AD Genetic Risk Tied to Changes in Microglial Gene Expression
Quick Links
Geneticists have found dozens of loci that associate with Alzheimer’s disease, but the functional variants and even the genes that underlie these associations often remain elusive. Now, researchers led by Alison Goate and Edoardo Marcora at the Icahn School of Medicine, Mount Sinai, New York, describe a method for quickly finding causal genes and variants in risk loci of myeloid cells. Starting with 16 AD risk variants located in enhancers, the DNA regions that influence transcription, the researchers linked each locus to changes in expression of a downstream gene that correlated with AD risk. Nine of the genes had not been associated previously with AD. For eight of the 16 enhancers, the researchers pinpointed the specific genetic change that affected expression. All were point changes that disrupted binding of transcription factors. The paper is available in preprint form on bioRxiv.
- AD risk variants predominantly occur in enhancers of myeloid genes.
- In eight enhancers, the authors pinpointed the functional variant for a GWAS risk locus.
- Altogether, enhancer data nominated 16 causal genes, nine of them new to AD.
“I think this is the first study where we have been able to go from the AD GWAS signal and comprehensively show what genes are affected and how,” Goate told Alzforum. The next step will be to investigate how these changes in gene expression alter microglial function, she added.
Others agreed that the method represents an advance. “It’s exciting that they were able to map a relatively large number of loci associated with AD risk using this approach. This is elegant work that moves the field forward,” Carlos Cruchaga at Washington University in St. Louis told Alzforum (see comment below). He suggested applying the same methodology to other brain cell types such as neurons and oligodendrocytes. Because many putative AD risk genes are highly expressed in microglia, Goate and colleagues limited their investigation to myeloid cells, which comprise microglia, macrophages, and monocytes. “We need to do more of these studies to map GWAS signals to genes, and those genes to pathways,” Cruchaga said.
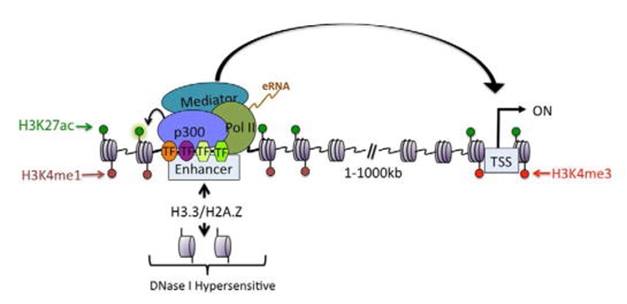
Chromatin Flags Active Enhancers. Modifications to histone proteins (green and red dots) mark active enhancers, where a transcription complex (colored circles) assembles on a chromosome. DNA then loops over (arrow) to contact an active promoter (gray rectangle) and initiate transcription. [Courtesy of Calo and Wysocka, 2013.]
In the case of myeloid cells, the new genes primarily relate to the endolysosomal system, highlighting the idea that the risk for Alzheimer’s may depend on how effectively microglia respond to amyloid and brain damage and clean up debris (Apr 2019 conference news). “This study is an important step toward extracting meaningful biology from genomic studies of Alzheimer’s disease. The prioritization of variants and genes will accelerate the generation of disease-relevant models, ultimately leading to a better understanding of pathogenesis,” Matthew Hill at Cardiff University, U.K., wrote to Alzforum (full comment below).
Goate and colleagues became interested in the microglial contribution to Alzheimer’s when they identified a polymorphism in AD GWAS data that lowered expression of the microglial master regulator PU.1 and delayed disease onset (Jun 2017 news). Because PU.1 controls the expression of numerous microglial genes, including many linked to AD, the results suggested that wholesale microglial gene expression could influence Alzheimer’s pathogenesis.
If so, AD risk variants might preferentially occur in regulatory regions, such as enhancers and promoters, that are active in these cells, the authors reasoned. To find these regions in myeloid cells, first author Gloriia Novikova examined human datasets such as ENCODE that catalog chromatin modifications in different cell types (ENCODE Project Consortium, 2012; Schmidt et al., 2016). Chromatin modifications point toward open regions of DNA where proteins can bind to initiate transcription. Novikova integrated these structural data with GWAS results from the International Genomics of Alzheimer’s Project (IGAP). She found that active myeloid enhancers contained a disproportionate number of AD risk loci. In particular, AD GWAS hits were associated with binding sites for PU.1 or its partners MAF, SMAD, and USF.
What genes did these enhancers control? The authors used two different approaches to find them, the first based on chromatin structure, the second on expression.
In the first, they made use of the fact that enhancer regions physically bind promoters, the DNA regions that initiate gene transcription (see image above). This brings different parts of the DNA strand together. Those DNA interactions can be teased out with a technique called High-throughput Chromosome Conformation Capture, or Hi-C, which basically cross-links adjacent pieces of DNA, isolates, and sequences them (van Berkum et al., 2010). In this manner, Hi-C can identify the target genes of each enhancer.
Luckily, others have already cataloged Hi-C datasets from various cells, including peripheral blood monocytes and macrophages, but not microglia (Javierre et al., 2016). Novikova and colleagues searched these data for myeloid enhancers that contained AD risk variants that associated with changes in expression of downstream genes. The analysis turned up 14 such genes. Seven of these were previously linked to AD, including SPI1, which makes PU.1, BIN1, MS4A, ABCA7, PTK2B, PILRA, and TREM2. The seven other genes were ZYX, TP53INP1, AP4E1, RIN3, APBB3, RABEP1, and CASS4. All the new genes are highly or exclusively expressed in microglia (Gosselin et al., 2017).
In the second method, the authors investigated whether genetic variants known to affect enhancer activity, or histone modification quantitative trait loci (hQTLs), co-localized with AD risk loci. For those that did, they linked the hQTL to expression changes in a downstream gene, and then examined whether that expression change associated with AD risk. If so, it was likely the causal gene at this risk loci. Again using monocyte data, the authors found 10 genes that fit the bill. Eight had been found by the first method. The two additional genes were CD2AP and GPR141. “We’re encouraged that there’s a lot of overlap between the gene lists that come out of the two approaches,” Goate said.
Notably, most of the new genes pertain to endolysosomal function. For example, ZYX is found in early endosomes and helps assemble actin filaments that propel movement. RIN3 loiters in endosomes along with BIN1, while RABEP1 facilitates endosome trafficking and fusion. AP4E1 sorts APP and other transmembrane proteins from the Golgi to endosomes, and APBB3 helps internalize APP from the cell surface. TP53INP1 is involved in phagocytosis.
While the endolysosomal system has long been of interest in AD research, most previous studies focused on neurons. “They are putting together two things that have never been side by side: microglia and lysosomal dysfunction,” Cruchaga said. Goate believes the findings can guide research into potential therapeutics. If scientists can figure out how risk and protective alleles change the endolysosomal system in microglia, they might be able to develop drugs that protect against AD, she suggested.
Although these approaches identified candidate causal AD genes, they still did not pinpoint the functional genetic variants that directly affected risk. To find these, the authors focused on eight loci where associations between enhancer activity, gene expression, and AD risk would have enough statistical significance to identify the functional variant. For each of the eight loci—SPI1, BIN1, MS4A, ZYX, TP53INP1, AP4E1, RABEP1, and GPR141—the authors found one or more SNPs in the myeloid enhancer region that disrupted transcription factor binding and correlated with differences in gene expression. These SNPs accounted for the entire GWAS signal at each loci, indicating they were the functional variants. That suggests that these genes exert their effect on AD pathogenesis solely through microglia or macrophages, not neurons, Goate noted.
The authors chose one variant to analyze in depth. An SNP in the enhancer region of MS4A disrupted binding of the transcription factor CTCF, abolishing the looping of the DNA strand to engage target genes. CTCF binding compacts chromatin and represses gene expression, so its absence would lead to higher expression of MS4A. The SNP, rs636317-T, boosts the risk of AD. However, other data conflict on whether lower or higher MS4A expression promotes risk (Apr 2019 conference news).
“The data are very exciting, and already pinpoint specific mechanisms altered in myeloid cells. We now need to understand how this translates into a chronic neurodegenerative process, whether the myeloid dysfunction is sufficient to drive disease, and how this relates to the main pathological hallmarks present in the disease, Aβ and tau,” Diego Gómez-Nicola at the University of Southampton, U.K., wrote to Alzforum (full comment below).—Madolyn Bowman Rogers
References
News Citations
- Expression, Expression, Expression—Time to Get on Board with eQTLs
- Microglial Master Regulator Tunes AD Risk Gene Expression, Age of Onset
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
Paper Citations
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why?. Mol Cell. 2013 Mar 7;49(5):825-37. PubMed.
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012 Sep 6;489(7414):57-74. PubMed.
- Schmidt SV, Krebs W, Ulas T, Xue J, Baßler K, Günther P, Hardt AL, Schultze H, Sander J, Klee K, Theis H, Kraut M, Beyer M, Schultze JL. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res. 2016 Feb;26(2):151-70. Epub 2016 Jan 5 PubMed.
- van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, Lander ES. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp. 2010 May 6;(39) PubMed.
- Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, Burden F, Farrow S, Cutler AJ, Rehnström K, Downes K, Grassi L, Kostadima M, Freire-Pritchett P, Wang F, BLUEPRINT Consortium, Stunnenberg HG, Todd JA, Zerbino DR, Stegle O, Ouwehand WH, Frontini M, Wallace C, Spivakov M, Fraser P. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell. 2016 Nov 17;167(5):1369-1384.e19. PubMed.
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JC, Sajti E, Jaeger BN, O'Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DD, Levy ML, Ransohoff RM, Gage FH, Glass CK. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017 Jun 23;356(6344) Epub 2017 May 25 PubMed.
External Citations
Further Reading
News
- Do Microglia Finish Off Stressed Neurons Before Their Time?
- Deleting CD33 Benefits Mice—If Their Microglia Express TREM2
- Down to Sex? Boy and Girl Microglia Respond Differently
- TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
- Gently Used: Can Recycled Microglia Receptors Prevent Plaque?
- Cut Loose, Soluble TREM2 Beckons Microglia to Mop Up Plaques
- CD22 Suppresses Microglial Phagocytosis—A New Therapeutic Target?
Primary Papers
- Novikova G, Kapoor M, TCW J, Abud EM, Efthymiou AG, Cheng H, Fullard JF, Bendl J, Roussos P, Poon WW, Hao K, Marcora E, Goate AM. Integration of Alzheimer’s disease genetics and myeloid cell genomics identifies novel causal variants, regulatory elements, genes and pathways. 2019 Jul 6. bioRxiv. BioRxiv.
Annotate
To make an annotation you must Login or Register.
Comments
University of Southampton
These findings provide strong mechanistic association of a dysfunctional myeloid compartment with AD, adding to the existing evidence arising from GWAS studies. The data provided is very exciting, and already pinpoints specific mechanisms altered in myeloid cells. We now need to understand how this translates into a chronic neurodegenerative process, and whether the myeloid dysfunction is sufficient to drive disease, and how is this related to the main pathological hallmarks observed present in AD, Aβ and tau.
The case for the role of neuroinflammation, and more specifically microglia, in Alzheimer’s grows at an unprecedented pace, but still lacks clinical validation. The field is in need of validation of this hypothesis in experimental medicine studies, and hopeful that for once an approach will provide benefit for this devastating disease.…More
Cardiff University
Novikova et al. report results from integrating Alzheimer’s disease GWAS with multiple functional genomic data sets generated from myeloid cells, including microglia. The path from identification of common variant risk loci to actionable biology has proven challenging. For complex disorders, most of the associated alleles reside in noncoding regions of the genome and are inherited along with non-functional alleles. Nominating causal variants, genes, and tissue types is therefore necessary in order to conduct appropriate, disease-relevant, investigations of mechanism.
Consistent with previous work, they show that AD risk variants are enriched at DNA regulatory elements active in monocytes, macrophages and microglia (Gagliano et al., 2016; Gjoneska et al., 2015; Tansey et al., 2018). They then link AD risk variation to target genes through studies of gene expression and chromatin interactions. For many risk loci they now propose causal genes and provide additional support for previously nominated targets. Those genes supported by multiple lines of evidence, chromatin conformation, and gene expression should be considered as high-priority targets for future biological investigation. Many of these have not been previously been considered in the context of Alzheimer’s disease but do appear to be related to the endolysosomal system.…More
This study is an important step toward extracting meaningful biology from genomic studies of Alzheimer’s. The prioritization of variants and genes will accelerate the generation of disease-relevant models, ultimately leading to a better understanding of pathogenesis. Although the authors have used state-of-the-art methods and data sets, it is important to note that many of the findings rely on studies of peripheral immune cells. As similar data becomes available for microglia it will be vital to evaluate the specific contributions of each cell linage to the genetic risk mechanisms of Alzheimer’s disease.
References:
Gagliano SA, Pouget JG, Hardy J, Knight J, Barnes MR, Ryten M, Weale ME. Genomics implicates adaptive and innate immunity in Alzheimer's and Parkinson's diseases. Ann Clin Transl Neurol. 2016 Dec;3(12):924-933. Epub 2016 Nov 4 PubMed.
Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015 Feb 19;518(7539):365-9. PubMed.
Tansey KE, Cameron D, Hill MJ. Genetic risk for Alzheimer's disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018 Feb 26;10(1):14. PubMed.
Washington University School of Medicine
Clearly GWAS studies have been very successful in identifying genetic regions associated with AD risk. But GWAS, in general, not only for AD, have two major problems: One is that the GWAS do not identify genes but regions, and second that the effect size of those regions are normally small. This makes it very difficult to functionally follow up GWAS studies. Additional studies that identify the functional gene or variant are needed.
This study is a very good example of how, using additional genomic approaches, it is possible to identify not only the functional gene, but the specific variant that drives the GWAS association. These results are instrumental to really understand the biology of AD and identify novel targets.…More
Recent studies have highlighted the importance of microglia in AD. For this reason Novikova et al. decided to use myeloid-specific epigenomic and transcriptomic data. As their data shows very nicely, they were able to resolve a good number of loci. This also highlights the role of microglia in disease. At the same time, we know other cell types are involved in AD, and similar studies with neuron-specific or oligodendrocyte-specific analyses should able to resolve more loci. In any case, this study includes a very large amount of work, with functional validation. Very elegant work.
Van Andel Institute
This is an interesting follow-up from the authors’ previous study showing the role of PU.1 in microglial AD risk gene networks. Now the authors extend their analyses to functional variants in monocyte and macrophage enhancers that alter the expression of AD genes, and they nominate candidate functional variants in several GWAS loci.
Interestingly, many of these new nominated genes are functionally associated with the endolysosomal system, indicating a significant role for myeloid endolysosomal pathways in Alzheimer's disease.
The specific enrichment of AD alleles in myeloid enhancers is striking, making these a very relevant cell type to study. It will also be interesting to extend these studies to other cell types relevant to disease. This will become feasible and easier with the generation of single cell sequencing data for AD. It will be important to see from AD-specific data if the same biological pathways associated with disease are relevant in all cell types and to start mapping the relevance of pathways for AD across different types of cell.…More
Make a Comment
To make a comment you must login or register.