogliosis
Could GFAP, that oldest of astrocyte markers, hand researchers a plasma surrogate for astrocytosis in Alzheimer’s disease, as well? Only astrocytes seem to produce this structural filament protein in the central nervous system and, like other protein markers such as Aβ, phospho-tau, and NfL, GFAP wends its way into the cerebrospinal fluid and even into the blood stream. Benedet correlated plasma and CSF GFAP with amyloid and tau PET as measured by the same tracers that Villemagne used, namely NAV4694 and MK6240. Benedet, a postdoc in the labs of Kaj Blennow and Henrik Zetterberg at U Gothenburg, used data from 171 volunteers in the TRIAD biomarker study run by Pedro Rosa-Neto at McGill University in Montreal.
In her ADPD talk, Benedet reported that cognitively unimpaired, amyloid-negative older volunteers had more GFAP in their blood than young, healthy controls, showing it rises somewhat with age alone. Amyloid-positive but cognitively healthy older adults had even more. Those with mild cognitive impairment and a positive amyloid test had higher levels still, and people with AD had the highest—about double the cognitively unimpaired Aβ-negative group. The same pattern was seen in CSF, but the differences between groups were neither as stark, nor statistically significant.
Nonetheless, both plasma and CSF GFAP significantly correlated with amyloid as a continuous variable based on global retention of NAV4694 in the brain. Once again, the plasma marker performed best. Drilling down into the data, Benedet found that CSF levels seem to plateau as amyloid accumulates, while plasma GFAP continues to climb. “We are not sure what might explain this pattern,” said Benedet.
For his part, Martins was surprised but found the pattern intriguing. “To my knowledge, no other marker behaves this way,” he told Alzforum. One possibility is that GFAP more easily leaks through the blood-brain barrier than other markers, but to Martins this makes little sense. He suggested testing CSF collected in the AIBL cohort, saying “We need other studies to confirm this pattern.” Still, that Hansson’s group sees plasma GFAP correlating more tightly with brain amyloid than CSF GFAP strengthens the idea that this marker somehow behaves differently than other markers that leave the brain and enter the body’s fluids.
Despite these nuances, plasma GFAP looks promising. In this TRIAD sample, it predicted amyloid positivity with an AUC, i.e., accuracy, of 0.83. It performed slightly better than plasma p-tau181, which posted an AUC of 0.81. CSF GFAP predicted brain amyloid with an AUC of 0.75.
Martins, and also Teunissen, reported similarly tight correlations of plasma GFAP with brain amyloid in their recent papers. “I was surprised how well plasma GFAP performs compared to p-tau,” said Martins. “It is on par with both p-tau181 and p-tau231.”
Martins and colleagues measured plasma GFAP in 100 healthy older people who enrolled in the Kerr Anglican Retirement Village Initiative in Ageing Health, located around Perth, Western Australia. Called KARVIAH, the cohort excludes people with cognitive impairment, though some participants did express concern about their memories, i.e., fell into the category of subjective cognitive complaint. Their average age was 78. As judged by 18F-florbetaben retention in the brain, 33 of them tested positive for amyloid.
They had significantly more GFAP in their plasma than the amyloid-free participants. First author Pratishtha Chatterjee and colleagues found that plasma GFAP predicted brain amyloid with an AUC of 0.80, slightly better than the 0.78 derived from a basic model based on age, sex, and APOE genotype, and better than the AUC of 0.67 for the plasma Aβ42/40 ratio (Chatterjee et al., 2021).
The best prediction of brain amyloid, AUC 0.92, came from combining the basic model with plasma GFAP and Aβ42/40, but this was only marginally better than the basic model plus GFAP alone (AUC 0.91).
Martins collaborates with the Zetterberg/Blennow group in Gothenburg and with Teunissen’s in Amsterdam. He told Alzforum that both had measured GFAP in the same sample and got the same results. All are using a Simoa-based assay from Quanterix. “The assay is very robust,” said Martins. “The coefficient of variation is so low that we only need to measure in singlicate. This makes it cost-effective.”
Teunissen’s group was the first to detect an uptick in GFAP in AD plasma. They measured the marker in 252 people in the Amsterdam Dementia Cohort. Of these, 70 voiced subjective cognitive complaints, 50 had mild cognitive impairment, and 132 had been diagnosed with dementia. All had been scanned for brain amyloid by PET using one of the four commonly used tracers. Again, co-first authors Inge Verberk, Elisabeth Thijssen, and colleagues found that the plasma GFAP concentration, on average, was significantly higher in the 176 people whose amyloid scans were positive than in the 76 amyloid-negative folks. Even among the 70 volunteers with subjective cognitive decline, GFAP was higher among amyloid-positives, suggesting the plasma marker ticks up early in the disease process (Verberk et al., 2020).
In this population, plasma GFAP best predicted amyloid positivity, with an AUC of 0.81, compared to AUCs of 0.73 and 0.71 for plasma Aβ42/40 and plasma NfL, respectively. Among the participants without dementia, the predictive values of all three markers was lower. But even in them, GFAP, with an AUC of 0.76, outperformed both Aβ42/40 and NfL, which had AUCs of 0.67 and 0.63, respectively. Of the three markers, GFAP most strongly associated with global cognitive decline and with decline on specific cognitive domains, including memory, language, attention, and executive function.
In this cohort, too, Teunissen found the best panel for predicting brain amyloid to comprise age, APOE genotype, plasma Aβ42/40, and plasma GFAP. This model returned an AUC of 0.88.
“The field is definitely headed toward algorithms based on multiple markers and risk factors,” said Martins. “I don’t think we yet have a biomarker that can stand alone. We need a good combination to get us across the finish line.”—Tom Fagan
Buoyed by data crediting amyloid removal with ever-so-slightly slowed cognitive decline, many scientists are now optimistic about the prospects for anti-amyloid immunotherapy. Four “-mabs”—of the ganteneru-, lecane-, aducanu-, and donane- kinds—have proven to dramatically clear plaque, while also nudging downstream tangle pathology. Three of these have data showing they can tap the brakes on mental slippage, while high-dose gantenerumab has not yet read out on cognition.
The 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, featured updates on all these programs. Mark Mintun of Eli Lilly presented donanemab’s Phase 2 trial data (Mar 2021 conference news). Luka Kulic of Roche reviewed a strategy to amplify how much gantenerumab reaches its target when delivered via a “brain shuttle” technology, and showed that it appeared safe in a first-in-human trial. Chad Swanson of Eisai presented amyloid PET data from the first year of the lecanemab open-label extension. Meanwhile, Samantha Budd Haeberlein of Biogen shared new analyses of the same Phase 3 data for aducanumab, which awaits regulatory decisions in the U.S., the EU, and Japan on its pending licensing applications.
Overall, researchers were encouraged that signals from different molecules, approaches, and trials seem to be converging. “There are very similar efficacy findings in clinical measures and even downstream biomarkers,” Swanson said. Budd Haeberlein agreed, saying, “Reproducing effects across different molecules is a strength.” Mintun believes the totality of the data indicate that attacking amyloid does slow progression. “It’s been exciting for the field to see the amyloid cascade hypothesis get more support every year,” he said. Speaking to Alzforum, Randall Bateman, Washington University, St. Louis, said he, too, was struck by the commonalities across the different antibodies. “When you remove the majority of amyloid in mildly symptomatic people, that is when you see movement in other soluble markers, tau PET, and even cognitive/clinical outcomes,” he said. “The effects are in the range of 20 to 30 percent, individual secondary outcomes are weaker, and there is variance within and across studies. But when put together, it is a signal,” Bateman added.
Passing Through. Roche’s “brain shuttle” (orange/yellow) allows gantenerumab (green) to bind transferrin receptor (blue) and hitch a ride into the brain, where it binds Aβ. [Courtesy of Roche.]
Brain Shuttle Sneaks Gantenerumab Past the Blood-Brain Barrier
Initially, gantenerumab was nearly scuttled when low doses failed to budge endpoints in the Phase 3 Scarlet RoAD and Marguerite RoAD trials. Later data showed that fivefold higher doses mopped up plaque (Dec 2017 conference news; Aug 2018 conference news). The drawback to such high doses, besides the cost projections of manufacturing expensive biologics for millions of patients, is more frequent occurrence of amyloid-related imaging abnormalities (ARIA) that reflect swelling or microhemorrhages in the brain.
If antibodies better penetrated the brain parenchyma, they could be given at lower doses. Scientists at Roche and elsewhere have been working on a method to ferry large molecules into the brain. Roche’s approach conjugates a cargo to a Fab antibody fragment that recognizes transferrin receptor. These receptors sit on endothelial cells lining the brain’s blood vessels. They take up transferrin floating by in the blood and pass it through into the brain. By hijacking this system, researchers were able to boost brain uptake of a generic anti-amyloid antibody 50-fold in mice (Jan 2014 news; Jan news 2018).
At AD/PD, Kulic showed data for the hybrid molecule RG6102. It consists of the Fc portion of gantenerumab, i.e., its tail, conjugated to the Fab shuttle (see image above). In mice, a mouse version of RG6102 entered the brain in 12-fold higher quantities than unbound gantenerumab, penetrated brain tissue more evenly, and cleared plaque at lower doses. Immunofluorescence staining showed the mouse RG6102 suffused throughout the whole brain four hours after dosing, as compared to a few scattered dots near blood vessels for the unbound antibody. In non-human primates, RG6102 brain exposure exceeded that of unbound antibody by 6 to 42-fold, depending on the parameter and brain region assessed.
For Phase 1, Roche recruited 36 healthy young men to receive one infusion of RG6102. In this single-ascending-dose study, the first six volunteers received 0.1 mg/kg RG6102 or placebo, in a 4:2 ratio; the second six 0.4 mg/kg. For the higher doses, researchers infused six volunteers with RG6102 and two with placebo, testing 1.2, 3.6, and 7.2 mg/kg in turn. These doses are lower than the 1,020 mg, or around 15 mg/kg, given subcutaneously in the ongoing Phase 3 GRADUATE trials of gantenerumab, but cannot be directly compared because of the different bioavailability of drugs given by these two different routes.
The brain shuttle-gantenerumab construct behaved as expected, with a half-life of three to six days in plasma, and a linear relationship between the concentration in plasma and cerebrospinal fluid. The CSF/plasma ratio was 0.8 percent, indicating eight molecules out of every 1,000 entered the central nervous system. This compares to the 0.1 percent ratio for gantenerumab seen in previous studies. In other words, the brain shuttle amplified CNS exposure eightfold, similar to the effect seen in animals. “This is the first evidence for a brain-shuttling effect to the CNS compartment in people,” Kulic told Alzforum.
The researchers saw no ARIA in these volunteers, but did not expect to, either. In older people with brain amyloid, could higher amounts of gantenerumab in the brain lead to more ARIA? Kulic noted that ARIA is believed to be caused by the interaction of antibodies with vascular amyloid. Because the brain shuttle carries the antibody deeper into tissue and away from blood vessels, it may allow fewer opportunities for this interaction. “The different distribution of Brain Shuttle gantenerumab could potentially result in a different ARIA profile compared to monoclonal antibodies,” Kulic wrote to Alzforum. In addition, because more of the antibody gets into the brain, they may be able to use lower systemic doses to achieve the same effect on plaques.
Another risk is that, because transferrin ferries iron from blood to brain, hijacking the system could interfere with this physiological function. In answer to an audience question, Kulic noted that the volunteers developed no anemia or hematology-related adverse events. The researchers did see transient effects on immature red blood cells, which Kulic said they would monitor closely in later trials. Based on their modeling, they do not expect these effects to cause anemia.
Roche is now recruiting for a multiple-ascending-dose Phase 1b/2a trial of RG6102. They plan to enroll 120 people with prodromal to moderate AD and an amyloid PET scan above 50 centiloids. Participants will receive doses ranging from 0.2 to 3.6 mg/kg, given intravenously every four weeks and compared with placebo. The trial will focus on safety and tolerability, but also examine immunogenicity, pharmacokinetics, and changes in amyloid PET. It is expected to run until 2024.
“This study will inform us as to potential future clinical efficacy trials in AD,” Kulic said. “Our primary goal is that RG6102 will result in superior target engagement, faster Aβ clearance, and improved efficacy.” Roche is also exploring the potential of this technology to deliver other therapeutic cargoes to the brain.
How Fast Does Amyloid Leave? Depends on How Much Is There
Eisai/Biogen’s lecanemab, previously BAN2401, vanquished plaque in an 856-person Phase 2b study (Jul 2018 conference news). Participants who had completed this trial were invited back for an open-label extension after a gap of about two years. When they re-enrolled, Eisai scientists realized that during the gap, brain amyloid had resumed its accumulation in people who had been treated during the original trial, and it did so at the same slow rate seen in the placebo group during the blinded period. This added 0.1 to 0.2 SUVR per year (Dec 2019 conference news).
At AD/PD, Swanson extended this OLE baseline observation with new, one-year data. The OLE enrolled 180 people from 56 sites. All receive the highest tested dose of 10 mg/kg every other week, and are followed with plasma biomarkers and clinical assessments every six months. A subset of 39 people undergo florbetapir amyloid PET at six-month intervals; 10 of them had been on placebo previously, 19 on a 10 mg/kg monthly dose, and 10 on 10 mg/kg biweekly.
In all OLE participants, amyloid measurements nosedived during a year on lecanemab. Curiously, the higher a person’s starting plaque load, the faster it fell. The former placebo group, starting with a 1.36 SUVR at OLE baseline, dropped by 0.31. Those who had been on 10 mg/kg monthly in the original trial started the OLE at 1.19 and dropped by 0.12, while the 10 mg/kg biweekly group started OLE at 1.08 and fell only 0.05.
The upshot was that all three groups ended up between 1.03 to 1.07 SUVR on average. Around 80 percent of the OLE amyloid PET subgroup were amyloid-negative, defined as an SUVR below 1.17, after one year. “Amyloid burden seems to approach a floor,” Swanson summed up. Swanson did not present CSF or cognitive data from the OLE.
On Aducanumab, a Little More Analysis, Not Much Change
Aducanumab stumbled through Phase 3, with a futility analysis crash followed by a U-turn after more data were analyzed (Oct 2019 news; Dec 2019 conference news). Its path through the FDA’s licensing process is no less rocky. After blistering criticism from its advisory committee, the agency, under opposing pressures from various advocacy groups, requested more data from Biogen and delayed making a decision (Nov 2020 news; Feb 2021 news).
At AD/PD, Budd Haeberlein presented a dollop of new data. She discussed a tertiary outcome, the neuropsychiatric inventory NPI-10. It followed the pattern of the other clinical outcomes. In the EMERGE study, billed as positive, decline on the NPI-10 slowed by 87 percent in the high-dose group, significant at p=0.02, while in the low-dose group, it nudged down nonsignificantly by 33 percent. In the ENGAGE study, considered negative, decline on the NPI-10 accelerated a tad in the high-dose group, by 8 percent, while it slowed 83 percent in the low-dose group, significant at p=0.04. This matches the other clinical measures, where the ENGAGE low-dose group seemed to respond better than the high-dose group. Biogen attributes this to a higher percentage of fast progressors in the latter.
The NPI-10 includes a measure of caregiver burden. Budd Haeberlein said caregivers of patients in the EMERGE high-dose group reported 84 percent less burden compared to those looking after patients on placebo, but did not report caregiver burden numbers for the low-dosage group, or ENGAGE.
Alzheimer’s researchers have requested sensitivity analyses for these trials. These examine how changes in statistical methodology or assumptions affect trial outcomes, essentially as a way to determine how robust a trial’s findings are. Budd Haeberlein showed some of that data at AD/PD. She reported that sensitivity analysis found little effect from missing data due to patient withdrawals or the trial’s early termination, with all outcomes in EMERGE still favoring aducanumab. Tests that take into account the “normality” or distribution of the data gave results similar to the primary analysis. Excluding data gathered after ARIA onset made no difference in the outcomes, suggesting unblinding was not a factor, Budd Haeberlein said.
Budd Haeberlein presented a smidgen of new biomarker data. She had previously reported positive results on a medial temporal composite in a tiny tau PET substudy, comprising 12 patients on placebo, 14 on low dose, 11 on high. The tau PET signal rose slightly on placebo, and dropped slightly on the high dose. At AD/PD, she said the temporal and frontal composite measures followed the same pattern, rising on placebo and nudging down on the high dose. EMERGE and ENGAGE participants were pooled.
Budd Haeberlein also showed a new analysis of CSF biomarkers that suggested a correlation between a person’s degree of change from baseline and his or her cumulative dose. This relationship was stronger in EMERGE than in ENGAGE. The EMERGE CSF substudy comprised only 28 people on placebo, 33 on low dose, and 17 on high; the ENGAGE CSF substudy, 15 on placebo, 20 on low dose, and 18 on high.
While the field awaits the FDA’s decision on aducanumab, it seems certain that research on anti-amyloid antibodies will continue, regardless of whether the agency approves aducanumab this June or requires a confirmatory trial.
“This is the beginning, not the end. Twenty to 30 percent slowing is better than zero, but it’s not nearly where we need to go. Patients need much more than that,” Bateman said.—Madolyn Bowman Rogers
N-Terminal Tau Antibodies Fade, Mid-Domain Ones Push to the Fore
With anti-amyloid antibodies now consistently hitting their target, tau immunotherapy represents the next frontier. In Alzheimer’s disease, tau tangles correlate far more closely with cognitive decline than plaques do, and tau aggregates are the main pathology in many related disorders. As with amyloid, however, initial trials of anti-tau antibodies have been beset by failures. Already, several antibodies that bind the N- or C-terminus of tau have been scuttled after not doing recipients any good. Meanwhile, preclinical evidence suggests that antibodies that go after the protein’s mid-section, particularly its microtubule-binding region (MTBR), may be better at preventing aggregates from spreading. Several such antibodies have now entered Phase 1 or 2.
At the 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, researchers discussed a number of these programs. Kristin Wildsmith of Roche offered a first look at biomarker data from the negative Phase 2 trial of the N-terminal-targeting antibody semorinemab. Other speakers touted MTBR-binding antibodies. Wendy Luca of Pinteon Therapeutics showed preliminary Phase 1 findings for PNT001, while Günter Höglinger of the Technical University of Munich presented on UCB’s bepranemab, also in Phase 1. Prothena’s MTBR-binder PRX005 is still preclinical, but the company’s Philip Dolan offered mechanistic data on how it might inhibit the transfer of pathological tau.
Time will tell if this newest crop can perform in the clinic. Wildsmith believes the field is making progress in figuring out how to target the protein. She is encouraged by cerebrospinal fluid data from WashU’s Randall Bateman and colleagues that link CSF MTBR tau with tangles, and specific tau phospho-species with plaques (Mar 2020 news; Dec 2020 news). “That’s really exciting for us as a field. We’re learning so much more about this target,” Wildsmith said in an AD/PD panel discussion.
Tau Strung Out. Researchers are zeroing in on tau’s proline-rich mid-domain and four repeat domains (R1-R4) as the best region for antibodies to target. The microtubule-binding region spans from residues 224–369. [Courtesy of Horie et al., 2021, Brain.]
N-Terminal Strategy: All Duds So Far
Initially, tau’s N-terminus was the favored target, because it produces the highest-affinity antibodies. However, antibodies that bind this region have had no success in Phase 2 trials to date. Biogen’s gosuranemab and AbbVie’s tilavonemab both posted negative findings for progressive supranuclear palsy (PSP) and have been discontinued for that disorder, but remain in trials for Alzheimer’s disease (Jul 2019 news; Dec 2019 news). Biogen presented data at CTAD 2020 showing that gosuranemab hit its target in PSP patients, lowering unbound N-terminal tau in CSF, but had no effect on downstream markers of disease severity.
Similarly, Roche previously announced negative topline results for the Phase 2 TAURIEL trial of semorinemab for AD. Semorinemab targets residues 6–23 in tau’s N-terminus. It did not budge clinical endpoints, and had no effect on tangle accumulation by tau PET (company press release). No safety issues cropped up.
The semorinemab data Wildsmith presented at AD/PD provide another glimpse at the effects of an N-terminal tau antibody on soluble tau and downstream biomarkers. The 16-month TAURIEL trial enrolled 422 people who had prodromal to mild AD and brain amyloid. Participants received either placebo or 1,500, 4,500, or 8,100 mg of semorinemab by infusion once a month. Fewer than a third, or 123 people, participated in the CSF subgroup at baseline. This shrank to about half that number at 49 weeks and about a third at 73 weeks.
First, the good news. As expected, plasma semorinemab rose with dose, with a half-life of 32 days. CSF data showed that about 0.3 percent of the antibody got into the central nervous system, similar to other antibodies. In the two higher-dose groups, N-terminal tau increased in CSF at 49 and 73 weeks, indicating that semorinemab was finding its target. Meanwhile, CSF total tau, as measured by an Elecsys assay that recognizes a mid-domain region, and phosphorylated tau dropped at all doses of drug compared to the placebo groups. This was consistent with mouse data, and demonstrates that the antibody altered tau processing, Wildsmith said. Lower CSF tau is associated with less pathology in AD. Curiously, however, total tau rose in plasma in tandem with semorinemab dose. This trial included tau PET, but Wildsmith did not show that data.
Now, the bad news. The antibody did not budge downstream CSF markers of degeneration and inflammation, such as NfL, neurogranin, S100B, IL-6, and sTREM2. The inflammation marker YKL-40 went the wrong way, increasing in people on semorinemab at 49 and 73 weeks. This astrocytic protein rises as AD progresses, and is linked to brain shrinkage and other deleterious effects (Dec 2020 news). It is unclear why this happened, and Wildsmith did not speculate.
While there have been several N-terminal tau antibodies, far fewer target tau’s C-terminus. One that did, Roche’s RG7345, was dropped after Phase 1. Lundbeck’s LuAF87908, which binds the C-terminus, is completing Phase 1 and should read out soon.
Hot Zone. Prothena’s antibody screen found that those binding the shaded yellow region, spanning tau’s repeat domains R1 and R2, best blocked tau uptake into cells. [Courtesy of Prothena.]
Mid-Region: Up and Coming
The new hot tickets in the field are antibodies targeting tau’s middle, especially the MTBR. This region drives tau aggregation, and in cell culture, antibodies that bind here best prevented the spread of misfolded forms (von Bergen et al., 2005; Apr 2018 conference news). Earlier this month, the DIAN-TU trial chose Eisai’s E2814, which recognizes a motif in the MTBR, for its anti-tau arm (Mar 2021 news).
Several more such antibodies are in trials. Janssen has an MTBR-targeting antibody, JNJ-63733657, that has just begun Phase 2. Biogen’s mid-domain BIIB076 completed Phase 1 in AD in 2020, but no results or future plans have been announced. The antibody remains listed on Biogen’s pipeline. Lilly’s zagotenemab will read out for Phase 2 in AD later this year. It was derived from the research antibody MC1, which is reputed to bind amino acids 312–322 in tau’s mid-region. However, Lilly previously reported that zagotenemab recognizes a conformational epitope involving the N-terminus and is selective for aggregated tau, leaving it unclear in what category it belongs (Alam et al., 2017).
AD/PD showcased three more mid-tau antibodies. Pinteon Therapeutics’ PNT001 is the furthest along, having completed Phase 1. It recognizes phosphoThr231 in the MTBR. Luca noted that it blocked tau aggregate seeding in lysates from AD frontal cortex and hippocampus by 88 percent. In the Tg4510 mouse model of tauopathy, five months of dosing lowered insoluble tau and serum NfL, a marker of degeneration, while improving performance in the Morris water maze.
The Phase 1 single-ascending-dose study enrolled 49 healthy volunteers at three U.S. study sites, who received either 33, 100, 300, 900, 2,700, or 4,000 mg. In each dose group, six people got PNT001 and two placebo, each as a 30-minute infusion. Participants were followed for four months, donating CSF at days three and 28. The antibody was well-tolerated, Luca reported.
Serum levels of PTN001 increased with dose as expected. In CSF, too, the antibody concentration reflected the dose given, and stayed constant through day 28. For doses of 900 mg and above, the CSF concentration topped 45 ng/ml. This is the minimum concentration necessary for this antibody to bind tau well, Luca noted. Pinteon will take these top three doses into Phase 2. The company is still analyzing serum and CSF tau, as well as CSF markers of inflammation. Meanwhile, Pinteon has started a multiple-ascending-dose trial for traumatic brain injury, and is planning a similar trial in AD.
UCB’s bepranemab is in Phase 1 for PSP. This antibody, now being developed in partnership with Roche, recognizes residues 235–250 in the MTBR. This stretch overlaps the end of the proline-rich region and the beginning of the first repeat. At AD/PD, Höglinger showed cell culture data indicating that as little as 0.3 nM of bepranemab prevented uptake of tau aggregates. In the same assay, antibodies against the N-terminus required more than 10 times that concentration to have an effect.
The Phase 1b study enrolls 24 people with PSP at sites in Belgium, Germany, Spain, and the U.K. Eighteen participants take bepranemab, six placebo, for one year. Researchers will collect CSF to assess whether the antibody hit its target and how it was metabolized. The trial is expected to complete this year, after which participants will have the option to go on to an open-label extension, or a 16-week follow-up to collect safety data. No safety concerns have arisen so far, Höglinger noted.
UCB initially planned to run a Phase 3 PSP trial, dubbed auTAUnomy, but put it on hold and is now prioritizing Phase 2b for AD, Höglinger said. More money and more patients are available for AD research, and such a trial will still allow UCB and Roche to evaluate safety and efficacy in a tauopathy. The sidelined PSP trial would have run for two years and included mortality as an outcome measure, as amyotrophic lateral sclerosis trials do. It would have been the first PSP trial to use a combined assessment of function and survival, known as CAFS (Berry et al., 2013). Höglinger believes such a design makes sense. “It would be a good step forward in getting meaningful outcomes in clinical trials, because it focuses on a longer observation period and incorporates time to the most meaningful milestone to patients, which is survival,” he said.
Werner Poewe of Innsbruck Medical University, Austria, agrees that one-year trials are too short for many neurodegenerative diseases, and longer trials would serve the field better. The effects of a drug may accrue over time, and the magnitude of change may be too small to see in a short timeframe, he suggested. “I am worried that we are throwing away valid drugs and targets,” he said at AD/PD.
Why Is Mid-Tau Better? Preclinical Data for PRX005 Offers Clues
Pass the Tau, Please. Tau (red) needs to bind sulfated proteoglycans (brown twigs) to exit and enter cells.
Prothena’s Dolan showed more data on the merits of targeting tau’s middle. His team screened a panel of antibodies, and found that those that bound repeat regions 1 and 2, near the beginning of the MTBR, worked best to block internalization of tau aggregates in cell culture. They selected one such antibody, PRX005, that bound at the start of R1 (see image above). This antibody binds all splice forms of tau, as R2 is the one that gets spliced out in 3-repeat tau. PRX005 recognizes phosphorylated and unphosphorylated forms with equal affinity, Dolan said. It also lit up neurofibrillary tangles and dystrophic neurites in AD brain tissue.
Dolan noted his company’s choice of antibody was also influenced by Bateman’s CSF data on MTBR tau. “That solidified for us that it was a region worth pursuing. It’s mechanistically distinct and, in our experiments, was a bit of a hot spot,” he said at AD/PD.
Curiously, PRX005 inhibited tau binding to heparan sulfate proteoglycans. HSPGs on the cell surface are believed to both secrete and take up tau (Mar 2013 conference news; Katsinelos et al., 2018; image above). In culture, even picomolar quantities of PRX005 disrupted tau-HSPG binding. This helped cells; PRX005 protected rat cortical neurons from tau toxicity. Dolan did not discuss whether PRX005 might interfere with HSPG binding of other, physiological cargo.
The antibody worked in animals, too. The researchers treated PS19 mice, which express P301S tau, with PRX005 starting at 6 months of age. Four months later, treated mice had less ptau199/202 and ptau212 in the brainstem, and were able to hang longer from a grid, indicating improved grip strength. In a different model, mice injected with AD lysate, weekly antibody injections likewise prevented tau aggregates. Dolan noted that they used both models because some experiments indicate tau internalization may happen differently in each.
Höglinger asked if PRX005 would block tau aggregation. “We think it probably would,” Dolan said. He questioned how much that would help, given that most tau aggregation is intracellular, and most antibodies stay outside cells. This may help with safety, since antibodies that enter cells could interfere with tau’s physiological functions. Dolan believes the risk of intracellular effects is small, and PRX005’s main mode of action will be to stop cell-to-cell propagation. He did not discuss a timeline for clinical trials.—Madolyn Bowman Rogers
Plasma phospho-tau took the Alzheimer’s field by storm in 2020. Paper after paper described how p-tau181, p-tau217, and p-tau231 specifically and sensitively picked up the disease, especially in its presymptomatic phase. So: Are we ready for a simple phospho-tau blood test for AD? At this year’s virtual ADPD meeting last month, the collective answer was: Not quite.
That said, a clearer picture did emerge of how these biomarkers may be used clinically before too long. For older people without cognitive problems, p-tau appears insufficient to identify who might have presymptomatic AD. Even for people who have been diagnosed with some form of cognitive impairment, p-tau markers cannot stand alone. Instead, researchers are building and testing out algorithms based on several different biomarkers and other risk factors, such as age and APOE genotype.
This approach assumes that the impressive correlations between plasma p-tau isoforms and AD pathology seen in selected cohorts hold true in the general population. “We are just really at the beginning of this field,” said Michael Weiner, University of California, San Francisco, in a session on biofluid markers. “We need bigger, more epidemiologically sampled populations that better represent the community than do clinical trial populations. We need a lot more studies, and we need more data to compare the different tests,” said Weiner. He stressed that most of the studies to date have come from “samples of convenience,” i.e., highly educated Caucasians.
P-tau, You’ll Never Walk Alone?
In various memory center cohorts, p-tau181, p-tau217, and p-tau231 distinguish Alzheimer’s disease from controls with such high accuracy that some have suggested these markers are good enough to become AD diagnostics by themselves. Not all are on board with this idea, even based on the cohorts that exist today. At ADPD, Cliff Jack, Mayo Clinic, Rochester, Minnesota, argued that these markers are not quite up to snuff. “For this notion of a stand-alone, one point of caution is that these markers should differentiate amyloid-positive from -negative in individuals at all points along the disease spectrum. Studies cast doubt on this,” he said.
One such study comes from ADNI. Weiner, ADNI's principal investigator, reported that among people in that cohort who are cognitively impaired, plasma p-tau181 distinguishes amyloid-positives with a moderate AUC of around 0.67, much lower than the AUCs of 0.77 to 0.91 reported in some memory clinic cohorts (Janelidze et al., 2020; Karikari et al., 2020; Thijssen et al., 2020).
This new data comes from a study led by Duygu Tosun at UCSF and was reported in February in the journal Brain Communications.
Much Overlap. In ADNI, people with brain amyloid (brown) have only moderately higher plasma p-tau181 than people without (green). The difference is smaller among the cognitively unimpaired (CU) than the impaired (CI). Group differences are highly statistically significant but individual levels show much overlap. [Courtesy Tosun et al., 2021.]
Tosun found that among cognitively unimpaired ADNI participants, plasma p-tau181 distinguished 109 amyloid-positives from 224 -negatives with even less accuracy—AUC of 0.5. That does not bode well for a stand-alone marker for the general population.
Likewise, in a small cohort at Washington University, researchers led by Nicolas Barthélemy and Randall Bateman found that plasma p-tau181 distinguished 20 cognitively normal amyloid-positive people from 31 amyloid-negative people with an AUC of only 0.67 (Barthélemy et al., 2020).
In the larger Mayo Clinic Study of Aging, a prospective population-based cohort, Michelle Mielke at Mayo in Rochester, Minnesota, had previously found that this tau marker identified 72 amyloid-positives among 172 cognitively unimpaired with an AUC of 0.7. At ADPD, Mielke reported that in a larger sample of 892 cognitively unimpaired people, p-tau181 performed only slightly better, with an AUC of 0.78.
Is p-tau181 the wrong species? While the few studies that have analyzed p-tau217 and p-tau231 thus far suggest that these may prove more accurate, they still may not be accurate enough, Mielke said. She reported that p-tau217 identifies amyloid-positives among the MCSA cognitively unimpaired participants with an AUC of 0.83. In his much smaller sample, Barthélemy calculated 0.86 for p-tau217.
After all the excitement for blood tests, what explains the different accuracies now being reported in additional cohorts for p-tau181? Mielke has begun to study the effects of various health parameters on plasma levels of p-tau181 and p-tau217. In her ADPD presentation, she reported that among the cognitively unimpaired, a higher score on the Charlson comorbidity index correlated with higher levels of both tau isoforms in the plasma. Chronic kidney disease by itself had an even greater effect. Mielke thinks this is because of a failure to clear the isoforms from the blood. On the other hand, high body mass index associated with lower levels of the two markers, possibly because there is more blood to dilute them.
Even among memory clinic populations, i.e., those “convenience samples,” plasma tau markers may be insufficient by themselves to serve as a diagnostic. Christopher Clark, University of Zurich, reported p-tau181 and neurofilament light (NfL) data from 221 people recruited from a memory clinic there. Among the 91 who were cognitively normal, p-tau181 could not identify those who had been diagnosed with AD based on CSF markers.
Enter Algorithms
What about combining p-tau with other markers? Clark reported that adding plasma p-tau181 to a reference AD prediction model that marries age, sex, years of education, and ApoE increased the model's AUC from 0.82 to 0.86. Most other groups have come to similar conclusions. Mielke reported that adding the age and ApoE4 genotype of cognitively unimpaired people increased the accuracy of plasma p-tau181 to identify amyloid-positives among them from AUC 0.78 to 0.84. For p-tau217, it upped the AUC to 0.86. By the same token, Tosun found that adding clinical information for the cognitively unimpaired, including age, sex, years of education, APO4 status, and global cognitive test scores, boosted p-tau181's AUC from 0.55 to 0.80.
Individualized Prediction. An online research tool allows users to test how various parameters, such as age, cognition, or plasma p-tau, increase a person's four-year risk of Alzheimer’s. [Courtesy of Oskar Hansson].
Researchers in Oskar Hansson’s group in Lund University, Sweden, are testing different plasma tau-based prognostic algorithms to see which one best predicts AD over a four-year period. They previously reported that in Sweden's BioFinder cohort, baseline plasma p-tau217 in people with mild cognitive impairment predicted deterioration to dementia with an AUC of about 0.88. In ADNI, plasma p-tau181 performed less well, but when the scientists added baseline NfL into the mix, the results were on par with BioFinder (Nov 2020 news on Cullen et al., 2020). This data led Niklas Mattsson-Carlgren at Lund to develop an online research tool based on age, sex, cognitive score, and plasma or CSF level of p-tau181 and NfL. It predicts deterioration to dementia over four years.
But what about people who do not already have cognitive impairment? In their ADPD presentations, Hansson and Sebastian Palmqvist, also from Lund, described how they are building algorithms from a broader range of measures that are sensitive in the preclinical phase of AD. They include plasma markers, scores in different cognitive domains, structural MRI, and demographics. They started with plasma p-tau217 and then added variables such as age, sex, APOE genotype, cortical thickness, memory, executive function, verbal recall, and plasma NfL until hitting upon the most predictive combination. The best model had six ingredients: plasma p-tau217, plasma NfL, APOE genotype, cortical thickness, delayed recall for memory, and Trail Making Test B for executive function. This pegged conversion to AD within two, four, or six years with AUCs of 0.91, 0.92, and 0.94, respectively. This trajectory indicates that this algorithm will predict AD even better over a longer time frame.
An algorithm that relied on only four ingredients—plasma p-tau217, APOE, memory, and executive function—performed nearly as well, with AUCs only 0.01 lower. Interestingly, this algorithm still did a much better job than did dementia clinicians at baseline, whose own AUC for AD in four years was 0.72. “Clearly, this algorithm is very powerful at predicting AD dementia,” said Hansson.
Next, the researchers tested their algorithms in the ADNI dataset. Here, the best model was slightly different. It included sex and years of education, plasma p-tau18 but not plasma NfL. (In ADNI, p-tau217 was not measured). Still, the AUC for a four-year prediction reached 0.91, and the four-parameter model worked almost as well, again yielding AUCs only 0.01 lower.
Finding Cutoffs
The different plasma p-tau measures—217 in one cohort and 181 in the other—presented Palmqvist with the quandary of how to directly compare algorithms for the BioFinder and the ADNI datasets. To solve it, he treated each p-tau marker as either positive or negative based on a cutoff, rather than treating each as the continuous variable that it truly is. While this reduced the accuracy, yielding AUCs of 0.89 and 0.86 for BioFinder and ADNI, respectively, now the two algorithms were directly comparable. Anyone can try out these research algorithms using an online tool.
For real-world deployment of any plasma test, scientists will have to set cutoff values for plasma p-tau species that indicate a person's risk for dementia, much like cholesterol values do for cardiovascular disease and insulin for diabetes. Setting cutoffs is yet another task of applied science that commands no glory but is oh-so-important for any test to work robustly at your local doctor's office. AD researchers have begun testing these waters.
Clark determined that a cutoff of 9.6 pg/mL for p-tau181 improved the prediction of an algorithm based on age, sex, years of education and APOE, bumping up its AUC from 0.82 to 0.87.
For her part, Mielke has tested various cutoff values using data from a clinical trial population. Plasma p-tau181 and p-tau217 values of 1.56 pg/mL and 0.25 pg/mL, respectively, or greater, predicted amyloid-positivity with specificities of 92 percent and 96 percent, respectively. Alas, sensitivities were low, around 50 percent, meaning the algorithm missed half of the people who did have brain amyloid. Reducing the cut points to 1.31 and 0.23 pg/ml for p-tau181 and p-tau217, respectively, improved the sensitivities by 15 percent and 7 percent, without compromising specificity much.
Still, these numbers are meh. “A lot of work is going to be needed to identify the best cut points for screening, or perhaps diagnostic purposes, and that can be replicated across cohorts,” Mielke said.
Case in point: the sixfold difference in cutoff values that Clark and Mielke settled on. Clark told Alzforum that he opted for a value that would maximize his ability to predict “cerebral AD” in cognitively impaired subjects. By cerebral AD, he means a CSF p-tau181/Aβ1-42 ratio of greater than 0.0779. “One must remember that the cut-offs are dependent on the model in which they are used, including confounders, but also on the cohort.” Clark believes it is difficult to harmonize cut-offs since many parameters differ between studies. “Nonetheless it is encouraging that defining these is plausible, because it opens up the possibility of efficient and reliable diagnosis and risk assessment in the clinic,” he said.
Mielke cautioned about co-morbidities, such as BMI and kidney disease, that influence plasma p-tau levels. “This will be important when interpreting levels for clinical patients, especially in the general population, and also in the development of reference ranges,” she said. “Additionally, as studies examine levels across race and ethnicity, it is very important to consider these variables in the interpretation of any notable differences that could be due to comorbidities and health disparities, and therefore not to overinterpret results as solely due to race or ethnicity.”
All told, researchers agree that much work remains to be done before these exciting markers are ready for prime time. Besides cut-offs, they are beginning to address questions such as which isoform, assay, and platform might be best, how reproducible those assays are, and how they might work in different populations and for what purpose. In one bit of good news, Mielke reported that over three consecutive tests 15 months apart, p-tau181 and p-tau217 levels were within 92 and 95 percent agreement in the same person, respectively, suggesting that at least intra-individual variability is low. Likewise, in his presentation, Kaj Blennow, University of Gothenburg, reported that the coefficient of variation for plasma p-tau181 over 24 months was about 7 percent in cognitively unimpaired groups, a bit higher in those with mild cognitive impairment, and 12 percent in people with AD. This, too, is far less than what was seen with, for example, CSF Aβ42 or total tau tests early in these markers' “careers.” “This low variability suggests that p-tau181 could be used to monitor drug effects on tau pathology in clinical trials of this duration,” he said.
Hansson thinks so, too. He showed data indicating that people whose plasma p-tau217 rises more steeply over six years decline faster on cognitive tests over the same period of time, suggesting the marker could be a good readout of disease progression.
Along those lines, Hansson thinks these markers are attractive for trials to use to select participants whose likely rate of progression can be estimated better than in years past. This may well happen before their rollout in broad clinical practice. His group reported that among people in BioFinder who were deemed cognitively normal, performance on the PACC declined much faster in those who tested positive for plasma p-tau217 as per the research group's internal cut off.
As has been done before with new biomarkers, Hansson calculated that a trial's sample size could be shrunk by 70 percent by prescreening candidate participants with a combination of plasma Aβ42/40, p-tau217, and NfL. The PACC was designed to detect subtle changes in cognition among people who still appear normal on standard cognitive batteries such as the ADAS-cog, which is most sensitive at a later, more clearly symptomatic stage of AD (Jun 2014 news).
Will plasma p-tau replace brain imaging? This remains to be seen. Researchers are indeed toying with this idea not just for tau, but for all three biomarker criteria in AD—amyloid, tangles, and neurodegeneration. “I think most people would agree that it’s pretty simple,” said Jack. “If plasma is ultimately as accurate as CSF/imaging, then plasma will supplant imaging biomarkers. But if plasma turns out not to be as accurate as imaging, then it is reasonable to expect that plasma will be used for screening, and CSF and imaging will be used for people who pass certain screening criteria.”
Hansson was optimistic on this score. “I think within five years we will have these assays set up on fully automated platforms, and we will be using them in specialized clinics to replace CSF and PET analysis,” he said. He cautioned that appropriate use criteria have to be defined. He is even optimistic that these tests will be used in primary care settings. “But we need to do a lot more studies in those populations,” he agreed.—Tom Fagan
Clinicians from Developing Nations Discuss Dementia in Their Populations
Most dementia and Alzheimer’s disease epidemiology research has focused on people in developed countries, e.g., the United States, Canada, Australia, Japan, Taiwan, and Europe. However, in many low- and middle-income countries, life expectancy is rising, and by 2050, it is estimated that most of the world’s elderly will live in developing nations (Shetty, 2012). With an aging population comes an increase in people with dementia, making diagnosis, management, and treatment of these diseases a pressing concern.
At the virtual AD/PD conference held last month, the pharmaceutical and diagnostics company Roche sponsored a symposium on AD perspectives from around the world, where clinicians presented case studies, local challenges, and best practices in their cities or countries. One hope is that connecting dementia leaders in this way will improve diagnoses and help people and families suffering from AD across the globe. Another hope on the part of the sponsor might be to develop market interest in such countries for diagnostic blood tests this company and others anticipate selling before too long.
The Lancet Commission's most recent report on dementia prevention, intervention, and care stated that, globally, low- and middle-income countries are home to about two-thirds of all people with dementia. It named 12 modifiable risk factors that increase the risk of dementia in both developed and developing nations (Aug 2020 conference news).
Many patients in developing nations lack access to adequate health care, including dementia treatment. In Nigeria, for example, an estimated 4.9 percent of people age 60 and older have dementia (Adeloye et al., 2019). However, people in rural areas live far away from the nation's few centers with necessary diagnostic tools like neuroimaging, said Adesola Ogunniyi from University College Hospital in Ibadan, Nigeria.
In Brazil, a 2013 study projected that country's prevalence to be around 7.9 percent by 2020 among people 65 years and older, which would place Brazil among the highest age-standardized population prevalences of AD and other dementias in the world (Burlá et al., 2013; Feter, Leite, and Rombaldi, 2020). Its residents face a similar problem in that PET scans and cerebrospinal fluid (CSF) biomarker tests are only available in large cities and are not covered by the public health system or private medical insurance, according to Paulo Caramelli from the Federal University of Minas Gerais, Belo Horizonte, Brazil. Amyloid PET scans exist in only two academic medical centers across this country of 211 million people. In both Nigeria and Brazil, older women, widows, and people with few years of education, or who are socially isolated, are at a higher risk of dementia.
In China, around 5.6 percent of people age 65 and older have dementia; indeed, the country fields roughly 25 percent of all dementia patients worldwide (Jia et al., 2020). Yet there is little public awareness of the disease, and 70 to 80 percent of all dementia patients receive no formal treatment, said Jiong Shi from the Cleveland Clinic. Shi has disclosed a relationship with Roche in a recent scientific paper.
Are fluid tests the way forward? Fluid biomarkers may offer an affordable, easy-to-use tool to help diagnose dementia across large populations. Henrik Zetterberg, University of Gothenburg, and other clinicians in Sweden have been using fluid biomarkers in clinical neurology practice for some time. CSF Aβ42, for example, has been shown to differentiate healthy people from those with AD, and NfL is useful in diagnosing a range of neurodegenerative conditions (Mar 2015 news; Jun 2019 news). Research on plasma biomarkers—raising hope for simple blood tests—has made tremendous strides in the past year alone (Apr 2021 conference news). For a meta-analysis survey of fluid biomarker research in Alzheimer's disease, see AlzBiomarker database.
By adopting plasma markers in particular, physicians in developing nations will be able to offer a more precise approach to AD diagnosis. But there are challenges. For one, these fluid biomarkers are not ready for prime time, and they must be used in conjunction with other AD diagnostic measures, such as neuroimaging or cognitive/clinical assessments, Zetterberg noted during the symposium.
For another, many of these countries’ health care systems are not designed to care for people with chronic illnesses, especially for the comorbidities that tend to afflict the elderly. Until recently, developing nations, owing to poverty and high child-mortality rates, have had younger populations, hence their health care systems are structured around treating single, acute diseases such as infections.
Moreover, dementia is poorly understood in many of these nations, and the result can be stigmatization and minimal care. In Nigeria, for example, some people still ascribe dementia to possession by evil spirits or to witchcraft, causing families to conceal their medical problems, according to Ogunniyi. Superstition in Colombia has long called Alzheimer's "La Bobera," or "the foolishness."
Many medical professionals are not trained in dementia. In China, lack of education on dementia is a significant problem, Shi said. As a result, dementia diagnoses are made differently in academic hospitals than they are in non-academic and county hospitals, resulting in wrong and missed diagnoses. To properly use fluid biomarkers, education is key, Zetterberg told his colleagues. “There is a need to be able to understand when to test and what the results mean with respect to Alzheimer’s disease pathogenesis,” he said. Practitioners also need training on how to communicate biomarker results to their patients.
This doesn’t mean fluid biomarkers aren’t a viable option in low- and middle-income nations. “Nigeria can afford to do biomarker assays both as a research tool and as part of routing assessment of cases,” Ogunniyi wrote to Alzforum. “Patients who receive care at tertiary care centers, and in big cities like Lagos and Abuja, can afford such tests when adequately educated on the benefits and clinical utilization.”
For anyone in these nations—as indeed in developed countries—the speakers recommended lifestyle choices, such as a healthy diet, physical activity, limiting alcohol consumption, practicing social engagement, and avoiding air pollution, as much as possible. “Those are the goals of our care at the moment, as that would help prevent dementias according to the Lancet Commission on dementia,” wrote Ogunniyi.
Beyond lifestyle recommendations, Zetterberg hopes that research-grade assays for phosphorylated tau and other biomarkers in plasma can be standardized and certified, and then adopted into clinical practice in the near future. Blood tests are less costly than PET scans and less invasive than CSF tests.
Zetterberg envisions these biomarkers being distributed to, and used by, general practitioners and other doctors who aren’t dementia experts. Hopefully, this means the tests will reach larger populations and rural areas in developing nations, thus helping millions of people with dementia who currently lack care. In addition, biomarkers will make it easier for scientists to stratify people with dementia into subgroups for clinical trials, thus increasing the trial's ability to predict how fast people will progress, according to Carla Abdelnour from Fundació ACE in Barcelona, Spain.—Helen Santoro
Jia L, Quan M, Fu Y, Zhao T, Li Y, Wei C, Tang Y, Qin Q, Wang F, Qiao Y, Shi S, Wang YJ, Du Y, Zhang J, Zhang J, Luo B, Qu Q, Zhou C, Gauthier S, Jia J, Group for the Project of Dementia Situation in China.
Dementia in China: epidemiology, clinical management, and research advances.
Lancet Neurol. 2020 Jan;19(1):81-92. Epub 2019 Sep 4
PubMed.
New Mouse Models Better Mimic Tauopathy, Alzheimer's
More clinically relevant mouse models for neurodegenerative diseases are sorely needed as tools to study disease progression and to develop future therapeutics. At last month's virtual International Conference on Alzheimer’s and Parkinson’s Disease (AD/PD), two separate research groups labs offered some new options.
Michael Koob, University of Minnesota, Minneapolis, presented data on targeted replacement mice in which he swapped out the mouse MAPT tau gene against the human MAPT gene. His group also produced five strains into which they incorporated a pathogenic variant of the human MAPT gene, enabling scientists to mimic the genetics of different human tauopathies. Adrian Oblak and colleagues at Indiana University, Indianapolis, and at Jackson Laboratory (JAX), Bar Harbor, Maine, used a multi-gene approach to generate mouse models of late-onset AD, aka LOAD, which afflicts people 65 or older. The mice were fed a high-fat Western diet to examine the impact of some environmental influences on disease progression.
Targeted Replacement of MAPT
In 1998, several research groups first discovered pathogenetic variants in the MAPT gene on chromosome 17 as the underlying cause of certain tauopathies (Hutton et al., 1998; Dumanchin et al., 1998; Spillantini et al., 1998; Clark et al., 1998). Twenty-three years later, dozens more pathogenic tau mutations have been reported; however, exactly how any of them leads to dementia remains unknown.
Mouse models incorporating some of these mutations (see Alzforum Research Models database) have focused primarily on disease phenotypes, above all the signature protein deposits, not on the molecular genetic modifications that lead to these traits. “When you model toward the phenotype, you miss a lot of the subtleties,” Koob told Alzforum. In humans, disease phenotypes take 40 to 60 years to develop. “If you model having dementia when the mouse is 4 months old, you probably did something very different than what happens in a human, because that’s just not how that works,” Koob said
To address this problem, the NIH in 2019 funded the AD-related dementias gene replacement project, of which Koob is the PI. It aims to evaluate the impact of MAPT mutations within the context of the genomic sequence. Koob and colleagues developed a gene-replacement technology that allows them to take out the mouse MAPT gene and substitute in the entire human MAPT gene.
They produced two different wild-type versions of the mice. In H1.0, all 190,000 base pairs of the human sequence are of the H1 haplotype; in H2.1, the first 23,000 base pairs are H1, and the next 167,000 base pairs are of the H2 haplotype. The H1 haplotype block is more common, and carries more risk of neurodegeneration than the H2 haplotype (e.g., Mar 2019 news; Sep 2005 news). To induce a disease phenotype in these wild-type mice, the scientists inflicted a mild traumatic brain injury. After one week, they saw an accumulation of phosphorylated tau near the TBI site.
In addition, the scientists generated five pathogenic variant lines with single nucleotide changes in exon 9, exon 10, and intron 10. In people, these mutations lead to diseases including frontotemporal dementia and Pick’s disease.
Previously, researchers at the University of North Carolina, Chapel Hill, had developed a mouse model with targeted replacement of the endogenous murine ApoE gene with each of the human ApoE alleles. These mice have proven useful, for example in studying an anti-ApoE antibody that removes plaques from both the mouse brain's parenchyma and blood vessels without causing ARIA (Feb 2021 news). The mouse model out of Koob’s lab is the first to replace MAPT in a similar way.
Koob plans to make these mice available without restrictions to the research community as soon as possible. For this, Koob’s lab partners with JAX, which already has the H2.1 model available.
“The mouse models expressing human tau developed in the Koob lab will be a great resource for the AD and ADRD research effort,” Mike Sasner wrote to Alzforum. Sasner directs the Model Organism Development & Evaluation for Late-Onset AD, aka MODEL-AD, program. “While transgenic tau models—particularly Peter Davies’ ‘hTAU’ model—have been widely used for many years, the genome replacement mice should be much better models to study the underlying pathophysiology of AD and FTDs and are more translationally relevant for preclinical studies,” Sasner wrote.
Modeling Late-Onset Alzheimer's
Another limitation of current mouse models is that many reflect autosomal-dominant, familial AD, which accounts for fewer than 1 percent of people with Alzheimer's. Many of the older models overexpress their transgenes, causing artifactual phenotypes. To address these problems, the NIH started the MODEL-AD consortium (Oct 2019 news; Oblak et al., 2020).
As part of the MODEL-AD project, Oblak at IU and her team developed several LOAD knock-in mouse strains. Their LOAD1 mice have human ApoE4 and the TREM2 R47H variant knocked in; the LOAD2 mouse expresses knocked-in human ApoE4, TREM2 R47H, and humanized Aβ42.
Using these mice, Oblak and colleagues can ask such questions as how AD progression is influenced by environmental and lifestyle factors such as diet. A high-fat Western diet has been shown to boost TREM2 in microglia (Mar 2016 news). The scientists fed the LOAD1 and 2 mice a 45 percent high-fat and sugary chow from 2 to 12 months of age, collecting fecal and blood samples along the way. At AD/PD, Oblak showed that by 12 months of age, blood glucose and cholesterol levels were up in both mouse strains. The LOAD2 mice also had higher blood concentrations of the proinflammatory cytokines TNFα and IL10, and of neurofilament light chain, than did LOAD1 mice.
Combining risk variants for LOAD AD with environmental exposures such as a high-fat diet can produce a more relevant model for preclinical testing, Oblak said during the conference's live discussion. She believes that just because these models don’t have an overt neuropathological phenotype doesn’t mean they aren’t representative of the human condition.
“These predementia steps can be modeled really well in mice,” said Koob. “But this hasn’t been appreciated, because reviewers are focused on dramatic changes.” He hopes more groups will use the LOAD model.
With ever more models to choose from (peruse at Alzforum Research Models database), the question on many scientists’ minds is: Which one is the best? “There is never going to be one perfect model,” Frank LaFerla, University of California, Irvine, said during the live discussion. “What specific questions you are seeking to address determines which model you would pick.” LaFerla is a member of the MODEL-AD consortium.—Helen Santoro
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P.
Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17.
Nature. 1998 Jun 18;393(6686):702-5.
PubMed.
For α-Synuclein Immunotherapy, Is Going Later the Key?
α-Synuclein immunotherapy is in its infancy, with few trials posting results yet. The first two completed Phase 2s, of Biogen’s cinpanemab and Prothena and Roche’s prasinezumab, were both negative. However, while the cinpanemab study was fully negative, prasinezumab posted positive results on some secondary motor measures. At the 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, Gennaro Pagano of Roche gave researchers a first look at prespecified subgroup analyses from this study, called PASADENA. It turned out that subgroups of participants whose disease progressed fastest benefited more from prasinezumab, with more slowing of motor decline. Pagano believes this is because the signal-to-noise ratio in these subgroups was greater, allowing the small effect of prasinezumab to be discerned more readily in this slow disease.
“A key takeaway from PASADENA is that, in order to demonstrate the beneficial effect of an intervention designed to slow the course of a disease, there must be adequate disease progression to measure,” Pagano told Alzforum. Based on these data, Roche has started a new Phase 2b study in a more advanced, symptomatic population.
Werner Poewe of Innsbruck Medical University, Austria, agreed that patient selection is important. “Almost all trials to date, with the exception of those that target genetic forms of Parkinson’s, treat the disease as a single entity. It certainly is not,” he said in an AD/PD talk. He urged the field to develop biomarkers to distinguish different forms.
Signal-to-Noise. The slight slowing of motor decline by prasinezumab (left) was more evident in subgroups of patients whose disease progressed faster, such as those on MAO-B inhibitors (middle) and those with a more aggressive form of PD (right). [Courtesy of Roche.]
Prasinezumab recognizes the C-terminus of α-synuclein, and binds preferentially to aggregated forms. In Phase 1, the antibody entered the central nervous system but did not affect monomeric, physiological α-synuclein there (Jun 2018 news). The Phase 2 trial enrolled 316 people who were newly diagnosed with PD, with mild symptoms of slowness, rigidity, or tremor. None of them were on dopamine replacement therapy, and about one-third took monoamine oxidase-B inhibitors such as rasagiline or selegiline. For the first year, one-third of the participants received 1,500 mg of prasinezumab as a monthly infusion, one-third received 4,500 mg, and the rest placebo. Roche previously reported no effect of this yearlong intervention on the primary outcome measure, the MDS-UPDRS, but positive signals on secondaries (Apr 2020 conference news).
At AD/PD, Pagano elaborated on these findings. On the MDS-UPDRS as a whole, prasinezumab slowed decline by 14 percent. This fell short of statistical significance. However, in a planned secondary analysis, the researchers also examined each of the three parts of the MDS-UPDRS separately. Part I measures non-motor symptoms, part II activities of daily living, and part III motor symptoms. Pagano said that in early symptomatic PD, most of the decline occurs in motor symptoms; non-motor symptoms are already advanced, and ADLs are still strong. In the PASADENA cohort, participants on placebo worsened by only one point on part I over one year, compared with about 2.5 points on part II and five points on part III.
Prasinezumab had its greatest effect on part III, slowing decline by 25 percent and reaching statistical significance. Because motor abilities were scored by clinicians at each site, potentially introducing site-to-site variability, the videotaped motor assessments were also sent to a central group of raters. This prespecified central analysis returned a larger drug effect, saying prasinezumab slowed motor symptom decline by a third. This finding again implied that a higher signal-to-noise ratio rendered a treatment effect more detectable.
For all these analyses, both prasinezumab dose groups were pooled. Pagano noted that the two dosages had similar effects, and even the low dose was expected to achieve a high enough concentration in the CNS to saturate its target, based on Phase 1 data.
Digital More Sensitive? Motor decline tracked by smartphone revealed a slowing of the slope of decline on prasinezumab. [Courtesy of Roche.]
Parkinson’s is a notoriously variable disease, with patients doing better on some days than others. This means that MDS-UPDRS assessments done infrequently at a clinic are inherently noisy. For this reason, the PASADENA trial added digital motor assessments taken continuously at home using a smartwatch and smartphone. These measures were expected to be more sensitive and less variable (May 2017 conference news). Smartphone use produced a wealth of information, since there were 17 separate measures tracked either daily or every other day. With participants completing 93 percent of the requested tests, this resulted in 966,000 bits of data. For simplicity, the researchers combined several of the digital measures that assessed slowness of movement and resting tremor into a single motor score. They chose the measures expected to change the most in early PD, based on data from the Parkinson’s Progression Markers Initiative.
Prasinezumab performed similarly on this combined digital motor score as on the MDS-UPDRS part III, slowing decline by 25 percent in the pooled treatment group (see image above). In a separate talk, Kirsten Taylor of Roche broke down the details of this digital measure, noting that measures of arm and hand dexterity showed the greatest difference after treatment. For example, people taking prasinezumab were able to flip their hands from side to side more quickly and tap on their smartphone screens more accurately than people on placebo. Their smartphones also scored their hand movements throughout the day as being stronger than those of the control group.
If the effects of prasinezumab require a high signal-to-noise ratio to be visible, then subgroups of patients who decline faster should reap more easily apparent benefits. Prespecified subgroup analyses supported this, Pagano said. In one such analysis, researchers divided the cohort into people who are on concomitant MAO-B therapy and those who aren’t. Because MAO-B inhibitors are prescribed to Parkinson’s patients with more symptoms, this group is likely to have more advanced or faster-progressing disease. Indeed, the subgroup of 115 participants taking these medicines worsened by 7 points per year on the MDS-UPDRS part III, rather than 5. Prasinezumab slowed their decline by 40 percent.
Parkinson’s disease can also be classified by its severity. People whose decline is in the lower 75th percentile on all measures are considered to have “mild motor-predominant” disease, while those with several motor and non-motor scores in the top 25th percentile are labeled as having “diffuse malignant disease.” Everyone else falls into the intermediate category. In the PASADENA cohort, the 15 participants with diffuse malignant disease who were on placebo declined by 12 points on the MDS-UPDRS part III. For the 44 with diffuse malignant disease who received prasinezumab, this decline slowed by 64 percent.
“Based on these findings, we decided to invest further in this compound,” Pagano said. Roche has started the Phase 2b PADOVA study, which will enroll 575 people who are on levodopa therapy, indicative of more advanced PD. In the PASADENA trial, if a participant worsened enough to start dopamine therapy, their subsequent data was not included. The PADOVA trial will run for 18 months, with the primary outcome measure being how long it takes each participant to worsen by 5 points on the MDS-UPDRS part III. A change of 5 points is considered clinically meaningful, Pagano noted.
There is more data to come from the PASADENA trial, as well. In its second year, all participants went on prasinezumab, including those who were formerly on placebo. Data collection from that portion of the trial recently wrapped up, and the findings are being analyzed now, Pagano said. These data will help answer the question of whether prasinezumab slowed underlying disease progression, or merely reduced symptoms. If the former, people who were on drug in the first part of the study would be expected to maintain an advantage over those who were on placebo.
Other researchers welcomed the data from PASADENA. Tim Bartels of the U.K. Dementia Research Institute at University College, London, was encouraged by what the positive signals on secondary measures might mean for antibody development. “The results are very promising, and I think there is huge potential for improving [them],” he wrote. Bartels believes researchers may be able to refine the antibodies they use, based on ongoing investigations into what forms of α-synuclein are the most toxic in human brain. To Tiago Outeiro of University Medical Center Göttingen in Germany, the negative findings on primary imply that either prasinezumab did not target the correct toxic species, or that α-synuclein is not central to PD pathogenesis. “Future studies will be important to further document the actual effect of the antibody,” he wrote (full comment below).
What about other antibodies? In February, Biogen announced the discontinuation of cinpanemab development (see industry news). This antibody targeted α-synuclein’s N-terminus. Some preclinical data suggest that antibodies targeting the C-terminus, as prasinezumab does, may prove more effective (Masliah et al., 2005; Masliah et al., 2011; Games et al., 2014).
This may soon be tested, as other C-terminal-targeting antibodies are in the works. AstraZeneca’s MEDI1341 is in Phase 1, and Lundbeck’s LuAF82422 in Phase 1b. AbbVie had an α-synuclein antibody in Phase 1, ABBV-0805, but stopped the trial and has not announced future plans. The antibody is still listed in their pipeline. Meanwhile, UCB has just started Phase 1 with its α-synuclein antibody UCB7853.
Beyond antibodies, other PD drugs are in trials, targeting diverse pathogenic pathways (Apr 2018 conference news; Apr 2020 conference news). “The number of drugs in development has never been as large in the history of Parkinson’s,” Poewe noted at AD/PD.—Madolyn Bowman Rogers
Part 1 of a two-part story. Click here for Part 2.
As COVID-19 drove yet another conference online, it was also the topic of discussions there—held from the dull safety of researchers' home screens. At the 15th International Conference on Alzheimer's and Parkinson's Diseases, held virtually March 9–14, clinicians grappled with neurological symptoms they see in people with the disease, and they discussed what COVID complications could mean for people with preclinical dementia. Researchers pointed to activated microglia as the culprit behind COVID delirium. Trial investigators also described how they minimized lockdown disruptions and debated how to include people who had COVID in future trials (see Part 10 of this conference series).
Information continues to come in about what SARS-CoV-2 does to the brain. Previously, scientists reported that it could impair short- and long-term memory, increase brain-damage markers in the cerebrospinal fluid and blood, and, in severe cases, cause transient brain or vascular damage (Jan 2021 news). Genetic risk plays a role, such that people homozygous for ApoE4 are more likely to be infected with this new coronavirus and die from COVID-19 than noncarriers (Jan 2021 news). What else have researchers learned since then?
COVID Worsens Neurological Problems
On March 31st, scientists at the CDC's National Center for Health Statistics published U.S. mortality data for the year 2020 (Ahmad and Anderson, 2021). In addition to the 345,323 deaths from COVID-19 itself, there were 133,382 deaths attributable to Alzheimer's disease in 2020, compared to 121,499 deaths from AD in 2019, and 122,019 in 2018. At 9.7 percent, this represents a far larger year-on-year increase than in the years between 2015 and 2019. Among the country's 11 leading causes of death, only the “unintentional injury” category, which includes drug overdoses, rose as much as Alzheimer's in 2020 compared to the year before.
Amid this terrible toll, how do COVID-19 and Alzheimer's interact? It's not clear but at AD/PD, scientists reported that COVID-19 can cause serious neurological symptoms in some people. Why is that? Rocksy Situmeang, a neurologist at Siloam Hospitals Lippo Village, Tangerang, Indonesia, presented observations from her site. Her team collected medical records, laboratory tests, and brain scans of 22 people ages 26 to 98 who had COVID-19 with neurological symptoms and were referred to neurologists at the Siloam Hospitals Mampang, South Jakarta, from April to July 2020. Thirteen were women; the average age was 60. Eighteen had a history of health problems: six had hypertension; four had ischemic stroke or cancer, respectively; three, diabetes. Two each had Alzheimer's, Parkinson's, or sepsis. One person each had atrial fibrillation, pulmonary embolism, a heart attack, myocarditis, a ruptured aneurysm, or a broken leg.
After contracting SARS-CoV-2, half the participants complained of altered mental status, six of weakness on one side of the body. Five had tremors, four were in pain. Blood tests revealed that 11 participants had increased neutrophil-to-lymphocyte ratios, a marker of inflammation; 10 were anemic; nine had a high white-blood-cell count, another marker of infection or inflammation; six had low blood sodium, and four had low potassium levels. At Situmeang's clinic, six participants were newly diagnosed with ischemic stroke, three with electrolyte imbalances that affect brain function, three with AD, three with parkinsonism, and two with metastatic brain tumors. Where available, CT scans confirmed the strokes and tumors, and revealed chronic brain bleeds in one. Three participants had normal brain CT scans.
"I was not surprised that stroke and ischemic complications were the most frequent neurological issues," Dietmar Thal, KU Leuven, Belgium, told Alzforum. "Low blood oxygen seen in some COVID patients, coupled with existing small-vessel disease or cerebral amyloid angiopathy, may easily affect susceptible brain areas." Roger Nitsch, Neurimmune, Switzerland, agreed. "Virus-induced small-blood-vessel damage on top of CAA can have detrimental effects on blood supply in the brain," he told Alzforum.
Situmeang believes that neurological symptoms appear because inflammation caused by the virus aggravates pre-existing conditions. For example, people with dementia were 2.8 times more likely to catch COVID-19, 2.6 times more likely to become severely ill, and 2.6 times more likely to die, she recently reported (Hariyanto et al., 2021).
If people with dementia fare worse, could the virus affect the trajectory of the disease in people with presymptomatic Alzheimer's? "People with tau and amyloid pathology may experience worse symptoms due to inflammation triggered by COVID-19," Thal said, and Nitsch agreed, noting that brain damage, such as that caused by a stroke, is known to trigger rapid decline in people with mild or moderate dementia. "I would not be surprised if the same happens in COVID-19 patients, especially those with existing small vascular disease and hyperinflammation," he said.
Situmeang noticed that people who suffered a stroke while sick with COVID-19 had increased blood levels of D-dimer, a clotting marker, suggesting that they may be at risk of multi-infarct dementia caused by many small strokes. "We do not know how widespread small-vessel blood clots are in the brains of COVID-19 patients, but it is plausible if there are lots of clots," Johannes Attems, Newcastle University, U.K., said.
Delirium, Dementia—Are Microglia the Instigators? Tino Emanuele Poloni, Golgi Cenci Foundation, Milan, Italy, and colleagues reported other serious consequences of the virus in people with dementia. As did other groups, they identified that delirium precedes better-known COVID-19 symptoms, such as trouble breathing. Some patients were only delirious, never developing the telltale COVID symptoms. People who were delirious deteriorated faster and were more likely to die than those without delirium (Poloni et al., 2020; commentary by Wang, 2020; Kennedy et al., 2020). In older people hospitalized with COVID-19, those with dementia were three times as likely to be delirious as cognitively normal people (Harb et al., 2021). "People with dementia can suddenly become delirious, lethargic, and have rapidly worsening memory, which caregivers may just attribute to their dementia, not COVID-19," James Galvin, University of Miami, said at AD/PD.
What could be causing the delirium? Poloni searched for answers in brain tissue. At AD/PD, he described differences in autopsy samples from older people with or without COVID-19; some had also had dementia. The scientists obtained brain tissue from six non-COVID cases in the Abbiategrasso Brain Bank (Poloni et al., 2020) and from nine COVID-19 autopsies completed between April and June 2020 at the Institute of Legal Medicine, University of Pavia.
"Autopsies on COVID-19 patients have been discouraged due to the risk of spreading disease, but these cases were selected for forensic autopsies ordered by the state prosecutor," Poloni told Alzforum. COVID safety protocols included immediately fixing the brain in formalin and freezing only a small sample of fresh tissue for biochemical analysis. The researchers looked at the brain stem, cerebellum, frontal cortex, temporal and parieto-occipital cortices, and hippocampus. Four COVID-19 cases and three non-COVID cases had moderate to severe AD pathology.
The COVID-19 brains were swollen and inflamed, with CD68-positive activated microglia and GFAP-positive reactive astrocytes flooding the basal ganglia and hippocampus. Ameboid microglia were seen in the pons, frontal cortex, midbrain, medulla oblongata, and olfactory bulb, as well. The pathologists spotted abnormal, tuft-like capillaries in the pons and T- and B-cells surrounding blood vessels.
This matches what others have found. In a larger series, autopsies of 184 COVID-19 brains showed activated microglia in 43 percent, acute hypoxic changes in 30 percent, and dead tissue around blood clots in 21 percent of cases (Lou et al., 2021). "Poloni's autopsy findings and Situmeang's report of ischemic complications in COVID-19 cases fit well with one another and with other studies," Thal noted.
Poloni also found evidence linking activated microglia to delirium. He saw more of these cells in the hippocampi of the five cases with delirium before they died than the four cases without. Tau neuropathology showed a trend whereby people with more tangles were more likely to have become delirious early on in their bouts of COVID. Those with few or no tangles tended not to have been delirious.
One case stood out. Brain tissue from a young man who had died in an accident turned out to be positive for SARS-CoV-2. To Poloni's surprise, his pons and frontal lobe lit up with active microglia, even though the man had had no COVID symptoms and an otherwise healthy brain.
Many questions remain. "There is so little COVID-19 autopsy data that it is not always clear if the findings are related to the disease or to pre-existing conditions," Thal explained. Ronald Petersen, Mayo Clinic, Rochester, Minnesota, added, "A key unresolved point is whether COVID's neurological effects are due to direct viral infection of the brain, blood-brain barrier breakdown, or secondary effects of systemic inflammation."
In his series, Poloni found traces of the virus in lower brainstem tissue from only one COVID-19 case, leading him to doubt that it was actively replicating in the central nervous system. Likewise, other researchers found the virus and its spike protein in the lower brainstems and cranial nerves from 21 of 40 people who died of COVID-19 (Matschke et al., 2020). This hints that the virus or viral antigens may pass from the respiratory tract through the lower cranial nerves and inflame the lower brainstem. Definitive evidence for direct viral invasion of the brain is lacking (Boldrini et al., 2021).
Meddling Microglia
COVID delirium is the latest addition to the plethora of problems caused by activated microglia. In response to cell damage or infection, the inflammasome protein complex assembles in microglia, triggering them to crank out cytokines. This complex has been linked to AD by prompting tangle and plaque formation (Dec 2017 news; Nov 2019 news).
Could activated microglia in the COVID-19 brain be using the inflammasome to cause damage? Michael Heneka, German Center for Neurodegenerative Diseases (DZNE), Bonn, did not discuss COVID-19 directly at AD/PD, but thinks this is entirely possible. "If COVID-19 aggravates microglia, maybe it can trigger damage via the inflammasome, such as exacerbating tau pathology over time," Thal agreed.
Heneka mentioned two studies that will look at the immune system and lingering cognitive effects of COVID-19 in people with "long-COVID." The NIH-funded U.S. study, based at the University of Massachusetts Medical School, Worcester, will follow long-COVID patients for 18 months, testing their cognition and collecting MRI scans and blood samples. A similar study at the University of Bonn will track long-haulers for 12 months. "The U.S. study has not begun enrolling yet, and the German study is working its way through ethics approval," Heneka told Alzforum. In February, the NIH announced a major initiative to study long-COVID. It will provide $1.15 billion in funding over the next four years.— Chelsea Weidman Burke
Kennedy M, Helfand BK, Gou RY, Gartaganis SL, Webb M, Moccia JM, Bruursema SN, Dokic B, McCulloch B, Ring H, Margolin JD, Zhang E, Anderson R, Babine RL, Hshieh T, Wong AH, Taylor RA, Davenport K, Teresi B, Fong TG, Inouye SK.
Delirium in Older Patients With COVID-19 Presenting to the Emergency Department.
JAMA Netw Open. 2020 Nov 2;3(11):e2029540.
PubMed.
Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, Mushumba H, Fitzek A, Allweiss L, Dandri M, Dottermusch M, Heinemann A, Pfefferle S, Schwabenland M, Sumner Magruder D, Bonn S, Prinz M, Gerloff C, Püschel K, Krasemann S, Aepfelbacher M, Glatzel M.
Neuropathology of patients with COVID-19 in Germany: a post-mortem case series.
Lancet Neurol. 2020 Nov;19(11):919-929. Epub 2020 Oct 5
PubMed.
Clinical Trials in COVID Era: How To Keep Moving Forward
Part 2 of a two-part story. Click here for Part 1.
The COVID-19 pandemic has touched every aspect of health, from neurologic disease broadly (see Part 1 of this story) to clinical research. At the 15th International Conference on Alzheimer's and Parkinson's Diseases, held virtually March 9–14, clinical investigators talked about how they tried to prevail despite interruptions and preserve their trials. Looking to the future, how they will handle people who had COVID-19, or develop it during a trial, remains unclear.
"Many more questions were raised than answered," Ronald Petersen, Mayo Clinic, Rochester, Minnesota, told Alzforum. One thing is certain—the research community will feel the effects of trial delays for a long time. "The pandemic has delayed clinical research by three or four years," Tomislav Babic, Worldwide Clinical Trials, U.K., estimated.
COVID-19 threw a wrench into clinical research, including for people with dementia (Mar 2020 news; Jun 2020 news; Jan 2021 news). How have trials dealt with the crisis?
At AD/PD, Alison Searle, who works in clinical operations at Roche, described what her company did to continue Phase 3 trials testing whether the plaque-binding antibody gantenerumab slows cognitive decline in early AD. Gantenerumab is injected under the skin and was designed to be given at home, either by the participant or a home nurse. Serendipitously, this allowed researchers to ramp up at-home visits once social distancing became necessary.
In March 2020, Roche expanded home nursing capacity to almost twice as many participants at twice the number of sites. Home visits were allowed earlier in the trial, at week 12 versus week 16, which opened up home administration for most participants. Local study investigators had discretion about whether and how to continue visits based on the impact of COVID-19 in their region. There were temporary interruptions in dosing and study visits at certain sites due to the local impact of the pandemic. In total, about half of the possible home visits were completed during the first COVID peak in April 2020. The other visits were either done in the clinic or delayed by lockdowns. On average, five times more monthly home visits were made in the COVID era than before.
As COVID-19 vaccines roll out, Roche will not track who has been vaccinated, Searles said. However, it will space out vaccination and gantenerumab dosing to ensure side effects can be attributed correctly.
For its part, Eisai/Biogen also implemented home administration—in this case infusions given by a nurse—and remote assessments during trials for the anti-amyloid antibody BAN2401 (Nov 2020 news). While this change has allowed the gantenerumab and BAN2401 trials to proceed through the pandemic, other trials may be unable to do the same. "Home nursing feasibility varies by country and is not a universal solution right now," Searles noted.
How have other researchers adapted? Roy Alcalay, Columbia University, New York, described how the PD Generation (PDGENE) trial of genetic testing in Parkinson's transitioned from in-person counseling and testing to remote visits via phone. The scientists made this change because of the pandemic, and were then pleasantly surprised to discover that losing the geographic constraint of clinic location opened their trial to more participants across the country and sped up recruitment.
Mark Espeland, Wake Forest School of Medicine, Winston-Salem, North Carolina, described how the World-Wide FINGERS study group navigated through the lockdowns (Röhr et al., 2021). This network of 13 lifestyle intervention trials involves 27 countries (Kivipelto et al., 2020). Espeland and colleagues discussed three, each at a different stage. Japan's J-MINT is recruiting, the U.S. POINTER study is recruiting and delivering interventions, and Germany's AgeWell.de is delivering interventions and follow-up. Each had to adapt differently to stay the course (see image below).
Going with the Flow. In three FINGERS trials, steps were delayed to accommodate lockdowns (grayed areas) and waves of COVID-19 cases (gray bars). [Courtesy of Röhr et al., 2021, Alzheimer's & Dementia.]
J-MINT halted recruitment from late February to late May 2020, restarting in person with social distancing, masking, and other familiar safety measures. Slowed recruitment has delayed the trial by more than six months.
Just as interventions were taking off in Germany, the first lockdown grounded the AgeWell.de study. Like their Japanese counterparts, the Germans instituted COVID safety measures to resume in-person meetings as quickly as possible. They also offered telephone visits for participants reluctant to meet in person. To gauge how debilitating social distancing and closures were, the researchers mailed participants a survey during the first lockdown. They will do so again to track the pandemic's impact over time.
During its March-to-July pause, the U.S. POINTER team tried to keep its participants hopeful and motivated with regular phone and video check-ins. Virtual intervention meetings continued after the trial resumed.
The Path Forward
How to bridge the data gaps created by lockdowns is a question that haunts statisticians who are currently searching for solutions. It also plagues internal review boards as they adjust protocols. "Missing dosing and data collection appointments means that primary endpoints are not measured at the planned intervals, causing dramatic protocol deviations," Roger Nitsch, Neurimmune, Switzerland, explained. The FDA is trying to support trialists and has issued guidance on this problem.
What about people who had COVID-19? "Will someone still be eligible for a study if they were sick or treated with an experimental drug?" Petersen asked. To avoid potentially muddied results from unknown long-term effects of the virus, researchers only include people free of the disease, for now, Babic said. "Trials may specifically enroll people who had COVID-19 to study effects on neurological diseases and dementia, but trials likely will not mix COVID and non-COVID people," he explained. Researchers are just beginning to study lingering COVID symptoms, termed “long-COVID” (see Part 1 of this story).
What if a participant contracts COVID-19 during a trial? "Should you make subgroups for them, or should they be excluded?" Petersen asked. "We have not stratified people if they catch the flu while in a trial, but we may treat COVID infection differently, especially based on its severity," he added. Babic agreed, saying that researchers should not automatically drop people who develop mild COVID-19. Dietmar Thal, KU Leuven, Belgium, suggested that trials note cases of COVID-19 and monitor but do not automatically exclude these participants. "If in a few years we know that having COVID is not a big problem regarding pathophysiology and treatment of AD, then those patients could still be included in the trial," he said. Nitsch proposed trying to match how many people contract COVID-19 in each arm of a study, though this may be tricky to control.
There is no one-size-fits-all solution. In exploratory trials, counting COVID-19 as an adverse event and excluding participants who develop it during the trial may be best, Babic explained. This may not be necessary for confirmatory studies.
There is at least one upside to the pandemic—the research community has learned to adapt and become more resilient. "Clinical trial planning can now incorporate changes made during the pandemic, such as home visits and drug administration, to be better prepared for the future," Babic said.—Chelsea Weidman Burke
Drop of Hope? No Cognitive Worsening on BACE Inhibitor
Can BACE inhibitors be resurrected as an Alzheimer’s therapy? Data presented at the 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, suggested that the possibility remains. Alas, the path to get there looks rocky.
Michelle Gee of Eisai went over data from the terminated Phase 3 MISSION AD trials of Eisai and Biogen’s elenbecestat. In a bit of good news, the full dataset for this compound showed none of the cognitive decline seen with previous BACE1 inhibitors. However, neither did elenbecestat help cognition. Plus, it brought many of the other worrisome side effects seen with this class of drugs at the initial, high doses used—volume loss in the brain, weight loss, skin rashes—as well as new ones, with a few participants having abnormal liver readings and dwindling white blood cell counts. It was this safety profile, along with the lack of efficacy, that led to the early trial termination. “The results support a lack of benefit for elenbecestat in this specific, early AD population,” Gee concluded.
Meanwhile, basic science research continues to unravel the complex effects of BACE inhibition on the body. Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases in Munich identified a new BACE1 substrate, the cytokine receptor glycoprotein 130 (Gp130). BACE1 inhibition boosts signaling through Gp130, possibly leading to pro-inflammatory or synaptic effects, Lichtenthaler said. Another study cast doubt on the idea that lowering the dose of inhibitors to avoid side effects would fix everything. Gerhard Multhaup of McGill University, Montreal, argued that BACE1 plays a role in degrading toxic Aβ42, as well as forming it. At low doses of inhibitor, BACE1’s protective role is more curtailed than its harmful one. That might leave but a narrow therapeutic window, Multhaup suggested (see Part 12 of this series).
Some see the odds for BACE inhibitor therapy as long. “BACE inhibitors have a very high hurdle [to overcome] if they’re going to progress as a treatment for AD,” Gee said at AD/PD. Others were encouraged. “I do think BACE1 inhibitors are still viable,” Robert Vassar of Chicago’s Northwestern University told Alzforum. To Lichtenthaler’s mind, the elenbecestat data confirm that BACE can be inhibited without causing cognitive decline. “With this new data on elenbecestat, there is a convincing reason to continue using BACE1 as a drug target. … We now need comparative studies of different BACE inhibitors, ranging from studies on synaptic deficits in mice to detailed plasma and CSF biomarker analytics, including different BACE1 substrates,” Lichtenthaler wrote to Alzforum (full comment below).
Cognitive Readout: No Harm, But No Gain Either
During the great clinical reckoning for late-stage BACE inhibitors, elenbecestat was the last one to fall (Sep 2019 news). Since then, pharma researchers have been comparing data in hopes of gaining insights into what causes the adverse effects seen with this class of drugs, and whether they can be avoided (Dec 2019 conference news; Dec 2020 news).
At AD/PD, Gee provided the first detailed look at the final MISSION AD dataset. Together, the two trials enrolled 2,209 people with early symptomatic AD and an average age of 72. They had a mean MMSE of 25.7, indicating mild impairment. Half the participants, 1,101 people, took 50 mg elenbecestat as a pill once daily. This strongly suppressed BACE1 activity, lowering Aβ40 and Aβ42 by 70 percent.
In the final dataset, 1,472 participants had completed one year in the study, while only 189 had made it out to two years. Decline on all cognitive measures—comprising the CDR-SB, MMSE, ADCOMS, ADAS-Cog11, ADASCog14, and the word list from the ADASCog14—was identical in both groups at every time point except at six months. At that point, people on elenbecestat performed slightly worse on the MMSE and the three ADAS tests, with the difference reaching statistical significance on the ADASCog11 and the word list. This difference had disappeared by 12 months and did not reoccur.
Preliminary data from the MISSION AD studies, taken before data lock when there were only about 40 people with two-year data, had suggested a deficit in the CDR-SB at this final time point in people on elenbecestat; however, this did not hold up in the full analysis (Dec 2019 conference news). A functional measure, FAQ, was identical in both groups throughout the study.
It is unclear why elenbecestat did not cause the cognitive worsening of other BACE inhibitors. Elenbecestat is more selective for BACE1 over BACE2 than some of the other compounds, but so was umibecestat. That drug was terminated after subtle cognitive decline was seen at the three- and six-month time points (Jul 2019 conference news). At the time, some scientists privately grumbled that they would have liked to have seen the trials continue, even if just to learn.
Gee noted that in animal studies, high doses of elenbecestat did not reduce dendritic spine density, in contrast to other BACE inhibitors. This could indicate a lesser effect of this drug on the substrates that are suspected to cause cognitive worsening, such as SEZ6 and CHL1, Lichtenthaler speculated. SEZ6 is the leading candidate for this synaptic effect, since deleting this protein in mice prevents spine loss after BACE inhibition (Oct 2016 conference news). CHL1, on the other hand, guides axons to their targets, and BACE1 inhibition of its cleavage comes with stunted axons in the hippocampus (Dec 2013 conference news; Sep 2018 news).
Toward a side-by-side comparison of these drugs, umibecestat did not harm axon length, suggesting that this drug leaves CHL1 cleavage alone, yet it still harmed cognition (May 2019 conference news). There was no data on elenbecestat on axonal health.
So is SEZ6 the likely culprit? This remains unclear, but Novartis is analyzing CSF from the umibecestat trials to find out which BACE1 substrates correlate most with cognitive worsening (Aug 2020 conference news).
Mixed Message on Biomarkers: Less Plaque, More Shrinkage
While elebecestat did not affect cognition in the Phase 3 trials, biomarkers did show differences between the treatment groups. On amyloid PET, people on placebo gained an average of 8 centiloids of plaque over two years, while those on elenbecestat lost 5. This difference is much smaller than that seen with anti-amyloid antibodies, yet it was statistically significant, with a p value below 0.001.
On volumetric MRI, both treatment groups showed brain shrinkage, but people on elenbecestat lost more volume in the whole brain, hippocampus, and on a measure of cortical thickness. The differences were significant at p=0.001. These data match findings from the other four BACE inhibitors that entered late-stage trials—verubecestat, atabecestat, lanabecestat, and umibecestat—all of which have been found to accelerate brain volume loss (Sperling et al., 2021). Previous analyses do not chalk this up to neuron death or plaque removal. The leading contenders are fluid changes due to reduced inflammation, or synapse loss (Sur et al., 2020).
Fluid biomarker data were more ambiguous. The neurodegeneration marker plasma NfL was slightly worse at six months in people taking elenbecestat than controls, but Gee said NfL levels did not correlate with cognitive scores at this time point. At other time points, plasma NfL was identical in both treatment groups. In cerebrospinal fluid, there were hints of a benefit on elenbecestat, with total tau rising less than in the placebo group, and the neurodegeneration marker neurogranin trending down on drug while rising on placebo. However, Gee cautioned that these were sub-studies, and there were too few CSF samples to draw conclusions from these data. Lichtenthaler bemoaned the fact that the early trial termination deprived the field of this biomarker data.
Safety at High Doses Signals Caution
As did other BACE inhibitors, elenbecestat caused people to lose some weight. Participants on elenbecestat dropped an average of 1.4 kilograms, compared with 0.2 in the placebo group. Perhaps more concerning, about 6 percent of people on elenbecestat developed skin rashes and 5 percent reported abnormal dreams, also matching findings from other drugs in this class. Gee mentioned an increase in neuropsychiatric events as well, another common side effect, but did not show those data.
Other adverse effects were more specific to elenbecestat. The most troubling were a transient drop in white blood cell count, seen in 6.5 percent of people on drug, and elevation of several liver enzymes and markers of liver injury, occurring in 9.5 percent of the elenbecestat group compared with 4 percent of controls. Liver enzyme changes were what brought down atabecestat. Gee noted that the prevalence of these effects was low, with most people tolerating the drug well.
While these safety signals by themselves did not knock out elenbecestat, the lack of any signal for a treatment benefit prompted the safety monitoring board to pull the plug. “One commonality across [BACE inhibitor] trials is that we haven’t yet seen evidence of clinical effectiveness in early [symptomatic] AD,” Gee noted.
Some researchers think that if these drugs still hold potential, it will be as a preventative treatment in people at very early preclinical stages of Alzheimer's disease. Vassar, Lichtenthaler, and others argue for running 13-week trials of low-dose BACE inhibitors in people at risk of AD, while collecting plasma and CSF. “If cognitive worsening is avoided, but at least 30 percent Aβ lowering achieved, and beneficial effects on p-tau and NfL observed, then a large Phase 2/3 trial at the safe doses may be undertaken to see if conversion to MCI/AD could be delayed,” Vassar wrote to Alzforum (full comment below). If BACE can be inhibited safely, these drugs could also be used after anti-amyloid immunotherapy to keep Aβ from building up again, Vassar added.
“BACE1 remains a validated target, but the side effects are a concern. We need to understand these side effects and be able to control them,” Lichtenthaler said at AD/PD.—Madolyn Bowman Rogers
What BACE Hits: New Substrates Create New Headaches
While the Alzheimer’s field absorbs full Phase 3 data from elenbecestat, the last of the terminated batch of BACE inhibitors (see Part 11 of this series), scientists are also digging deeper into the myriad biological effects of BACE1 itself. They are hoping to unearth clues as to how BACE inhibitors affect the body, and which substrates might be responsible for the cognitive and other adverse effects seen in the trials conducted thus far.
At the 15th International Conference on Alzheimer’s and Parkinson’s Diseases, held virtually March 9–14, Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases in Munich implicated the cytokine receptor glycoprotein 130 (Gp130) as a new BACE1 substrate that might mediate the synaptic effects seen after inhibition. Meanwhile, Gerhard Multhaup of McGill University, Montreal, made a case for a protective effect of BACE1 in degrading Aβ42 to the harmless intermediate Aβ34. This effect might complicate inhibitor dosing, since in Multhaup’s studies low doses hampered BACE1’s protective effects more than its harmful ones. Together, the data underscore the complexity of BACE inhibition as a therapeutic strategy, suggesting researchers might have to thread a needle to achieve the desired effect.
Another Substrate Muddies the Water
Lichtenthaler teamed up with Matthew Kennedy at Merck to identify BACE1 substrates in CSF. Merck provided CSF data taken from three rhesus monkeys that had received a single 100 mg/kg dose of the experimental BACE inhibitor MBI-4 24 hours earlier. An analysis of more than 600 proteins found a handful that dropped with BACE inhibition. Some of these were known BACE1 substrates, such as APP and CHL1. Most changes were small, but two proteins stood out. CSF concentrations fell by more than half for the known BACE1 substrate SEZ6—and a new one, Gp130.
Cytokine Signaling. Gp130 (yellow ovals) blanket an IL-6 complex (blue and green) to activate transcription through a JAK-STAT pathway. New evidence suggests BACE1 can clip Gp130, preventing IL-6 signaling. [Courtesy of Gottschalk et al., Frontiers in Immunology.]
Lichtenthaler confirmed these findings in human CSF samples from Merck’s verubecestat trials. Thirty hours after participants took that drug, soluble SEZ6 had plummeted by three-fourths, and Gp130 by two-thirds. Lichtenthaler noted that this rapid response to verubecestat is likely due to both proteins’ short half-life in CSF, meaning they fluctuate rapidly in response to processing changes. He believes the proteins might make useful target engagement biomarkers to track BACE1 inhibition.
Gp130 normally binds to membranes, and its soluble version was previously believed to arise through alternative splicing, Lichtenthaler said. The new data instead suggest that it is a product of BACE cleavage. Lichtenthaler confirmed this in cell-free assays and primary neuron cultures, where suppressing BACE1 lowered soluble Gp130. He noted that some previous cell culture studies reported an increase in sGp130 upon BACE1 overexpression, strengthening the case for this relationship (Hemming et al., 2009; Stützer et al., 2013).
In its membrane-bound form, Gp130 participates in cytokine signaling. Extracellular IL6 forms a complex with the soluble IL6 receptor, and this complex binds a Gp130 dimer, activating intracellular signaling and gene transcription through the JAK-STAT pathway (see image above). Lichtenthaler noted that Gp130 is expressed in neurons (Watanabe et al., 1996). Its signaling is believed to be pro-inflammatory, with some evidence that it affects synaptic transmission.
For example, chronic IL-6 signaling amped up excitatory neuronal transmission in the mouse cerebellum, but suppressed excitation in cerebral cortex (Nelson et al., 1999; D’Arcangelo et al., 2000). Another study concluded that IL-6 shifts the balance between synaptic excitation and inhibition in rat cortex to favor the former (Garcia-Oscos et al., 2012). IL-6 signaling also has been linked to autism (Wei et al., 2016).
BACE1 cleavage of Gp130 could lower this signaling, while BACE1 inhibition would increase it. Supporting this, inhibiting BACE1 in cultured cells doubled the amount of activated STAT, Lichtenthaler found. It is not yet clear if this causes any harmful effects. Some previous research links alternative APP processing with heightened IL-6 signaling (Dos Santos Guilherme et al., 2019). IL-6 has been found to suppress neurogenesis, hinting at negative effects on cognition (Dec 2003 news).
In future studies, Lichtenthaler will examine whether BACE1 activity affects Gp130 signaling in vivo. He will also analyze whether the amount of sGp130, or any other BACE1 substrate, in the CSF of people taking verubecestat correlates with the cognitive deficits that develop on the drug. Such a relationship could provide clues to the mechanism behind this worsening.
BACE1 Makes (and Breaks?) Aβ. BACE1 starts the cascade that generates Aβ species including 40 and Aβ42, but it may also help to degrade these peptides into harmless Aβ34. [Courtesy of Liebsch et al., Nature Communications.]
Too Much BACE1 Inhibition, or Too Little?
Lest the solution be simply dialing down BACE1 inhibitor levels, Multhaup introduced another layer of complication. Previously, while working in Christian Haass’ lab at Ludwig-Maximilians University in Munich, Multhaup, who now chairs the department of pharmacology and therapeutics at McGill, found that BACE1 not only snips APP to produce the β-CTF fragment that is later cleaved into Aβ40 and Aβ42, but also chops off the end of these peptides to generate Aβ34 (Fluhrer et al., 2003; see image above).
Aβ34 is not toxic. It does not aggregate. Biochemistry and cell-based experiments flagged it as a marker for Aβ degradation and clearance, Multhaup noted (Hernandez-Guillamon et al., 2015). Human data from Randall Bateman and colleagues at Washington University in St. Louis confirmed that higher amounts of CSF Aβ34 correlate with increased turnover of longer Aβ species, as measured by stable isotope labeling kinetics (SILK), in people who have plaque buildup (Patterson et al., 2015; Liebsch et al., 2019).
How would changes in BACE1 affect Aβ34 production? Multhaup found that BACE1 knockout mice produce almost none of the peptide. Heterozygote mice, with half the normal amount of BACE1, have half the normal amount of Aβ34. In fact, Aβ34 tracked BACE1 levels more closely than did Aβ40 or Aβ42. In cultured cells overexpressing β-CTF, rising amounts of BACE1 caused Aβ40 and Aβ42 to drop, while Aβ34 rose. Conversely, inhibiting BACE1 in cultured cells made Aβ34 fall, and Aβ40 and Aβ42 rise slightly. Multhaup believes that this relationship helps explain a paradox in the literature, where overexpressing BACE1 in mice or human cell culture lowered Aβ40, rather than raising it as expected (Lee et al., 2005; Chiocco et al., 2004; Scholz et al., 2018).
What about in people? Multhaup found that Aβ34 tracked BACE1 activity in human brain as well, with both proteins being twice as high in AD as in control brain samples. The amount of Aβ34 varies with disease stage, however. Its levels peak at Braak stages I to III and then fall, perhaps hinting at a failure of clearance mechanisms later in disease. Multhaup had previously reported a similar pattern in CSF, where Aβ34 is high in people with mild cognitive impairment, and a high ratio of Aβ34/Aβ42 serves as a biomarker of preclinical AD (Liebsch et al., 2019).
Does BACE1 inhibition slow Aβ clearance in people? Possibly, Multhaup believes, as there have been independent reports of a drop in CSF Aβ34 after BACE1 inhibition (Mattsson et al., 2012).
To Multhaup’s mind, this hints at trouble for developing a safe and effective BACE1 inhibitor therapy. Multhaup’s group tested two such inhibitors in the same cultured cell line that overexpress β-CTF, and found that Aβ40 tended to rise at low inhibitor concentrations. This suggests that, in this cell line, inhibitors impair BACE’s amyloidolytic activity at lower concentrations than they impair BACE’s amyloidogenic activity, Multhaup said. The exact range of concentrations where this happened depended on the particular inhibitor’s affinity for BACE1.
These findings could complicate attempts to titrate the dosing of BACE1 inhibitors at a low level to avoid deleterious side effects, Multhaup noted. Too low, and the inhibitors might interfere with the clearance of toxic Aβ peptides. Too high—and we have already seen what happens. “Our data imply a partial BACE inhibition might not provide therapeutic benefits,” Multhaup said at AD/PD.
Others are not convinced. Vassar thought this was unlikely to be a problem for inhibitor therapy. “In my view, the main effect of a BACE1 inhibitor is to reduce the production of Aβ in the first place, which would trump any negative effect stemming from secondary inhibition of Aβ42 degradation to Aβ34,” he told Alzforum. Lichtenthaler agreed, pointing out that in clinical trials, low doses of the BACE inhibitor umibecestat did not raise Aβ40 or Aβ42 in CSF (Neumann et al., 2018). “I cannot rule out the possibility that Gerd [Multhaup’s] effect is seen upon single dosing or a short time after dosing, but it is not an issue upon chronic dosing,” he wrote to Alzforum.—Madolyn Bowman Rogers
If enhancing memory with light and sound seems futuristic, then welcome to the future. Or so some scientists say. Results from four early stage clinical trials on mild Alzheimer’s disease were presented at the AD/PD 2021 conference, held virtually March 9 to 14. The studies used two closely related approaches to modulate brain waves. Both reportedly synchronized neuronal firing activity in the gamma frequency range, strengthened neuronal connectivity—and perhaps strengthened memory by a bit.
Li-Huei Tsai, Massachusetts Institute of Technology, Boston, and colleagues some years ago turned light and sound into a therapy. They called it GENUS, short for gamma entrainment using sensory stimuli. Gamma brain waves, thought to be a neurophysiological correlate of attention and sensory processing, weaken in people with AD and in mouse models of amyloidosis (Dec 2016 news). Flashing a 40 Hz light at mice for an hour daily for a week enhanced and synchronized the mice’s gamma rhythms, which in turn rallied microglia to mop up plaques in their visual cortices. Adding a 40 Hz buzzing sound spread the benefits across the brain, cleared plaques and tangles, and improved the mice’s memory (Mar 2019 news; May 2019 news).
Other researchers have partially replicated these results. Sylvain Williams, McGill University, Montreal, and colleagues saw restored hippocampal gamma waves and slightly better spatial memory in amyloidosis mice after 40 Hz flashes using optogenetic stimulation, but plaques did not budge (Etter et al., 2019). Researchers led by Shuzo Sakata, University of Strathclyde, Scotland, U.K., actually saw plaques grow, also after optogenetic stimulation, but they did not assess changes in memory (Wilson et al, 2020).
Gamma Gains. In mice, light and sound induce gamma oscillations in the brain. These senses stimulate neurons, glial cells, and blood vessels, possibly clearing debris and improving synaptic function and memory. [Courtesy of Li-Huei Tsai, MIT.]
At AD/PD, Tsai presented new mouse data by her graduate student Mitchell Murdock. In 5xFAD amyloidosis mice, GENUS widened brain capillaries, increasing blood flow. It also spurred the glia-mediated lymphatic, aka “glymphatic,” system to flush the brain with cerebrospinal fluid and clear waste. How would sensory stimulation trigger CSF movement? Lined by contractile cells, blood vessels pulse not only to the rhythm of the beating heart, but also to a rhythm of their own making, and this shuffles fluid along (Iliff et al., 2013). Murdock found that after 40 Hz stimulation, arteries pulsed faster than veins, and Tsai believes this could explain the increased CSF clearing.
Could this work in people? To find out, Tsai and colleagues began a Phase 1 trial looking for data on safety, tolerability, and target engagement, i.e., entrainment. They recruited 46 healthy adults ages 18 to 85 and 26 older people with mild AD. The goal is 80 participants, but COVID temporarily stalled recruitment. Each sat in front of a panel of LED lights with a tablet in the center and a speaker that pulsed either 40 Hz light, sound, or both for one hour (see image below). The researchers played with the settings, either showing a movie on the tablet or not, and tweaking the device’s brightness and loudness, to make sure the stimulation entrained gamma waves. The researchers gauged participants’ brain activity using an electroencephalogram (EEG).
Entrainment Entertainment? Volunteers watched hundreds of flashing LED lights mounted on a four-square foot panel, and heard a high-definition sound bar buzz, both at 40 Hz. A center tablet entertained them to help them sit through an hour of this. [Courtesy of Li-Huei Tsai, MIT.]
While a one-time treatment of either light or sound alone boosted people's brain waves, the combination engaged the most brain areas in all participants. The flashing lights and humming sounds triggered no headaches, vision or hearing changes, or seizures, Tsai reported.
EEG measures mostly surface activity. Could GENUS stimulate coordinated neuronal firing deep within the brain? Tsai collaborated with Aaron Boes at the University of Iowa, Iowa City. He asked two people who had electrodes implanted into their brains because of their epilepsy to sit through a light and sound session. The researchers picked up more gamma power from the patients’ amygdalae, hippocampi, gyrus rectus, and posterior insulae during stimulation, according to Tsai’s plenary at AD/PD.
To find out what happens when people sit for this “show” regularly, the MIT scientists recruited 15 people with mild AD into a Phase 2A study. Before assigning treatment, the researchers used EEG to confirm that the optimized device settings from Phase 1 entrained gamma waves in all participants. Eight were stimulated with 40 Hz flashes and buzzes; seven received constant light and white noise as the control. Surprisingly, Tsai said, people could not tell which treatment they were receiving, possibly because they did not know what 40 Hz light looked like. “TV and computer screens are displayed at 60 Hz, so perhaps people did not expect 40 Hz to flicker that much,” she said.
Participants watched and listened for one hour daily over six or nine months, at home. To monitor compliance, the researchers recorded participants as they watched, logging how long the device was on and tracking their eye movements. Participants wore a clinical-grade Fitbit every day to track their activity and sleep. Researchers tested participants’ memory at baseline, one month, and three months, and captured structural and functional MRIs at baseline and three months. Three months into this trial, pandemic lockdowns halted data collection and delayed its follow-up timeline.
Here is what the scientists were able to glean thus far: Within one month, participants treated with flickers and hums slept better, and woke up less often during the night, than those treated with control settings. Functional MRI suggested three months of active treatment had strengthened connections in areas that process sight and in the posterior cingulate, a part of the default mode network involved in memory. Structural MRI suggested active treatment may have preserved tissue in the hippocampus, whereas control treatment did not slow atrophy. The eight people who received three months of flickers and buzz remembered more face-name pairs than the seven controls, and the difference correlated with stronger brain connectivity.
Participants in the active group were 6.4 years older than controls; they also had 5.4 more years of education than controls—the latter difference was statistically significant. Tsai said that years of education did not correlate with any outcomes, nor did it predict better performance on the face-name association test.
Regarding genetics, five treated people and four controls had one copy of ApoE4. They started the study with weaker connections in their visual networks than ApoE3 carriers, but all participants had a similar initial treatment effect regardless of their ApoE genotype.
In 2016, Tsai and her MIT colleague Ed Boyden co-founded Cognito Therapeutics in Cambridge, Massachusetts, to develop GENUS clinically. Rather than using the easel with the light panel and sound bar mounted to it, the company designed a wearable system dubbed GammaSense Stimulation (see image below). In 2018, it started three clinical trials in mild AD: Flicker, Overture, and Etude. The latter is expected to wrap up this year; the former two were presented at AD/PD.
AD Gearhead? The early version of Cognito’s apparatus combined black LED glasses with over-ear headphones. The next trials will use a sleek upgrade with a remote. [Courtesy of Annabelle Singer, Georgia Tech (left) and Kimberly Ha, KKH Advisors for Cognito Therapeutics (right).]
The Flicker Study
Also at AD/PD, James Lah, Emory University, Atlanta, and Annabelle Singer, Georgia Institute of Technology, Atlanta, presented results from the Phase 2 Flicker study, which wrapped up in February 2020. The primary endpoints were how people would tolerate daily stimulation, and if they would comply with using the device every day. The researchers recruited 10 people with mild AD from Emory’s Alzheimer’s Disease Research Center (ADRC). In their homes, five received an hour of daily double stimulation over eight weeks; the other five waited four weeks, then started their four weeks of stimulation. “This study was initially going to be placebo-controlled, but concerns about what an appropriate placebo was led us to choose a delayed start design instead,” Lah said in his presentation. This rendered Flicker an open-label trial.
First, the researchers set the flashes and buzzes from low to high intensity, asking participants to rank their comfort. Nine of the 10 tolerated the stimulation at full blast. For treatment, light and sound were then set at each person’s maximum tolerance level; for some, that was the highest intensity.
Were the strobes and sounds safe? Yes, the scientists contended at AD/PD. No one reported major side effects. That said, two volunteers experienced dizziness, two had headaches. Two people developed tinnitus, and one person's hearing loss worsened.
Participants stuck with the treatment, completing 95 percent of all sessions on average. The device automatically logged when it was on and for how long, and participants manually recorded their device usage. They stayed motivated with a study partner and weekly check-ins from a study researcher. “This was key to having such high adherence,” Singer noted. Nine chose to continue treatment in a 10-month, open-label extension.
Flicker In, EEG Out. A research volunteer receives 40Hz sensory treatment through a GENUS prototype apparatus while her brain activity is being recorded simultaneously via an EEG cap. [Courtesy of Annabelle Singer, Georgia Tech.]
Did their brain waves become entrained? The scientists recorded EEGs at baseline and after four or eight weeks of neuromodulation. At AD/PD, Lah reported that gamma waves matched the 40 Hz stimuli during each timepoint in an average of 49 of the 64 brain areas hooked up to the EEG. Gamma power stayed unchanged after four weeks of stimulation, and then it weakened after eight weeks. This stumped the researchers. “We expected to see stronger gamma waves, but the fact that we saw a difference in gamma activity at all was important” Lah said.
Singer and colleagues tracked brain connectivity using functional MRI captured at baseline, four, and eight weeks of treatment. Here, too, nothing changed after four weeks of treatment. After eight weeks, neuronal connections strengthened between the posterior cingulate and precuneus, two areas in the default network prone to disruption due to amyloid plaque buildup in people with AD (Sept 2020 news; Aug 2009 news).
As for neuroinflammation, Singer had recently published that, in wild-type mice, neurons jiving at 40 Hz thanks to gamma visual stimulation release cytokines, such as interleukin-4 and -6, that activate microglia to vacuum up debris (Garza et al., 2020).
How about in people? In Flicker, scientists measured 77 different cytokines in the CSF before and after eight weeks of daily stimulation. At AD/PD, Singer reported that about two-thirds decreased after eight weeks (see image below). She highlighted two in particular: macrophage inflammatory protein-1β (MIP-1β), which controls microglia, and TNF-like weak inducer of apoptosis (TWEAK), which upregulates NF-κB and other cytokines (e.g., Samidurai et al., 2020).
Other groups have shown that inhibiting TWEAK increases synaptic signaling in hippocampal slices from an amyloidosis mouse model, decreases microglial activation in an ALS mouse model, and attenuates neurological symptoms in a model of the auto-immune disease lupus erythematosus (Nagy et al., 2021; Bowerman et al., 2015; Wen et al., 2016). Singer thinks TWEAK may be regulating microglia and other cytokines. “It could be a key knob controlling many other immune factors,” she said.
Shifting Cytokines. After eight weeks of neuromodulation, CSF levels of two-thirds of 77 cytokines measured were lower. Some of them stimulate astrocytes (TGF-α), microglial proliferation (IL-5), and microglial motility (MIP-1β). Among the cytokines raised, some promote neural and glial cell differentiation (LIF), flag neurovascular injury (MMP-10), and recruit neutrophils (IL-8). [Courtesy of Annabelle Singer, Georgia Tech.]
What about amyloid and tau? In Flicker, amyloid PET, CSF Aβ, total tau, and phospho-tau 181 stayed the same after eight weeks. “Perhaps nothing changed because it was such a short and small study,” Singer said.
The Overture Study
This Phase 2A trial evaluated daily gamma stimulation for six months. In back-to-back presentations at AD/PD, Cognito’s Thomas Megerian and Suzanne Hendrix, Pentara Corporation, Salt Lake City, gave updates on the trial, which is slated to end in August 2021. It enrolled 76 people aged 50 and older across the U.S. who had mild to moderate AD; two withdrew before starting treatment. Under the watch of a caregiver at home, all participants wore the GammaSense device for one hour every day for six months. For 46 of them, the device emitted 40 Hz sound and light as active treatment; for 28, it was set to “sham” settings, which the company did not specify. To track compliance, the device records when and for how long it is used daily.
The control group was older and scored worse on memory tests, on average. Hendrix said she accounted for those imbalances during data analysis by including baseline memory test scores for each participant.
According to Megerian, participants tolerated the treatment well, and it was safe overall. That said, echoing the Flicker finding, people on active treatment complained of tinnitus more than those exposed to sham settings. In each group, 28 percent dropped out early by withdrawing their consent or having an adverse reaction that led them to quit. One of the three participants who left the treatment group due to an adverse reaction did so because they developed tinnitus. Others grew tired of the time commitment—people with moderate AD had a tougher time sitting for the daily hour than those with mild AD. Most dropouts happened within the first two months. “Once a participant hit two or three months, they stuck with it,” Megerian added. Forty-four of the 53 participants who finished the trial continued into a one-year open-label extension.
The researchers tracked brain changes with structural MRI before and after six months of treatment. Active treatment preserved 61 percent more tissue throughout the brain than control. “This was unexpected and very interesting,” Tsai remarked. Hendrix agreed. “It is fairly unusual to see large changes in the whole brain volume,” she said.
In AD, the hippocampus usually shows signs of atrophy early on, and in the Phase 2A study also reporting data at AD/PD (see above), GENUS appeared to protect this deep-brain area. In the larger Overture study, the hippocampi in active treatment and sham groups shrunk at similar rates, Hendrix reported.
Missed Primary. The cognitive clinical batteries ADAS-Cog14, CDR-sb, and MADCOMS remained unchanged after GENUS; functional and mental state tests showed less slippage. GENUS preserved whole brain volume, but not in the hippocampus specifically. [Courtesy of Suzanne Hendrix, Pentara Corporation, and Kimberly Ha for Cognito Therapeutics.]
What about cognition, though? At baseline, three, and six months of treatment, the participants sat for the ADAS-Cog14, CDR-sb, MADCOMS (a combination of questions from the two previous batteries), ADCS-ADL, and MMSE. The three former constituted the primary, the latter two, secondary endpoints. Memory and cognition did not differ between participants on active or sham treatment, with all three primaries falling far short of statistical significance (see image above). In contrast, daily functioning and mental status declined 84 and 83 percent more slowly in 40Hz-treated participants as measured by ADCS-ADL and MMSE, respectively. This was statistically significant.
Why some outcome measures showed no difference and some did puzzled researchers. “We need to carefully consider what aspects of cognition to measure,” Hendrix said. Future trials might explore how strengthening brain connections affects cognitive function more broadly, not just memory, Megerian said. Hendrix proposed comparing ADAS-Cog scores to measures of processing speed, executive function, or attention. “These tasks are not covered well by the ADAS-Cog, and we might be seeing effects in those areas,” she said. Megerian agreed. GENUS focuses on increasing broad neuronal connectivity that may improve functions not well examined by the ADAS-Cog, he said.
Hendrix wondered if GammaSense might help certain patients more than others, such as people with low or high brain amyloid loads. The researchers collected amyloid PET scans, CSF, and blood samples in this trial, but have yet to analyze them.
Even though Overture missed its primary endpoint, the researchers were hopeful. “The most important thing is that we see evidence of target engagement and some changes in imaging biomarkers and memory,” Lah said, adding, “These small studies warrant additional larger studies.”
Cognito is planning to start a Phase 3 trial later this year (company press release). Megerian said Cognito will also evaluate GENUS in other conditions, such as Down’s syndrome.—Chelsea Weidman Burke
It’s as if herpes were trying to push corona out of the limelight—it keeps popping up in the news. At the virtual AD/PD 2021 conference, Morgane Linard, University of Bordeaux, France, linked brain-tissue changes and a higher Alzheimer’s disease incidence to herpes simplex virus (HSV) infection, with ApoE4 carriers doing worse. Also at the meeting, Oliver Goldhardt, Technical University of Munich, tied AD biomarkers in the cerebrospinal fluid to herpes infection. Both teams measured herpes infection by detecting anti-HSV immunoglobulin G (IgG) in participants’ blood or CSF.
How about populations in France? Last year, Linard and colleagues saw much the same when they calculated the 10-year risk of developing AD in 1,037 people 65 or older from the long-standing Three-City cohort, of whom 178 had at least one ApoE4 allele (3C Study Group, 2003). ApoE4 carriers who had had frequent herpes flare-ups, as measured by higher anti-HSV antibody titers in their blood, were 3.3 to 3.7 times more likely to develop AD, whereas the risk in noncarriers was unaffected by herpes infection (Linard et al., 2020).
In her AD/PD presentation, Linard reported on the continuation of this work. Her team is probing whether herpes infection or ApoE status were associated with brain-tissue changes in old age. The scientists initially gathered data on 2,104 participants over 65 from the Bordeaux 3C cohort and 1,002 from the AMI cohort, a longitudinal study on elderly French farmers in rural areas (Pérès et al., 2012). Of those, they chose 1,599 on whom the study had collected both HSV serology and AD incidence data; 438 of those had had MRI scans. Half were women, 288 carried at least one ApoE4 allele, and the mean age was 77 years. The researchers corrected for confounding factors, such as age, sex, years of education, marital status, and cardiovascular risk factors.
Eighty-three percent of participants were infected with herpes, measured by the presence of HSV antibodies in the blood. Over an average follow-up time of 6.8 years, 293 participants developed dementia, of which 222 cases were AD.
Did herpes infection influence dementia development? In participants without an ApoE4 allele, it did not, Linard reported. However, infected ApoE4 carriers were 2.7-times more likely to develop AD than uninfected carriers. The risk correlated with antibody titers: Those with the lowest IgG concentrations were about twice as likely to develop AD, those with the highest IgG, four times as likely. Linard suspects that higher HSV antibody titers reflect more frequent viral reactivations over time.
What about brain tissue? The researchers extracted hippocampal volume in the scans of 330 participants, of whom 69 were ApoE4 carriers. Herpes infection itself did not affect hippocampal volume; however, the hippocampi of people with the highest herpes antibody titers were 1 percent smaller on average compared to the uninfected. This difference doubled in infected ApoE4 carriers compared to uninfected carriers, but hippocampal volume differs so much from person to person that this group difference was not statistically significant in this small sample. The scientists saw no links between herpes infection and hippocampal volume in noncarriers.
Linard and colleagues then checked the brain’s white matter. They used 243 participants in whom they had diffusion tensor imaging. DTI is an MRI method that tracks the direction and speed of water molecules moving through tissue as an indicator of the tissue’s microstructural integrity. Healthy white matter has high fractional anisotropy, a measurement of water molecules flowing along white-matter tracts, and low diffusivity, a measurement of the speed of water molecules diffusing locally. Compared to uninfected people, those with herpes infection had reduced fractional anisotropy and increased diffusivity in the cingulum and fornix, two axon bundles near the hippocampus.
DTI interpretation can be controversial (Wheeler-Kingshott et al., 2009). Still, Linard concluded from her data that infected people may have weakened white matter and she suspects that high diffusivity indicates neuronal damage. Participants’ herpes antibody titers did not track with DTI alterations, nor did their ApoE4 status.
If herpes simplex virus can distort brain tissue, does infection correlate with markers of brain damage in the CSF? At AD/PD, Goldhardt, of TU Munich, addressed this question using data from 117 older people with mild cognitive impairment or early AD, of whom 92 had herpes IgG antibodies in their CSF and 25 did not. The researchers measured Aβ40, Aβ42, the Aβ42/40 ratio, total tau, and p-tau in the participants’ CSF.
None of these biomarkers were different in infected people as a group versus uninfected people as a group. However, Goldhardt proposed that the ratio of herpes antibody titers in the CSF versus in the serum may be a way to gauge herpes virus activity in the central nervous system. A higher ratio would indicate more herpes antibodies in the CSF, possibly meaning more viral activity in the CNS than the periphery. Indeed, higher CSF-to-serum antibody ratios correlated with higher total tau and p-tau in the CSF. High CSF p-tau has previously been seen in older people with herpes encephalitis (Krut et al., 2013).
The presence of brain amyloid influenced the p-tau/herpes antibody correlation. CSF p-tau and CSF-to-serum anti-herpes IgG ratio tracked closely in people who had lower Aβ42/40 ratios, while this correlation was absent in those with higher ratios. “The association of CSF p-tau and herpes antibody ratio is weakened by higher Aβ42/40, which supports the idea that Aβ inhibits HSV,” Goldhardt wrote to Alzforum.
Previous cell-culture studies show that adding Aβ40 and Aβ42 to fibroblasts and neuronal cells muted HSV infection (Bourgade et al., 2015; Bourgade et al., 2016). In a mouse model of amyloidosis, some—but not all—studies have painted Aβ as protective against herpes infection (Jun 2018 news; Jan 2021 news).
Goldhardt hypothesized that HSV may more easily reactivate in people with AD pathology because they may have less soluble amyloid. Viral reactivation prompts a p-tau spike in the brain, perhaps reflecting a cycle of p-tau aggregation and inflammation. “I am not saying that HSV causes sporadic AD—we need herpes vaccination trials to clarify that,” Goldhardt noted in his presentation. Several treatment trials of valacyclovir are ongoing.—Chelsea Weidman Burke
Pérès K, Matharan F, Allard M, Amieva H, Baldi I, Barberger-Gateau P, Bergua V, Bourdel-Marchasson I, Delcourt C, Foubert-Samier A, Fourrier-Réglat A, Gaimard M, Laberon S, Maubaret C, Postal V, Chantal C, Rainfray M, Rascle N, Dartigues JF.
Health and aging in elderly farmers: the AMI cohort.
BMC Public Health. 2012 Jul 27;12:558.
PubMed.
Can electric current spark better memory in people with mild cognitive impairment? Possibly, according to researchers—at least short-term. At the 15th International Conference on Alzheimer's and Parkinson's Diseases, held virtually March 9–14, Alberto Benussi, University of Brescia, Italy, presented results from a neuromodulation pilot study. The complete findings were published in the March 21 Brain Stimulation. He and other researchers led by Barbara Borroni at U Brescia used transcranial alternating current stimulation (tACS), a low-intensity electric current therapy set at a gamma frequency of 40 Hz. After a one-hour session, people with MCI recalled more words and matched more names to faces than people who got sham treatment. Their acetylcholine signaling was restored to levels seen in healthy people, the scientists claimed. Whether gamma entrainment occurred will be tested in a larger trial.
This neurophysiological approach to therapy is similar, but not identical, to GENUS, which uses the sensory stimuli light and sound to coordinate neuronal firing via gamma entrainment (see Part 13 of this series). tACS stimulates specific brain areas with electrical current through electrodes on the scalp. Pulsing electricity at 40 Hz is thought to entrain gamma waves (reviewed by Strüber and Herrmann, 2020). Neuronal firing in the gamma band falls out of sync in people with MCI and Alzheimer’s disease (Koenig et al., 2005; Dec 2016 news).
Previously, other researchers had used tACS in cognitively normal older people to stimulate 4–8 Hz theta waves in the frontotemporal cortex. This synchronized participants' natural theta oscillations within 25 minutes of treatment, which in turn improved their working memory for at least 50 minutes afterward (Apr 2019 news).
For the GammAD pilot study, Benussi and colleagues recruited 20 people with mild AD from the Center for Neurodegenerative Disorders in Brescia. The researchers stuck one electrode to the top of each participant’s scalp and the other to his or her right shoulder (see image below). “Current flows from one to the other, so the second electrode guides the current from the skull through deep brain structures, such as the precuneus,” Benussi explained. They targeted the precuneus, an area in the default network, because amyloid plaques accumulate there early in AD (Aug 2009 news).
In at Red, Out at Black. During transcranial alternating current stimulation, low-intensity 40 Hz electricity flows from one electrode (red) to the other (black). The current is believed to travel through the precuneus deeply into the brain and out to the shoulder. [Courtesy of Benussi et al., 2021.]
All participants received both active and sham treatment in a crossover design, i.e., they got one, waited a week, then got the other. “We chose a crossover design to increase statistical power to tease out differences triggered by the treatment,” Benussi said. At the clinic, participants wore the tACS device for one hour. It emitted 40 Hz electric current for the entire hour during active treatment and for one minute during sham treatment. “After one minute, people cannot feel the current anymore, so they cannot tell if they receive pulses for longer,” Benussi wrote to Alzforum. All 20 participants completed the trial; none reported significant side effects.
Benussi and colleagues relied on the Rey auditory verbal learning test (RAVLT), before and after each treatment, to test the participants’ memory (see image below). Participants treated with tACS remembered 25 percent more words immediately, and twice as many after the 20-minute delay, than did controls. While participants were completing their last 20 minutes of treatment, they completed the face-name association task. People who got active treatment also remembered almost twice as many face-name pairs. “We were astonished to see clinical and neurophysiological changes after only one hour of stimulation,” Benussi said.
Crisscross. Participants received both tACS and sham treatment, separated by a week. They were evaluated with transcranial magnetic stimulation (TMS) and took the Rey auditory verbal learning test (RAVLT) before and after each session. During the last 20 minutes of treatment, participants matched faces to names. [Courtesy of Benussi et al., 2021.]
What caused this improvement—could it be synchronized gamma waves? The researchers did not record EEGs, so they do not know. “We are currently putting together a larger study where we will record EEGs to answer this question,” Benussi told Alzforum.
Benussi initially pursued a different concept, wondering if tACS might improve cholinergic transmission. People with AD have less acetylcholine than healthy people, which is why cholinesterase inhibitors were developed to slow its breakdown. This neurotransmitter deficit can be indirectly detected using transcranial magnetic (TMS)—not electrical—stimulation to inhibit short-latency afferent currents in cholinergic circuits. In this case, the authors placed the TMS coil on the scalp above the region of the motor cortex that controls primary hand movement. The TMS signal could then be used to block motor evoked potentials in the cortex. When there’s less acetylcholine around, this TMS-induced short-latency inhibition is weaker.
Benussi and colleagues previously used this strategy to detect transmission deficits in people with MCI that were not found in healthy people (Benussi et al., 2021).
Would tACS stimulation counter TMS-driven inhibitor of cholinergic circuits? Benussi reported that, in this pilot trial population, tACS stimulation restored short-latency afferent inhibition to levels seen in healthy people, suggesting there was more acetylcholine transmission (see image below). “Increasing cholinergic transmission could be one way this stimulation works,” Benussi wrote to Alzforum.
Back to Normal? In this pilot trial, people had less short-latency afferent inhibition of cholinergic circuits (right) after treatment (green bars) than after a placebo (blue bars). Inhibition was restored from baseline (left) to healthy control levels (blue line) as compared to levels typically seen in AD (red line). [Courtesy of Benussi et al., 2021.]
For next steps, Benussi and his colleagues want to define how much electric current is reaching the precuneus and if repeated treatment extends the benefit. They plan to see if ApoE status and baseline cognitive reserve affect treatment response. They will also study how long the short-term memory improvements last, since they only measured memory immediately after tACS in this pilot study.
To that point, another pilot study recently investigated longer-term effects of multiple sessions of gamma tACS in combination with cognitive training. Seventeen older adults with mild to moderate dementia performed cognitive training tasks in the lab for one hour daily, five days a week for four weeks; 11 also received simultaneous tACS. Participants sat for Wechsler Memory Scale (WMS-IV) testing at baseline, after four weeks of treatment, and one month after treatment ended. Both groups improved their scores after treatment, but the tACS recipients better sustained their improvement one month after treatment than did participants who did not receive tACS (Kehler et al., 2020).
TMS itself was being developed to treat AD by Neuronix, a company based in Yokneam, Israel. Their neuroAD chair apparatus paired repetitive TMS with simultaneous cognitive training (Apr 2017 conference news). In 2019, an FDA panel advised against its approval (Mar 2019 news). The company’s website is no longer active. Benussi’s group is not using TMS as a treatment, but rather as an indirect way to measure acetylcholine signaling.—Chelsea Weidman Burke

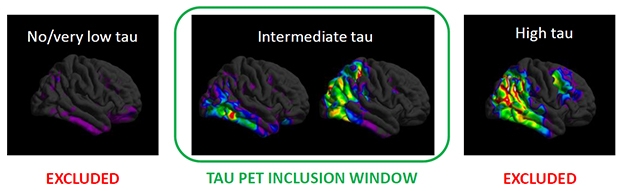

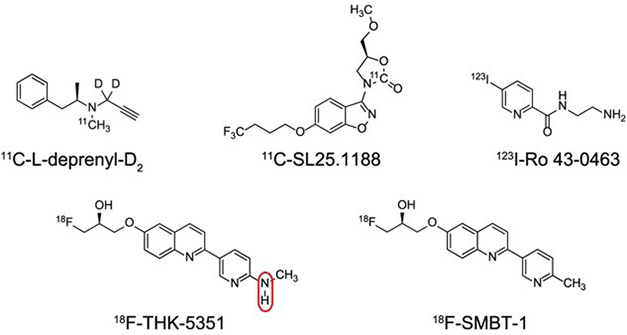

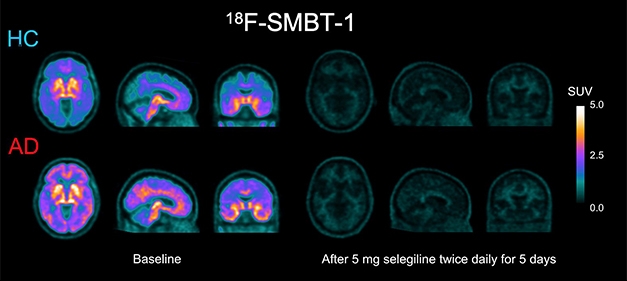



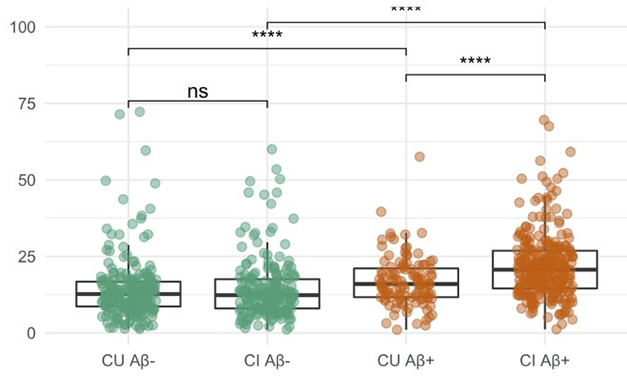





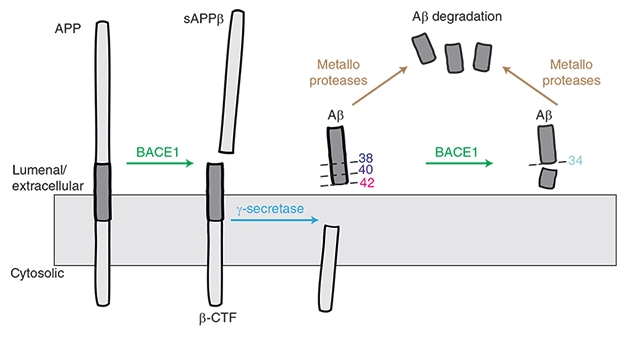





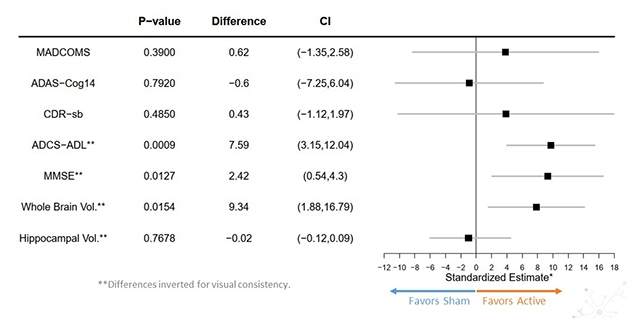



Comments
No Available Comments
Make a Comment
To make a comment you must login or register.