Microglia Inflammasome Stokes Tau Phosphorylation, Tangles
Quick Links
Microglia have been firmly linked to amyloid plaques, but scientists are only beginning to examine their relationship to tau tangles. In the November 20 Nature, Michael Heneka and colleagues at the German Center for Neurodegenerative Diseases in Bonn suggest that microglial inflammation incites tau toxicity, too. Activated microglia secrete proinflammatory cytokines that signal neurons to ramp up tau phosphorylation, the authors found. Keeping microglia quiescent by deleting their NLRP3 inflammasome blocked tangle formation in a tauopathy mouse model. Loss of inflammasome also prevented tau aggregation in response to injected Aβ.
- Activated microglia release cytokines that trigger tau phosphorylation in neurons.
- Ablating microglial NLRP3 inflammasome curbed tangles in tau mice.
- It also prevented tangles caused by injected Aβ seeds.
The findings add to the evidence that microglia propel Alzheimer’s pathology, and hint that they could link plaques and tangles in the amyloid cascade. “Aβ activates the innate immune system, and that drives tau phosphorylation,” Heneka said.
“These are exciting and dramatic results,” Marc Diamond, University of Texas Southwestern Medical Center in Dallas, told Alzforum. “It would be extremely interesting to see if similar effects result from inflammasome inhibitors, and whether those effects hold up across different mouse models of tauopathy and other proteinopathies.”
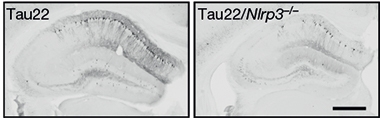
Inflammation Powers Tauopathy. Tau22 mice (left) accumulate phosphorylated tau (black) in their hippocampus; not so if they lack the NLRP3 inflammasome (right). [Courtesy of Ising et al., Nature.]
Previously, Heneka proposed a role for microglia in amyloid plaque seeding, proposing that activated microglia secrete protein complexes called ASC specks that accelerate Aβ deposition (Dec 2017 news). Other groups also blame microglia for amyloidosis, reporting an almost complete absence of parenchymal plaques when they are missing (Mar 2018 news; Sep 2019 news).
Links between microglia and tau pathology have been more tenuous. Virginia Lee’s group at the University of Pennsylvania, Philadelphia, implicated microglial activation in tangle formation in a tauopathy model (Feb 2007 news). Kiran Bhaskar’s at the University of New Mexico, Albuquerque, implicated microgliosis in tau pathology and spreading (Oct 2010 news; Maphis et al., 2015). Recently, David Holtzman’s at Washington University in St. Louis tied microglial ApoE to tangle formation and neurodegeneration (Oct 2019 news). Exactly how microglia promote tau pathology remained unclear, however.
To investigate, Heneka and colleagues turned to Tau22 mice, which express human tau carrying the P301S and G272V mutations. These mice develop tangles and gliosis at 3 months, memory problems at 6, and widespread pathology by 9 months of age. First author Christina Ising found that proinflammatory signaling in the cortex rose at 3 months. By 8 months, some microglia had assumed an activated, amoeboid shape. These cells had turned on the NLRP3 inflammasome, as seen by the release of ASC specks and the proinflammatory cytokine IL-1β, and they were adjacent to tau tangles.
Did this make tau pathology worse? Yes, Ising found. When the authors bred Tau22 mice with NLRP3 knockouts, the crosses accumulated only half as much hyperphosphorylated and aggregated tau by 11 months as the Tau22 mice. Phospho-tau was detected with antibody AT8, which recognizes phosphorylations at serine 202 and threonine 205. Mice without the inflammasome performed as well as wild-types in the Morris water maze.
The researchers used primary cell preparations to investigate how this might happen. Conditioned media from mouse microglia boosted phosphorylation at tau serines 396 and Ser404 in mouse neurons, pointing to soluble factors. Blocking the IL-1 receptor on neurons, or its downstream effectors, prevented p-tau. This suggested IL-1β might be a culprit. This cytokine appears to accelerate tau phosphorylation by affecting neuronal enzymes. In the hippocampi of Tau22 mouse lacking NLRP3, GSK-3β and CaMKII-α kinases were less active while the phosphatase PP2A was more active; together, this dialed down tau phosphorylation.
But what activates NLRP3 in the first place? In Tau22 mice, it might be tau itself, the authors report. In primary microglial cultures from these mice, 2 μM exogenous monomeric and oligomeric tau, but not fibrils, revved up NLRP3. Both wild-type and mutant human tau had this effect. The findings suggest a feedback loop, where tau accumulation kindles inflammation, which then fuels tau pathology.
What about AD? In this disease, Aβ likely kicks off the cascade that leads to microgliosis and tangles, conclude the authors. They injected brain homogenate from APP/PS1 mice into the hippocampi of 3-month-old Tau22 animals. Five months later, they had accumulated twice as much hyperphosphorylated tau in their hippocampi as Tau22 controls. NLRP3 knockouts were protected, however, with APP/PS1 homogenate having no effect on tau.
These data pry open the mechanistic link between plaques and tangles, Heneka believes. Researchers have long known that aggregated Aβ triggers tangle formation in mice (Aug 2001 news; Bolmont et al., 2007). Human imaging validated the mouse data, showing that the spread of plaques through the brain opens the floodgates for tangles to invade the cortex from the medial temporal lobe (Aug 2016 news; Jun 2017 news; May 2019 news). But the exact nexus between the two pathologies remains mysterious.
Now research is homing in on microglia as the missing link. A statistical analysis of human postmortem cortex found that microglial activation follows plaques but precedes tangles in the AD brain (Feb 2019 news). Genetics suggest that the key factor in whether someone develops Alzheimer’s is how their microglia respond to amyloid (Apr 2019 conference news; Aug 2019 news; Nov 2019 news). The present data strengthen the evidence for microglia being a linchpin of the cascade, after amyloid but prior to tau pathology and cognitive decline.
Heneka thinks of Alzheimer’s as a relay race, where a succession of different pathologies propel the disease. Amyloidosis is the first runner. Once it has passed the baton to microglia, targeting amyloid does no good, he said. Likewise, once microglia have initiated tangle formation, the window for dampening inflammation may have passed. This implies therapeutic interventions will have to be tailored to the disease stage. Complicating matters, different brain regions may be at different stages in this cascade at any given point in time, Heneka noted. And different microglia may be at different stages of activation, as well. A current analysis of human brain biopsy samples suggests a spectrum of microglial activation states that depend on a person’s age, location in the brain, and the type of trauma experienced (Nov 2019 news).
To try to figure all this out, Heneka helps run the DELCODE study at DZNE, which follows about 1,000 patients at different stages of AD with cognitive testing, fluid biomarkers, structural MRI, and PET. The goal is to link inflammatory markers in cerebrospinal fluid and blood to neurodegeneration in specific brain regions. “We want to build an inflammatory map of the entire disease trajectory in order to know which brain regions to treat at what stage of disease,” Heneka explained.
One possible treatment would be to inhibit the NLRP3 inflammasome. There are various compounds that do this, and Heneka noted that biotech and pharma companies are developing brain-penetrant inhibitors (Sep 2017 news; Nov 2019 news). Earlier this year, Novartis acquired IFM Tre, a subsidiary of IFM Therapeutics that has a systemic NLRP3 inhibitor in Phase 1 trials, and a brain-penetrant version in preclinical studies.
Microglial inflammation may help propagate other pathologies, too. Seung-Jae Lee at Seoul National University College of Medicine reported earlier this year that α-synuclein oligomers injected into mouse brain did not spread by templated misfolding alone. Instead, these oligomers stimulated microgliosis, which then released proinflammatory cytokines that triggered α-synuclein aggregation in nearby neurons (May 2019 conference news).—Madolyn Bowman Rogers
References
News Citations
- Do Microglia Spread Aβ Plaques?
- Wiping Out Microglia Prevents Neuritic Plaques
- Are Microglia Plaque Factories?
- Tau Toxicity—Tangle-free But Tied to Inflammation
- Paper Alert: Fractalkine Receptor Hits Aβ, Tau, in Opposite Ways
- In Tauopathy, ApoE Destroys Neurons Via Microglia
- Finally United? Aβ Found to Influence Tangle Formation
- Brain Imaging Suggests Aβ Unleashes the Deadly Side of Tau
- Analysis of PET Scans Suggests Link Between Amyloid and Tau
- Longitudinal Tau PET Links Aβ to Subsequent Rise in Cortical Tau
- In Pathology Cascade, Microglia Rev Up After Plaques but Before Tangles
- Expression, Expression, Expression—Time to Get on Board with eQTLs
- AD Genetic Risk Tied to Changes in Microglial Gene Expression
- Cell-Specific Enhancer Atlas Centers AD Risk in Microglia. Again.
- The Human Brain Hosts a Menagerie of Microglia
- New AD Target: Silencing the NLRP3 Inflammasome with Boron?
- Acetaminophen Derivative Tempers Microglia, Spurs Plaque Clearance
- Do Immune Cells Promote the Spread of α-Synuclein Pathology?
Research Models Citations
Paper Citations
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015 Jun;138(Pt 6):1738-55. Epub 2015 Mar 31 PubMed.
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007 Dec;171(6):2012-20. PubMed.
External Citations
Further Reading
News
- ApoE: Common Microglial Culprit in Aging, Alzheimer’s, and Tauopathy?
- Tau PET Aligns Spread of Pathology with Alzheimer’s Staging
- Imaging Clinches Causal Connections between Aβ, Tau, Circuitry, and Cognition
- Honolulu: The Missing Link? Tau Mediates Aβ Toxicity at Synapse
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
Primary Papers
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buée L, Heneka MT. NLRP3 inflammasome activation drives tau pathology. Nature. 2019 Nov;575(7784):669-673. Epub 2019 Nov 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
This paper takes the field closer to NLRP3 as a target for a therapeutic intervention in AD. The previous data from Michael Heneka's lab showing a role for NLRP3 in amyloid seeding already suggested it as a target (Venegas et al., 2017). The implication in this paper that IL-1β promotes tau pathology is consistent with previous work from Bruce Lamb’s lab and our own data (Bhaskar et al., 2010; Mancuso et al., 2019).
References:
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. 2017 Dec 20;552(7685):355-361. PubMed.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010 Oct 6;68(1):19-31. PubMed.
Mancuso R, Fryatt G, Cleal M, Obst J, Pipi E, Monzón-Sandoval J, Ribe E, Winchester L, Webber C, Nevado A, Jacobs T, Austin N, Theunis C, Grauwen K, Daniela Ruiz E, Mudher A, Vicente-Rodriguez M, Parker CA, Simmons C, Cash D, Richardson J, NIMA Consortium, Jones DN, Lovestone S, Gómez-Nicola D, Perry VH. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain. 2019 Oct 1;142(10):3243-3264. PubMed.
Michigan State University
The paper by Ising et al. is a very important contribution to defining the deleterious roles of microglial activation in promoting tau pathology. It adds to these authors’ prior data finding analogous influences of inflammasomes upon amyloid pathology in mouse models. Perhaps a key element is the indication that some of amyloid's influence upon tau pathology is mediated by inflammasome-dependent processes. Certainly this would elevate inhibition of inflammasome activity as a drug target for testing in Alzheimer's.
There are a number of key questions which follow from this work. As yet, it has not been demonstrated in humans when the innate immune system activation begins in the sequence of presymptomatic events leading to Alzheimer's dementia. These results would predict that at least a major increase would be expected after amyloid deposition and before tau deposition.
A second question regards how the inflammasome influences phagocytosis. It appears that under some activation states, microglia can clear amyloid deposits. How the activation of the inflammasome interacts with these beneficial outcomes of microglial activation is very important to understand.
A final surprise is that aging to 11 months results in changes in microglial gene expression that are similar in transgenic-tau-depositing mice and non-transgenic mice. This is especially surprising since an 11-month-old mouse is a middle-aged one, not an aged one. Given the concept that part of the effects of aging that contribute to AD might be senescence of beneficial microglial functions, learning how the senescent condition influence the inflammasome is critical.
Excellent and convincing work by the Heneka group.
Washington University School of Medicine
Washington University in St. Louis
Alzheimer’s disease is characterized by extracellular deposition of Aβ and intraneuronal accumulation of hyperphosphorylated tau protein. Previous studies showed that dementia correlates well with tau, but not with Aβ burden, indicating that tau aggregation might be the strongest driver of disease progression. However, amyloid pathology initiates decades before the first clinical symptoms of dementia, suggesting that Aβ, in combination with other factors, may eventually trigger tau pathology and neurodegeneration. This led to a very basic question: What is the connection between Aβ and tau, and how do they mutually affect each other?
This latest work by Ising and colleagues sheds some light on this dilemma. The same group had previously demonstrated that the inflammasome, a protein complex leading to the production of the inflammatory cytokine IL1β, is activated by Aβ (Heneka et al., 2013). Additionally, inflammasome components can be released into the extracellular environment, where they facilitate Aβ deposition, thus amplifying amyloid pathology (Venegas et al., 2017). In the brain, the inflammasome is expressed in microglia, which represent the main source of IL1β during brain diseases.
With the present work, Ising and colleagues show that the inflammasome is active in the brains of patients with frontotemporal dementia and Alzheimer’s disease. The same findings were replicated in a transgenic mouse model developing tau pathology with age. Next, the authors assessed tau pathology in the same mouse model, but deficient for crucial components of the inflammasome complex (namely NLRP3 and ASC). Interestingly, inflammasome-deficient mice were significantly protected, and displayed reduced tau burden and improved memory skills.
Ising and colleagues provide evidence that mice lacking the inflammasome fail to induce the protein kinase CaMKII-α, which plays a critical role in tau hyperphosphorylation and aggregation. By contrast, microglia-derived IL1β increases the levels of CaMKII-α and promotes tau aggregation in neurons. Similarly, tau monomers and oligomers, but not fibrils, induce IL-1β release in microglia. Lastly, the authors show that Aβ worsens tau deposition in mouse in an inflammasome-dependent manner.
Altogether, these data elucidate a novel link between Aβ and tau pathology. Aβ accumulation triggers inflammasome activation in microglia, with ensuing release of IL1β, which in turns activates kinases in neurons that induce tau hyperphosphorylation and aggregation. Future studies will determine whether inflammasome could be a promising therapeutic target for Alzheimer’s and tau pathologies.
References:
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434):674-8. Epub 2012 Dec 19 PubMed.
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. 2017 Dec 20;552(7685):355-361. PubMed.
University of Florida
Goizueta Institute @ Emory Brain Health
This manuscript by the Heneka lab dissects the influence of inflammasome activation on tau pathology in a mouse model of tauopathy. The notion that exacerbated inflammatory responses can accelerate tau pathology ties in with previous reports indicating that microglia may be necessary for neurodegeneration associated with tau aggregation (Shi et al., 2019). This study adds to a vast body of literature suggesting that dampening inflammation and/or the release of proinflammatory cytokines can ameliorate deficits in neuronal activity and cognitive decline in various mouse models of neurodegenerative diseases (Prokop et al., 2019).
As they have previously shown that inflammasome activation is also associated with Aβ aggregation (Heneka et al., 2013; Venegas et al., 2017), the authors come full circle to put Aβ and inflammation upstream of tau phosphorylation in an injection model of amyloidosis. This paper provides further evidence for the amyloid cascade hypothesis (Hardy, 2017) of Alzheimer’s disease (AD) pathogenesis, integrating Aβ deposition, tau pathology, and immune response into a complex picture of cascading events ultimately leading to neurodegeneration.
While this study provides important insight into the role of inflammatory processes in progression of tau pathology, there are some caveats with broadening the findings of this study.
First, the mouse model used relies on overexpression of a tau variant carrying two pathogenic tau mutations, and does not necessarily reflect the speed and pattern of tau aggregation observed in sporadic AD. Studies of the impact of inflammasome inactivation in other mouse models of tauopathy expressing wild-type tau and exhibiting longer progression times will be very informative moving forward.
Second, the effect of inflammasome inactivation was achieved using knockout animal models, which often develop compensatory signaling pathways due to the permanent absence of the deleted key messenger proteins. Studies with inflammasome inhibitors as treatments will therefore be a very important next step in validating this pathway for potential therapeutic interventions.
Third, most of the current genetic risk variants in microglia-associated genes are partial loss-of-function variants, making an important argument for immune activation, rather than immune inhibition as a potential therapeutic strategy moving forward (Golde, 2019). This only underscores the complexity of the system, where it will be of utmost importance to find the right balance between activation and inhibition of immune responses to address the human AD condition.
References:
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019 Nov 4;216(11):2546-2561. Epub 2019 Oct 10 PubMed.
Prokop S, Lee VM, Trojanowski JQ. Neuroimmune interactions in Alzheimer's disease-New frontier with old challenges?. Prog Mol Biol Transl Sci. 2019;168:183-201. Epub 2019 Oct 24 PubMed.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434):674-8. Epub 2012 Dec 19 PubMed.
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. 2017 Dec 20;552(7685):355-361. PubMed.
Hardy J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the "amyloid cascade hypothesis". FEBS J. 2017 Apr;284(7):1040-1044. PubMed.
Golde TE. Harnessing Immunoproteostasis to Treat Neurodegenerative Disorders. Neuron. 2019 Mar 20;101(6):1003-1015. PubMed.
Boston University Chobanian & Avedisian School of Medicine
"This implies therapeutic interventions will have to be tailored to the disease stage."
Until such therapeutic agents are actually available, advising at-risk patient populations to minimize their exposure to toxic agents, including diesel exhaust, that have the potential to activate microglia and promote NLRP3 inflammasome activation seems prudent, and does no harm.
References:
Roqué PJ, Dao K, Costa LG. Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. Neurotoxicology. 2016 Sep;56:204-214. Epub 2016 Aug 16 PubMed.
Hullmann M, Albrecht C, van Berlo D, Gerlofs-Nijland ME, Wahle T, Boots AW, Krutmann J, Cassee FR, Bayer TA, Schins RP. Diesel engine exhaust accelerates plaque formation in a mouse model of Alzheimer's disease. Part Fibre Toxicol. 2017 Aug 30;14(1):35. PubMed.
Wang BR, Shi JQ, Ge NN, Ou Z, Tian YY, Jiang T, Zhou JS, Xu J, Zhang YD. PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer's disease. J Neuroinflammation. 2018 May 2;15(1):132. PubMed.
Levesque S, Surace MJ, McDonald J, Block ML. Air pollution & the brain: Subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J Neuroinflammation. 2011 Aug 24;8:105. PubMed.
Make a Comment
To make a comment you must login or register.