2021—A Turning Point for Alzheimer’s Research and Therapy?
Quick Links
Therapeutics
Alzforum's review of therapy development for 2020 began with "The drama of aducanumab was the year's big story." In 2021, the drama of aducanumab was, once again, the year's big story. That is true even without the latest plot twist, i.e., the January 11 CMS proposal to cover Aduhelm, and the three other anti-amyloid antibodies in its wings, only as part of clinical trials, i.e., under coverage with evidence development (see news).
The story exploded in June, when the U.S. FDA overruled its internal statisticians and external advisory committee and conditionally approved aducanumab. This garnered some cautious praise, but also unleashed a torrent of criticism that kept on coming throughout the year. Some took the long view, welcoming a new era of mechanism-based treatment and believing that data showing the drug works would come in time. Others, including neurologists and geriatricians, stood aghast at aducanumab's skimpy efficacy data, its broad label, and cost ($56,000/year plus MRI and other fees)—all without a peer-reviewed publication on the Phase 3 data.
Six months hence, aducanumab has received some backup. The FDA's claim of a “reasonably likely” treatment benefit based on amyloid removal drew support from a CTAD presentation reporting that aducanumab had reduced plasma p-tau181 in the Phase 3 trials, i.e., further data toward disease modification. The label has been narrowed to early stage patients like those in the trials, Biogen has halved the price, and Phase 3 safety—though still not efficacy—data is published. The field's leaders issued appropriate-use recommendations. An open-label observational study of a representative patient population was announced, as were a national registry to track real-world treatment data, and a May 2022 start for the required Phase 4 trial.

Aduhelm Dunks Tau. Plasma p-tau181 fell in the Phase 3 EMERGE (left) and ENGAGE (right) trials, with higher doses (blue) having a greater effect than low (green). In people on placebo (gray), p-tau rose. [Courtesy of Biogen.]
More broadly, aducanumab's approval occasioned a shift in the field, by spurring investigators to incorporate it—or a different anti-amyloid antibody—into their treatment trials. They will use an anti-amyloid antibody either as one of two drugs in combination trials (e.g., the DIAN NexGen study of both lecanemab and an anti-tau drug), in head-to-head comparison to a competitor's drug (e.g., Lilly trialing donanemab against aducanumab), or, in the future, as a background therapy to the investigational agent at hand.
That said, the aducanumab approval has damaged both Biogen and the FDA. The U.S. federal government is investigating the appropriateness of their interactions prior to June 2021. Scrutiny of the agency's accelerated approval pathway in general has intensified. Biogen's Al Sandrock was forced out, and the FDA's Janet Woodcock was sidelined from contention for her agency's top job. The country's Medicare and Medicaid programs are grappling with how aducanumab will consume their respective budgets, even as independent health economists put the drug's value below its current price. Aducanumab is cited in policy debates calling for drug price-control legislation, and in published critiques of approving drugs based on need more than efficacy. Large health care providers and insurers have declined to administer or cover the drug for the time being, and few patients are receiving it thus far. European Union and Japanese health regulators rejected Biogen's marketing license application, and leading clinicians in Canada called on their agency to do the same.
In December of 2021, formal publication of the extent of ARIA in the Phase 3 trials, together with an ARIA-related death and a report that a majority of Medicare recipients have vascular comorbidities with their AD, revived concern about how patients will fare once aducanumab infusions ramp up beyond the expert ARIA care at academic AD research centers and into community settings across the country.
Despite the controversy, aducanumab's approval has opened the door at the FDA—if not at the CMS—for three other anti-Aβ antibodies. In the fall, both Eisai/Biogen's lecanemab and Lilly's donanemab teams requested accelerated approval, using a rolling admission scheme whereby they submit portions of the application as they complete them. For lecanemab, which appears to cause less ARIA than aducanumab and donanemab, 2021 data includes Phase 2 open-label and post hoc analyses showing drastic amyloid removal, change in the desired direction of the plasma Aβ42/40 ratio and p-tau217, and, most importantly, a slowing of cognitive decline. The data supporting the application are published, the confirmatory Phase 3 trial Clarity will read out in the fall of 2022, and a secondary prevention trial is enrolling at 99 sites worldwide.

Lecanemab. During a Phase 2a trial's core period (blue field on left), the ratio of Aβ42/Aβ40 increased in the two treatment groups (green, blue lines) but not the placebo group (black line). During a treatment gap (orange field), the ratio fell; it rose again once treatment resumed (green field, right). [Courtesy of Eisai.]
For donanemab, 2021 brought similar news. In Phase 2, cognitive and functional decline slowed slightly. Amyloid plaques just about vanished, accompanied by a dent in both plasma p-tau217 levels and tangle growth as per PET. Also, the data supporting Lilly's FDA submission are published, and Phase 3 trials include a fully enrolled confirmatory study in early symptomatic AD and a fledgling secondary prevention study. The main differences to the lecanemab program are in trial designs; the treatment trial tests a shorter treatment course that ends once amyloid plaques are gone, while the prevention trial uses a decentralized approach that does away with most in-person visits to trial sites. In 2021, the cutting edge of innovation has moved to how best to deploy fluid-based markers, and to exploring cognitive and digital markers in hopes of slashing the high failure rate and cost of screening by PET.

Donanemab. Plasma p-tau217 plummets in response to donanemab treatment (right), as does amyloid plaque burden (left). [Courtesy of Eli Lilly.]
Gantenerumab made few headlines in 2021, but all the while was quietly inching forward. Its Phase 3 trials evaluate a formulation that gets injected under the skin—a simpler and quicker way of receiving a drug than infusion—and will start reading out this coming September. Roche eschewed accelerated approval in favor of filing for full approval based on the Phase 3 data. The subcutaneous antibody was designed to be given at home, which helped the trials navigate the COVID-19 turmoil without too much attrition. The DIAN trials unit (DIAN-TU) published results of its first treatment trial, which is continuing to administer gantenerumab with good tolerability in its open-label extension. Moreover, DIAN-TU chose gantenerumab for its upcoming primary prevention trial in people as young as 18, who have an autosomal-AD mutation but little or no brain amyloid yet. Finally, a version of gantenerumab engineered to shuttle larger amounts of it into the brain was reported to have done just that in a Phase 1 trial.
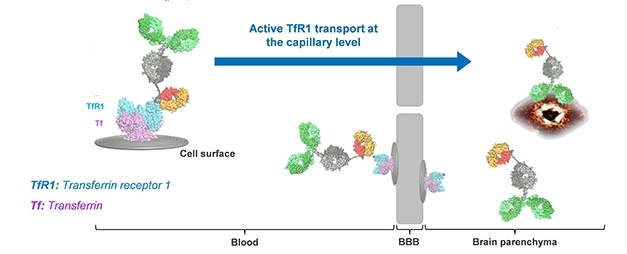
Passing Through. Roche’s “brain shuttle” (orange/yellow) allows gantenerumab (green) to bind transferrin receptor (blue) and hitch a ride into the brain, where it binds Aβ. [Courtesy of Roche.]
All three antibodies have FDA breakthrough therapy status, expediting review. Barring unforeseen setbacks, their approval is widely expected because the FDA's aducanumab decision has essentially made brain amyloid removal the core requirement for accelerated approval.
- Advisory Committee Again Urges FDA to Vote No on Aducanumab
- Aducanumab Approved to Treat Alzheimer’s Disease
- Fallout Continues After Aducanumab Approval
- Aducanumab: Will Appropriate-Use Recommendations Speed Uptake?
- Seeking Real-World Data on Whether Aducanumab Works
- Flurry of Investigations Besets Aducanumab
- Aduhelm Approval Reverberates Through Research
- Will Insurance Cover Aducanumab? Jury is Out
- Aduhelm Administration Remains a Trickle, ARIA a Concern
- Aduhelm Lowers Tau; Registry to Track Real-World Performance
- Aduhelm Phase 3 Data: ARIA Is Common, Sometimes Serious
- Donanemab Confirms: Clearing Plaques Slows Decline—By a Bit
- On Donanemab, Plaques Plummet. Off Donanemab, They Stay Away
- Lecanemab Post Hoc: Is Continual Treatment Required for Cognitive Benefit?
- Lecanemab Follows Aduhelm’s Path to Accelerated Approval
- Lecanemab Sweeps Up Toxic Aβ Protofibrils, Catches Eyes of Trialists
- In Phase 2, Donanemab Curbs Cognitive Decline in Early Alzheimer’s
- Can Donanemab Prevent AD? Phase 3 TRAILBLAZER-ALZ3 Aims to Find Out
- Donanemab Phase 3 Puts Plasma p-Tau, Remote Assessments to the Test
- Clinical Trials in COVID Era: How To Keep Moving Forward
- Brain Shuttle Could Halve Amount of Gantenerumab Needed
- Shuttle Unloads More Gantenerumab Into the Brain
Tau Therapies
In the arena of anti-tau therapeutics, a shakeout finally happened. It had been building since the year before, when four antibodies directed against tau's N-terminus started to wobble. Three fell; one is teetering on the edge. Lilly's zagotenemab exited the ring defeated by a negative 360-person Phase 2 study. Abbvie's tilavonemab stepped down after a negative 453-person trial. Alas, an odd thing happened with the other two contenders. While zagotenemab and tilavonemab were both ineffective yet safe, the remaining two split on this score. In its Phase 2 study of 654 people with early AD, Biogen's gosuranemab worsened scores on the ADAS-cog13, whereas a smaller Phase 2 trial of Genentech's semorinemab punched above expectations. It posted an apparent benefit on one of its two co-primary endpoints; an ongoing open-label extension will show whether the signal has staying power.
In the wake of these losses, the field's searchlight has swung toward targeting tau's mid-region. This is where 2021 showed much activity, though no results yet. At least six such antibodies are wending their way through the clinical pipeline. The farthest along are Janssen's JNJ-63733657 and UCB's bepranemab, which are both in sizeable, international Phase 2 studies. Nipping at their heels, Eisai's E2814 has completed a Phase 1 trial and is starting to be evaluated, by itself and concurrently with lecanemab, in DIAN-TU's Phase 1/2 NextGen trial. Of the three antibodies still in Phase 1, PNT 001 in 2021 reported data from an initial Phase 1 study and started a second trial, Lundbeck's AF 87908 spent 2021 recruiting, and Biogen's BIIB076 had Phase 1 data at CTAD, but no subsequent trials on the docket yet.
Beyond immunotherapies, scientists are attacking tau with small molecules and genetic therapies. On the former front, Lilly advanced its O-GlcNAcase enzyme inhibitor into a 330-person Phase 2 trial, and the startup Asceneuron moved ASN51 into Phase 1. Both drugs are to keep tau from aggregating by boosting its glycosylation. On the latter front, Ionis/Biogen's antisense oligomer BIIB080 slashed CSF tau levels by up to half, without serious adverse effects, in a Phase 1/2 trial that is continuing in open-label and expected to be followed with a Phase 2 study this year.
This data renewed hope for ASO treatments of neurodegenerative diseases after a particularly disheartening setback in March 2021, when an independent review board stopped dosing in Phase 3 with the huntingtin ASO tominersen for lack of benefit. This has not stopped ASO development. In 2021, at least six ASOs targeting expression of C9ORF72, SOD1, LRRK2, and FUS, were in Phase 1 to 3 trials for ALS or Parkinson's disease. (Earlier this week, Roche announced it had learned from its phase 3 debacle and would resume evaluating tominersen in a different trial).
Overall, drug development in Alzheimer's is increasingly pursuing targets identified by genomic studies that are fleshing out the pathways underpinning late-onset AD. Besides ApoE, these include targets in lipid metabolism, endosomal/lysosomal/autophagy and, of course, neuroinflammation. Much of that work remains preclinical. Notable clinical examples include therapeutic antibodies activating Trem2 signaling, antibodies targeting sortilin, and attempts to boost progranulin levels via gene therapy or small molecules.
Last, but not least, the old idea of repurposing, for AD, approved drugs that were originally made for other diseases drew new support in 2021, buoyed by government and private funders that push for diversification beyond Aβ and tau. Current attempts include bosutinib, dasatinib, atomoxetine, or liraglutide. None have posted robust benefits thus far.
- N-Terminal Tau Antibodies Fade, Mid-Domain Ones Push to the Fore
- More Tau Antibodies Bid Adieu; Semorinemab Keeps Foot in Door
- Aiming at the Tangle’s Heart? DIAN-TU Trial to Torpedo Tau’s Core
- Lecanemab Sweeps Up Toxic Aβ Protofibrils, Catches Eyes of Trialists
- Antisense Therapy Stifles CSF Tau in Mild Alzheimer’s Disease
- Target or Decoy: Are Drug Developers Chasing the Right Thing?
- Can an Old Diuretic Drug Disarm APOE4, Prevent Alzheimer’s?
- FTD Trials: The Now and the Future
Biomarkers
With regulatory approval for additional amyloid immunotherapies under consideration, blood-based markers could not have come at a better time. Evidence grew last year that Aβ42/40, various phospho-tau, and other plasma proteins will be valuable, if not as stand-alone diagnostics, then as screening tools to enrich trial populations and to limit reliance on expensive PET scans. That’s not to discount brain imaging. These direct measures of brain pathology continue to reveal new insights, particularly about disease progression.
For fluid markers, mass-spec assays came out on top in head-to-head testing for plasma Aβ42/40. Scientists also reported that Precivity, C2N’s mass-spec test, detected change in the Aβ42/40 ratio in blood years earlier than in CSF, suggesting that slowing Aβ42 clearance from the brain might be one of the earliest signs of AD. There is debate about how robust a marker this ratio can be, given the small difference between amyloid-positive and -negative people. Even so, Precivity is being used in screening for the ongoing AHEAD 3 and AHEAD 45 secondary prevention trials.

Ruled Out. A plasma Aβ42/40 cutoff (vertical line) could screen out amyloid-negative people, enriching clinical trial populations for participants with brain amyloid pathology. [Courtesy of Christina Rabe, Genentech.]
On the tau front, new data suggests that baseline levels and change in plasma p-tau181 predict not only amyloid and brain atrophy, as had been found previously, but neurodegeneration and cognitive decline as well. Scientists are actively dissecting tau to document all its forms and fragments that get secreted into the blood; at this point in time, it appears that p-tau231 may be the first to tick up. Tau alone appears insufficient to diagnose AD, but researchers have begun to pile various plasma markers and demographic variables into diagnostic algorithms. Glial markers come in here, too. Levels of GFAP and YKL40 were found to rise in CSF and plasma early on, maybe even before p-tau231. A preliminary analysis suggested a panel of 19 plasma proteins could predict AD with up to 97 percent accuracy. If this holds, it would be the best blood diagnostic to date.

Only if Aβ-Positive. Brain atrophy as per MRI (left) associated with plasma p-tau181, but only in cognitively unimpaired, amyloid-positive people, implying that blood p-tau181 reflects brain Aβ plaques. [Courtesy of Moscoso et al., JAMA Neurology, 2021.]
Of course, no experimental marker will be used for diagnosis without robust, validated, and certified assays. On that score, the Bio-Hermes initiative made news last year. This collaboration of 10 companies will assess the accuracy of low-cost, rapid blood tests for amyloid and cognitive decline, as well as of digital biomarkers. App-based cognitive tests and artificial intelligence-based analysis of behavioral patterns are gaining traction as potential diagnostics.
On the brain-imaging front, the predictive power of MRI and PET shone last year. Scientists found that waning MRI signals from the locus coeruleus (LC), a speck in the brainstem packed with noradrenergic neurons, correlate with plaque and tangle load and might represent an early warning of impending AD. The LC is one of the first regions of the brain to accumulate tangles, which evade detection by PET because this cluster of neurons is so small. On the amyloid front, scientists found that once plaques have crossed a certain threshold, they accumulate at the same steady rate in everyone, like clockwork. In other words: A single amyloid PET scan can extrapolate when a person will begin to develop symptoms of dementia. As for how that dementia will proceed, here tau imaging stood out. This surprised no one, since tau pathology correlates better with cognition. When compared directly with Aβ PET and structural MRI, tau PET best predicted falling MMSE scores.

Amyloid Clock. Taking each person’s amyloid accumulation trajectory versus age from a spaghetti plot (left) and lining them up using years since the person reached SUVR of 1.2 on PiB PET (right), reveals that plaques grow at the same rate in everyone. [Republished with permission, © 2021 American Academy of Neurology.]
- In Side-by-Side Test of 8 Blood Aβ Assays, Mass Spec Shines
- Plasma Aβ—First Sign of AD, But Tough to Measure Prospectively?
- Plasma P-Tau181 Predicts, Monitors Alzheimer’s Progression
- Earliest of Them All: Blood P-Tau231 Assay Flags Pre-Amyloid Alzheimer’s
- Where to Now, Phospho-Tau?
- Algorithm Bests Humans in Predicting Alzheimer’s
- Astroglial Markers Poised for Stardom?
- Mirror, Mirror on the Wall, Who’s the Earliest of Them All?
- Stunning Diagnostic Accuracy from a Plasma Protein Panel?
- Bringing Alzheimer’s Detection into the Digital Age
- Bio-Hermes: Making Alzheimer’s Diagnosis Fast, Cheap, Accessible
- Is a Waning Locus Coeruleus an Early Sign of Alzheimer’s
- Can a Single Amyloid PET Scan Predict Time to Symptom Onset?
- Tau PET Best Predicts Short-Term Decline in Early Alzheimer’s
Genetics
If you thought geneticists were slowing down, think again. For one, they found a new APP variant that causes autosomal-dominant AD. Dubbed Uppsala, this multi-codon deletion clips six amino acids from the middle of Aβ, speeding its production and boosting its propensity to aggregate.

Nip and Tuck. The Uppsala mutation, circled red, snips out residues 690-695 of APP.
For another, a massive GWAS meta-analysis fished out 42 new risk loci for late-onset AD. Most fell into known pathways, such as APP processing, endosomal/lysosomal function, and innate immunity, in keeping with the cellular phase of the disease being marked by dysfunctional protein trafficking and microglial activation.
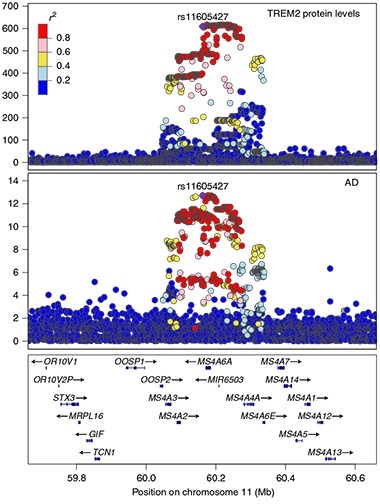
TREM2 Connection. Bubbling up from the MS4A gene cluster (bottom) are many variants linked with AD (middle). Variants at this locus associate with reduced plasma sTREM2 (top). [Courtesy of Ferkingstad et al., Nature Genetics, 2021].
Newer approaches in search of functional risk variants merged GWAS with proteome- or transcriptome-wide association data. One such PWAS x GWAS turned up 10 new AD genes, while a TWAS x GWAS spat out 11 Parkinson’s disease loci, four of them new. Studies of protein quantitative trait loci (pQTL) have started to pay dividends, as well. One linked protein levels in brain parenchyma, cerebrospinal fluid, and plasma to 433 gene loci, tying 20 proteins to AD risk. Similarly, a pQTL study of 35,000 Icelanders identified more than 18,000 loci that will help connect the dots between risk alleles and disease, starting with a link between the two known AD risk loci MS4A and TREM2.
Given that a person might carry myriad variants that each increase or decrease his or her likelihood of getting AD by a few percent, what do they all amount to? Looking to centenarians for clues, scientists realized that some live longer than most of us not because they got stuck with fewer risk alleles, but because they are blessed with rare variants that protect against age-related diseases, including AD and diabetes. Polygenic risk scores will help explain why. Last March, the U.K. company Cytox Ltd. introduced genoSCORE™-LAB, a test that types not only AD variants that pass thresholds for significance in GWAS, but also variants that just miss the mark yet might be meaningful. The polygenic AD score tallies risk from 114,000 such loci and is sold to physicians in the U.S., U.K., and EU.
- Double Whammy: APP Uppsala Deletion Ups Aβ and Its Aggregation Propensity
- Massive GWAS Meta-Analysis Digs Up Trove of Alzheimer’s Genes
- PWAS x GWAS? Proteome Analysis Nets 10 New Alzheimer’s Genes
- TWAS x GWAS? Transcriptome Analysis Finds 11 Parkinson’s Genes
- Paper Alert: pQTLs Pin GWAS Loci to Tissue Proteins, Drug Targets
- Massive Proteomics Study Connects Genes, Proteins, Disease
- In Oldest Old, Rare Longevity Variants Suppress Common Pathogenic Ones
- Just What the Doctor Ordered? Polygenic Test Gauges Your Alzheimer's Risk
Microglia
Scientists are slowly beginning to understand the wild and varied world of microglia. In 2021 a subtype emerged that regulates synaptic plasticity. Another mops up fraying myelin; dubbed white-matter-associated microglia or WAM, it seems to let plaques grow. More evidence strengthened the idea that yet other microglia restrict plaque growth by using lysosomes as trash compactors to compress Aβ, which they then spit out as dense core plaques. This process may protect the brain from more toxic, diffuse amyloid. If that isn’t enough, microglia may drive formation of tau aggregates according to a PET study that correlated microglial activation with the subsequent arrival of nearby tangles.
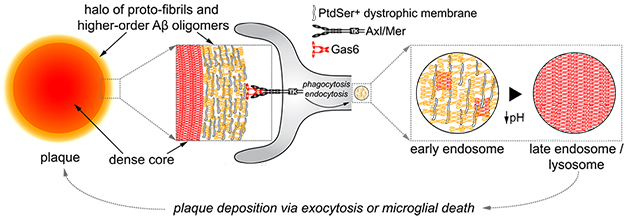
Pack It Tight. Microglia around plaques take up loose amyloid (yellow fuzz) using TAM receptors, and condense it (red cross-hatches) in their lysosomes. Then they expel this material, building dense-core plaques. [Courtesy of Huang et al., Nature Immunology.]

Hello Microglia? PS2APP amyloid-laden mice (left) take up plenty of FDG (orange). Killing off their microglia (middle) drops the signal below that of wild-type (right). [Courtesy of Xiang et al., Science Translational Medicine.]
Still, much remains unknown about these polymorphic cells, particularly what drives them to disease-associated or inflammatory phenotypes. Besides amyloid, one trigger might be signals from nearby astrocytes, or simply age, which seems to send these cells into a metabolic tailspin. Along the way, AD-associated microglia may replicate their DNA and divide so many times that they enter a toxic, senescent state. One provocative study even suggested that microglial activation accounts for much of the well-known FDG PET signal in early stage AD brain.
A new APP knock-in mouse model may help scientists better understand microglia. In addition to developing rampant plaques, these mice mobilize microglia that adopt a transcriptional profile similar to that seen in AD. The glia surround plaques, and they fill up with Aβ and lipids. Levels of the microglial receptor and AD risk factor TREM2 also spike in the animals’ brains.
- Not Just Alzheimer's: Microglia Sculpt the Brain in Health and Disease
- WAM! New Microglial Subtype Mops Up Moribund Myelin
- Flipping the Script: Could Myelin Degeneration Drive Amyloidosis?
- Microglia Build Plaques to Protect the Brain
- PET Firms Up Amyloid Cascade: Plaques, Inflammation, Tangles
- Ingested Aβ Trips Transformation in Microglia
- In Triculture Model, Astrocyte-Microglia Cross Talk Spurs Inflammation
- Myeloid Metabolic Crisis May Trigger Cognitive Decline in Aging Brain
- DAMned to Death? Microglia May Proliferate to Senescence
- What FDG PET ‘Sees’ in AD: Angry Microglia, Not Just Neurons
- Striking Microgliosis in New APP Knock-in Mice
Astrocytes / ApoE
Used to taking a back seat to microglia in the lab, these most numerous cells in the brain came to the fore last year. Research offered several explanations for how astrocytes turn toxic in neurodegeneration. One study found that when their lysosomes stop working, astroglia dump the organelles and their contents into the extracellular space, where they kill nearby neurons. Another attributed astrocyte neurotoxicity to long-chain fatty acids, as removing a lipid elongase called ELOVL1 reined in reactive astrocytes. Scientists also discovered a tau-driven path to toxicity. Astrocytes exposed to tau oligomers entered a senescent state, releasing a protein that propagates senescence to nearby cells, exacerbating tau toxicity and accelerating its spread. A consensus effort among astrocyte and neurodegeneration scientists called for nuanced, multivariate characterization of reactive astrocytes, and proposed a more consistent terminology (Escartin et al., 2021).
And of course, glial cells talk to each other. Adding to this theme, scientists last year reported that a subset of astrocytes make interleukin-3, a cytokine attributed to immune cells. They showed that, in AD, microglia make more IL-3 receptors, and that the IL-3 released by astrocytes strengthens signaling downstream of TREM2, a microglial cell surface receptor needed for clearing Aβ.
Mention of astrocytes requires a shout out to ApoE, as they produce the lion’s share of it. Astrocytes expressing this genetic risk factor for AD accumulate unsaturated fatty acids as lipid droplets. How the droplets might factor in AD is unclear, but adding the lipid precursor choline restored lipid metabolism back to normal. In mice expressing human ApoE isoforms, deleting ApoE4, but not ApoE3, specifically in astrocytes protected against tau toxicity and neurodegeneration. This, too, seems to require glial cross-talk, because taking astrocyte ApoE4 out of the picture dampened microglial phagocytosis of synapses.

Speed Traffic, Lose Plaque? Amyloid (white) in 1-year-old APP knock-in mice (left) was nearly prevented by knocking out the gene for the endosomal proton leak channel NHE6 (right) when the animals were 2 months old. Ablating NHE6 reduced endosome pH and increased ApoE recycling. [Courtesy of Pohlkamp et al., eLife.]
Boosting ApoE recycling through the endosome/lysosome system might be a therapeutic avenue worth exploring. Scientists reported that ApoE4 becomes sticky at pH 6.4, the precise acidity of early endosomes. Lowering the vesicle pH a smidgen in neurons was enough to restore ApoE4 trafficking, and relieve amyloidosis in mice. Alternatively, upping expression of the low-density lipoprotein receptor, which binds ApoE, dramatically lowered levels of the apolipoprotein in mouse brain, tempered microglia, and reduced toxicity in a mouse model of tauopathy.

LDLR: Guardian Angel? Synapses (red) are dense in the wild-type mouse hippocampus (left), lost in P301S mouse hippocampus (center), and partially preserved by LDLR overexpression (right). [Courtesy of Shi et al., Neuron.]
- Reactive Astrocytes Boot Basic, Dysfunctional Lysosomes
- ELOVL Hurts—Enzyme Makes Lipids That Turn Astrocytes Toxic
- Astrocytes Are Just Dying to Spread Tau
- Old Cytokine, New Tricks? Astrocyte IL-3 Pokes Microglia in AD
- Droplets of Unsaturated Fats Burden Human ApoE4 Astrocytes
- Squelching ApoE in Astrocytes of Tau-Ravaged Mice Dampens Degeneration
- Could Juicing Up Trafficking Abolish ApoE4’s Alzheimer’s Risk?
- Taming ApoE Via the LDL Receptor Calms Microglia, Slows Degeneration
Immune Cells at the Brain's Border
Last year brought the stunning revelation that the mammalian brain has its own collection of adaptive immune cells patrolling its border. Scientists found skull bone marrow brimming with monocytes and B cells. These squeeze through narrow channels in the cranium into the meningeal membranes that surround the brain. From there, the cells infiltrate the parenchyma or spinal cord in response to inflammatory signals, all without ever seeing the inside of a blood vessel, let alone the spleen or thymus, where B and T cells typically mature. Some think this “private stock” of adaptive immune cells arose to distinguish the brain as “self” and safeguard its privilege. Others doubt the role of the skull bone marrow. They report that the meninges themselves generate brain B cells. Single-cell RNA-Seq analysis found niches of mature and immature B cells in the dura matter.

Marrow to Meninges. The bone marrow (BM) contains B cells (green) that cross physical channels into the dura (left). The transiting B cell (arrowhead) expresses no IgM (right), indicating it is still developing. [Courtesy of Brioschi et al., Science, 2021.]
Do these newly discovered immune cells play a role in disease? This will likely be an area of intense research, and Alzforum will monitor news from these “border” tissues. One idea is that as mice age, T cells accumulate in their meninges, where they enter an immunosuppressive state. This appears to pose a plethora of problems, including with glymphatic flow, microglial function, plaque accumulation, and memory decline. The meninges themselves also turned out to boost clearance of amyloid from the mouse brain by passive immunotherapy.
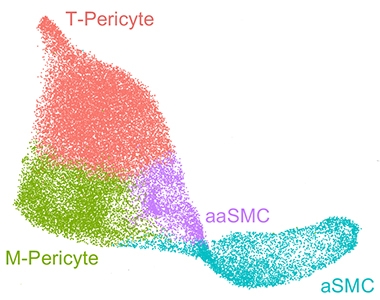
Atlas of the Vasculature. Mural cells on blood vessels in the human brain fall into four distinct groups: arterial smooth muscle cells (aSMC), arteriolar SMCs (aaSMC), matrix-specialized and transport-specialized pericytes (M-pericytes, T-pericytes). [Courtesy of Yang et al., 2021.]
Lest we forget about peripheral immune cells, evidence continued to build that they can invade the brain, with potentially disastrous consequences. For example, T cells that recognize α-synuclein were seen surrounding Lewy bodies in the brain and flood these areas with the inflammatory cytokine IL-17A.
How peripheral cells squeeze through the blood-brain barrier remains a bit mysterious. High-resolution expression maps of the human brain vasculature should help address this and other questions about blood vessels and neurodegeneration. Last year, scientists found a way to isolate vascular and perivascular cells from postmortem brain tissue that was compatible with single-nuclei RNA-Seq, dubbed VINE-Seq. Besides identifying new types of pericytes and fibroblasts, this method revealed that vascular cells express 30 of the top 45 AD risk genes, implying that their role in this disease is underappreciated. A similar, single-nuclei spatial expression analysis found that, as mice age, inflammation ramps up in their choroid plexus, the network of vessels that surround the brain's ventricles and produce the cerebrospinal fluid.
- Private Stock—Brain Taps Skull Bone Marrow for Immune Cells
- More Evidence for Meningeal B Cells
- As Mice Age, T Cells Traipse Around Their Meninges. Mayhem Ensues
- Does Anti-Amyloid Immunotherapy Need the Lymphatic System?
- Intruder Alert: Inflammatory T Cells Lurk Near Lewy Bodies, Neurons
- Map of Human Vascular Expression Highlights its Potential Role in Alzheimer’s
Proteinopathy
Despite decades of work, scientists still lack a firm grasp of how plaques, tangles, and other types of amyloids form and spread in the brain. Last year, new clues came from using stable isotope labelling kinetics to study the formation of plaques in real time in mice. Aβ42 formed dense cores first, then Aβ38 added itself to the plaque's periphery. Whether plaques form this way in the human brain remains to be seen.

Folds of a Feather Flock Together. Dendrogram of a proposed structure-based classification of tauopathies. Colors denote microtubule-binding domains R1 to R4, arrows denote β-strands. Non-protein entities are in black. [Courtesy of Shi et al., Nature 2020.]
As for tangles, scientists discovered that they are far from inert tombstones, as had been thought. Instead, tau drifts in and out, with a half-life of about a week. Plus, there may be more to plaques and tangles than Aβ and tau. Other amyloidogenic proteins and peptides insinuate themselves into both, and their expression patterns might explain why some neurons are more prone to forming amyloids. A form of RNA even gets into the mix: methylated RNA and its protein partner, HNRNPA2B1, were reported to bind tau and pile up in tangles as AD worsens.
Scientists have long wondered whether the spread of amyloid seeds from region to region, or de novo emergence of amyloids in vulnerable cells, explains the progression of AD and related diseases. A kinetic model of tangle growth came down on the side of the latter. It suggested that replication of tau seeds sets the pace for tangle accumulation, and that spread plays but a negligible role.

Double Spiral. Protofibrils of TDP43 taken from two people who had ALS/FTD adopt the same fold. [Courtesy of Arseni et al., Nature, 2021.]
Then what makes cells vulnerable? On this old question, a single-nucleus RNA-Seq study pointed at subsets of excitatory neurons found close to tangles that express the transcription factor RORB, whereas a subset of astrocytes in the same vicinity may exacerbate the situation by failing to protect the neurons.
In 2021, years of effort solving cryo-EM structures culminated in a family tree of tau protofibrils. Comparing new structures for a group of 4R tauopathies to previously reported ones from 3R- and mixed 3R+4R tauopathies, scientists found that the unique way tau contorts itself within a protofibril maps onto the neuropathological characteristics of each disease. In a surprise twist, the first high-resolution cryoEM structure for TDP-43 fibrils taken from the human brain showed that its core structure resembled no other amyloid.
- iSILK Tracks Growth of Mouse Plaques at Peptide Level
- Not Merely Tau Tombstones, Neurofibrillary Tangles Are Dynamic
- Methylated RNA: A New Player in Tau Toxicity?
- Doubling of Tau Seeds, Not Spread, Sets Pace of Tauopathy in Alzheimer's
- Amyloids Fibrillize, or Stay Solo, Based on Liaisons with Similar Proteins
- Selective Vulnerability News: RORB Neurons Are First Victims of Tangles
- Flock of New Folds Fills in Tauopathy Family Tree
- Double Spiral Sets TDP-43 Apart from Other Amyloids
Clues from Big Data
Big data comes with big promises, and fulfilling them can be easier said than done. Years of anticipation about leveraging artificial intelligence and other statistical packages to draw knowledge out of reams of transcriptomic, proteomic, metabolomic data have produced no major Eureka! moments yet. Still, scientists wrangling these large datasets are beginning to make progress. Large-scale surveys of proteomes and transcriptomes helped them distinguish, in greater detail than ever, the Alzheimer's brain from a healthy one. Bulk RNA-Seq analysis proposed three distinct types of Alzheimer’s. One looked like typical disease, with plaques, tangles, and neuroinflammation, while the other two featured tau pathology more prominently. A machine-learning analysis of the largest collection of tau PET scans to date predicted four subtypes of AD. And a single-nucleus RNA-Seq approach found unique clusters of cells in autosomal-dominant AD that distinguish it from sporadic AD.

Gene Drivers. Network analysis identifies key genes that are down (left) or up (right) in three molecular subtypes of AD. Two, B and C, can be further divided, for a total of five. [Courtesy of Neff et al., Science Advances/AAAS.]
Scientists are also using omics approaches to unravel early changes in the AD brain. Proteomics identified 53 proteins that are up- or downregulated at different stages of the disease. Proteins that function in endocytosis or synapses fluctuated in the preclinical stage. Reaching back further, scientists used massive gene-expression datasets to identify transcriptomic signatures in healthy people aged 45 to 70 that resemble transcriptomes of AD patients, perhaps explaining how aging increases risk for the disease in some more than others.
And in a tour de force, researchers combined electron, super-resolution, and fluorescent microscopy with mass spectrometry to map stubby and dendritic spines in exquisite detail. This nanoscopy approach could show scientists what happens when synapses are lost in AD and other neurodegenerative diseases. A new technique called SynTOF, aka synaptic mass spectrometry, enabled analysis of millions of individual synapses from AD and control brains. This study found that spines coated with CD47 were more likely to survive in people who had tau pathology. Usually associated with cancer, this immune receptor has not been studied much in the brain, but might act as a “don’t eat me” signal for microglia as they prune spines.
Anatomy of a Spine. Video model of an average mushroom spine shows tight packing of myriad cell surface and cytosolic proteins, structural elements, and organelles. [Courtesy of Helm et al., Nature Neuroscience, 2021.]
With large omics projects, replication becomes a problem for the field, especially without standards for data gathering and analysis. To help with that, last year the NIH announced the human induced pluripotent stem cell Neurodegenerative Disease Initiative, or iNDI. IPSCs are widely used and often get differentiated into neurons and glia, which then serve to model disease risk or progression. But with so many labs using their own in-house cell lines, comparing data across labs becomes daunting. iNDI will engineer healthy donor cells to carry one of 134 genetic variants associated with neurodegeneration, and will make differentiated cells and data available.
- Expression Analysis Uncovers Three Distinct Forms of Alzheimer’s
- ADAD and LOAD: At Cellular Level, They Are Not the Same
- Forget Typical Alzheimer's: AI Finds Four Types
- Proteomics Dates Endosomal, Synaptic Changes to Preclinical AD
- Do Gene Expression Signatures of Aging Signal AD?
- Peering Inside Stubby and Mushroom Dendritic Spines
- Single Synapse Mass Spec Snags CD47 as Alzheimer’s Resilience Factor
- iNDI Aims to Standardize Human Stem Cell Research
Viruses and dementia
We almost did it: write a 2021 roundup without mentioning the word we are all so tired of. COVID. Alas, we must. Besides the pandemic’s ongoing impact on research, the virus itself affects the brain in some people, and last year, clinicians realized that COVID can worsen existing neurologic conditions. Exactly how remains to be worked out, but people who are ApoE4-positive are more susceptible to coronavirus infection and severe illness.

Spiking Amyloid Transmission? Donor HEK cells (left) bearing aggregates (red) of the NM domain from the yeast prion Sup35 were co-cultured with Vero cells expressing soluble NM (green). If the donor cells expressed the Sars-CoV-2 spike protein (right), then NM aggregation in Vero cells accelerated. [Courtesy of Liu et al., Nature Communications, 2021.]
What’s more, SARS capsid glycoproteins, including spike, may promote release of extracellular vesicles that could spread amyloidogenic proteins. In an odd twist, scientists found that SARS-CoV-2 only infects cells that express the lysosomal receptor TMEM106B, whose gene has known risk variants for AD and FTD; whether those affect COVID is unclear. As for other viruses, the controversy about Herpes simplex and AD risk endures. One 2021 study found no consistent correlation across four European countries, while another, of more than half a million people in Sweden, linked untreated herpes infection to higher dementia risk, particularly in ApoE4 carriers.
- How Does COVID-19 Affect the Brain?
- COVID-19 Worsens Neurological Problems, Delirium
- APOE Tied to Increased Susceptibility to SARS-CoV-2
- Viral Proteins Help Shuttle Tau Aggregates Among Cells
- Sars-CoV-2 Virus Needs TMEM106B to Infect Human Cells
- Herpes Update—Virus Increases Dementia Risk in Sweden
- New Data Questions Herpes-Alzheimer’s Connection
- More Data on Herpes and Alzheimer’s Disease
If 2020 was the year Alzheimer’s researchers absorbed, and adjusted to, the shock of COVID-19, 2021 was the year they had settled into their new circumstances and made them work. Now, 2022, we expect better from you!—Tom Fagan and Gabrielle Strobel
References
Therapeutics Citations
- Aduhelm
- Leqembi
- Kisunla
- Gantenerumab
- Zagotenemab
- Tilavonemab
- Gosuranemab
- Semorinemab
- Posdinemab
- Bepranemab
- E2814
- PNT001
- Lu AF87908
- BIIB076
- Ceperognastat
- ASN51
- BIIB080
- PBFT02
- Ezeprogind
- Bosutinib
- Dasatinib + Quercetin
- Atomoxetine
- Liraglutide
News Citations
- Advisory Committee Again Urges FDA to Vote No on Aducanumab
- Aducanumab Approved to Treat Alzheimer’s Disease
- Aducanumab: Will Appropriate-Use Recommendations Speed Uptake?
- Seeking Real-World Data on Whether Aducanumab Works
- Flurry of Investigations Besets Aducanumab
- Aduhelm Approval Reverberates Through Research
- Will Insurance Cover Aducanumab? Jury Is Out
- Aduhelm Administration Remains a Trickle, ARIA a Concern
- Aduhelm Lowers Tau; Registry to Track Real-World Performance
- Aduhelm Phase 3 Data: ARIA Is Common, Sometimes Serious
- Donanemab Confirms: Clearing Plaques Slows Decline—By a Bit
- On Donanemab, Plaques Plummet. Off Donanemab, They Stay Away
- Lecanemab Post Hoc: Is Continual Treatment Required for Cognitive Benefit?
- Lecanemab Follows Aduhelm’s Path to Accelerated Approval
- Lecanemab Sweeps Up Toxic Aβ Protofibrils, Catches Eyes of Trialists
- In Phase 2, Donanemab Curbs Cognitive Decline in Early Alzheimer’s
- Can Donanemab Prevent AD? Phase 3 TRAILBLAZER-ALZ3 Aims to Find Out
- Donanemab Phase 3 Puts Plasma p-Tau, Remote Assessments to the Test
- Clinical Trials in COVID Era: How To Keep Moving Forward
- Brain Shuttle Could Halve Amount of Gantenerumab Needed
- Shuttle Unloads More Gantenerumab Into the Brain
- N-Terminal Tau Antibodies Fade, Mid-Domain Ones Push to the Fore
- More Tau Antibodies Bid Adieu; Semorinemab Keeps Foot in Door
- Aiming at the Tangle’s Heart? DIAN-TU Trial to Torpedo Tau’s Core
- Antisense Therapy Stifles CSF Tau in Mild Alzheimer’s Disease
- Target or Decoy: Are Drug Developers Chasing the Right Thing?
- Can an Old Diuretic Drug Disarm APOE4, Prevent Alzheimer’s?
- FTD Trials: The Now and the Future
- In Side-by-Side Test of 8 Blood Aβ Assays, Mass Spec Shines
- Plasma Aβ—First Sign of AD, But Tough to Measure Prospectively?
- Plasma P-Tau181 Predicts, Monitors Alzheimer’s Progression
- Earliest of Them All: Blood P-Tau231 Assay Flags Pre-Amyloid Alzheimer’s
- Where to Now, Phospho-Tau?
- Algorithm Bests Humans in Predicting Alzheimer’s
- Astroglial Markers Poised for Stardom?
- Mirror, Mirror on the Wall, Who’s the Earliest of Them All?
- Stunning Diagnostic Accuracy from a Plasma Protein Panel?
- Bringing Alzheimer’s Detection into the Digital Age
- Bio-Hermes: Making Alzheimer’s Diagnosis Fast, Cheap, Accessible
- Is a Waning Locus Coeruleus an Early Sign of Alzheimer’s Disease?
- Can a Single Amyloid PET Scan Predict Time to Symptom Onset?
- Tau PET Best Predicts Short-Term Decline in Early Alzheimer’s
- Double Whammy: APP Uppsala Deletion Ups Aβ and Its Aggregation Propensity
- Massive GWAS Meta-Analysis Digs Up Trove of Alzheimer’s Genes
- PWAS x GWAS? Proteome Analysis Nets 10 New Alzheimer’s Genes
- TWAS x GWAS? Transcriptome Analysis Finds 11 Parkinson’s Genes
- Paper Alert: pQTLs Pin GWAS Loci to Tissue Proteins, Drug Targets
- Massive Proteomics Study Connects Genes, Proteins, Disease
- In Oldest Old, Rare Longevity Variants Suppress Common Pathogenic Ones
- Just What the Doctor Ordered? Polygenic Test Gauges Your Alzheimer’s Risk
- Not Just Alzheimer's: Microglia Sculpt the Brain in Health and Disease
- WAM! New Microglial Subtype Mops Up Moribund Myelin
- Flipping the Script: Could Myelin Degeneration Drive Amyloidosis?
- Microglia Build Plaques to Protect the Brain
- PET Firms Up Amyloid Cascade: Plaques, Inflammation, Tangles
- Ingested Aβ Trips Transformation in Microglia
- In Triculture Model, Astrocyte-Microglia Cross Talk Spurs Inflammation
- Myeloid Metabolic Crisis May Trigger Cognitive Decline in Aging Brain
- DAMned to Death? Microglia May Proliferate to Senescence
- What FDG PET ‘Sees’ in AD: Angry Microglia, Not Just Neurons
- Striking Microgliosis in New APP Knock-in Mice
- Reactive Astrocytes Boot Basic, Dysfunctional Lysosomes
- ELOVL Hurts—Enzyme Makes Lipids That Turn Astrocytes Toxic
- Astrocytes Are Just Dying to Spread Tau
- Old Cytokine, New Tricks? Astrocyte IL-3 Pokes Microglia in AD
- Droplets of Unsaturated Fats Burden Human ApoE4 Astrocytes
- Squelching ApoE in Astrocytes of Tau-Ravaged Mice Dampens Degeneration
- Could Juicing Up Trafficking Abolish ApoE4’s Alzheimer’s Risk?
- Taming ApoE Via the LDL Receptor Calms Microglia, Slows Degeneration
- Private Stock—Brain Taps Skull Bone Marrow for Immune Cells
- More Evidence for Meningeal B Cells
- As Mice Age, T Cells Traipse Around Their Meninges. Mayhem Ensues
- Does Anti-Amyloid Immunotherapy Need the Lymphatic System?
- Intruder Alert: Inflammatory T Cells Lurk Near Lewy Bodies, Neurons
- Map of Human Vascular Expression Highlights its Potential Role in Alzheimer’s
- iSILK Tracks Growth of Mouse Plaques at Peptide Level
- Not Merely Tau Tombstones, Neurofibrillary Tangles Are Dynamic
- Methylated RNA: A New Player in Tau Toxicity?
- Doubling of Tau Seeds, Not Spread, Sets Pace of Tauopathy in Alzheimer's
- Selective Vulnerability News: RORB Neurons Are First Victims of Tangles
- Flock of New Folds Fills in Tauopathy Family Tree
- Double Spiral Sets TDP-43 Apart from Other Amyloids
- Expression Analysis Uncovers Three Distinct Forms of Alzheimer’s
- ADAD and LOAD: At Cellular Level, They Are Not the Same
- Forget Typical Alzheimer's: AI Finds Four Types.
- Proteomics Dates Endosomal, Synaptic Changes to Preclinical AD
- Do Gene Expression Signatures of Aging Signal AD?
- Peering Inside Stubby and Mushroom Dendritic Spines
- Single Synapse Mass Spec Snags CD47 as Alzheimer’s Resilience Factor
- iNDI Aims to Standardize Human Stem Cell Research
- How Does COVID-19 Affect the Brain?
- COVID-19 Worsens Neurological Problems, Delirium
- APOE Tied to Increased Susceptibility to SARS-CoV-2
- Viral Proteins Help Shuttle Tau Aggregates Among Cells
- Sars-CoV-2 Virus Needs TMEM106B to Infect Human Cells
- Herpes Update—Virus Increases Dementia Risk in Sweden
- New Data Questions Herpes-Alzheimer’s Connection
- More Data on Herpes and Alzheimer’s Disease
Series Citations
Paper Citations
- Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SH, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner IB, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021 Mar;24(3):312-325. Epub 2021 Feb 15 PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.