Aducanumab Still Needs to Prove Itself, Researchers Say
Quick Links
Will Biogen’s aducanumab become the first drug approved to slow progression of Alzheimer’s disease? Biogen’s licensing application for the biologic ran into headwinds at a U.S. Food and Drug Administration advisory committee meeting held November 6. It was led by the agency’s Billy Dunn, who directs development of new neurology treatments (Nov 2020 news). The agency’s internal biostatistical and neurologic reviews presented starkly opposing views of the efficacy data—the former critical, the latter glowing. This disconnect consternated both the committee and the many scientists listening to the public proceedings from around the country.
- The FDA advisory committee cautions that positive data from EMERGE could be a mirage.
- Experts say negative data from ENGAGE cannot be disregarded.
- The committee, and peers throughout, deems the evidence premature for approval, recommends confirmatory trial.
Unlike the FDA’s neurology review, the biostatistical review got no presentation slot during the day-long meeting. Even so, the independent experts who sat on the committee had read that review in the background materials uploaded to the FDA website, and they found it convincing. The experts agreed the aducanumab efficacy data were weak. They pointed to inconsistencies in the data, which came from two futility-stopped Phase 3 trials and one Phase 1b trial. They complained that the sponsor and FDA presenters “talked down the clock” and did not adequately answer their questions. They unanimously voted against approval. The decision now rests with the FDA.
What do Alzheimer’s researchers think? They still have many questions about aducanumab. Most believe that the antibody slows progression of the underlying amyloid pathology. But they doubt whether there is a meaningful clinical benefit, what aducanumab’s effects are on markers of pathology downstream of Aβ, and what is the best patient population to target. To settle those issues, they want one more trial. Some spoke on the record, but many others requested their names not be used for this story.
“The current aducanumab trials fail to prove efficacy,” Todd Golde at the University of Florida, Gainesville, told Alzforum, expressing the majority view. “There may be a very modest beneficial effect in the early clinical stage of AD, but given safety, cost and possible access issues, this is far from the home run we had all hoped for,” Golde said (full comment below). Likewise, David Holtzman, Washington University, St. Louis said he found Biogen’s post hoc analyses explaining away discrepant results to be speculative and uncertain. “I believe further assessments of aducanumab are needed,” Holtzman wrote (see more below).
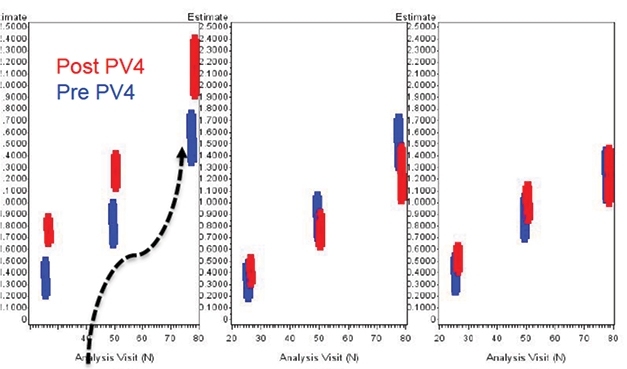
Placebo Decline. Did a random worsening in the placebo group (left) of the EMERGE trial after amendment 4 took effect create the illusion of a treatment effect? In low-dose (middle) and high-dose (right) groups, scores on the primary outcome measure did not budge from before the amendment (blue bars) to after (red). [From FDA website.]
Biogen’s licensing application touted positive findings from the aborted EMERGE trial, in which sustained dosing at 10 mg/kg slowed cognitive decline on the primary outcome measure, the CDR-SB, by 23 percent at 78 weeks. Decline on secondary cognitive and functional measures slowed by about a quarter as well. Biogen supported this finding with a dramatic drop in amyloid plaque as seen by PET, and with hints of a decline in tangles in tiny subsets of participants who underwent tau PET or lumbar puncture. However, the identical ENGAGE trial was negative. Biogen attributed this to a lower total exposure to the effective dose, 10 mg/kg. Maximum dosing for APOE4 carriers was raised midway through the trial, and in post hoc analysis, a subset of ENGAGE participants who received the most aducanumab responded similarly to EMERGE participants (Oct 2019 news; Dec 2019 conference news).
Caveats Raised for Positive Findings
The FDA’s advisory committee meeting cast doubt on this analysis. In their comments and questions, panelists repeatedly referred to the review from the FDA biostatistician, Tristan Massie (narrated slide presentation 3, available via FDA website). According to Massie, the data from EMERGE does not hold up to scrutiny. The positive signal in this trial could have arisen partly from an accelerated decline in the placebo group, and partly from inadvertent unblinding due to side effects from ARIA in participants on drug.
Both issues arise from how APOE4 carriers were treated. Carriers made up about two-thirds of participants. Because carriers are more prone to ARIA, the initial protocol called for them to titrate to a maximum of 6 mg/kg aducanumab, but this was revised in protocol amendment 4 to allow carriers to reach 10 mg/kg. Biogen researchers maintain that this increase in exposure generated the efficacy signal seen in the 10 mg/kg EMERGE group at 78 weeks.
In contrast, Massie’s analysis found little change in the overall rate of decline on the CDR-SB in the high-dose group after amendment 4 took effect. Rather, he found, the EMERGE placebo group declined more steeply post-amendment, from an average of 1.51 points in the pre-amendment population to 1.75 in the post-amendment group (see image above). Thus, he called the apparent efficacy signal “entangled” with variability in the placebo group. “The whole pattern of study outcomes could be explained by a randomly worsened placebo in study 302 [EMERGE] after the dose-increase amendment,” Massie suggested.
The advisory panel found these data troubling. In a question-and-answer session, Craig Mallinckrodt of Biogen disagreed with Massie’s conclusion, saying the company’s own analysis found the differences in placebo decline to have had a minimal effect. The discussion was cut short for time, and the panelists remained unconvinced.

Did Only APOE4s Benefit? On the clinical outcome measures CDR-SB, MMSE, ADAS-Cog13, and ADCS-ADL (top to bottom), APOE4 carriers (red) fared better, while noncarriers (blue) did no better than the placebo group (dotted line). [From FDA website.]
Massie’s analysis also focused on potential effects of unblinding. About one-third of the high-dose group experienced ARIA. This necessitated pauses in dosing, and MRI monitoring until the condition cleared up. Thus, affected participants became aware they were most likely on drug, as did some of their physicians. This may have affected ratings on the CDR-SB and activities of daily living (ADL) scales, which are somewhat subjective to begin with because they are based on information provided by the patient and his or her care partner. Additionally, Massie noted, the treatment effect in EMERGE was seen exclusively in APOE4 carriers, with noncarriers trending worse on drug than on placebo (see image above).
The lack of effect in noncarriers, who on the whole received more doses at the maximum 10 mg/kg, suggests either the treatment does not work for them, or the dosing issue was less critical than Biogen claimed, Massie said. That the APOE4 subgroup who had the greatest response to the drug also had the most unblinding raises the specter that this could have been a placebo effect.
Panelists, too, saw this as a problem. Madhav Thambisetty at the National Institute on Aging, Bethesda, Maryland, called it a “huge concern.” Mallinckrodt countered that excluding post-ARIA data from participants did not change the overall study results. However, Massie noted that excluding this data insufficiently corrects for the effects of unblinding, since the exclusion imbalances treatment groups.
The advisory committee had other concerns about the EMERGE data. Because of the trial termination prompted by the futility analysis, almost half of the participants did not have week 78 data. In fall 2019, Biogen and the FDA collaborated to simulate completion of the trial and imputed the missing data. However, such an approach magnifies the possibility of error. “A simulated completed trial is always more uncertain than a truly completed trial,” Massie said.
The cognitive benefit emerged at the final 78-week time point, and only in the high-dose group, Massie noted. Because only a single time point showed a change from placebo, the data cannot prove that the treatment slows disease progression, he said. Furthermore, the analysis plan specified that secondary outcomes were only meaningful if the low dose outperformed placebo. This kind of prioritization helps control Type 1 error. The low dose did not outperform placebo, hence findings on these secondary outcomes cannot be considered significant, Massie said.
Thambisetty and other members of the committee emphasized that the size of the claimed benefit was small, amounting to a difference of 0.40 points on the CDR-SB over 18 months. Panelists questioned whether this amount of slowing would be meaningful, especially given the cost and side effects of the treatment. On a voice vote, only one of the 11 panelists found the evidence from EMERGE convincing; eight rejected it, two were uncertain.
Negative Findings Can’t Be Dismissed
One of the committee’s biggest sticking points was a perceived attempt to sweep aside the negative findings from the ENGAGE study. Both Biogen’s application and the clinical review from the FDA’s neurologist Kevin Krudys (FDA presentation 1) argued that the negative findings could be explained by inadequate dosing, and should not detract from the positive EMERGE data. Panelists outright rejected this.
For a drug that did not work, there would be a 40 percent chance that two identical studies would be this discordant and show an apparent benefit in one, said biostatistician Scott Emerson at the University of Washington, Seattle. At best, the p value for EMERGE would have to be adjusted to take into account the negative study, Emerson suggested. Drug efficacy expert Caleb Alexander at Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, said, “You can’t look at EMERGE in isolation.” The other panelists concurred, with 10 saying ENGAGE cast doubt on efficacy, and one uncertain.
Biogen threw a second post hoc pitch for disregarding the ENGAGE findings, namely that the treatment groups in this trial contained a greater number of fast progressors, arguing that their more rapid decline masked a treatment effect in the rest of the group. The committee found this unconvincing, as well. Alexander countered that there were but a few fast progressors in a group of hundreds. “I just don’t see it. I don’t think the evidence is there,” he said. Alexander bemoaned the “extraordinary amount of explaining around the contrary [ENGAGE] findings.” Massie’s analysis found that excluding outliers with rapid decline did not change the overall findings for the ENGAGE high-dose group.
The Biogen application further argued that positive clinical data from the Phase 1b PRIME study could be considered supportive for approval. In that trial of 165 people with prodromal or mild AD, decline on the CDR-SB slowed after six months on drug, with the highest dose group of 10 mg/kg having the greatest benefit (Mar 2015 conference news; Sep 2016 news; Dec 2016 conference news). The study was not powered to detect cognitive benefits, and leading trialists have previously noted issues with double blinding in dose-escalation trials (Q&A in Nov 2015 conference news).
Massie determined that many of the findings in PRIME indeed were discordant with the Phase 3 trials. For example, in PRIME, APOE4 carriers fared worse than noncarriers, whereas in EMERGE they fared better. There was no clear dose-dependence in PRIME, with people on 3 mg/kg doing better than those on 6 mg/kg. Importantly, some participants started standard AD medications during the trial, which could have affected their cognitive scores. When those participants were excluded from the analysis, the results were no longer significant, going from a p value of 0.04 to 0.1, Massie calculated. The PRIME data do not demonstrate robust drug efficacy, Massie concluded. “There’s no reason this data could override a large, well-controlled failed trial,” Massie said.
Once again, the advisory committee agreed. Aaron Kesselheim, an expert on drug approval at Harvard Medical School, noted that the PRIME study wasn’t designed to demonstrate clinical efficacy, and so its data couldn’t be used to support the Phase 3 trials. Altogether, seven of the 11 panel members voted against considering these data as supportive, with the remaining four uncertain.
Biogen remains committed to bringing the drug to market now. “We are disappointed that the Advisory Committee recommendation disregarded clinical evidence and patient experience,” Samantha Budd Haeberlein wrote to Alzforum (full comment below). “We will respond promptly to the statistical questions raised by the Committee and bring clarification that is much needed for the community… We will continue to work with FDA as it completes its review of aducanumab.”
Who Benefits? And at What Cost?
The data left the panelists and listeners puzzled about which patients might benefit from aducanumab. Not only did the EMERGE benefit seem to be confined to APOE4 carriers, but there were also unexplained interactions with age, sex, disease stage, and even nationality. Massie’s analysis placed the primary clinical benefit in people older than 70, with younger participants not significantly different from the placebo group. Men benefited more than women. People with mild AD fared better than those at prodromal stages. This would appear to counter the concept that the earlier one treats, the better. Figuring out why only some participants do better on aducanumab, and what distinguishes them, will be critical for determining the drug indication, Emerson argued.

Data All Over the Place? On this forest plot of subgroup effects in EMERGE, it appears that aducanumab worked better in older people, men, APOE4 carriers, and those with more advanced disease. By country, it may have worked in Spain but not Italy, in the Netherlands but not Germany, the U.S. but not Canada. [From FDA website.]
Country differences were dramatic in the EMERGE data. Participants in Spain and Japan experienced large benefits, those in Belgium, Sweden, Finland, France, the Netherlands, and the U.S. had modest benefits, those in other countries, none (see image above). In the U.S., the error bars around the CDR-SB effect were tight, whereas in Finland, they ran the entire range of the forest plot. In ENGAGE, country differences were also pronounced but cut across different countries, i.e., the widest error bars were posted by centers in Taiwan.
These issues matter because aducanumab has side effects. Drawing attention to one that has not been discussed much thus far, Massie’s analysis found a 33 percent higher risk of falls in the EMERGE high-dose group. The reason for this is unclear.
Meanwhile, ARIA is common with treatment. It is often asymptomatic but can cause headaches and confusion. Researchers led by Lawren VandeVrede at the University of California, San Francisco, recently described a case study of an ENGAGE participant who was homozygous for APOE4 and developed severe headaches, hypertension, and epileptiform brain activity while on aducanumab. He had trouble reading, and his cognitive scores on the Montreal Cognitive Assessment plummeted. He was treated with steroids as well as anti-epileptics and hypertensive medication. After six months, his cognitive abilities returned to baseline (VandeVrede et al., 2020).
This participant, retired neurologist Daniel Gibbs at Oregon Health and Science University, Portland, told Alzforum that, if anything, he felt cognitively sharper after his ordeal. “I’m not really worried about the side effects. I believe my case of ARIA is the most severe that has been reported, and I fully recovered and may even have benefited,” he wrote. He has mixed feelings about aducanumab, and believes it will do best in a preventative paradigm, before neurons are lost. “I was disappointed but not surprised that the advisory committee did not recommend approval. I hope an aducanumab study using presymptomatic APOE4 subjects will be considered in the future,” Gibbs said (full comment below). Biogen had been planning an early stage trial but canceled it in 2019, to the chagrin of site leaders at the time. Gibbs has written publicly about his experience with developing Alzheimer’s (Gibbs, 2019).
And What Does the Field at Large Think of All This?
Most of the Alzheimer’s researchers Alzforum contacted sided with the advisory committee and agreed that aducanumab is not ready for clinical use. When asked directly, 18 out of 21 researchers Alzforum spoke with believed that more data are needed before aducanumab is approved; two were unsure. “The two trials taken as a whole do not, in my opinion, meet the level of scientific and statistical evidence required for approval,” Daniel Gillen, who chairs the statistics department at the University of California, Irvine, wrote to Alzforum (full comment below). “I would like to see the FDA require another well-designed and adequately controlled randomized clinical trial.”
Charles DeCarli at the University of California, Davis, thinks aducanumab could be a key drug for testing the amyloid hypothesis in secondary prevention studies, because of its dramatic effects on plaque. “But it hasn’t shown sufficient efficacy to be universally used for LOAD,” DeCarli noted.
Because aducanumab has to be infused, expense and logistics are also considerations. “We presently are woefully unprepared to incorporate any truly effective therapy into clinical practice,” John Morris at Washington University wrote to Alzforum. Clinics will need new resources and training to enable them to diagnose and treat patients. “If the drug doesn’t work, those resources will have been squandered,” Morris said (see comment below).
Speaking at the advisory committee meeting, Diana Zuckerman at the National Center for Health Research in Washington, D.C., noted that approving aducanumab now could make recruitment for other AD trials more difficult, since patients may prefer to take an approved drug rather than take a chance on a new one. In a recent snap poll of more than 1,000 people by a biopharma industry newsletter, 71 percent thought the agency should reject Biogen’s current application (Endpoint News).
On the FDA website, public comments submitted prior to and around the time of the meeting ran two to one against approval. Most of the critiques were from neurologists concerned about the sketchy data, while one relative of an AD patient also recommended rejecting the application because, “We need the next approved drug to be proven without a doubt to work.” The comments in favor came from AD patients and caregivers, primary care physicians, advocacy groups, and a medical imaging group who noted aducanumab approval would encourage the Centers for Medicare and Medicaid Services to offer reimbursement for amyloid PET scans.
In one such comment to the FDA, Lon Schneider at the University of Southern California, Los Angeles, wrote, “To represent aducanumab as substantially effective, and in this context safe, would be misleading and ultimately hurtful for many people with Alzheimer’s disease, not to mention wasteful in terms of economics and human resources.”
Likewise, David Knopman at the Mayo Clinic in Rochester, Minnesota, wrote to the FDA, “I desperately wish for a genuine therapy that substantially slows or reverses the disease. Yet the evidence that aducanumab is that therapy and has any benefits in persons with AD is terribly weak.” After noting the same objections as did Massie and the committee, he concluded, “Biogen needs to do a third trial with high-dose aducanumab. If the drug’s benefits are truly substantial, such a trial could recruit quickly and only several hundred patients would need to be randomized.” Knopman, together with Mayo colleague David Jones and Michael Greicius at Stanford, had published their concern days before the meeting (Knopman et al., 2020).
Others consider this assessment too harsh. “The FDA advisory committee meeting and its aftermath have been discouraging,” Paul Aisen at USC, San Diego, wrote to Alzforum (full comment below). “The tenor of the meeting and subsequent discussion have generally been dismissive of the important, positive signals in the data.” Marwan Sabbagh at the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas and Jeffrey Cummings at the University of Nevada, Las Vegas, noted in a response to Knopman’s article that approving aducanumab could encourage more investment in and development of AD drugs (Sabbagh and Cummings, 2020).
Some reached for compromise. Pierre Tariot at the Banner Alzheimer Institute in Phoenix said that perhaps the FDA could require a quick confirmatory trial, rather than a multiyear study. In the advisory committee meeting, Emerson, the UWashington biostatistician, picked up on testimony of trial participants and their carers, who claimed that they had maintained their cognition while on drug but slipped when they had to stop. Emerson suggested that using a randomized withdrawal-of-dose study could provide clearer evidence of benefit. In this study design, all participants start on drug, and those who do not benefit are dropped from the trial. Later, some participants are switched to placebo, and worsening then indicates there was a drug benefit. This design enriches for responders, making it easier to see a treatment effect in a heterogenous population, and it minimizes the time spent on placebo, aiding recruitment.
“I personally hope this treatment pans out,” Emerson added, noting his own experience caring for loved ones with Alzheimer’s.—Madolyn Bowman Rogers
References
News Citations
- FDA Advisory Committee Throws Cold Water on Aducanumab Filing
- ‘Reports of My Death Are Greatly Exaggerated.’ Signed, Aducanumab
- Exposure, Exposure, Exposure? At CTAD, Aducanumab Scientists Make a Case
- Biogen Antibody Buoyed by Phase 1 Data and Hungry Investors
- Paper Alert: Aducanumab Phase 1b Study Published
- Much ‘Adu’ About a Little: Phase 1 Data Feeds the Buzz at CTAD
- Outcomes, Outcomes: Cognition is Crux of New Alzheimer’s Trials
Paper Citations
- VandeVrede L, Gibbs DM, Koestler M, La Joie R, Ljubenkov PA, Provost K, Soleimani-Meigooni D, Strom A, Tsoy E, Rabinovici GD, Boxer AL. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement (Amst). 2020;12(1):e12101. Epub 2020 Oct 9 PubMed. Correction.
- Gibbs DM. Early Awareness of Alzheimer Disease: A Neurologist's Personal Perspective. JAMA Neurol. 2019 Mar 1;76(3):249. PubMed.
- Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021 Apr;17(4):696-701. Epub 2020 Nov 1 PubMed.
- Sabbagh MN, Cummings J. Open Peer Commentary to "Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE Trials as reported by Biogen December 2019". Alzheimers Dement. 2021 Apr;17(4):702-703. Epub 2020 Nov 1 PubMed.
Other Citations
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
We are disappointed that the Advisory Committee recommendation disregarded clinical evidence and patient experience. We will respond promptly to the statistical questions raised by the Committee and bring clarification that is much needed for the community.
We stand strong behind the integrity of our clinical data and statistical methods. The data provides substantial evidence of clinical effectiveness of aducanumab, as demonstrated by study 302 (EMERGE) and supported by study 103 (PRIME). We also stand behind the FDA’s assessment that partially discordant results of study 301 (ENGAGE) do not meaningfully detract from the persuasiveness of study 302. We will continue to work with FDA as it completes its review of aducanumab.
I am a retired neurologist with Alzheimer’s disease. I am an APOE-4 homozygote, and participated in the ENGAGE trial. I was in the placebo arm of the 18-month double-blind portion, and then entered the open-label continuation. I had no significant side effects except for mild headache until about two weeks after receiving the third monthly dose, when my headaches got worse and I started having mild trouble reading. A few days after the fourth dose, I had sudden onset of severe headache and encephalopathy. I was admitted to a local hospital ICU for blood pressure control with a working diagnosis of stroke vs PRES, but the MRI in the morning showed widespread areas of focal edema and micro hemorrhages consistent with amyloid-related imaging abnormalities (ARIA).
Although my blood pressure was brought under control, my encephalopathy and reading problems as well as MRI images worsened over the next month, requiring treatment with high-dose intravenous steroids. My symptoms rapidly resolved; my MRIs returned to baseline (except for residual hemosiderin from the micro hemorrhages) over the next five or six months.
Cognitively I felt better than at baseline, although I realize that an N of 1 is meaningless. A subsequent amyloid PET showed reduction of amyloid in the areas where the ARIA was worse, but a tau PET showed no improvement compared to a previous one three years before. In fact, the tau had progressed.
I have mixed feeling about aducanumab. I was disappointed but not surprised that the advisory committee did not recommend approval. Aducanumab is the most effective anti-amyloid treatment thus far in terms of reducing the β-amyloid burden, especially in APOE-4 carriers, but the effect on slowing cognitive decline is not nearly as robust.
Some have argued that this is nothing but a sign that the amyloid hypothesis is incorrect. This may be true, but I believe that it is more likely that anti-amyloid drugs will only be effective if used prior to the beginning of cognitive impairment, before neurons are lost. Fortunately there are at least two studies using presymptomatic subjects underway to test this hypothesis. I hope that in an aducanumab study using APOE-4, presymptomatic subjects will also be considered in the future.
I’m not really worried about the side effects. I believe my case of ARIA is the most severe that has been reported, and I fully recovered and may even have benefited. No one has died of ARIA to my knowledge. I think that ARIA may actually be necessary to open the blood-brain barrier and allow the MAB to engage with amyloid.
UCSF
It’s a tough situation, and I see both sides. As was beautifully articulated by the patients during the public comments period, there is an urgent need for therapies, and delaying access to a treatment that looks promising is incredibly disappointing. On the other hand, there are significant concerns about the strength of the data supporting clinical efficacy, most importantly the discordance between EMERGE and ENGAGE, which can only be definitively addressed by a third Phase 3 study based on the high-dose regimen.
I worry that FDA approval based on the current data would lead to confusion and uncertainty for clinicians and patients when facing decisions about treatment. Ultimately, I remain hopeful that we are on the cusp of disease-modifying treatments, but it seems like that day may arrive a bit later than we all would hope for.
Washington University School of Medicine
In 37 years of caring for persons with Alzheimer’s disease dementia, and in conducting clinical and translational research to advance the development of truly effective therapies for the disorder, I am very excited that a candidate mechanism-based drug, aducanumab, finally has been submitted by its sponsors to the FDA to consider for registration. If an effective anti-Alzheimer’s therapy indeed were available, it would represent a truly remarkable advance for the field and provide patients and their families with great hope for clinical benefit.
However, with the clear caveat that I have not reviewed the data from the ENGAGE and EMERGE trials of aducanumab that were sponsored by Biogen, it is difficult for me to reconcile that the drug’s approval should be based on apparently positive results from one of the trials when those same results were not realized in the identically conducted second trial. The seemingly appropriate next step would be to conduct at least one further rigorous trial to learn which of the two outcomes is, in fact, supported.
It is understandable that, given how desperately an effective therapy is needed, there will be sentiment to approve aducanumab even with the currently conflicting data. Should it turn out, however, that aducanumab is approved and yet proves to be ineffective, it could have many negative consequences. For example, because we presently are woefully unprepared to incorporate any truly effective therapy into clinical practice, enormous resources will be required to markedly increase the capacity to properly diagnose and treat patients in accordance with approved protocols. If the drug doesn’t work, those resources will have been squandered.
USC Alzheimer’s Therapeutic Research Institute
The FDA advisory committee meeting and its aftermath have been discouraging. The submitted data is highly challenging, with two interrupted Phase 3 trials yielding discordant results. But the tenor of the meeting and subsequent discussion have generally been dismissive of the important, positive signals in the data. EMERGE provides the most encouraging data to date in support of disease modification in AD. The primary analysis, despite the study interruption, indicates a modest beneficial effect of high-dose treatment on clinical progression, and is supported by the results on all key secondary measures, as well as biomarkers of downstream pathology. ENGAGE is negative, though this discordant finding can be partially explained by differences in study timing relative to the major amendment, and therefore reduced drug exposure.
As the FDA noted in its position statement, the many prior failures of anti-amyloid therapies are not grounds for dismissing the class. The failures have taught the field important lessons regarding target engagement, dosing, and study populations that greatly increase the likelihood of success. The aducanumab data, regardless of the committee vote or the eventual FDA decision, represents a major step forward.
Beneficial anti-amyloid therapy will reach the clinic, along with other effective therapies targeting the various pathological processes contributing to AD. We are on the right track.
Washington University
Like many physicians, physician-scientists, and scientists who work on Alzheimer’s disease, my major goals for our field are to develop treatments that prevent, delay the onset of, or slow the cognitive decline that occurs in AD. In the case of EMERGE and ENGAGE trials of aducanumab and the studies leading up to these trials, it is clear that aducanumab at high enough doses removes a significant amount of amyloid from the human brain. I believe that antibodies, and other treatments like this that target amyloid removal, have a lot of promise in delaying the onset of cognitive decline in individuals developing amyloidosis but before they are symptomatic. What was attempted in the EMERGE and ENGAGE studies was to treat individuals who already have some cognitive decline that is associated with underlying AD pathology.
In EMERGE and ENGAGE, individuals with very mild/mild dementia/mild cognitive impairment who have brain amyloid deposition were enrolled in randomized controlled clinical trials in which the primary outcome was the clinical dementia rating (CDR) sum of boxes (SB) in the 78-week trials. The primary outcome of EMERGE showed a statistically significant slowing of decline (22 percent) of the CDR-SB at the highest dose (10 mg/kg), whereas there was no clear effect on the primary outcome in the ENGAGE trial. Post hoc analysis of ENGAGE suggested the possibility that the negative result might have been due to lower exposure in the 10 mg/kg group in this trial, as well as possibly other factors.
For a very common, hugely important and impactful disease such as AD, it is key that RCTs are done and the primary outcome(s) shows clear, impactful results for patients and their families. While arguments are being made by some that suggest the possibility the negative primary outcome in ENGAGE might be explained by different factors such as total exposure to the antibody and other factors, to me, this is very uncertain and speculative.
The bottom line is that the EMERGE trial shows a very small but statistically significant effect at the highest dose of an important clinical outcome (CDR-SB). The ENGAGE trial is negative on the primary outcome.
I think it would be a big mistake to approve a treatment that literally millions of people would be eligible for with a cost just in the U.S. alone estimated to be well over $20 billion a year when it simply is not clear whether it is likely beneficial or not. I believe further assessments of aducanumab are needed to be sure, one way or the other, that it will or won’t benefit the population with very mild dementia/mild cognitive impairment due to AD.
I think similar criteria should be applied to any new treatment for such a population. Just as much as any physician/provider who sees patients with AD and knows its impact, I want to see treatments that have effects. We need to be sure that such new treatments have meaningful effects. This is not clearly the case with the results to date with aducanumab.
Goizueta Institute @ Emory Brain Health
I agree with published studies by Knopman and Schneider and colleagues that the current aducanumab trials fail to prove efficacy. I am worried that many stakeholders with good intentions are trying to push this forward, but this is a dangerous path. We need to rely on data. Overall, my “gestalt” is that there may be a very, very modest beneficial effect in this early clinical stage of AD, but given safety, cost, and possible access issues, this is far from the home run we had all hoped for. Perhaps it is an attempted bunt single with a very close call at first base (but with most saying the batter is out)?
I have additional concerns that are not yet clearly addressed in what I have seen. First, what is the relationship between the reduction in amyloid ligand binding and true pathological biochemical Aβ in the brain? If it is strong, then I think this drug should be evaluated in a rigorous secondary prevention study.
Second, I worry about the integrity of the blinding given the side effects on high dose, both from the evaluator side and the participant side. I have not seen this formally addressed.
Third, what was the long-term outcome of those who had strong reductions on PET of amyloid ligand binding in the Phase 1b? If it really had a big impact, those folks should have had a pretty substantially different clinical course of disease. This would be underpowered and on the anecdotal side, but if the E4 carriers were still doing well, I’d like to know. The targets for cognitive and ADL benefit for disease modification at 18 months to gain approval are pretty modest. A drug that hits those targets but failed to have a sustained effect would not benefit public health all that much. Again, smaller trials for longer time periods might actually tell us a lot more and essentially take the same amount of time due to easier enrollment.
This is an unfortunate, if predictable, outcome. For reasons I and others have stated in multiple published reviews, this data does not undermine the amyloid hypothesis in any way. It does say we need to think harder about whom we try to alter disease course in. I have believed and said for some years that we should stop using the term “early AD.” Yes, it is early symptomatically, but it is late pathologically. Symptoms mean the brain is starting to enter organ failure. Conceptualizing the symptomatic phase of AD as organ failure means a radically different approach to therapy in the symptomatic phase than what we are doing now.
University of Kansas
There are several layers to consider here. Given the contrasting Phase 3 study results, it’s hard to feel confident the active intervention attenuated the rate of decline. Had the negative study not been negative, it would still be important to consider what might account for any attenuation. A slight attenuation of the decline rate could reflect a true benefit to those receiving the active intervention, but it could also arise through the elimination of the most rapid progressors from the group.
The FDA briefing document does cite two papers as reporting that, in general, AD APOE4 carriers do not decline more rapidly than APOE4 noncarriers, although it was not indicated that one of those two papers actually reported APOE4 carriers with MCI actually do decline more rapidly, a finding also suggested by Figure 1C in Petersen et al., 2005.
To further drill down on whether shifts in APOE admixtures could create a picture of attenuated decline within a group, this issue came up previously with bapineuzumab, where the sponsor resolved the question by conducting separate Phase 3 trials that included just APOE4 carriers or just APOE3 carriers. Those trials, as we know, came to a negative conclusion. This issue also came up recently with BAN2401, and no doubt will once again as we are dealing with a class of drugs that strategically cause a significant adverse event in APOE4 carriers, and can also lead to the unblinding of APOE4 carriers.
References:
Barnes DE, Yaffe K. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005 Sep 1;353(9):951-2; author reply 951-2. PubMed.
Chiesi Farmaceutici S.p.A.
I agree with Tristan Massie regarding the issues he raised in the statistical section of the FDA/Biogen document, and with David Knopman and colleagues regarding the methodological and clinical issues they raised in their recent article published by Alzheimers & Dementia.
I see these major concerns for both trials:
1. Large number of patients not completing the trials, both for efficacy and for adverse events at highest doses.
2. Unblinding, especially at high doses, mostly due to ARIA.
3. Limited effect size that is inversely correlated with subjectivity of cognitive and clinical scales. Basically, the mean effect size in EMERGE is greater for those scales that have a higher number of subjective items evaluated by the rater, whereas the effect size is minimal for MMSE—an objective scale.
4. The presence of fast decliners in the 10 mg/kg group could be interpreted as indicating a detrimental effect of the drug at high doses in a subset of patients, similarly to what was observed with BACE1 and γ-secretase inhibitors.
We should consider the aducanumab story in the context of those of several other monoclonal antibodies in AD, including those claimed to target oligomeric Aβ species (crenezumab). Monomers, oligomers, protofibrils, and fibrils are in equilibrium, and any drug that mainly works on a specific Aβ species will influence the other species. This consideration can explain why clinical failures were seen with monoclonal antibodies with different affinity profiles for Aβ species.
If the FDA approves aducanumab now, it will be:
1) the first time FDA approves a drug based on two pivotal Phase 3 trials interrupted for futility,
2) the first time FDA approves a drug in open contrast with the agency’s own statistical assessor,
3) the first time FDA approves a drug in open contrast with the unanimous negative opinion of the entire advisory expert panel.
University of California, San Diego
I am a paid consultant for Biogen, so will not comment directly on the FDA’s deliberation of aducanumab. I raise here some general questions that may have an impact on AD therapeutics going forward as we are all taking stock of where we are with the data from the aducanumab trials and elsewhere.
In MCI and mild AD, if the CDR-SB is the primary outcome measure, then how much change in slope is meaningful? Retention of cognitive and functional abilities for longer is likely to be perceived as worthwhile by patients (even if they remain with impaired memory) and their families, but there is no consensus on what magnitude of clinical change is meaningful. How high should the bar be set?
To have the potential to reveal greater effect sizes, should trials in this population go for longer than 18 months? The popular 18-month design is an arbitrary duration that seems to have arisen out of habit rather than natural history studies. And: Is it time for new cognitive tests to replace the ADAS-cog in these early stages of AD?
ARIA unblinds the people who provide CDR and ADL ratings. Some FDA reviewers thought this was a major problem but, according to the briefing document on the aducanumab trials, omitting all post-ARIA ratings did not change results. More discussion is needed, because ARIA is a potentially unblinding feature for any antibody that clears amyloid.
Since amyloid burden does not correlate with measures of cognition in AD, the extent of removal should not be expected to correlate directly with clinical outcomes. We do not understand what happens to the brain after deposited amyloid is removed, and how long after removal people should be followed to assess whether this may show a clinical benefit. There are biomarkers that could be deployed to assess whether amyloid removal impacts neurodegeneration, and CSF data from the DIAN-TU gantenerumab trial supports this contention. It would be of great interest to see what plasma P-tau and NfL biomarkers look like in the aducanumab trials, and the advent of plasma biomarkers will have a dramatic effect on trials going forward.
The advisory panel mentioned surrogate markers several times, but amyloid PET alone will not serve as a surrogate marker in trials. It is tempting to propose that tau PET or CSF or plasma P-tau biomarkers, synaptic biomarkers, etc., could serve in this regard, but it is more conservative to think of these as markers of downstream effects of target engagement in an anti-amyloid trial rather than as surrogates. So the need to show clinical benefit in trials in symptomatic patients along the AD spectrum remains.
University of California, Irvine
Multiple issues in this clinical program make assessing aducanumab’s efficacy difficult. Many of these are highly technical from a statistical and clinical perspective. I won’t go into all of them, but will focus on two major points that were also brought up during the advisory committee meeting.
The first obvious issue is that the two parallel Phase 3 trials, 301 and 302, were discrepant in their results. Generally speaking, FDA approval requires two independent confirmatory trials. This usually entails one being statistically significant and the other offering supportive evidence of efficacy that yields a point estimate for the treatment effect that is in line with that of the significant trial. In cases where there is an unmet need, which this qualifies as, the FDA is often willing to give expedited approval based upon a single confirmatory trial if the estimated treatment effect is clinically meaningful and the result highly significant (say, a p-value <.001).
The issue with aducanumab, beyond the fact that study 302 was stopped early for futility then later showed a beneficial effect, is that the two trials showed discrepant results and, when taken as a whole, did not show compelling evidence of effectiveness. During the meeting, the FDA asked the committee members in the first discussion point to consider only study 302. This was viewed as an inappropriate stance by many on the committee members, and I fully agree with them.
One cannot simply pretend that the results of 301 do not exist when trying to evaluate the scientific evidence for efficacy afforded by 302. The studies were very similar in nature and, in my opinion, any good scientist should not “cherry pick” the results of 302 without accounting for those of 301. The FDA statistician’s report correctly pointed out that the results of 302 should be interpreted in light of 301 as well. If one does this, the two trials taken as a whole do not, in my opinion, meet the level of scientific and statistical evidence required for approval in either of the two situations I described above.
In an attempt to provide “an independent study, providing supportive evidence of effectiveness,” the sponsor brought forth the results of study 103. The issue with this argument is that study 103 was an earlier trial that preceded 301 and 302 and informed their designs. Thus the undertaking of studies 301 and 302 were almost certainly conditional upon study 103 results. Thus, in my mind study 103 cannot, in any way, be considered an independent confirmatory trial that provides the supportive evidence to justify approval.
For the two main reasons above, and for multiple others, I do not think approval of aducanumab is warranted at this time. I am very aware of the unmet need to treat this terrible disease and have dedicated much of my professional time to trying to discover safe and effective therapies for the prevention and treatment of AD. I do, however, believe that we should have convincing evidence of the safety and efficacy of any intervention before approval. The consequences are too great not to.
I would like to see the FDA require another well-designed and adequately controlled randomized clinical trial that yields supportive evidence of the 302 result. If the sponsor is willing to conduct such a trial and can provide supportive data at its conclusion, then I believe approval would be warranted, but not until then.
Acumen Pharmaceuticals
The FDA advisory committee meeting held on November 6, 2020, yielded an overall negative view by the committee members of the aducanumab submission. The meeting itself, however, was not able to discuss adequately the most pertinent issues given the process utilized. The sponsor, as might be expected, presented a positive view of the submission. Somewhat surprisingly, the clinical review from FDA in many ways echoed or even exceeded the enthusiasm of the sponsor. The statistical review from FDA, which concluded that approval should not be granted and another study was needed, was not presented during the meeting. This left the advisory committee members in an awkward position of trying to balance the FDA clinical review and statistical review in a relatively short period of time in a virtual meeting. Thus, the process itself, despite thoughtful discussions from the advisory committee members, may not have adequately addressed the most relevant questions.
The sponsor and FDA clinical reviewers made a strong case that the positive Study 302 should be considered without considering results from the negative Study 301. They indicated that the Phase 1b Study 103 should be viewed as supportive evidence. The FDA statistical reviewer made a traditional frequentist argument that with one positive and one negative Phase 3 trial, a third trial should be required and that the results from Study 103 should be ignored. In my view, neither of these arguments is correct. Certainly, the advisory committee should not be expected to “unsee” Study 301 and consider Study 302 only. Similarly however, although Study 103 was not designed as an efficacy study, those results should not be ignored.
The sponsor, FDA clinical reviewers, and the FDA statistical reviewer all made various post hoc analyses intended to support their positions. Discussion of these post hoc analyses unfortunately required a great deal of time during the advisory committee meeting. While these analyses may be of some interest, the fact that post hoc analyses are hypothesis-generating rather than hypothesis-testing was largely ignored during the discussion. An example of such a discussion is the effect size assessed by country. The idea that the drug is effective in Spain but not in Italy has little biological plausibility, just as would a post hoc analysis showing that people born on a Tuesday benefited from treatment while people born on a Wednesday did not. Thus, the discussions of changes in placebo group rate of decline, ApoE differences, subgroups with more consistent dosing, subgroups without rapid decliners etc. should all be taken with a large grain of salt. Since these analyses are hypothesis-generating, the findings might guide the design of future clinical trials, but their utility in answering the most important questions for the advisory committee regarding the aducanumab submission are limited.
The argument has been made that approval of aducanumab would make the development of future disease-modifying drugs difficult or impossible. This is unlikely to be the case. There was some discussion of delayed withdrawal designs of future studies, but limitations in delayed withdrawal designs have been appreciated since publication of the DATATOP study in 1993 (Parkinson Study Group, 1993). A delayed start design provides clear advantages. One is that study participants understand that they will be randomly assigned to drug or placebo for the first study period, but subsequently all participants will be given active treatment. Many if not most potential study participants would accept the possibility of placebo treatment for 18 or 24 months with the promise of active treatment thereafter with a drug with potentially better efficacy and safety than aducanumab. Thus, the approval of aducanumab is unlikely to have substantial effects on enrollment in subsequent thoughtfully designed trials incorporating more promising drugs.
The discussion that likely would have been most useful for the committee would be the value of a more Bayesian approach rather than a traditional frequentist approach applied to only two pivotal trials. There now appears to be a broad consensus that not all Aβ/amyloid-targeting therapies should be grouped together, especially when considering secretase inhibitors and monoclonal antibodies. Broadly speaking, the monoclonal antibody solanezumab showed small but consistent numeric changes in the EXPEDITION, EXPEDITION2, and EXPEDITION3 Phase 3 studies using what is now considered a low dose. The Phase 2 study results from BAN2401, with all the caveats of Phase 2 studies, appear promising. Even these findings relating to other monoclonal antibodies should influence the prior probability assigned to aducanumab trials. Given whatever is the assigned prior probability for aducanumab, the question remains what is the posterior probability that aducanumab has zero efficacy, given the results of Studies 103, 301, and 302 taken together. A discussion of the probability of zero efficacy should influence any discussion of regulatory approval.
Of note, the following paragraph in the FDA statistical review was not fully discussed by the committee: “Conditional power based on all ITT data with censoring after March 21, 2019 of meta analysis being positive if non-OTC patients by the time of futility were somehow able to complete is estimated to be .61 as follows. This kind of pooled analysis was the basis for the interim treatment estimate on which the conditional power calculation was based. Perhaps, this calculation does not adequately take into account heterogeneity between studies, but it summarizes all the available phase 3 evidence and follows the spirit of the sponsor’s original plan to use a pooled treatment effect estimate at the interim. Based on a Bayesian meta analysis of study 301 and 302 the posterior probability of the alternative hypothesis is 0.62 and a corresponding 0.38 probability for the null hypothesis.” Additional discussion of that analysis and the probability of zero efficacy would have substantially improved the depth of the discussion at the advisory committee meeting.
References:
Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. The Parkinson Study Group. J Biol Chem. 1993 Jan 5;268(1):608-12. PubMed.
Brown University
Early termination of studies 301 and 302 and a dose increase midway through the trial led to an incomplete dataset and clearly complicate the interpretation of the results. However, the totality of the data provides evidence of dose-related lowering of amyloid plaques across all three clinical trials and clinically meaningful benefits for aducanumab on the CDR-SB and other clinical measures in two of the three clinical trials.
We are at a critical juncture in AD treatment and I understand the difficult decision that the FDA faces regarding aducanumab. In order to have the best-in-class drug, we must have the first-in-class medication. Approval of aducanumab would provide a treatment foothold we can build on, akin to the approval of AZT, which, despite its limitations, energized HIV research, leading to powerful new treatments. Approval of aducanumab would be associated with many firsts for Alzheimer’s disease: the first drug approved in 17 years, the first approval for mild cognitive impairment due to AD, and the first treatment targeting a core pathology of the disease.
I recommend that approval of aducanumab be closely aligned with the clinical and biomarker criteria utilized in the Phase 3 trials, be managed by a dementia expert, and require a careful Phase 4 post-marketing surveillance program. Patients and families urgently need to preserve time and quality of life and delay the disability of Alzheimer’s disease. For them, the potential benefits clearly outweigh the risk of certain decline if the drug is not approved. Access should be provided to this medication so that patients can decide with their physician if aducanumab is right for them.
Alzheimer Center Amsterdam; Head EQT Life Sciences Dementia Fund
Amsterdam UMC Dept. of Neurology/Alzheimer Center and Brain Research Center Amsterdam
Much has been said on aducanumab already and, as in any scientific debate, opinions differ widely. Unfortunately, as was evident from the FDA hearing, the debate has become polarized, as if it were politics. This is never beneficial to any debate.
This scientific debate reflects ambiguity around the data and the road that the development of aducanumab traveled up till now. Based on a brilliant discovery from our colleagues in Zurich, very promising Phase 1 data showed clear dose-dependent amyloid plaque removal on amyloid PET imaging, with some hints of clinical efficacy upon one year treatment, although the study was not powered to find such an effect (Sevigny et al., 2017).
The most reasonable and standard drug development step then would have been to design and conduct a proper Phase 2 study looking for the optimal dose, and powered to establish clinical efficacy upon which large Phase 3 trials could have been based. Instead, Biogen skipped Phase 2 and immediately jumped into two large Phase 3 trials, which were unfortunately stopped for futility. There is another lesson here about not stopping too early when you are dealing with a disease that is already present 10-15 years before patients are entering the trial.
Biogen ended up with two “unfinished” trials, of which one seems to be positive and one negative. Despite many sensible and reasonable post hoc analyses on why both trials did not show the same result, the fact remains that study 301 did not meet its primary endpoint. As Eric Siemers rightly pointed out in his comment, such post hoc analyses are for hypothesis generation, not testing.
As can be judged from the many comments on Alzforum, one can look at the “aducanumab glass” as being half empty or half full. On the positive side, we feel that the fact cannot be disputed that aducanumab, for the very first time in the history of AD drug development, demonstrated substantial dose-related lowering of amyloid plaques across all three clinical trials. This by itself is groundbreaking.
The lack of translation of this true disease-modifying effect into clinically meaningful outcomes in both Phase 3 trials can be seen as a matter of time (too short) or lack of sensitivity of the outcome measures.
In other fields of neurology, this has not stopped the FDA. The agency has approved treatments that have clear biomarker effects but ambiguous, or hardly clinically relevant, outcomes on crude scales, simply because the need was so huge and the biomarker effects so compelling. Take multiple sclerosis. The first approval of betaferon came in 1993, on the basis that “it affected underlying disease.” The rest is history. If one looks at how the MS field has succeeded since then at improving and developing MS therapies, one might wonder, how is this different from Alzheimer’s?
If we follow this reasoning, and combine it with the huge unmet need, the clear and present desire of the field to have a treatment for AD, knowing that amyloid lowering will never be enough by itself and combinations with other approaches will need to be developed, then we would argue for a “conditional approval” of aducanumab. This is an instrument the EMA has used in the past, as well.
Experts in the field, working at expert centers, could be charged with the responsibility to use this drug in practice in selected patients—perhaps only ApoE4 carriers?— who have NIA-AA biomarker-proven AD, using stringent ARIA and clinical monitoring. The highest dose could be used for a long duration of time with regular biomarker and efficacy monitoring and with continuous gathering and sharing of data with the authorities. It could be done a bit like the "rolling review" that is happening in the development of COVID-19 vaccines (see Reuters news).
In our opinion, this could be the solution for our field. We could have a treatment approved without having to wait more years—time that is so precious to our patients and should not be lost.
References:
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O'Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A. Addendum: The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature. 2017 Jun 21;546(7659):564. PubMed.
UsAgainstAlzheimer’s
With not a single disease-modifying therapy available today, the millions of people experiencing Alzheimer’s have an intense interest in the safety and efficacy of aducanumab.
An Advisory Committee’s input is important to the evaluation process. However, as the article above alludes to, many questions went unanswered due to time constraints. We encourage the FDA to address those questions in its continuing evaluation, recognizing that the FDA has access to more information and more evidence after years of work on Alzheimer’s drugs than could possibly be presented at a single Advisory Committee meeting. The FDA has considerable regulatory discretion, and exercising that discretion has sparked major advances in caring for patients with HIV, cancer, and MS. People experiencing a loss of self from Alzheimer’s deserve no less.
Many Advisory Committee members noted the discordance between the clinical review and the statistical review. Rather than exploring the differences in search of the best interpretation, they extolled the merits of the statistical review and discounted the clinical review. We note that people are more than a p-value.
If the analysis is a close call, we believe the FDA must decide in favor of giving patients and their physicians the choice of using a treatment option where today there is none. Post-approval collection of real-world evidence will improve our understanding of aducanumab’s effectiveness. We are prepared to be partners in that work.
For families living with the disease, every single day matters. Several thousands of those in our community invested 18 or more months in clinical trials to help not just ourselves but future generations. The results of those trials provided evidence that this drug reduces beta amyloid pathology in our brains. We cannot wait years for better data on aducanumab or for a more perfect drug even were the current data viewed as less than perfect.
Our nation needs a first-in-class drug before there can be any hope of a best-in-class drug. And those who suffer deserve a process that does not substitute unanswered questions and unexamined doubts for a rigorous, independent regulatory decision on the merits of aducanumab.
Make a Comment
To make a comment you must login or register.