The Alzheimer’s Association International Conference, held July 28 to August 1, drew 8,200 attendees to the beautiful if distressed city of Philadelphia. Thousands more watched online. The growth reflects expansion in both research topics and commercialization ranging from drugs to mobile testing apps to head-only desktop MRI scanners. The big news? Blood tests are getting really good. Amyloid immunotherapy gained ground with eight-year treatment data in DIAN and three-year treatment data for lecanemab, while regulators around the world are deciding whether to approve. New clinical trial data were sparse but research from human genomics to nodal biology strutted its stuff.
Are Alzheimer’s Blood Tests Ready for Primary Care?
“I can feel the ground move under the podium.”
No, not an earthquake. At this year’s Alzheimer Association International Conference, held July 27 to August 1 in Philadelphia, Stephen Salloway, Brown University, Providence, Rhode Island—along with many others—felt a slight sense of dizziness at the speed at which blood-based biomarkers for Alzheimer’s disease are racing forward. The newest data suggest that plasma p-tau217 knocks the socks off clinical assessment for amyloid pathology.
“The acceleration has been amazing,” said Gil Rabinovici, University of California, San Francisco. “Amyloid PET took 20 years to be approved. The first p-tau paper came out in 2020, and here we are talking about using it in primary care.”
At AAIC, data showed that several different tests perform well in head-to-head comparisons. A fragment of tau in the blood appeared exquisitely accurate at identifying people who have neurofibrillary tangles. Even a mail-in test for Alzheimer’s disease could be in the cards. Before long, a teeny drop of dried blood might be all it takes. Alzforum will summarize findings on each of these points in the days to come. For the first—just how robust are these tests—read on below.
Over the last four years, blood measures of tau phosphorylated on serine 217 have consistently distinguished amyloid-positive from amyloid negative people, at least in memory clinics and carefully recruited cohorts. The question on clinicians’ minds was whether it would work equally well in primary care. Based on several presentations in Philadelphia, the answer seems to be a resounding yes.
Nailed It. In primary care, C2N’s APS2 test (orange) and the %p-tau217 (gray) in plasma pegged people with amyloid pathology with high accuracy, specificity, and sensitivity. Positive predictive values (PPV) ran between 88 and 91 percent; NPVs between 87 and 92 percent. [Courtesy of Palmqvist et al., 2024.]
In a highly anticipated talk of the meeting, Oskar Hansson, University of Lund, Sweden, reported that mass spectrometry tests for %p-tau217 in the plasma, that is, the ratio of fragments of phosphorylated versus unphosphorylated at that amino acid, identified people with amyloid pathology with high accuracy in primary and secondary care settings in Sweden. Randall Bateman, Washington University, St. Louis, subsequently showed data from the Seabird trial, which evaluates blood markers in a representative sample of the St. Louis population. Here, too, plasma %p-tau217 identified amyloid-positive volunteers with specificities and sensitivities akin to those seen in memory center cohorts.
It’s not just the C2N test. Nicholas Ashton, Banner Alzheimer’s Institute, Phoenix, offered a snapshot of how the fully automated ADx Neuroscience Lumipulse assay for plasma p-tau217 performs in secondary care. This runs on a desktop machine that renders results on the same day, whereas the C2N test requires shipping of the sample to a mass spec lab for analysis. Measured onsite at four centers across three countries, it identified AD with accuracies upwards of 92 percent. “I think we are in desperate need of a fully automated version of this biomarker to meet the needs of the general population,” Ashton said.
Markers for Primary Care
As reported in JAMA on July 28, Hansson, first author Sebastian Palmqvist, also at Lund University, and colleagues evaluated how well C2N’s tests for plasma %p-tau217 alone, or in combination with the plasma Aβ42/40 ratio, i.e., their APS2 test (see Dec 2023 news), identified people with amyloid pathology or Alzheimer’s disease. From 1,213 volunteers, they tested samples in two ways—storing then analyzing samples in one large batch, as is typically done in cohort studies, or prospectively analyzing individual samples as they were collected over four years. The former tends to be more accurate since all samples are measured simultaneously, reducing variability due to handling, instrument accuracy, and other systematic errors. The latter, however, is how routine clinical care works, hence useful tests must perform reliably in this setting, too.
In this study, both types of analysis seemed to work equally well. “I was surprised to see that there was really no drop in performance in the prospective analysis,” Hansson told Alzforum. “The assays are very robust.”
How accurate were these tests? Among 515 primary care subjects in Sweden who had cognitive symptoms, 276 were deemed to have amyloid pathology based on a positive CSF Aβ42/40 test or an amyloid PET scan. Batch analysis of 307 of 515 plasma samples with the APS2 and the %p-tau217 tests correctly identified AD pathology in 91 and 86 percent of cases, respectively, when a single cutoff point was used (image above). As in a previous analysis by this group, the cutoff was chosen to ensure a specificity of 90 percent (Barthélemy et al., 2024). In this study, that equated to an APS2 score of 36 and a %p-tau217 ratio of 3.26. Based on those numbers, positive and negative predictive values rang in at 91 and 85 percent, respectively, with the APS2 tests performing slightly better on most metrics. Importantly, in prospective analyses of 208 of the 515 samples, the two tests worked just as well as in the batch analysis.
Nailed It Again. The assays performed as well in secondary care as in primary. [Courtesy of Palmqvist et al., 2024.]
Among 698 volunteers from secondary care sites, the tests identified the 344 amyloid-positive cases just as well as they had in primary care, with PPVs and NPVs hovering around 90 percent. Again, it was a dead heat between the batch and prospective analysis (image at right).
Using a two-cutoff approach improved accuracy. Hansson and co-author Suzanne Schindler at Washington University proposed doing this to improve assay performance. Values above the upper cutoff would indicate with a high degree of certainty that a person has AD pathology, values below the lower cutoff would rule out AD with equal certainty, while anyone who fell in the middle “gray” area would need further testing via PET or CSF (see Nov 2023 news). In the study shown in Philadelphia, Palmqvist and colleagues set the lower and upper limits for the APS2 at 31 and 62, respectively, and 3.93 and 5.18 for %p-tau217 ratio. This improved the PPVs to around 95 percent or higher in the batch analysis, and only slightly less in the prospective analysis (image below).
Two Beats One. Using upper and lower cutoffs made the APS2 and %p-tau217 tests more accurate in primary and secondary care cohorts (top). Fewer people fell into the intermediate zone using the %p-tau217 test (bottom). [Courtesy of Palmqvist et al., 2024.]
One caveat to this approach is that if many people land in the gray zone, that would necessitate so many follow-up tests as to negate the advantage of a blood test. At least in these Swedish cohorts, this did not happen. With the two APS2 cutoffs, 11 to 15 percent fell into the intermediate category; for the %p-tau217 cutoffs, 4 to 8 percent did (image above).
Douglas Galasko, University of California, San Diego, thinks the assays show great promise in both primary and secondary care settings. He cautioned that the two cohorts seemed quite alike. “Characteristics of patients who had clinical screening/assessment and then biomarker evaluation were remarkably similar in primary care and in the BioFINDER secondary care cohort, including age, sex, and APOE e4 genotype. The last of these may have been an important aspect that contributed to the high diagnostic accuracy/predictive value in the primary care cohorts,” he wrote to Alzforum (comment below).
Are these assays good enough for real-world applications? Hansson believes some further testing is required, especially in more diverse populations and in other countries. University of Pennsylvania’s David Wolk agreed. “I wonder how much we really know about the PPV and NPV across much wider clinical practice,” he said during a session on translating biomarker into practice. “It is very much based on the prevalence of the disease, and there are likely big differences in prevalence and the ability to classify with these blood-based biomarkers.”
On that note, Palmqvist and colleagues found that in people with subjective cognitive decline, these tests predict amyloid pathology less well, with PPVs around 75 percent and 83 percent for the one- and two-cutoff approaches, respectively. This might be a disadvantage in identifying people with asymptomatic AD to include in clinical trials. Negative predictive values, however, still clocked in at above 90 percent, suggesting that these tests might be most helpful at ruling out AD in people whose symptoms are very subtle.
As for people with mild cognitive impairment or dementia—still the largest group of people presenting at clinics today—the tests beat clinical assessment hands-down. In the primary care setting, physicians correctly diagnosed AD pathology in only 63 percent of cases. In secondary care, namely the memory clinics at Skåne University Hospital and Ångelholm Hospital, both in Sweden, specialists only got it right 73 percent of the time.
“This study makes the case convincingly that highly sensitive blood measures of Alzheimer’s disease can be integrated into the clinical decision-making process, including in the primary care setting,” wrote Salloway, Christopher Rowe, Florey Institute of Neuroscience and Mental Health, Melbourne, Australia, and Jeffrey Burns, University of Kansas Medical Center, Lawrence, in an editorial in JAMA. “Accurate and early diagnosis of Alzheimer disease is increasingly important because of the new era of monoclonal antibodies targeting amyloid reduction in the brain.” This means that more people will get diagnosed with early Alzheimer’s in primary care, from where they will get referred to secondary care for treatment and side effect monitoring, adding pressure to these systems, Rowe told Alzforum. “We need to train care navigators and staff to handle this,” Salloway said.
The Seabird data Bateman presented, and the secondary care data Ashton described, support this. Bateman and colleagues designed Seabird to test how well plasma %p-tau217 and the plasma Aβ42/40 ratio reflect amyloid pathology in a cohort that was representative of the greater St. Louis area. The trial, which enrolled 1,122 people aged 60 and older, largely met its goal for recruiting across diverse demographics. Bateman reported tight correlation between plasma %p-tau217 and amyloid PET SUVR values, which was minimally affected by race, sex, and comorbidities. This was true, as well, for chronic kidney disease, which can skew the level of p-tau217 in the blood (Aug 2022 conference news), The data suggest that using the p-tau217/217 ratio, rather than the absolute levels of these fragments, can largely account for effects of comorbidities, as has been seen by others (Janelidze et al., 2023).
Clifford Jack, Mayo Clinic, Rochester, Minnesota, referring to intense discussion about the effects of race on plasma markers, was intrigued that it didn’t seem to affect the correlation between the %p-tau217 and amyloid. Bateman said the trial enrolled enough white and black Americans to detect an effect. “Race did not affect ratios, but it can affect levels,” he said. “We would not need different cutoffs among races for ratios, but we might need it for absolute values,” he said.
The secondary care study Ashton reported tested how well absolute levels of p-tau217, measured with the fully automated ADx Lumipulse immunoassay, identify people with Alzheimer’s. It recruited more than 1,500 participants from four sites in Sweden, Italy, and Spain, and plasma was analyzed in Gothenburg, Brescia, and Barcelona. Using a two-cutoff approach, accuracies reached 92 to 94 percent, and PPVs as high as 96 percent. “This meets the criteria for clinical implementation,” said Ashton.
Among the participants, 12 to 17 percent fell into the intermediate zone between the cutoffs. This is higher than in Hansson’s study using C2N’s mass spectrometry assay. Ashton thinks the cohorts explain some of difference, acknowledging that comorbidities, including chronic kidney disease, can affect fully automated tests for absolute p-tau levels. “We need to understand this quickly,” he said.
One option might be to account for a person’s glomerular filtration rate, a commonly used lab test. After all, chronic kidney disease is not difficult to diagnose. In her talk, Alicia Algeciras-Schimnich, from the Mayo Clinic in Rochester, showed how plasma p-tau217 levels rise as filtration rates fall in amyloid-negative people. Half of those with an eGFR of 35 mL/min/1.73m2, which indicates moderate to severe kidney disease, had p-tau17 levels above the cutoff for positivity. “Knowing the eGFR could help avoid false positives and signal the need for an alternate test,” she said.—Tom Fagan
This study combines data from a group of primary care practices and from the BioFINDER study to analyze the value of plasma %p-tau217 (measured as part of the C2N Precivity2 assay and compared against the p-tau217 percentage occupancy alone) in improving diagnostic accuracy, with a prespecified single cutoff, or range of cutoffs, that optimized sensitivity and specificity at the expense of a “gray area” of indeterminate results. Plasma %p-tau217 showed great promise in both settings. The results are highly informative and promising for demonstrating feasibility and utility of plasma %p-tau217 as an aid to AD diagnosis.…More
The characteristics of patients who had clinical screening/assessment and then biomarker evaluation were remarkably similar in primary care and in the BioFINDER secondary care cohort, including age, sex and APOE e4 genotype. The last of these may have been an important aspect that contributed to the high diagnostic accuracy/predictive value in the primary care cohorts. There is a higher prevalence of APOE4 in Northern European countries than elsewhere, and it would be interesting to see comparable studies from countries or populations where APOE4 is less common. The primary care cohorts had higher rates of comorbidity than the secondary clinic cohort, which is more representative of a general population.
The accuracy of the primary care clinicians’ suspicion of AD was only 58% compared to %p-tau217, therefore the blood-based biomarker could make a difference to clinical judgment and have an impact on appropriateness of referrals. It would be interesting to know how (and whether) the primary care physicians diagnosed the categories of subjective cognitive decline, MCI or dementia, and what additional history or data may have been available besides the MMSE.
Also, the percentage of patients with SCD, MCI, and AD in primary care who were biomarker positive were not reported. Methods of initial history and cognitive screening could have an impact on how often a primary care physician might order a screening blood test, and the PPV will be lower if it is ordered in populations with lower prevalence APOE e4 or among people with low pretest likelihood of having AD. There may be room for improvement beyond the MMSE as a screening test in primary care, to increase the accuracy of pretest suspicion of AD.
It will be interesting to see further studies of how primary, and secondary, care clinics use and interpret blood-based biomarkers, especially in populations with diverse ethnic populations and among older patients, where multi-etiology dementia is common. How results are communicated to patients and whether blood tests can serve as stand-alone biomarkers that may rule a patient in or out as a candidate to receive therapy, will require further study. The present study makes important advances in addressing some of these questions.
Living Among Us: People Whose Alzheimer’s Is Already Being Prevented
At early symptomatic stages of Alzheimer’s disease, amyloid immunotherapy taps the brakes on cognitive decline, but does not halt the disease. Many researchers think the full promise of plaque removal lies in prevention. If plaques were abolished before they could kick off downstream pathologies such as tangles, would the disease be stopped in its tracks?
First glimmers of this were on display at the Alzheimer’s Association International Conference, held July 28 to Aug 1 in Philadelphia. Randall Bateman of Washington University in St. Louis presented data from the Dominantly Inherited Alzheimer Network that showed knocking down plaque halved the odds that cognitively healthy people who carry an autosomal-dominant AD mutation would develop memory problems over a 10-year period. In some individuals, the effects were dramatic, with their brains free of plaques and tangles and their memories normal as many as 12 years past their expected age of disease onset.
The numbers are small. Still, Bateman believes the data prove the principle that preventing Alzheimer’s is possible. This group of people has taken plaque-removing drugs for longer than anyone else in the world. Therefore, their data are leading the way in showing what is possible with amyloid immunotherapy.
At the Dominantly Inherited Alzheimer Disease Family Conference, held before AAIC, Teresa Buracchio of the U.S. Food and Drug Administration agreed with this view. “DIAN efforts have been groundbreaking in advancing the science,” she said. Buracchio told participating families that their data inform trial design in sporadic AD, determining parameters such as what biomarkers to use and how long trials should be. They will also aid the FDA in interpreting trial results. “This improves our ability to approve drugs,” Buracchio said.
Delayed Symptom Onset
The first trial in the DIAN cohort, dubbed DIAN-TU-001, enrolled 144 mutation carriers, as well as 50 noncarrying relatives, who received either gantenerumab, solanezumab, or placebo for as long as seven years. After a one-year gap, about half the mutation carriers entered an open-label extension of high-dose gantenerumab for up to three years. That ended last year when Roche stopped development of gantenerumab due to insufficient Phase 3 efficacy in late-onset AD.
Preliminary analysis of the OLE data, presented at the 2023 Clinical Trials on Alzheimer Disease conference, suggested a lower risk of clinical progression in people who had been on gantenerumab throughout. A few developed symptoms six years later than their expected year of onset (EYO) (Nov 2023 conference news).
Further analysis has now firmed up the numbers. In Philadelphia, Bateman said 22 initially asymptomatic mutation carriers received gantenerumab throughout the double-blind and OLE periods. These periods added up to an average of eight years of exposure. These 22 were compared with 74 untreated people; they comprised participants who received placebo in the double-blind period as well as matched controls from the DIAN observational study.
By the end of the OLE, the gantenerumab group had half the odds of developing symptoms as did controls. The finding missed statistical significance at p=0.07. Fleshing out the control group by adding the 12 participants who switched from placebo to gantenerumab strengthened the finding, making it nominally significant at p=0.03. Meanwhile, separate analyses of people who took gantenerumab only during the OLE showed no difference from controls.
The lesson? In order to make a difference, robust amyloid removal needs to start early and continue for a long time, Bateman concluded.
He noted caveats. Not only was the study small, but using external controls from the DIAN observational trial also weakens the conclusions. OLE data are subject to selection bias, as healthier participants tend to stay in. The blinded portion of the trial initially contained 53 people on gantenerumab, meaning more than half discontinued before the end of the OLE. Indeed, the family meeting welcomed care partners who had been accompanying their affected spouses for many years, but were no longer able to bring them as the spouse’s dementia had advanced beyond the point where they could continue in the trial, or travel. These care partners are now coming with their young adult children, who are hoping for primary prevention.
In addition, the study population was not uniform in terms of disease stage. This first DIAN trial had a range of presymptomatic and symptomatic carriers at baseline. This was done to learn, and also to offer a treatment trial to loved ones of family members who were giving time and effort to the DIAN project.
Even Incomplete Plaque Removal Arrested Tangles
From another point of view, however, the findings are remarkable, given the limited plaque-busting power of the low-dose gantenerumab used for much of this trial. Dosing started at 225 mg monthly, was quintupled to 1,200 mg halfway through the placebo-controlled period, and then nearly tripled again, to 1,500 mg biweekly, for the OLE’s last two years as scientists learned more about gantenerumab’s safety and required dosing. As a result, most plaque clearance happened late, according to data presented by Tammie Benzinger at WashU. During the placebo-controlled period, people taking the drug cleared only 10 centiloids of their plaque on PiB PET, while those on placebo or solanezumab accumulated 20 more centiloids. Familial AD mutations produce rapid, massive plaque deposition, unlike the slower accumulation seen in late-onset AD.
During the gap year, plaque rose similarly in all groups, by 5 to 10 centiloids. At the start of the OLE, participants had an average of 62 centiloids of plaque in their brains. For context, this is not far below the 76 centiloids that symptomatic late-onset AD patients started with in the lecanemab Phase 3 trial. It also tops a 60-centiloid threshold used to predict who in that cohort had higher tangle loads (Nov 2023 conference news).
During the first year of the extension, participants cleared only five centiloids. After the dosage was upped, they cleared about 12 centiloids per year, for a total removal of 29 centiloids in the OLE. This meant participants ended the trial with an average amyloid load of 33 centiloids, still above the positivity threshold of 24. Overall, only a third of participants became amyloid-negative by the end of the OLE, Benzinger said. In sporadic AD trials, cognitive benefits only emerge when plaque is completely removed (Nov 2023 conference news).
In essence, plaque removal was “underdosed” relative to what scientists have since learned from lecanemab and donanemab Phase 3 LOAD trials. Even so, the treatment helped check tangles. During the placebo-controlled period, the flortaucipir tau PET signal rose about 0.5 SUVR in people taking solanezumab or placebo, but stayed relatively flat over three years in those on gantenerumab. Looking a bit closer, tau PET actually rose slightly during the first two years on very-low-dose gantenerumab, but fell back to baseline levels once the dose went up. During the gap year without treatment, tau PET rose by 0.2 SUVR in people previously on gantenerumab or solanezumab, and shot up 0.6 SUVR in those previously on placebo. In the OLE, tau PET flattened again for the previous gantenerumab and solanezumab groups, but continued to rise in the former placebo group.
Why did people previously on solanezumab respond more robustly to gantenerumab, compared with those who started on placebo? DIAN researchers did not speculate. They did say the data were consistent, in that the same pattern showed up with cerebrospinal fluid biomarkers Aβ42/40 and p-tau217, according to Laura Ibáñez at WashU.
Underlying these averaged group results lay great variation in tau PET outcomes at the individual level. Some people remained tau-negative throughout the study. Others started the OLE tangle-positive and in some the signal went down with treatment, whereas in others it rose. It is not yet known what factors influenced this. For case reports of this trial, and more, continue on to the next story.—Madolyn Bowman Rogers
First Success Stories From Alzheimer’s Secondary Prevention Trial
Often in medicine, case studies, their lack of statistical power notwithstanding, tell a more dramatic tale than summary data. In the first secondary prevention trial run by the Dominantly Inherited Alzheimer Network Trials Unit, overall findings were hopeful, but not a home run. As discussed at the Alzheimer’s Association International Conference, held July 28 to August 1 in Philadelphia, nearly two dozen mutation carriers who took gantenerumab for eight years had half the risk of developing symptoms as did untreated controls (see previous story in this series). Breaking open these group averages, however, the study showcases that at least in a few pioneering people, AD prevention is currently happening.
In Philadelphia, Tammie Benzinger of Washington University in St. Louis highlighted the stark effects immunotherapy can have on amyloid and tau PET scans. Benzinger contrasted two participants. One started the double-blind study while being amyloid- and tau-negative, but received placebo. By the beginning of the OLE, this person had become positive on both scans and, even on high-dose gantenerumab, plaques and tangles continued to worsen. The other person started the double-blind study with a low amyloid load and no tangles, and received gantenerumab. By the beginning of the OLE, this person had fallen below the amyloid positivity threshold. During the OLE, sub-threshold plaques vanished, and tau PET remained negative.
Some people’s memories improved on treatment. Alireza Atri of the Banner Sun Health Research Institute in Sun City, Arizona, discussed one person who started the trial on gantenerumab. During the gap year, they reached their family’s estimated year of onset (EYO), and began to have memory problems, with CDR going from zero to 0.5. After resuming treatment with gantenerumab, the CDR fell back to zero. At the end of the OLE, the person was six years past EYO with normal cognition, even though their final amyloid load was slightly higher than at the start of the trial 10 years earlier.
Consider this case: Another person started the OLE almost 10 years past EYO with a CDR of 0.5. For this participant, too, CDR dropped back to zero during the OLE and has remained there, now at almost 12 years past EYO. Though the person’s amyloid load has fallen, it remains above the positivity threshold. Atri did not show tau PET scans for these cases. Other studies have closely correlated tangles with cognitive decline.
At the Dominantly Inherited Alzheimer Disease Family Conference held before AAIC, personal stories added poignancy to these data. They reveal how, for the trailblazers to plunge into these trials, small twists of fate can lead to heartbreakingly different outcomes. One woman said her husband remained cognitively healthy throughout the placebo-controlled portion of the trial, though he was six years past EYO by its end. Then during the gap year, he declined rapidly, and had to enter a care facility. He no longer qualifies for antibody treatment. His wife believes he was on gantenerumab in the trial and that it granted him an extra six years of quality life. She mourns that he lost ground so fast when the study ended.
Other participants continue to do well. One woman, now two years past EYO and still cognitively healthy, said she hadn’t realized how much dread was weighing on her until she passed that milestone. With no idea how much time she may have left, she is preparing for future memory problems. “I’m in stoppage time,” she quipped, using a soccer term. Another woman has paid for private amyloid PET scans to track her progress because DIAN does not disclose research results. Now past her EYO, she is amyloid-negative and remains healthy. She and others now live to see how much time treatment has bought them.
Some DIAN participants remain skeptical of how much trials will help, but are stepping up anyway. Many do not yet know their mutation status. In DIAN trials, the placebo-controlled portion does not require genetic testing, and noncarriers are put on placebo. Alas, subjecting noncarriers to the side effect risk of amyloid antibodies is unethical, hence open-label treatment is only for carriers. Some agonize over whether they are ready to learn their status. “I’m torturing myself about getting tested,” one man said, explaining, “I know how the movie ends.”
Helping to navigate these issues is one of the goals of Youngtimers, a nonprofit founded to support families with autosomal-dominant AD. The organization provides practical resources, such as information about genetic testing, long-term care insurance, the use of IVF to avoid passing on the mutation, and grief counseling. It offers emotional support as well, creating a space for people to connect with others going through the same things. Currently, Youngtimers is raising funds to expand mental health support for participants at the network’s sites, as ADAD is known to wreak havoc in families struggling to cope. In addition, the organization advocates for patients, so their concerns can improve clinical trial design. “Youngtimers gives a platform for families to have a voice,” said co-founder Lindsay Hohsfield, a research professor at the University of California, Irvine.
Ultimately, many DIAN participants are looking beyond their own welfare. As one man still shy of his EYO said, “I hope to be part of the solution for ending Alzheimer’s.”
Testing Prevention Paradigms
How much more time could treatment buy people who inherit an AD mutation? Randall Bateman of WashU presented two possible models in Philadelphia. If Alzheimer’s worsens at a steady rate, then a 50 percent slowing in decline at presymptomatic stages would grant people five more years prior to dementia. However, if instead there is a threshold at which degeneration takes off beyond the reach of anti-amyloid therapy, a 50 percent delay in reaching this tipping point could result in 15 more years of cognitive health. Recent data support the latter hypothesis, tying an inflection point to the spread of tangles into the neocortex. This has been dubbed the “cataustrophe” by Keith Johnson at Massachusetts General Hospital, Boston (Apr 2022 news; Apr 2023 conference news).
Future trials will show which, if any, of these models is correct. They will also answer questions such as what happens with continued treatment after plaques are gone, and what switching from one anti-amyloid therapy to another will do. Three trials are now underway in the DIAN cohort.
For former DIAN-TU-001 participants, the researchers offer the Amyloid Removal Trial. ART is a five-year, open-label study of lecanemab. It will test whether plaques can be fully removed in mutation carriers, and how that affects downstream biomarkers and longer-term disease progression. ART will also examine long-term safety and rates of ARIA (see upcoming conference story). The first ART participant was dosed in June, and the trial will be fully enrolled by early 2025.
The Tau NexGen trial evaluates combination amyloid and tau treatments in DIAN participants who are within 10 years of their EYO in either direction (Nov 2021 conference news). The trial is ongoing. Its first arm, testing Eisai’s anti-tau antibody E2814 along with lecanemab, is challenging for its participants, as both antibodies require infusions. Even so, it is fully enrolled, with 197 participants. At the family meeting, several people spoke of their dedication to making this their “full-time job” in order to save their children. Two additional tau drugs for the other arms have yet to be selected. Enrollment for these portions of the trial remains open.
For mutation carriers more than 10 years younger than EYO, the DIAN-TU is starting up its much-delayed primary prevention trial. DIAN personnel previously announced they had chosen remternetug as the drug (May 2024 news). A successor to donanemab, remternetug has fewer side effects and clears plaque faster, an advantage in dominantly inherited AD where amyloid production is in overdrive (Apr 2023 conference news).
In Philadelphia, Eric McDade of WashU offered new details on the study design. The trial will enroll 240 mutation carriers and noncarriers, who can be as young as 18. These younger ages create tough choices for some, because whether remternetug affects pregnancy has not been studied. Women will have to defer starting a family while in the trial. Youngtimers raised this issue, and McDade listened. As a result, the placebo-controlled portion of the trial will last only two years, enabling more young women to participate and still have time to start their families.
This trial is easier on the participants than Tau NexGen. They will inject remternetug under their skin once every three months. The primary outcome will be amyloid PET, with fluid biomarkers as secondaries. After the placebo-controlled portion ends, participants will have the option to enter a four-year open-label extension, which will require learning their mutation status. In this portion, cognition will be added as a secondary outcome. The trial will begin dosing next year in the U.S., Canada, U.K., and Europe, and in 2026 in Australia and Latin America.
Outside of McDade’s talk, AAIC this year featured no news on remternetug, whose separate LOAD Phase 3 trial is enrolling.
Janice Smith at Roche, the maker of gantenerumab, noted that the DIAN data has been “hugely informative” for the company, helping it to select biomarkers for other trials. The company has been heartened by the signs that dementia can be delayed or perhaps prevented. “These data encourage us to go early,” Smith said in Philadelphia. For its part, Roche is betting on gantenerumab’s successor trontinemab, a Phase 1/2 antibody that looks to quickly remove plaques without causing ARIA. It also has a Phase 2 γ-secretase modulator that may one day become interesting for dominantly inherited Alzheimer’s, too (see Part 5 of this series).—Madolyn Bowman Rogers
Liraglutide Trial Was Negative Four Years Ago, Still Negative Today
Among the hundreds of studies presented at this year’s Alzheimer’s Association International Conference, held July 27 to August 1 in Philadelphia, one of the few that made a splash in the news was not news at all. Perhaps spurred by a press release, media outlets reported that tantalizing results from a small trial suggested that the GLP-1 agonist liraglutide might shield the brain from dementia. The results were featured by Reuters, The Guardian, STAT News, USA Today, Forbes, CNN, and various national news channels.
Alas, the findings shown in Philadelphia were the same ones shared at the Clinical Trials for Alzheimer’s Disease conference back in 2020, after the trial had concluded in 2019. This information is available both at clinicaltrials.gov and, in chronologically narrated form, in Alzforum’s online therapeutics database. The same findings were also presented at AAIC in 2021, but were never published in a peer-reviewed journal.
Importantly, results from this Phase 2b trial, which compared liraglutide to placebo among 204 people with mild AD who did not have diabetes, were nothing much to write home about.
“This was a negative study,” said Paul Aisen of the University of Southern California in San Diego. Aisen added that the secondary and exploratory measures presented at AAIC did not look particularly encouraging.
Glucagon-like peptide analogues form the class of drugs including Ozempic, Mounjaro, and others, which are experiencing soaring use for the treatment of diabetes and to help people lose weight.
As shown (again) by Paul Edison of Imperial College London at this year’s conference, the trial missed its primary endpoint of curbing decline in brain glucose metabolism as gauged by FDG-PET scans, over the yearlong trial period. Both the press release and Edison’s subsequent talk focused largely on some of the trial’s secondary and exploratory outcomes.
Here, too, the findings fell flat. According to the published trial protocol, clinical secondary outcome measures included change on the ADAS-Cog-Exec z score—which contains the ADAS-Cog and the executive function portion of the neuropsychological test battery—as well as the Clinical Dementia Rating Scale Sum of Boxes and the AD Cooperative Study—Activities of Daily Living (Femminella et al., 2019). In Philadelphia, Edison reported that on the ADAS-Cog-Exec, participants on liraglutide declined less precipitously over the 12-month trial relative to those on placebo, and that, at p<0.01, the effect was statistically significant.
However, cognitive trajectories of the treatment and placebo groups largely overlapped, as did the error bars. Any difference between the groups was small, at best. Edison declined permission for Alzforum to include the plots in this news story.
In his Philadelphia talk, Edison designated the CDR-SB and ADCS-ADL as exploratory outcomes. He noted that the study was not powered to detect a benefit on these measures, and didn’t find one.
For MRI, Edison reported data from exploratory endpoints. They were based on 83 participants on liraglutide and 75 on placebo who underwent MRI scans at baseline, 24, and 52 weeks. He said participants on liraglutide lost significantly less gray matter across the brain, as well as independently in the temporal, parietal, and frontoparietal lobes. No meaningful difference between the groups was apparent in the plots presented. Edison told the audience the effects were statistically significant. Again, the curves he showed were close together and their error bars largely overlapped.
Edison reported that the drug curbed gray matter atrophy by 50 percent—a statistic then widely reported in the news. What was it based on? Participants in the placebo group lost a total of 13,500 voxels, or 1mm3 cubes, of cortical gray matter throughout the trial, while those taking liraglutide lost about half as many. To put those losses in perspective, both groups started the trial with some 555,500 voxels, meaning placebo and treatment groups lost roughly 2.4 and 1.2 percent of their cortical gray matter, respectively.
Other secondary and exploratory measures, including change in microglial activation as gauged by TSPO-PET, and changes in amyloid- and tau-PET, were part of the trial, but Edison reported no results for those.
Lon Schneider of the University of Southern California in Los Angeles noted that not only were the effect sizes of the presented exploratory outcomes too small to be meaningful, but the p values of such exploratory measures calculated by the investigators have no value, particularly when in the context of other primary, secondary, and exploratory outcome measures that were negative.
“In short, these findings do not demonstrate clinical benefit on these outcomes,” he said.
According to an Alzheimer’s Association spokesman, the authors have submitted their trial to Alzheimer’s & Dementia for review.
While this trial was negative, other trials of GLP-1 agonists in AD are ongoing. One evaluates liraglutide’s successor semaglutide, which Novo Nordisk markets as Ozempic for Type 2 diabetes and Wegovy for weight loss. The company is running two Phase 3 trials in people with MCI or mild AD. Both are fully enrolled and expected to finish in 2026. Other trials test Lixisenatide and Exenatide.
At an AAIC session on GLP-1 analogues in neurodegenerative disease, speakers elaborated on the concept and possible underlying mechanisms. Additional talks suggest GLP-1 manufacturers are exploring how to expand their drugs into these indications.
A recent review reported no evidence thus far of effects on core Alzheimer’s biomarkers or cognition, but possible metabolic or neuroprotective effects (Liang et al., 2024).—Jessica Shugart
How Presenilin Mutations Hobble γ-Secretase Predicts Onset, Progression
The γ-secretase enzyme complex—abandoned as a drug target after candidate molecules proved toxic—is getting another look. New research has correlated the degree to which presenilin mutations affect the enzyme’s propensity to churn out long forms of Aβ with the age at which the mutation causes both Alzheimer's biomarker changes and symptoms.
The findings were published 26 July in Lancet Neurology. They are good news for scientists developing next-generation γ-secretase modulators. GSM drugs are designed to help the enzyme cut the amyloid precursor protein (APP) into shorter forms of the Aβ peptide, which are less likely to aggregate into amyloid plaques (Jan 2022 news; Feb 2022 news). Recent drug development has focused more on removing amyloid than decreasing its production. Even so, “I think that now [targeting production] will start again, thanks to these modulators,” Bart de Strooper, KU Leuven, Belgium, told Alzforum.
In an editorial accompanying the Lancet Neurology paper, Lucía Chavéz Gutiérrez, also at KU Leuven, Marie-Claude Potier, Institut du Cerveau–Paris Brain Institute, and Harald Steiner, Ludwig-Maximilians–University Munich, wrote that the data “support further investigation of γ-secretase complex modulators as potential Alzheimer’s disease therapies” that could prevent or delay the onset of the familial disease and reduce toxic Aβ levels in sporadic AD.
The role of γ-secretase in amyloid production “is really the best-studied aspect of Alzheimer's,” said de Strooper, who organized a panel on the topic at the Alzheimer's Association International Conference (AAIC) in Philadelphia last month. γ-Secretase has long fascinated researchers because the genes for two of its four component proteins, PSEN1 and PSEN2, are associated with familial Alzheimer's disease. Alzforum’s Mutations database lists more than 300 different PSEN1 variants. Many of them cause AD, but scientists are still studying why individual mutations cause symptoms across such a wide age range. Most mutations do so in a carrier’s 40s and 50s, but the spectrum reaches from the 20s to the 70s.
As scientists dug into these mutations and their links to AD, γ-secretase turned out to be a “funny enzyme,” said the paper’s senior author, Jasmeer Chhatwal, Massachusetts General Hospital, Boston. Over the course of two decades and amidst fierce debates, researchers learned that it holds on to its substrate for a long time and can cut it four or five times before releasing it. Each sequential chop results in a shorter Aβ peptide, and the shorter the peptide, the less likely it is to aggregate into plaques.
This suggested that mutations that weaken γ-secretase’s grip on its substrate allow long Aβ peptides to be released before the enzyme is finished with them. While studying 25 PSEN1 mutations, Chavéz Gutiérrez and her colleagues had found previously that the ratio of short to long Aβ peptides produced in each person’s cells predicted how early they would develop the disease (Petit et al., 2022; Apr 2022 news).
GSC Predicts Onset. A PSEN1 mutation’s γ-secretase composite (GSC) score correlates with its age of symptom onset. Lower scores mean earlier symptoms. Purple squares represent variants in the DIAN observational cohort, green triangles do not.
To study this in a larger group, lead author Stephanie Schultz and colleagues turned to the Dominantly Inherited Alzheimer's Network observational study (DIAN-OBS), which has been collecting clinical and biomarker data from people with PSEN and APP mutations for 15 years (Nov 2008 conference news, Daniels et al., 2024). They identified 190 people with one of 56 different PSEN1 mutations, put each mutation into otherwise identical cultured cells, and compared directly how each variant processed APP, without interference from the rest of each carrier’s genome.
Following a system developed by Chavéz Gutiérrez’s group, Chhatwal’s lab measured the ratio of short to long Aβ peptides produced by each PSEN1 variant. This served as a proxy for each enzyme’s activity level, allowing them to then compute a ratio known as the γ-secretase composite (GSC) between the variant’s activity and the wild-type PSEN1 activity level. While the variants all produced similar total amounts of Aβ, more active γ-secretase variants produced more short peptides.
When the scientists compared these in vitro ratios to each person’s clinical profile and family history, they found a direct relationship between the GSC and age of symptom onset, as well as how fast the disease would progress. Mutations with GSC ratios below 1 brought on symptoms sooner, and their carriers had greater biomarker changes such as CSF phospho-tau and Aβ42/40 ratios.
Shorter Is Better. γ-Secretase enzyme variants with low GSCs cleave their APP substrates fewer times, spewing longer Aβ peptides and leading to more amyloid deposition.
The researchers confirmed this trend with 105 more variants described in people not enrolled in DIAN. “It seems like the enzyme function itself is shaping the clinical and biomarker course of the disease,” Chhatwal said.
The same correlation appears to occur with PSEN2 mutations. In a preprint posted to bioRxiv on June 28, Chhatwal’s group looked at 74 PSEN2 variants and found that γ-secretase’s activity level similarly predicted age of onset, although carriers tended to develop Alzheimer's more than 20 years later than those with equivalent PSEN1 mutations (Liu et al., 2024). Chavéz Gutiérrez, who presented her similar findings on PSEN2 in a smaller study at AAIC, told Alzforum this suggests PSEN1 is the more important contributor to toxic Aβ production.
The GSC screening method could help scientists determine which of the many Alzheimer’s-associated PSEN mutations are actually pathogenic. “We think we could almost recharacterize [some variants’] pathogenicity based on the cell-based measure,” Schultz said. The researchers plan to put all their data online for other scientists to use. “We see it as being a resource for researchers who study autosomal-dominant Alzheimer's disease,” she said.
That a given mutation’s GSC predicts age at onset could help with estimating the prognosis of individual mutation carriers. It could also help scientists make the call of whether a newly discovered mutation is likely to be pathogenic. “I think [this method] provides a tool to do fairly fast screening of a lot of different mutations,” Chhatwal says.
More Shrinkage. People whose PSEN1 variants score lowest on the GSCs (red) have earlier and faster hippocampal atrophy than those in the highest tertile (blue).
“This is not only smoke, this is the gun and everything,” de Strooper said. He added the tight correlation between γ-secretase activity and age of symptom onset shows that—at least in dominantly inherited Alzheimer's—amyloid production is enough to cause the disease. “From a scientific point of view, [γ-secretase modulation] is really the best path to do something about amyloid,” he said.
DIAN trials codirector Eric McDade, Washington University, St. Louis, said the paper “captures all the data we've been collecting in the DIAN study for years.” He said that understanding the gradient of Aβ37 to Aβ43 production could allow scientists to test different drugs or doses in people with different PSEN variants and monitor a given drug’s effect on the ratio of long and short Aβ peptides.
These treatments could start decades before a person’s disease is expected to develop. “If [long Aβ peptides] are not produced, then they will not accumulate, even if amyloid clearance gets impaired during aging,” Chavéz Gutiérrez said. The DIAN trials unit has started a primary prevention trial, where drugs will be evaluated for their ability to head off amyloid deposition in mutation carriers (Aug 2024 conference news).
Getting more drugmakers onboard could be difficult. For about a decade, several companies developed γ-secretase inhibitor drugs, but toxicity ended those trials, likely because blocking γ-secretase prevents it from cutting its dozens of other substrates (Dec 2012 news; Aug 2010 news). A first generation of γ-secretase modulators—which do not block the enzyme but shift its second, processive cleavage toward spitting out more Aβ37 and 38 and less Aβ42 and 43—ran into different toxicity in Phase 1 (Aug 2008 conference news; Dec 2008 conference news; Apr 2011 conference news, EVP-0962, PF-06648671).
Despite all that, “a couple of groups continued doing the hardcore basic research trying to understand [γ-secretase modulation],” de Strooper said.
Then, F. Hoffmann-La Roche, Basel, Switzerland, surprised many last year when it moved a new GSM into Phase 1, where so far it appears safe. Called RG6289, it does not seem to interfere with the enzyme’s ability to process other substrates (Nov 2023 conference news). RG6289 seems to stabilize the bond between γ-secretase and its APP-derived substrate C99, causing the enzyme to cut it into even smaller Aβ fragments that don’t generally occur in nature.
At AAIC, Roche’s Irene Gerlach presented data showing that the drug lowered the concentration of long forms of Aβ by up to 70 percent and increased those of short forms in plasma samples from healthy volunteers in a Phase 1b trial. The company is now testing the drug in 245 people at risk of Alzheimer's or in the disease’s prodromal stage, tracking Aβ ratios and biomarkers such as p-tau for 18 months. Gerlach said these Phase 2 results are expected soon.
Other researchers have developed GSMs that they plan to test in clinical trials beginning next year. At AAIC, Kevin Rynearson, University of California, San Diego, presented preclinical data on compounds being developed by Acta Pharmaceuticals.
De Strooper says the data so far is promising. “I hope in the future we will get a little bit more risk-taking in this field and that we can do a prevention trial,” he said, adding that it will take many years to prove that GSMs can prevent the disease.
Even if they do, McDade added, they may only weakly affect symptomatic AD, where plaques have formed years ago. GSMs may need to be tested in combination with anti-amyloid immunotherapies, or as maintenance therapies once immunotherapy has removed most plaques.—Sara Reardon
Sara Reardon is a freelance writer in Bozeman, Montana.
Two New Deaths on Leqembi Highlight Need to Better Manage ARIA
With two anti-amyloid antibodies now in clinical use, improving the safety of these treatments is front and center on clinicians’ minds. In the year since lecanemab was approved by the Food and Drug Administration, and gained insurance coverage, its use in clinical care has grown slowly, with perhaps 3,000 Americans now on the drug. Clinics had reported few safety problems—until now. At the Alzheimer’s Association International Conference in Philadelphia, Lawrence Honig of Columbia University in New York reported the recent death of an APOE4 homozygote on lecanemab. According to scuttlebutt at the conference, another similar death previously occurred elsewhere. These are the first two fatalities reported for amyloid immunotherapy in routine care. “Serious outcomes can occur even with the best monitoring,” Honig cautioned.
Stephen Salloway at Butler Hospital in Providence, Rhode Island, told Alzforum that in about three-quarters of deaths on amyloid immunotherapy to date, there were warning signs that might have lessons to teach about how to prevent this worst outcome. Other talks at AAIC sought to identify these clues, as clinicians explored gray areas around when to treat with anti-amyloid antibodies and when to discontinue treatment due to ARIA. There were no easy answers. One theme that emerged was the importance of using multiple different MRI sequences to check for vascular risk factors in cases that fall into that gray area.
In some cases, doctors deem higher risks acceptable. In younger people who carry an AD mutation, clinicians have been able to relax exclusion criteria and treat patients with more vascular issues at baseline, without serious consequences so far. For more on this, and possible mechanisms behind ARIA, see next story.
First Deaths in Specialty Care
In clinical trials, lecanemab triggered the edema known as ARIA-E in about 12 percent of participants. Most cases were asymptomatic, but occasional severe reactions occurred, including three deaths in the open-label extension (Jan 2023 news; Jan 2024 news). On donanemab, as well, five deaths were reported in the trial or open-label extension (Jun 2024 news). According to Salloway, most deaths on amyloid immunotherapy fall into one of three categories: an intracerebral hemorrhage due to underlying cerebral amyloid angiopathy; ARIA-E that mimics a stroke and prompts thrombolytic treatment; or severe ARIA-E resembling CAA-related inflammation.
The two new deaths seem to be of the third category. At baseline, the APOE4 homozygote at Columbia had one microhemorrhage on MRI, and no signs of CAA, and appeared to be a good candidate for the therapy, Honig said. After the third infusion, however, the patient developed severe ARIA-E. After five days of steroid treatment, it began to clear up. Nonetheless, the patient experienced uncontrolled seizures and died. According to hallway talk, the previous death was similar, also in an APOE4 carrier who developed inflammatory ARIA-E and succumbed to seizures. Details could not be confirmed.
The Food and Drug Administration’s adverse event reporting system (FAERS) bears this out, attributing two deaths on lecanemab to ARIA-E. One occurred in May in an 80-year-old man, the other in December 2023, in an 84-year-old man. A third death in FAERS, in a 79-year-old man taking lecanemab, was caused by a hemorrhagic stroke; it is unclear if this was related to the drug. Four other deaths listed in FAERS were considered due to falls or cardiovascular disease and unrelated to lecanemab.
Lawren VandeVrede of the University of California, San Francisco, told Alzforum that some APOE4 homozygotes can have an “explosive” response to amyloid immunotherapy. This response is unpredictable at present, as other homozygotes do well on the drugs. Homozygotes may need additional MRI scans at baseline to look for risk factors before prescribing immunotherapy, he suggested.
Mild ARIA-E. A patient with two APOE3 alleles and mild edema (arrow); doctors continued dosing with lecanemab, the edema went away, and the patient did well. [Courtesy of Stephen Salloway.]
Fewer ARIA Cases Than Expected
Honig noted that aside from this death, safety issues at his clinic have been less frequent than in the trials. At Columbia, 122 patients have now been treated with lecanemab, receiving an average of 10 infusions per person. Patients are 73 years old on average and mostly white. Of the 86 percent who agreed to APOE genotyping, half were E4 heterozygote, 16 percent homozygote.
Honig said this real-world memory clinic population was more medically diverse than the lecanemab trial cohort, as is typical when new drugs enter routine care. Trials excluded participants who had white-matter changes on MRI, a history of strokes or seizures, or other brain disorders. At Columbia, physicians only excluded patients who had more than four microhemorrhages on their baseline MRI. Two patients had recovered from previous strokes, and three had vascular malformations. So far, none have been on anticoagulants. This broad usage is in keeping with the FDA label, which lists only one contraindication for lecanemab treatment—known hypersensitivity to the drug.
In the first year, 7 percent of patients at Columbia developed ARIA-E, 6 percent ARIA-H. These rates are about half those in the clinical trial. One patient developed superficial siderosis, i.e., iron deposits on the surface of the brain that can indicate bleeds. None had macrohemorrhages. All the ARIA-E cases occurred within the first three months of dosing and, other than the death, were asymptomatic. Eight people stopped taking the drug, four of them due to ARIA.
Lecanemab use in a memory clinic is manageable and well-tolerated overall, Honig concluded. Other clinicians at AAIC agreed that patients with a wide variety of brain health conditions can be safely treated. Tammie Benzinger at Washington University in St. Louis noted that her clinic has given anti-amyloid antibodies to people with a history of subdural hematomas or encephalopathy. “Those patients have done really well. I’ve been surprised,” Benzinger said.
Salloway said people with small, benign meningiomas, tumors that form in the membranes around the brain, are eligible for lecanemab. Some level of white-matter disease at baseline is also allowable, Benzinger said, noting that in older people, white-matter hyperintensities on MRI are the norm. “If we excluded those, it would be a lot of patients,” she said.
Varied Views. Using multiple MRI modalities can help find microhemorrhages, as some are better distinguished from background on gradient echo sequences (upper right, circle) than with susceptibility-weighted imaging (upper left). Others (bottom, circles) show up better on SWI than GRE. [Courtesy of Tammie Benzinger, WashU.]
Making Difficult Calls on Eligibility
How, then, can physicians decide which patients run too high a risk? In Philadelphia, researchers debated how best to make those calls. In a teaching symposium for clinicians, VandeVrede presented hypothetical cases, and the audience voted on whether they would treat or not. This group process resembles decision-making at many clinics, where a multidisciplinary board discusses cases and comes to a joint decision.
VandeVrede said that at UCSF, physicians have developed guidelines based on the FDA label, clinical trial data, and the published Appropriate Use Recommendations for lecanemab (Apr 2023 conference news). Beyond excluding anyone with more than four microhemorrhages at baseline, the guidelines recommend not treating anyone with more than one small area of superficial siderosis, more than two lacunar infarcts, any territorial infarcts or intracerebral hemorrhages, or severe white-matter changes, VandeVrede said. The UCSF guidelines require APOE genotyping, and suggest excluding anyone on anticoagulants, or switching them to another medication such as an antiplatelet agent.
While these rules seem clear, in practice, decisions become tricky. Consider an APOE4 homozygote with three microhemorrhages at baseline. Even though this patient technically meets criteria for treatment, the higher risk for homozygotes gave many clinicians pause. Half the audience said they would treat, half not. Salloway noted that the three microhemorrhages could be evidence of CAA, warranting caution. VandeVrede agreed there is no right answer. “Entire centers have decided not to treat homozygotes. At UCSF, we want homozygotes to be at the earliest [disease] stage possible,” he said. Higher baseline amyloid loads are associated with a higher risk of ARIA.
One attendee asked whether lecanemab would be preferred over donanemab for E4 homozygotes, because the latter had twice the rate of ARIA in trials. Salloway noted that the rate of serious ARIA is comparable for both antibodies, at about 1.5 percent.
What about someone without an E4 allele, but with six microhemorrhages at baseline? Most said they wouldn’t treat. However, Benzinger noted that the number of visible microhemorrhages varies depending on the MRI protocol used. “At WashU, we wouldn’t exclude just for that,” she said. She suggested using different MRI modalities to fully assess risk. WashU recommends a minimum sequence consisting of fluid-attenuated inversion recovery and diffusion-weighted imaging, which reveal different types of lesions, as well as susceptibility-weighted imaging and gradient echo sequences to find hemorrhages. The scan takes 10 minutes.
Stroke specialist Jeffrey Saver at the University of California, Los Angeles, said clinicians should fully inform patients and their families of the risks and make the decision together. If a higher-risk patient wants to move forward, clinicians should monitor them closely.
Mild versus Severe ARIA-H. A single new microhemorrhage (left, arrow) does not require stopping, but multiple new indications of bleeding (right, arrows) do. [Courtesy of Tammie Benzinger, WashU.]
Avoiding Bad Outcomes
If ARIA develops, knowing when to suspend dosing and when to stop treatment altogether is a thorny issue. At UCSF, warning signs to stop permanently include more than 10 new microhemorrhages, any intracerebral hemorrhage, severe or symptomatic ARIA, or more than two episodes of ARIA, VandeVrede said. ARIA-E often triggers the microhemorrhages and siderosis known as ARIA-H. Benzinger noted that ARIA can fluctuate, and stressed the importance of allowing ARIA-E and H to fully stabilize before making decisions about continuing treatment.
Saver offered guidelines for handling stroke-like symptoms in an immunotherapy patient, where ARIA-E can masquerade as ischemia. If a CT scan does not reveal blockage in cerebral arteries, and stroke symptoms are mild, then no treatment is needed, he said. When CT shows a blockage in a large artery, then mechanical thrombectomy, rather than meds, is the treatment of choice. The most difficult cases are when stroke symptoms are moderate to severe, and CT detects no blocked artery. A follow-up MRI scan may reveal a clot in a smaller artery that could be removed by thrombectomy. Only when a clot is inaccessible, or MRI cannot find one, should doctors consider administering thrombolytic drugs, Saver said.
He described a case at his center where a patient developed trouble walking and speaking, and a CT scan showed a region of the brain with low blood flow. Alas, the scan revealed no blockage, and the patient was past the three-hour window for thrombolytic treatment. A follow-up MRI revealed ARIA-E as the cause of low blood flow. Doctors administered high-dose steroids, and the person recovered. “We need to be ready to off-road patients from stroke protocols,” Saver said.
Many clinics have put safety guidelines into place to prevent accidental prescription of thrombolytics to immunotherapy patients in emergency rooms. These include alerts in a patient’s medical record, or a bracelet patients can wear.
Benzinger recommended entering ARIA data into the ALZ-NET database to help other clinicians learn from these early experiences. ALZ-NET is hosted by the American College of Radiology, and collects real-world data on anti-amyloid antibody use (Aug 2022 conference news).—Madolyn Bowman Rogers
Implicated in ARIA: Perivascular Macrophages and Microglia
As more people are treated with lecanemab and donanemab in clinical care, the risk of serious reactions due to ARIA grows. At AAIC, held July 28 to August 1 in Philadelphia, clinicians discussed how to avoid those by screening patients better. The first report of deaths due to lecanemab use in primary care, in two APOE4 carriers who developed inflammatory ARIA-E and seizures, only made this more urgent (see previous story).
Risk assessment is complicated, and not the same for all groups. Participants in the Dominantly Inherited Alzheimer Network carry AD mutations that spur massive plaque deposition and lead to memory problems in mid-life. Randall Bateman of Washington University in St. Louis, who leads the DIAN Trials Unit, said clinicians were able to relax the entry criteria for middle-aged participants in the DIAN secondary prevention trial, relative to the more stringent standards used in late-onset AD trials. Alireza Atri of the Banner Sun Health Research Institute in Sun City, Arizona, said the decade-long DIAN trial taught scientists a great deal about ARIA, including that it can occur late in treatment and recur multiple times in the same patient.
Others in Philadelphia focused on the mechanisms underlying ARIA. Inflammation is emerging as a key culprit, with microglia and perivascular macrophages both playing a role. The latter pump out free radicals and inflammatory cytokines that damage blood vessels, leading to the leakiness that causes edema and brain bleeds. While scientists are trying to develop ways to target these cells, so far steroids remain the only treatment for severe ARIA. Some clinicians argued for using these drugs even in mild cases to prevent worse outcomes.
Younger Patient, Sturdier Brain?
Unlike older AD patients, many of whom have cerebrovascular pathology and other co-morbidities, DIAN participants tend to have overall healthy brains and blood vessels. Perhaps for this reason, they tolerate amyloid immunotherapy particularly well.
Scientists know that in LOAD trials, the biggest risk for ARIA came from APOE4, more than one microhemorrhage, or superficial siderosis on baseline MRI (Nov 2023 conference news). These trials excluded people with more than four microhemorrhages. Appropriate Use Recommendations for lecanemab stress this restriction (Apr 2023 conference news). Microhemorrhages can be a sign of cerebral amyloid angiopathy, which is closely linked with ARIA.
For the DIAN trial, clinicians did enroll people who had more than four microhemorrhages. These families, who have watched many of their relatives die of dementia in middle age, are willing to accept more risk to find a treatment. They supported tripling the dose of gantenerumab from that in LOAD trials to try to curb their rapid plaque growth, even though higher doses hike the risk of ARIA (see related AAIC news).
Despite this, DIAN participants had about the same ARIA incidence as seen in LOAD trials. In Philadelphia, Jorge Llibre Guerra of WashU said 22 of the 73 participants, or about one-third, developed ARIA-E. This was mostly occipital and cleared up after two months on average. Eight people had recurrent ARIA episodes. These were typically asymptomatic, with six of 32 total episodes causing noticeable symptoms. ARIA-H was seen in 37 percent of people, more commonly as disease progressed. No one in the trial had a macrohemorrhage, and no one died due to ARIA. Importantly, neither type of ARIA was associated with greater cognitive decline. Nick Fox of University College London called these clinical data reassuring.
Atri offered some detail by showing case studies. One participant had six recurrent episodes of ARIA-E over six years. The first one was mildly symptomatic, with dizziness and balance problems, the others were asymptomatic. Three of the episodes were moderately severe on MRI, involving multiple foci. Each time, dosing was suspended until the edema went away. The participant stayed in the study and is still cognitively well, with a CDR of 0 and MMSE of 30, six years past his or her estimated year of symptom onset.
In another case, a participant developed a single episode of mild, asymptomatic ARIA after a year and a half on drug. This shows that ARIA can pop up late in treatment, Atri said, cautioning clinicians to be on the watch for this. Indeed, about 10 percent of the ARIA in this study occurred after one year, he added.
Atri believes ARIA should be treated with steroids more often than is current practice. He described another case where the participant had severe ARIA on MRI, with seven foci, and complained of headaches and brain fog. Clinicians treated the patient with IV steroids but tapered off quickly, causing the edema to come back. They moved to high-dose oral steroids with a long taper to completely clear up ARIA, and the participant remains cognitively healthy. Atri believes the aggressive treatment potentially headed off worse problems. “We should have a lower threshold for giving steroids,” he said.
Are Bam Bad? In one model of ARIA, Aβ released from plaques acts on perivascular macrophages (lilac), a type of border-associated macrophage. BAMs then release free radicals and inflammatory cytokines that damage blood vessels, causing leakage. [Courtesy of Costantino Iadecola.]
Homing in on Inflammatory Pathways
Other speakers in Philadelphia focused on why steroids may help, presenting new evidence of inflammatory involvement. Steven Greenberg of Massachusetts General Hospital, Boston, summarized the two main hypotheses for what causes ARIA. Initially, scientists thought that Aβ, mobilized from plaques, floods the perivascular space, clogging it and worsening CAA. This is plausible, but still lacks a clear mechanism for how this leads to ARIA, Greenberg said. ARIA representing an inflammatory response to CAA is a newer idea that has caught on in the field (Aug 2023 conference news). One sign of this: Pathology from fatal ARIA cases resembles that from CAA-related inflammation (CAA-ri) (Jan 2024 news).
Fabrizio Piazza of the University of Milano-Bicocca, Italy, is a proponent of this latter hypothesis. He previously reported that in patients with CAA-ri and parenchymal amyloid plaques, microglia activate in regions with ARIA-E. Curiously, microglia did not activate in areas of edema in people with CAA-ri alone (Piazza et al., 2022). What explains this?
In Philadelphia, Piazza noted that people with CAA-ri make auto-antibodies to Aβ, and the concentration of these antibodies correlates with ARIA-E. He believes that, just as in amyloid immunotherapy, these auto-antibodies mobilize Aβ from plaques to blood vessels, thickening the CAA. At the same time, Aβ-antibody complexes trigger microglia, prompting inflammation and ARIA-E. Steroid treatment calms microglia, and this correlates with lessened ARIA-E. He believes this is similar to what happens with antibody therapy.
If microglia activation helps drive ARIA, then this may explain why antibodies against the microglial receptor TREM2 were associated with ARIA in a recent trial, Piazza speculated (Aug 2023 conference news).
Because the leaky vessels of ARIA-E can cause brain bleeds, aka ARIA-H, early steroid treatment may head off this vascular damage, Piazza said. Like Atri, he believes steroids should be given even for mild ARIA cases.
Costantino Iadecola of Weill Cornell Medical College, New York, points the finger at a different brain immune cell, the perivascular macrophages that nestle up to small blood vessels. Iadecola previously reported that APOE4 from perivascular macrophages slows cerebral blood flow (Apr 2023 conference news). In parallel, Aβ acts directly on these cells via their CD36 receptor, instigating them to spew free radicals and inflammatory cytokines that damage blood vessels and make them leaky (May 2017 news).
CAA Away. In Tg2576 mice (left), blood vessels (pink) are coated with amyloid (blue), but when the CD36 receptor is deleted from perivascular macrophages (right), this vascular amyloid goes away. [Courtesy of Uekawa et al., 2023, Molecular Neurodegeneration.]
In Philadelphia, Iadecola added functional data, reporting that knocking out CD36 only in perivascular macrophages of 15-month-old Tg2576 mice rescued cerebral blood flow. The vessels looked healthier, CAA cleared up, and the mice’s memories returned to normal, even though parenchymal plaques did not budge (Uekawa et al., 2023). “Perivascular macrophages are a hub for free radical production and neuroinflammation, and may be a therapeutic target in CAA and ARIA,” Iadecola said. Others have come to similar conclusions. Scientists at Eli Lilly reported that antibody treatment in amyloidosis mice activated perivascular macrophages to express matrix remodeling genes such as matrix metalloprotease 9, resulting in leakier vessels (Taylor et al., 2023).
Do microglial and perivascular macrophage dysfunction go hand in hand? Addressing this question, Donna Wilcock of Indiana University in Indianapolis reported at AAIC that within 24 hours after injecting an anti-amyloid antibody into Tg2576 mice, microglia released TGFβ, which signaled to other immune cells such as perivascular macrophages. Next, Wilcock will examine the effects of chronic immunotherapy on this cell-to-cell communication, and what its consequences are for blood vessels.
This mechanistic research may eventually yield better biomarkers of CAA and ARIA, and potentially treatments to prevent or resolve the latter. Other approaches are trying to circumvent ARIA entirely. Roche’s new antibody trontinemab, which conjugates the business end of gantenerumab to a brain shuttle, allows the antibody to enter the brain through capillaries, bypassing most vascular amyloid. In Phase 1/2, the highest dose tested, 3.6 mg/kg, cleared amyloid completely in three months, with no ARIA in the eight patients who have reached this timepoint so far (Mar 2024 conference news). In Philadelphia, Geoffrey Kerchner of Roche presented these data again, sparking excitement among attendees.
Kerchner promised new data at the upcoming Clinical Trials on Alzheimer’s Disease conference, to be held October 29 to November 1 in Madrid.—Madolyn Bowman Rogers
I’m Open! Exposed Enhancers Reveal Masters of Microglial Moods
When confronting a threat, microglia rapidly splinter off into a menagerie of transcriptional states. What purpose do they serve? And who’s in charge of orchestrating them? These are difficult questions for scientists trying to understand these shifty cells, or to target them with therapeutics.
At the Alzheimer’s Association International Conference, held July 27 to August 1 in Philadelphia, researchers reported finding clues within short DNA sequences nestled within enhancer regions, which are fleetingly exposed in response to a microglial encounter. By investigating these unveiled sequences, scientists can infer which master transcription factors might be calling the shots. For example, in response to a TREM2-activating antibody, IRF3 and E2F emerged as potential master regulators of microglial transitions into interferon-producing and proliferative states, respectively. Yet, after a brief spell of transcriptional upheaval, the cells settled back into their calm, “undosed” state within days.
Because many AD risk genes are active in microglia, the tiny immune cells of the brain are thought to play an outsize role in AD. The cells are intimately involved in every aspect of AD pathogenesis. They often behave in seemingly contradictory ways, such as building and containing Aβ plaques, or curtailing tau pathology in one context while egging it on in another (Apr 2021 news; May 2023 conference news).
How the various transcriptional states of microglia relate to these different functions, and whether each is helpful or harmful, are hotly investigated questions at the present (Oct 2022 conference news). Making matters devilishly complex, microglia in one region of the brain might behave differently than microglia in another. Worse still, their states shift as the disease wears on (Apr 2024 conference news). This multidimensional diversity makes the cells tricky targets for drug developers, to say the least.
Christopher Glass of the University of California, San Diego, is taking an epigenetic approach to understanding what makes microglia tick (Jul 2016 conference news; reviewed in Balak et al., 2024). In Philadelphia, he told the audience that tools such as single-cell RNA sequencing have allowed scientists to identify microglial phenotypes with a high degree of resolution. “Yet, we still lack an understanding of how the cells achieve these different phenotypes,” he said. “We need to decipher those mechanisms to more effectively advance approaches for prevention and treatment that target these cells.”
To that end, Glass focuses on enhancer regions, where the epigenetic rubber hits the road. In these stretches of DNA, a combination of lineage- and signal-dependent transcription factors pile on to specific sequence motifs. This triggers the enhancer to loop itself into physical proximity to the promoter of the gene it’s about to turn on. By investigating which of these enhancer motifs are exposed before and after a given stimulus, scientists can infer—with the help of machine learning—which transcription factors are orchestrating gene expression changes (image below).
Enhanced Landscapes. Enhancers host a combination of lineage-determining (green, blue) and signal-dependent (yellow, red) transcription factors, which cooperate to regulate gene expression in a cell type-specific manner. Exposed enhancer motifs suggest transcription factors involved in a response. [Courtesy of Christopher Glass, UCSD.]
Glass’ group is looking at this so-called “enhancer landscape” under different conditions, including amyloidosis. In collaboration with Oleg Butovsky’s group at Brigham and Women’s Hospital in Boston, Glass lab scientist Johannes Schlachetzki identified 11 enhancer sequences that were differentially exposed in microglia from amyloid-ridden 5xFAD mice relative to those in wild-type animals. These motifs were predicted to host some 25-30 transcription factors. For example, several different transcription factors are able to bind the C/EBP motif, which is uniquely exposed in microglia in the presence of amyloid.
Still, Glass and other scientists have demonstrated that mouse and human microglia mount remarkably different transcriptional responses to amyloid. To zero in on the human-specific response, Glass teamed up with researchers in Matthew Blurton-Jones’ lab to make use of a xenotransplantation model (Aug 2019 news). When Glass analyzed the enhancer landscape of transplanted human microglia isolated from the brains of 5x-MITRG mouse hosts, he was in for quite a surprise. Nine of the 11 enhancer motifs implicated in the amyloid response in human microglia were the same as those found in their mouse counterparts (image below).
Same Regulators, Different Genes. Enhancer motif sequences identified in human (left) and mouse (right) microglia inferred a similar cast of transcription factors, ranked in order of significance. Lines indicate commonalities between human and mouse. [Courtesy of Christopher Glass, UCSD.]
How do these similarities square with the known differences between mouse and human microglia in their responses to amyloid? Glass reported that although similar enhancer motifs, and therefore a similar cast of transcription factors, orchestrated the microglial response to amyloid across the two species, the genes they targeted were mostly different. This makes sense, as many amyloid-activated enhancers identified in the mice had no functional counterpart in humans. “So the transcription factors may be the same, but their target regions in the genome are different,” Glass said.
Notably, many of the activated enhancer regions that Glass uncovered in the human microglia are known to activate AD risk genes, such as HLADR8 and ApoE.
Glass noted that this initial work may point to master regulators of microglial responses to pathology. His group is also looking at enhancer landscapes in hopes of inferring the controllers of individual signaling pathways, such as TREM2 signaling.
Along those lines, Kathryn Monroe of Denali Therapeutics in Philadelphia presented findings from a collaboration with Glass to dissect how microglia respond to treatment with a TREM2-activating antibody, ATV:4D9. The molecule is an engineered antibody with Fab regions that recognize mouse TREM2, and an Fc region that adheres to the human transferrin receptor. When infused into transgenic mice expressing the human transferrin receptor, the antibody binds receptors that line the blood-brain border, gaining access into the parenchyma, where its target awaits on the surface of microglia. When bound, the antibody encourages TREM2 clustering on the microglial surface, which triggers microglial proliferation within the mouse brain.
In Philadelphia, Monroe first reported single-nuclei RNA sequencing data on microglia isolated from hTfR mice at one, seven, 14, and 28 days after treatment with ATV:4D9. Already on the day after dosing, microglia had undergone a dramatic shift in transcriptional states relative to untreated mice, ditching homeostatic signatures in favor of a cycling/proliferative profile, two disease associated microglia (DAM) states, and an interferon-producing state.
By day seven, the cells had largely returned to their control state. The exception? A persistent “pDAM” state, which was still detectable at 14 and 28 days. These findings demonstrated that TREM2 agonism triggers a rapid, yet largely reversible, transformation of microglial states.
There and Back Again. Relative to microglia in mice treated with a control antibody (top), microglia from mice injected with a TREM2-activating antibody transformed from homeostatic states to interferon, cycling, and DAM profiles within 24 hours. Some DAM persisted (pDAM) at later time points, but most cells returned to control state. [Courtesy of Kathryn Monroe, Denali.]
Monroe also presented preliminary snRNA-Seq findings in microglia from APP knock-in mice treated with the antibody. So far, this data comes from only two time points—before dosing and 24 hours later. Monroe reported that in these amyloid-burdened mice, the antibody instigated a massive shift away from the homeostatic state, and toward metabolic, interferon, and cell cycling states. Unlike its effect in mice without amyloid, the antibody reduced numbers of microglia in a DAM-like state.
Shifting Flocks. Relative to APP knock-in/hTfR mice treated with a control antibody (left), those dosed with a TREM2-activating antibody (right), shifted states dramatically. (Teal=homeostatic; pink=DAM/immune activity; purple=metabolic; yellow=interferon; green=cycling.)
Why would the TREM2 antibody shape microglial responses differently in the presence of amyloid? Monroe said that because TREM2 is an immunomodulatory receptor, how it steers microglia likely varies based on the state of the cells upon TREM2 agonism. Case in point, microglia were found to respond differently to antibody treatment depending on how much TREM2 they expressed at the time of agonism, according to a recent study that Denali scientists did in collaboration with Christian Haass at the German Center for Neurodegenerative Diseases in Munich (Feiten et al., 2024). “We interpret that to mean that TREM2 levels and, accordingly, microglial state, can be toggled in different directions based on the condition in which they exist,” she told Alzforum. More on this study below.
To find out what controls these dynamic responses, Monroe teamed up with Glass, who mapped the enhancer landscapes of the cells one and seven days after hTfR mice were treated with ATV:4D9. At 24 hours post-dose, the researchers detected 1,166 activated and 340 repressed enhancers in microglia from mice treated with ATV:4D9 relative to a control antibody. Among the most significantly exposed enhancer sequences in response to the antibody treatment were a combination of microglia-specific TFs, including PU.1 and AP-1, as well as signal-determining TFs. These included IRF3, which Monroe suspects might dictate the microglial shift into the interferon-responsive state, and E2F, which could push the cells into a cell cycling state.
Which master regulators are responsible for pushing microglia into an enhanced metabolic state is still unanswered, Monroe said. She suspects coordination among different transcription factors may turn out to push microglia into various states, rather than a single one for each. NFkB-p65—another TF that cropped up from the analysis—could be one of these coordinators.
Remarkably, seven days after treatment with the antibody, these revealing changes to the enhancer landscape had all but vanished. “The microglia had relaxed back to their naïve, undosed state,” Monroe said.
She told Alzforum that she interprets this finding as a good thing, as it suggests that treatment with a TREM2 antibody won’t irreversibly alter microglia, for better or worse. In Philadelphia, an audience member asked what the fleeting nature of this response might imply about how often people would need to be dosed with a TREM2 activating antibody. Monroe considers this an important question. She said answering it will require a deeper understanding of how well the persistence of changes at the epigenetic and transcriptional levels translate into staying power of functional effects.
Along those lines, Monroe and other Denali researchers teamed up with Haass and Astrid Feiten in his lab to investigate how levels of TREM2 expression might steer the downstream effects of TREM2 activation in microglia. To that end, the researchers generated a TREM2 reporter mouse, in which the mKate2 fluorophore reflects TREM2 expression. They were able to identify—and sort—microglia expressing low, medium, or high levels of TREM2 under different conditions. In the absence of amyloid, most microglia expressed low to medium amounts of TREM2, but in APP/PS1 mice with amyloid, TREM2-high microglia emerged; they were primarily spotted huddled around plaques.
When the researchers sorted microglia based on TREM2 expression levels and disease condition and compared their transcriptomes, they uncovered both TREM2- and amyloid-induced signatures. In short, the findings indicated differential transcriptional responses as a result of TREM2 expression level that were further shaped by exposure to amyloid plaques, the authors wrote. What’s more, they discovered TREM2 expression correlated with an uptick in glucose uptake and energy metabolism, as well as improved cholesterol handling in microglia, regardless of the presence of amyloid.
How would a TREM2 agonist antibody change this picture? To find out, the researchers administered monthly intravenous doses of ATV:4D9 to their TREM2 reporter, amyloidosis mice for four months. Then, a month after the last dose, they compared metabolomic and lipidomic profiles of microglia along with their levels of TREM2 expression. Based on these profiles, microglia expressing low levels of TREM2 mounted minuscule responses, whereas cells expressing intermediate or high levels of TREM2 changed dramatically. Strikingly, cells expressing moderate amounts of TREM2 were most responsive to the antibody, revving up metabolites related to glucose uptake as well as antioxidants. Some of these metabolites plummeted among cells expressing the most TREM2, suggesting a ceiling effect.
“These findings may have important consequences for the current design of clinical trials using TREM2 agonists,” the authors wrote. “Based on our findings, one must carefully monitor TREM2 expression in patients, to determine the optimal time point for interference.” They suggested that soluble TREM2 in the CSF as a potential biomarker for this purpose.
At this point in this fast-moving research area, modulating microglia via TREM2 looks like it comes with much complexity. In Philadelphia, Monroe interpreted the body of data collected so far to indicate that even a brief transcriptional shake-up might be capable of promoting a more durable functional responses in microglia.
For an example of how epigenetic changes to microglia can steer the role of ApoE isoforms in AD pathogenesis, see next story.—Jessica Shugart
Microglial Epigenetics Hints at How ApoE Toggles Alzheimer’s Risk
At the AAIC conference held 27 July to 1 August in Philadelphia, microglial epigenetics stood in the limelight. Microglia are infamous for their dynamic and varied responses to different threats they encounter within the brain environs, and epigenetic approaches hold promise for learning what controls these shifty cells.
At AAIC, scientists reported that the “landscape” of exposed enhancer sequences helps them infer master transcription factors in charge of orchestrating responses to amyloid and to signaling via the TREM2 receptor (see prior story). Other groups used xenotransplant models to decipher epigenetic responses of human microglia to amyloid in their mouse hosts. They discovered stark differences by ApoE genotype, whereby ApoE4 microglia mounted largely pro-inflammatory responses to amyloid, with many of their dysregulated genes overlapping with AD risk genes. ApoE2 microglia, on the other hand, stayed calm and collected, enhancing their signaling via the anti-inflammatory vitamin D receptor.
Sarah Marzi of King’s College London presented these findings. Drawing from a hefty epigenetic toolbox in her lab, Marzi is scouring the microglial genome for both chromatin accessibility and acetylation. She is looking to spot regions that show active versus repressed gene expression, and she corroborates those epigenetic measures with transcriptomics. For her current study, Marzi wanted to know how ApoE genotype influences the way microglia respond to amyloid. To that end, she teamed up with Bart de Strooper of KU Leuven and Renzo Mancuso of the University of Antwerp, who have developed xenotransplantation models to study human microglia respond to threats they encounter in the mouse brain (Oct 2022 news).
Many of the findings Marzi presented are now posted in a preprint (Murphy et al., 2024).
When Di Hu and Kitty Murphy in Marzi’s lab applied their omics techniques to human microglia isolated from the amyloid-ridden brains of 1-year old-APP knock-in mice, they found widespread epigenetic and transcriptomic differences based on microglial ApoE genotype. One standout was CHCHD2. This gene encodes a mitochondrial protein involved in myriad functions, including cell motility and the response to hypoxia. In ApoE2 and ApoE3 microglia, CHCHD2 transcripts were abundant, and the gene’s promotor was exposed in open chromatin. The opposite was true in both ApoE knockout and ApoE4 microglia. In them, CHCHD2 transcripts were nonexistent, and chromatin around the gene’s promotor was closed up tight.
This same finding between ApoE4 and ApoE knockouts went beyond CHCHD2. In fact, Marzi saw considerable overlap in the epigenetic and gene expression changes mounted by both their microglia in response to amyloidosis. Notably, many genes with variants implicated in AD risk were among those turned down in both ApoE4 and ApoE knockout microglia. That ApoE4 spurred some similar gene expression changes as did ApoE knockout suggests that E4 exerts some of its influence on AD risk via a loss of protective function. This manifests in the downregulation of beneficial genes.
In Philadelphia, Marzi noted that the convergence between ApoE4 genotype and expression of other AD risk genes argues against the recently proposed idea that ApoE4 homozygosity is a distinct form of AD (May 2024 news). “What we see here at the transcriptional level would suggest that is not true, because the changes associated with ApoE4 are mimicked by the overall genome-wide heritability of AD,” Marzi said.
ApoE2 and ApoE4 microglia also mounted qualitatively different inflammatory responses to amyloid. Microglia expressing ApoE4 revved up a host of pro-inflammatory cytokines, including CCL4L2, CCL3, CCL3L1, whereas cells expressing ApoE2 ramped up CXCL16, which reportedly has an anti-inflammatory effect.
Soothing Sunshine? Epigenetics suggest a boost to the vitamin D receptor response in ApoE2 microglia, whereby activation of the nuclear VDR enhances expression of target genes including the IL-10 receptor, which quells pro-inflammatory signaling via SOCS3. [Courtesy of Sarah Marzi, King’s College London.] [Courtesy of Sarah Marzi, King’s College London.]
Another example in support of ApoE2’s anti-inflammatory prowess came from an enhancer motif analysis similar to those conducted by Christopher Glass, University of California, San Diego, and Katherine Monroe, Denali (see previous story). The London scientists hunted for differentially exposed enhancer sequences in open, hyper-acetylated stretches of chromatin within the isolated microglia. What was the top discriminator between ApoE2 and ApoE4 microglia? A DNA-binding motif that binds the vitamin D receptor, which is a ligand-inducible transcription factor that orchestrates responses to vitamin D. In particular, regions with greater chromatin accessibility in ApoE2 microglia were strongly enriched for this motif.
Did these exposed motifs translate into a stepped-up vitamin D response? Indeed, by referencing the transcriptomic data they collected from the same samples, the researchers found an uptick in expression of genes known to be upregulated by VDR signaling in ApoE2 microglia, but not in ApoE4 ones. One such gene was the alpha subunit of the IL-10 receptor, which mediates anti-inflammatory signaling. Together, the findings mesh with other data casting ApoE2 and ApoE4 as anti- and pro-inflammatory mediators, respectively. They also jibe with reported associations between vitamin D deficiency and AD, Marzi noted.
“Vitamin D acts via binding to VDR, and enrichment of VDR in regions with increased chromatin accessibility in APOE2 may therefore enable these microglia to be more responsive to vitamin D, regardless of serum levels,” the authors wrote in their preprint.—Jessica Shugart
Clinicians may soon have a blood test for neurofibrillary tangles. At AAIC 2024, held July 27-31 in Philadelphia, Randall Bateman, Washington University, St. Louis, reported that a fragment of tau containing the microtubule-binding region, called eMTBR-243, can be detected in plasma and that it identifies people who have neurofibrillary tangles in their brains. When combined with a plasma marker of amyloid, such as p-tau217, it could help confirm when a person has AD.
“We can’t be assured that just because someone has amyloid plaques, their symptoms are due to Alzheimer’s disease,” noted Bateman. “This is where eMTBR-243, tau PET, or another measure of tau tangles will help.”
Thomas Karikari, University of Pittsburgh Medical Center, said the data was exciting. “The field has been missing a good marker for tau aggregates, and it will be great to have a blood-based assay that correlates.”
A multiplex blood test indicating both the presence of amyloid plaques in the brain, such as with p-tau217, and the presence of tau tangles in the brain, such as with eMTBR-243, would essentially meet the definition of Alzheimer’s disease set out by Alois Alzheimer. It would also reflect, during life, neuropathology-based criteria of AD (Hyman et al., 2012, Jack et al, 2024).
Henrik Zetterberg, University of Gothenburg, Sweden, told Alzforum that the eMTBR-243 data looked very promising. “It will be interesting to see if this marker increases in neurodegenerative diseases that are not amyloid-dependent, such as primary tauopathies,” he said.
Bateman and colleagues had previously reported that MTBR-243 in cerebrospinal fluid tracks with tangles (Dec 2020 news). Would this fragment, like various p-tau fragments found in the CSF, also turn up in the blood? After all, if it gets laid down in intraneuronal tangles, it might not. Sure enough, Kanta Horie at WashU, who had discovered the CSF marker, found it in plasma.
Alas, its levels there did not budge when tangles accumulated in the brain. Horie told Alzforum that the extraction protocol was the problem. The mass spectrometry-based test for CSF MTBR relies on tryptic digestion. Lo and behold, when Horie skipped this step and used a proteomic approach to find endogenously cleaved fragments, he found an MTBR-243 fragment in plasma that did track with tangles. The lab calls this endogenously cleaved marker eMTBR-243 to distinguish it from the CSF version.
In Philadelphia, Bateman reported that plasma eMTBR-243 showed extremely high specificity and sensitivity at identifying people who had tangles. In 108 people from the Swedish BiofFinder2 cohort, it boasted an AUC of 1.0 at distinguishing the 41 whose tau PET scan was positive. Horie told Alzforum that this high accuracy surprised him, and that he plans to test other and larger cohorts. In the present analysis, eMTBR-243 outperformed plasma p-tau217 and the p-tau217/217 ratio, which had AUCs of 0.87 and 0.89, respectively.
Looking more closely at tangle load, Bateman reported that eMTBR-243 correlates much more tightly with Braak stages III to VI than with Braak I and II. Among people who had brain amyloid, Spearman correlations between eMTBR-243 and tangle load, as judged by regional tau PET, was 0.54 in Braak stages I to III, 0.79 in stages III to IV, and 0.86 in V to VI. In keeping with this, among 55 volunteers at the Knight Alzheimer’s Disease Research Center at WashU, those with a clinical dementia rating of 1.0 had much higher levels of eMTBR-243 in their plasma than did people with a CDR of 0.5, who, in turn, had just slightly more eMTBR-243 in their blood than cognitively normal people who had brain amyloid. “This work indicates that eMTBR-243 greatly increases as people transition from very mild to mild dementia,” said Bateman, “namely CDR 0.5 to CDR 1.0.”
Horie told Alzforum that, in this regard, the marker behaves differently from CSF MTBR-243, which, like p-tau217, ticks up gradually as disease progresses. “Plasma eMTBR-243 shows an exponential increase after symptom onset, so it is much more specific to later-stage tau aggregation,” he said. Karikari thinks this may not be unusual. “If we expect the signal to come from the brain, then we should see less in the blood, and it may take more time and be dose-dependent,” he said.
Plasma eMTBR-243 could be useful to help stage or rule out AD, said Bateman. “If p-tau217 is elevated and eMTBR-23 is completely normal, then I would question whether that person’s symptoms are due to AD, and would consider another cause,” he said.
What about those other causes? Could plasma eMTBR-243 help identify other tauopathies? Horie said that CSF MTBR-243 appears to be a specific marker of tangles containing tau isoforms with three and four repeats of the microtubule-binding region. In addition to AD, some forms of primary tauopathy, including frontotemporal dementia caused by the R406W mutation in the tau gene, have 3R/4R tangles. “I have high confidence that we can differentiate 3R/4R tauopathies with CSF MTBR-243,” said Horie.
Testing of other tauopathy cohorts for eMTBR-243 is ongoing. Zetterberg thinks there’s a slight chance eMTBR-243 might be amyloid-dependent, like p-tau217. “It will be interesting to see what they find,” he said.—Tom Fagan
By Setting Standards, Experts Aim to Tame a Wild West of AD Blood Tests
Mass spectrometry tests, immunoassays, Aβ42/40, various p-taus, their ratios … the number of blood tests for amyloid pathology in the last seven years has blossomed into a hopeful but confusing mess. Some are CLIA-certified—meaning the tests perform as claimed—but as yet, none are approved by the U.S. Food and Drug Administration or any other regulatory agency. How’s a primary care physician or neurologist to choose?
“It has been a Wild West for AD blood tests,” said Suzanne Schindler, Washington University, St. Louis. “That has brought significant risk,” she added. “If clinicians start using these tests and some perform poorly, this can diminish trust in all these biomarkers.” At AAIC last month in Philadelphia, Schindler argued that performance standards are necessary to help ensure accurate and timely diagnosis of Alzheimer’s disease as these tests leave the confines of AD research clinics and start to enter routine clinical care. Schindler co-wrote a Consensus Statement article on the topic in the July 2024 Nature Reviews Neurology with Oskar Hansson, Lund University, Sweden, and a panel of experts.
The good news is that several immunoassays and mass spec tests for amyloid can meet these standards. The twist? Test accuracy depends on the patient’s pretest likelihood of having amyloid pathology. If that sounds circular, read on …
Performance Standards: Check
Getting a timely diagnosis has become ever more urgent with the rollouts of lecanemab and donanemab. Anticipating these approvals, the Global CEO Initiative in 2022 convened a blood biomarker (BBM) workgroup to develop minimum performance standards for amyloid pathology tests. Co-lead by Schindler and Hansson, the group included dementia biomarker specialists, clinicians, and stakeholders from academia, industry, private foundations, and advocacy groups. Companies that sell AD BBMs or immunotherapies are in it, including Biogen, Cambridge, Massachusetts; Biomarkable BV, Gent, Belgium; C2N Diagnostics, St. Louis; Eisai Inc., Nutley, New Jersey; Eli Lilly, Indianapolis; Prothena Biosciences Inc., Brisbane, California; Quanterix Corporation, Billerica, Massachusetts, and Roche, Basel, Switzerland. “The Global CEOi brought people together who don’t normally talk to each other,” Schindler told Alzforum. “They got everyone to agree.”
Agree to what? Here’s where the detail comes in. The consensus performance recommendations the workgroup issued depend on the reason a given test will be used. For example, to confirm a suspected diagnosis of AD, the recommendations stipulate that the test should perform as well as approved tests of cerebrospinal fluid, namely having a specificity and a sensitivity of 90 percent. For a triage test, aka a test that will be confirmed with follow-up amyloid PET or CSF analysis, the sensitivity should still be 90 percent. As for its specificity, greater than 85 percent would suffice in primary care. For secondary care, that specificity could be even less stringent, at 75 percent, depending on what further testing is available (table below).
The paper goes into considerable depth explaining how specificity, sensitivity, positive and negative predictive values, and area under the curve are calculated and what they mean.
Context Matters. This performance matrix for secondary and primary care shows that even when a test surpasses the minimum acceptable performance, its positive and negative predictive values (PPV, NPV) depend on how prevalent amyloid pathology is in the patient’s age bracket. [Courtesy of Schindler et al., 2024.]
Cutoffs: Check
Those numbers, in turn, depend on cutoff values used to distinguish positive from negative test results. Cutoffs are tricky business. Set them too high, and there will be many false negatives, meaning delays in care or misdiagnoses. Set them too low, and there will be many false positives, a grave mistake.
Even when the cutoff is “just right,” some results come awfully close to the line, creating uncertainty. For this reason, the workgroup opted for two cutoffs. Values above the upper one indicate a positive test with high certainty, those beneath the lower would almost certainly be negative, and anything in between would need clarification via PET or CSF. This approach has already improved accuracy in dementia centers and in primary and secondary care research cohorts (Nov 2023 news; Part 1 of this series). No more than 15 to 20 percent of results should land in this “gray” zone. The Global CEOi workgroup reported that around that number of CSF test results do.
Additionally, in most medical testing, a test’s positive and negative predictive values depend on the prevalence of the tested condition in the population at hand. For AD, this means that among 80-year-olds who are cognitively impaired, a positive test is much more likely to be a true positive than in a 60-year-old who occasionally forgets where the car keys are. “Many people think that a test will give the answer they are looking for, but for that you have to have a high pretest probability of being amyloid-positive, because prevalence affects interpretation of the results,” cautioned Schindler. The lower a population’s pretest likelihood of amyloid pathology, the higher the specificity of the test itself needs to be for a positive result to be true. Ergo, both the test’s specificity and amyloid prevalence should inform interpretation of a given person’s results.
Where does all this leave the primary care physician who may not be fluent in biomarker stats? Schindler thinks that depends on the physician. “There is a lot of information out there on how to understand the accuracy of tests,” she said. Other common lab tests work similarly. Metabolic panels use patient age and sex to estimate their glomerular filtration rate from blood creatinine level. Cholesterol, lipoprotein, and triglyceride levels are used to calculate cardiovascular disease risk scores. “It may not be much of a reach that we could do something similar for amyloid testing,” said Schindler.
Still, it might help if the predictive value was calculated for physicians. To that end, Schindler has created an app that calculates PPVs and NPVs based on a patient’s age, MMSE score, and the given test’s specificity and sensitivity. The consensus statement also provides guidance for interpretation of test results in primary and secondary care (table below).
Guiding Interpretation. In primary (top) and secondary care (bottom), a person’s pretest likelihood of having AD, and the test’s accuracy, together determine its predictive values and how it should be read. [Courtesy of Schindler et al., 2024.]
For people whose pretest likelihood of having brain amyloid is high, a test with 90 percent specificity would have extremely high predictive value. In fact, Schindler has started patients on lecanemab who have both very high scores on the Precivity AD2 test and a very consistent clinical picture of AD, even without a follow-up CSF test or PET scan. “Many people don’t realize the current CSF test for amyloid pathology only has about 90 percent specificity and sensitivity,” she said. “If you are using a high-performing blood test, then it makes no sense to do a follow-up CSF. Essentially, that would mean running a lower-performing CSF test that carries some risk from a lumbar puncture.”
Which blood tests should clinicians be using? The consensus statement focused squarely on standards any candidate test should meet. It does not recommend specific tests. To date, the different tests currently being sold have been evaluated largely in separate cohorts and by different academic and commercial research groups. To guide clinicians in their choice, then, direct comparisons are in order. For AAIC news on those, see next story.—Tom Fagan
This offers recommendations for the acceptable performance of blood biomarkers that predict abnormally high levels of cerebral amyloid plaques.
I note a few key points:
▶ The recommended specificities and sensitivities are only for this intended use—to identify individuals with an abnormally high amyloid load, regardless of their current or future symptoms, and using amyloid PET (and possibly CSF) as the reference standard. The specificity and sensitivity of a biomarker will vary significantly depending on its intended use. For example, the 18F-florbetapir PET scan predicts neuritic Aβ plaques abnormal level at autopsy with 92 percent sensitivity and 100 percent specificity (Clark et al., 2012), while it predicts MCI patients who will develop AD dementia clinical symptoms with 67 percent sensitivity and 71 percent specificity, suggesting that 18F-florbetapir PET scans cannot be recommended for routine use in clinical practice to predict the progression from MCI to AD dementia (Martinez et al., 2017). …More
▶ When Schindler et al. reference a confirmatory test, it should be understood as a test to confirm an abnormally high level of cerebral amyloid plaques, not as a test to predict clinical conversion to AD dementia symptoms or to diagnose AD. This is clearly outlined in the FDA dossier for 18F-florbetapir (202008Orig1s000) or the FDA De Novo dossier for the Lumipulse G Aβ42/40 ratio CSF test (DEN200072).
▶ In the context of initiating a treatment with significant side effects aimed at slowing the onset of AD dementia symptoms, blood tests predicting which patients would convert to AD dementia symptoms might be desirable. In this context, specificity should be preferred over sensitivity to avoid overdiagnosing individuals who will not develop symptoms of AD dementia, thus sparing them from side effects with little or no clinical benefit.
In conclusion, when discussing the specificity and sensitivity of a blood biomarker for Alzheimer's disease, it is crucial to understand the intended use. The performance of the same blood biomarker will vary significantly based on its intended use, whether it's predicting patients with abnormally high levels of cerebral amyloid plaques or identifying pre-demented patients who will develop clinical symptoms of Alzheimer's dementia.
Dr. Braudeau is correct that the performance of blood biomarkers may vary in cognitively impaired and cognitively unimpaired groups. However, this paper only considers use of blood biomarkers in determining the presence/absence of amyloid pathology in patients who are cognitively impaired at the time of the test. We did not consider cognitively unimpaired individuals in these recommendations.…More
In Head-to-Head Testing, P-Tau217/Tau217 Comes Out on Top. By a Hair.
With many different blood markers and assays for helping doctors diagnose Alzheimer’s disease, which are they to choose? Head-to-head studies can help answer this question, but few have directly compared blood tests on the same samples.
Last year at CTAD, Nick Ashton, then at University of Gothenburg, Sweden, presented data from a small round-robin that compared the performance of 26 different p-tau tests on 40 blood samples taken from people suspected of having AD. p-Tau217 came out on top (Nov 2023 conference news). Most of the tests were immunoassays.
Not represented was the p-tau217/tau217 ratio. The percentage of fragments phosphorylated at this residue is emerging as being perhaps a more robust marker than the absolute level of p-tau217. Now, two new head-to-head studies, one led by Suzanne Schindler of Washington University, St. Louis, the other by Oskar Hansson and NoëlleWarmenhoven at Sweden’s Lund University, support the idea that %p-tau217 is a more accurate marker, though the differences between tests are often marginal. The studies are described in manuscripts uploaded to medRxiv early in July and were recently presented at AAIC in Philadelphia.
Warmenhoven and colleagues compared mass spectrometry-based assays for %p-tau217 and p-tau217 run at Washington University with commercial immunoassays for p-tau217 from Lilly, Janssen, and ALZpath. They used plasma samples from 998 volunteers in the Swedish BioFinder-2 cohort, of whom 375 were cognitively healthy, the remainder having subjective cognitive impairment, MCI, AD, or dementia due to another cause. Almost all had had PET scans for neurofibrillary tangles; 694 had had amyloid PET scans.
The %p-tau217 test proved a tad more accurate in identifying people who were amyloid-positive. The p-tau217 mass spec test performed similarly to the Lilly and ALZpath p-tau217 immunoassays, and all three edged out the Janssen test. Most assays posted accuracies in the high 80s for identifying samples from tangle-positive volunteers, with the exception of p-tau181 (image below).
Testing, Testing. Plasma tests (purple) for p-tau217 identified amyloid-positive (left) and tangle-positive (right) people in BioFinder-2 with comparable accuracy. They are as good, if not better, than CSF tests (blue), including the FDA-approved Elecsys test for p-tau181/Aβ42 from Roche (dashed vertical line for reference). [Courtesy of Warmenhoven et al., 2024.]
Furthermore, the scientists compared how well each test correlated with amyloid and tangle load, as well as with presence of both pathologies, baseline cognitive scores, and decline from baseline. On all measures, the %p-tau217 correlated as well, or slightly better, than did the other tests, with Lilly’s immunoassay generally being the strongest of the immunoassays. Warmenhoven found a similar pattern in a smaller cohort of 219 volunteers from the Knight Alzheimer’s Disease Research Center at WashU.
The authors concluded that the %p-tau217 test might be considered as a stand-alone confirmatory test for AD, while the immunoassays might be better suited to triage. WashU’s Randall Bateman, who co-founded C2N Diagnostics, St. Louis, was a co-author on the paper. C2N sells PrecivityAD2, a mass spec-based test for AD that measures %p-tau217 and the Aβ42/40 ratio (Dec 2023 news).
Going Head-to-Head in ADNI
For her part, Schindlers’s medRxiv manuscript reports on an analysis commissioned by the Biomarker Consortium of the Foundation for the NIH. In a prior study, the consortium had compared six assays measuring plasma Aβ42/40 (Zicha et al., 2022). For the current one, they added the analytes %p-tau217, p-tau217, p-tau181, the glial marker GFAP, and the neurodegeneration marker NFL.
This was the biggest head-to-head study yet. It pitted these assays against each other: C2N’s Aβ42, Aβ40, p-tau217, and tau217; Fujirebio Diagnostics’ Lumipulse tests for p-tau217 and Aβ42 and Aβ40; ALZpath’s Quanterix p-tau217; Janssen’s LucentAD Quanterix p-tau217; Roche Diagnostics’ NeuroToolKit assays for p-tau181, Aβ42/Aβ40, GFAP, and NfL; and Quanterix’s Neurology 4-Plex tests for p-tau181, Aβ42/Aβ40, GFAP, and NfL. Most of these tests are commercially available.
At AAIC, first author Kellen Petersen, WashU, reported that the scientists ran each of these tests on aliquots of 392 samples from the ADNI cohort. They first appraised each test for its ability to detect people with brain amyloid. The authors then correlated each test with each participant’s tau PET status, cortical atrophy, and cognitive impairment.
Here, too, the %p-tau217, as measured by C2N, generally outperformed the immunoassays. It identified amyloid-positive people with an accuracy of 0.87. The ALZpath and Fujirebio p-tau217 assays came in a close second, with accuracies of 0.84 and 0.83, respectively, followed by Janssen’s 0.82. All did much better at detecting brain amyloid than did tests of p-tau181, GFAP, or NfL (table below).
The same pattern emerged when correlating the tests with the burden of amyloid, not just positivity as defined by a specific cutoff. Including the Aβ42/40 ratio where available, i.e., the C2N and Fujirebio platforms, did not significantly improve performance.
P-tau217. Again. Among 392 individuals tested in the ADNI cohort, plasma %p-tau217 most accurately identified who had brain amyloid. Immunoassays for p-tau217 came in a close second. [Courtesy of Schindler et al., 2024.]
In general, the tests did better in plasma from a subgroup of those 192 volunteers who were cognitively impaired, i.e., people who would qualify for biomarker testing in clinical practice under current guidelines. Here, adding the plasma Aβ42/40 data did improve things slightly, with the Fujirebio and C2N tests both posting accuracies of 90 percent. “This means that when trying to determine if someone who is cognitively impaired has Alzheimer’s, the immunoassays will work similarly to mass-spec assays,” said Schindler. In prevention studies, or a clinical trial that enrolls cognitively unimpaired people, mass spec tests may perform better.
Curiously, including plasma Aβ42/40 boosted accuracies most in people who were amyloid-negative on PET. “It seems the plasma Aβ42/40 ratio helps very early on in the disease trajectory, when there is a low level of amyloid,” said Schindler. Then as p-tau217 begins to rise and the change in Aβ42/40 plateaus, the latter becomes less informative.
For all the other outcome measures—tau PET, cortical thickness, and dementia severity—correlations were strongest with the %p-tau217 and p-tau217 tests (image below). “This is notable because, right now, we have different markers for ‘ATN’, or amyloid, tau, and neurodegeneration,” said Schindler (see Nov 2023 conference news on diagnostic criteria). “Why would you use Aβ42/40, or NfL, when p-tau217 is better?”
One Size Fits All. Rather than having separate markers for “ATN” the amyloid, tau, and neurodegeneration of AD, could just p-tau217 do the job? In ADNI, it best correlated with amyloid, tangles, atrophy, and cognitive impairment. [Courtesy of Schindler et al., 2024.]
Adieu Lumbar Punctures?
What does all this mean for CSF testing? In Schindler’s view, given how well some of the plasma tests perform, it is getting harder to justify lumbar punctures to collect CSF. “The major reason clinicians still do it is because blood tests are not reimbursed,” she told Alzforum.
C2N and Fujirebio have submitted applications to the FDA; others may soon follow. Given the current momentum in the AD field, some scientists at AAIC were hopeful that approval for plasma tests might come as early as next year. “In the not-too-distant future, we will be doing blood tests on most people,” Schindler said. Others at AAIC shared this view.—Tom Fagan
Zicha S, Bateman RJ, Shaw LM, Zetterberg H, Bannon AW, Horton WA, Baratta M, Kolb HC, Dobler I, Mordashova Y, Saad ZS, Raunig DL, Spanakis EM, Li Y, Schindler SE, Ferber K, Rubel CE, Martone RL, Weber CJ, Edelmayer RM, Meyers EA, Bollinger JG, Rosenbaugh EG, Potter WZ, Alzheimer's Disease Neuroimaging Initiative (ADNI), Foundation for the National Institutes of Health (FNIH) Biomarkers Consortium Plasma Aβ as a Predictor of Amyloid Positivity in Alzheimer's Disease Project Team.
Comparative analytical performance of multiple plasma Aβ42 and Aβ40 assays and their ability to predict positron emission tomography amyloid positivity.
Alzheimers Dement. 2022 Jul 12;
PubMed.
Further Reading
No Available Further Reading
A Finger-Prick Test for Alzheimer’s Disease?
Jab a finger, draw up a spot of blood with a test strip, let it dry, then mail it off to your doctor. Could testing for Alzheimer’s disease become that simple? Quite possibly. Modern immunoassays are so sensitive they can detect markers in that way.
At AAIC 2024, held last month in Philadelphia, Nicholas Ashton, Banner Alzheimer’s Institute, Phoenix, reported that p-tau217 is one such protein. Measuring this tau fragment in dried plasma spots identified people with Alzheimer’s disease about as well as did using venous blood, Ashton showed.
Henrik Zetterberg, University of Gothenburg, Sweden, works with Ashton on a project called Drop-AD to evaluate this methodology. He said it can measure other markers as well. Sebastian Palmqvist, University of Lund, Sweden, told Alzforum that the finger-prick analysis could be hugely helpful in countries where there are large rural populations with limited access to centrifuges needed to process venous blood draws.
For now, one dried spot is needed to test each marker, but new technology coming down the pike can measure more than 100 from a single finger prick. Called NULISA, it uses proximity ligation of nucleic-acid-tagged antibodies to amplify signals in biofluids in multiplex analysis. It is catching on in multiple labs (see next story).
Drop-AD
At AAIC, Ashton and Hanna Huber, University of Gothenburg, presented how their project analyzed blood from 327 volunteers at five centers across Europe and one in the United States. Of these, 165 were tested for amyloid pathology and 289 for tau pathology using previously established cutoffs in venous blood for the Aβ42/40 ratio and p-tau217, respectively. Each person also had a finger pricked to draw a few dozen microliters of capillary blood into test strips that either dried the blood as is, or separated out the red blood cells first, since protein markers are generally more stable in plasma.
Scientists at the various centers then shipped the cards, at ambient temperature, to Gothenburg, where Huber measured p-tau217 in the dried spots using the commercially available ALZpath immunoassay.
Drop It! Some microliters of blood from a finger prick contain enough p-tau217 to be measured on commercial immunoassay platforms. [Image courtesy Nick Ashton.]
At AAIC, Ashton showed that the concentrations of p-tau217 in capillary blood, as measured on the dried spots, were about 40-fold lower than in venous blood. That was not a big problem, though, because the two correlated tightly. Importantly, dried blood spot (DBS) p-tau217 identified people with amyloid pathology with a specificity and sensitivity that almost equaled that of venous p-tau217. Huber showed that the DBS assay distinguished amyloid-positive and p-tau217-positive people from their respective controls.
All told, the data indicate that it may well become possible to use such a simple finger-prick sampling approach for in-home testing. Much work remains. The method has to be tested in other cohorts, and in real-world settings, as these samples were taken in research clinics. “We need to see how these tests perform when people collect their own samples unsupervised,” Ashton said.
Zetterberg told Alzforum that the dried blood spots already work well for p-tau181, p-tau231, GFAP, and NfL. “We can measure these with excellent correlation with venous blood levels,” he said (Huber et al., 2024; Kolanko et al., 2024).
One downside? Each test strip can only be used once to measure one marker. Hence no multiplexing. Here’s where NULISA can help. This new type of assay can measure more than 100 central nervous system proteins simultaneously—in one dried plasma spot, and with sensitivities 10- to 20-fold higher than single molecule array (SIMOA) immunoassays currently used to measure AD markers (see next story).—Tom Fagan
NULISA—A New Proteomic Method to Revamp Biomarker Analysis
A new multiplex diagnostic method appears poised to resculpt the biomarker landscape. NULISA, aka NUcleic acid Linked Immunosorbent Assay, can detect Aβ peptides, p-tau isoforms, and other potential markers of neurodegeneration with sensitivities 10- to 20-fold greater than some single-molecule array (SIMOA) immunoassays currently used to measure AD markers, according to Henrik Zetterberg, University of Gothenburg, Sweden.
As reported at AAIC last month in Philadelphia, and in recent publications, Alzheimer’s disease labs have already started using this technology. So far, the results look promising. NULISA identifies people with brain amyloid as well as commercially available plasma tests do. Since it can detect hundreds of targets simultaneously in one sample, it is already starting to identify new potential markers.
The “NU” Proteomics. NULISA uses antibody pairs to detect minute quantities of antigen in fluid samples. Tails on the antibodies allow for a two-step capture and purification process. Proximity ligation amplifies the signal. [Courtesy of Alamar Biosciences.]
NULISA brings into apposition pairs of nucleic-acid tagged antibodies that recognize different epitopes of the same target. Enzymes ligate the tags, which can then be quantified by polymerase chain reaction or next-generation sequencing amplification. Specific tag sequences identify specific antibodies, allowing multiple targets to be quantified simultaneously. The technology was developed by scientists at Alamar Biosciences, Fremont, California (Feng et al., 2023). Because the antibodies have polyA or biotinylated tails, the antibody-antigen complexes can be captured sequentially on polyT or streptavidin matrices, allowing for a two-step purification.
“The upshot is you get really pure complexes and exquisite sensitivity,” said Zetterberg.
NULISA measures protein targets down to the attomolar level. That’s 18 zeroes, or 10-18 molar; for those of us who are not used to dealing in such infinitesimal quantities, that equates to just over 1 molecule in a 0.2 mL sample.
What Else Can NULISA Do?
Scientists have begun to use this technology to measure known AD markers in multiplex fashion—and to search for new ones. At AAIC, Xiao-JunMa and colleagues at Alamar introduced a suite of antibodies that quantify 120 proteins previously linked to neurodegenerative diseases. They include Aβ peptides, p-tau isoforms, and a range of other targets involved in immune responses, microglial signaling, and synapse maintenance, to name a few (table below). Most of these are present above the limit of detection in plasma, and more than 85 percent are detectable in dried blood spot samples (see previous story). In a small pilot study of 13 people with AD and 13 controls, Ma found 30 markers upregulated in AD and four downregulated, hinting at an AD proteomic fingerprint.
Targets Galore? In multiplex analysis, NULISA simultaneously detects almost 120 proteins linked to neurodegenerative diseases. [Courtesy of Alamar Biosciences.]
Also in Philadelphia, Andrea Benedet, U Gothenburg, reported on NULISA analysis of samples that had already been tapped for a previous round-robin comparison of different AD markers run by Ashton (see Nov 2023 conference news). These amply studied samples came from 20 people with AD and 20 controls.
As expected, NULISA detected more p-tau181, p-tau217, p-tau231, and GFAP in the plasma and/or CSF or the AD group than in controls, and less Aβ42. In fact, based on plasma p-tau217, NULISA identified people with amyloid pathology with the same accuracy as did the immunoassays and mass spectrometry assays used in the round robin.
In another head-to-head study, scientists at Oskar Hansson’s lab at Lund University, Sweden, have compared the NULISA p-tau217 tests against commonly used immunoassay- and mass-spectrometry-based tests in a subset of 463 samples from the BioFinder-2 cohort in Sweden, and 97 samples from the Knight ADRC at Washington University, St. Louis (see Part 12 of this series). In the former, NULISA p-tau217 was almost as good at distinguishing people who had amyloid in the brain as the %p-tau217 in plasma, yielding an AUC of 0.93 versus 0.96 for the latter. In the U.S. samples there was no difference.
Work from Pedro Rosa-Neto’s lab at McGill University, Montreal, supports the validity of this new technology. Yi-Ting Tina Wang and colleagues compared the performance of NULISA to that of SIMOA immunoassays for p-tau181 and p-tau231 run at UGothenburg, and commercial immunoassays for p-tau217 from Janssen and ALZpath. Among 397 volunteers in the Translational Biomarkers in Aging and Dementia, aka TRIAD, cohort in Montreal, NULISA analysis of plasma p-tau217 was the top performer, identifying people who were amyloid-positive with an AUC of 0.918. NULISA slightly outperformed the AlzPath and Janssen tests. Among the subset of people who were cognitively unimpaired, NULISA also did best. The Janssen test for p-tau217 best identified those who had positive tau PET scans, with NULISA a close second. The differences between these p-tau217 tests were negligible, hence NULISA appears about as accurate as immunoassays in discriminating amyloid- and tangle-positive individuals.
Benedet wants to use NULISA to find new markers. After all, the current crop captures only part of the pathology wrecking the AD brain. She studies later disease stages where additional markers might pop up. Among samples from TRIAD and BioFinder, she found more than a dozen changes in the CSF of people with known tau pathology. More abundant were the neurodegeneration marker neurofilament light, the fatty acid binding protein FABP3, the synaptic protein neurogranin, and the myeloid cell receptor TREM1. More depleted were synaptic neuropentraxins, interleukin 13, and interferon-γ. Some of these differences also emerged in plasma. Benedet said her pilot studies confirm that NULISA can detect known markers and highlight its potential to find and ones.
The Benedet lab has also begun to deploy NULISA in frontotemporal and Lewy body dementias. At AAIC, Joel Smirén reported more than a dozen proteins that are up in the plasma of 18 symptomatic progranulin mutation carriers compared to plasma of 20 noncarriers. They ranged from markers of neuronal injury, such as α-synuclein, neuropentraxin 2, SNAP-25 and NfL to markers of lysosomal dysfunction, such as SQSTM1, aka p62. The panel also spotted protein concentration increases in presymptomatic carriers, albeit without statistical significance. These will be re-examined in larger cohorts, Smiren said.
Meanwhile, Bárbara Gomes reported that in TRIAD, NULISA detected protein reductions in the CSF of 112 people who had Lewy body pathology as judged by seed amplification assay of α-synuclein protofibrils. Those included the transcription factor REST, which has been linked to neuroprotective responses (Lu et al., 2014), RUVBL2, a helicase involved in DNA repair, corticotrophin releasing hormone, the cytokine IL16, and the transmitter neuropeptide Y.
Other labs are also jumping on this new technology. Scientists in Thomas Karikari’s lab at the University of Pittsburgh are using Alamar’s CNS panel and an inflammation panel that measures 250 proteins. They are hunting for signs of neuronal injury, vascular damage, inflammation, and synucleinopathy in AD. As Xuemei Zeng of the University of Pittsburgh noted in her AAIC talk, these processes are recognized in the revised diagnostic criteria as important pathologies in AD, but no specific markers are yet available (Nov 2023 conference news).
Zeng has tested plasma from 113 volunteers in the Monongahela Youghiogheny Healthy Aging Team-Neuroimaging cohort. This population-based study characterizes mild cognitive impairment in economically distressed southwestern Pennsylvania. This population has a high level of comorbidities, such as diabetes, hypertension, and kidney disease. Participants had a clinical dementia rating of below 1 at baseline; at which point a quarter had amyloid pathology. Thus far, about half have had their second-year follow-up, by which point a third of them tested positive for amyloid.
In this MYHAT-NI cohort, too, NULISA levels of plasma p-tau217, p-tau181, p-tau231, NFL, and Aβ42 all correlated tightly with levels determined by SIMOA. Plasma p-tau217 and p-tau231 associated with amyloid, as did downregulation of superoxide dismutase 1, the tricarboxylic acid cycle enzyme MDH1, and the extracellular matrix protein TIMP3.
Could NULISA markers predict amyloid accumulation? Zeng correlated protein level at baseline with subsequent annual PET changes. As might be expected, the higher a person’s plasma p-tau217 at baseline, the more amyloid they accumulated. Interestingly, levels of the chemokines CCL13, CCL17, CCL26, CXCL1, and CXCL8 correlated with less amyloid accumulation over time. “This could mean that those chemokines are neuroprotective,” said Zeng.
She also found proteins that correlated with neurofibrillary tangles. Unsurprisingly, the former included plasma p-tau 181, 217, and 231, but also SFRPI, aka secreted frizzled-related protein 1, and 14-3-3g. SFRP1, an endogenous inhibitor of α-secretase, has been linked to amyloid pathology (Esteve et al., 2019). Zeng found it to be suppressed in tangle-positive volunteers, hinting that it might be contributing to the amyloid cascade.
14-3-3γ is a jack-of-all-trades that binds various signaling molecules, such as kinases, phosphatases, and membrane receptors. It was up in the blood of tau-positive people. Recently, in an unbiased proteomics analysis of ADNI, scientists reported that 14-3-3γ was the most upregulated in AD versus controls among more than 6,000 proteins (Guo et al., 2024).
At AAIC, 14-3-3γ, also called YWHAG, cropped up elsewhere, too. Scientists from Tony-Wyss Coray’s lab at Stanford University, California, reported that it is elevated in AD CSF. Among people in ADNI, at the Knight Alzheimer’s Disease Research Center at Washington University, and at Stanford, the higher the YWHAG:NPTX2 ratio, the faster a person’s cognition declined over the next one to 15 years. This ratio better predicted decline than did p-tau181/Aβ42, ApoE4, or level of baseline cognitive impairment. Markers of disease progression top many an Alzheimerologist’s wish list.
For her part, Zeng found that 17 markers in the NULISA panel changed over time in the MYHAT-NI cohort. Glial markers, including CHIT1 and CHI3L1, waned on follow-up in people who had tangles, as did the cytokines IL17A, SCF2, CXCL1, and markers of vascular integrity, such as PDGFRB and VEGFA. Curiously, no marker seemed to increase over that time. One marker, sequestosome 1, correlated with neurodegeneration over two years.
All told, NULISA and dried blood/plasma analysis (see previous story) appear set to reshape biomarker research analysis, and perhaps even routine clinical testing, for neurodegenerative diseases.
Some people at AAIC wondered if the time has come when scientists will finally find functional markers for cognitive decline. Benedet thinks the field is not quite there yet. So does Sterling Johnson, University of Wisconsin. “I think our field is getting there, but it will take more iteration,” he said. “What we saw with these new multiplex panels is pretty incredible. I think it is that kind of approach that is going to help.”—Tom Fagan
Feng W, Beer JC, Hao Q, Ariyapala IS, Sahajan A, Komarov A, Cha K, Moua M, Qiu X, Xu X, Iyengar S, Yoshimura T, Nagaraj R, Wang L, Yu M, Engel K, Zhen L, Xue W, Lee CJ, Park CH, Peng C, Zhang K, Grzybowski A, Hahm J, Schmidt SV, Odainic A, Spitzer J, Buddika K, Kuo D, Fang L, Zhang B, Chen S, Latz E, Yin Y, Luo Y, Ma XJ.
NULISA: a proteomic liquid biopsy platform with attomolar sensitivity and high multiplexing.
Nat Commun. 2023 Nov 9;14(1):7238.
PubMed.
Esteve P, Rueda-Carrasco J, Inés Mateo M, Martin-Bermejo MJ, Draffin J, Pereyra G, Sandonís Á, Crespo I, Moreno I, Aso E, Garcia-Esparcia P, Gomez-Tortosa E, Rábano A, Fortea J, Alcolea D, Lleo A, Heneka MT, Valpuesta JM, Esteban JA, Ferrer I, Domínguez M, Bovolenta P.
Elevated levels of Secreted-Frizzled-Related-Protein 1 contribute to Alzheimer's disease pathogenesis.
Nat Neurosci. 2019 Aug;22(8):1258-1268. Epub 2019 Jul 15
PubMed.
At the Edge of Spatial Omics, Cellular Response to Amyloid Comes into View
In the brain, location is everything. And the throngs of cells that live there are nothing without the billions of distinct connections between them. Although scientists can use single-cell transcriptomics to survey gene expression in individual cells throughout the brain, these methods don’t capture the spatial context of those cellular states. Spatial omics has arisen to home in on that. The AAIC held last month in Philadelphia featured the newest findings from the leading edge of this rapidly advancing field, which combines genomics, neuropathology, and high-resolution imaging to give various omes a physical address in the brain.
Drawing from a burgeoning toolbox of techniques, researchers investigated how Aβ plaques and tau tangles influence both composition and activity of cells stationed nearby. They tracked how cells responded as amyloid plaques formed and matured, and identified inflammatory flare-ups that could fuel amyloid-induced tau pathology. The activities of plaque-adjacent glial cells may explain how some people manage to fend off tau tangles and cognitive decline, despite harboring amyloid plaques.
“There’s nowhere in the body where the exact location of a cell is more important than it is in the brain,” said Bart de Strooper of KU Leuven in Belgium who, along with Lea Grinberg of the University of California, San Francisco, co-chaired a session devoted to emerging spatial omics findings at the meeting.
An early adopter of spatial transcriptomics, De Strooper’s lab had previously identified plaque-induced genes, aka PIGs, expressed by cells positioned near Aβ aggregates (Jul 2020 news). Since then, scientists there and elsewhere have sharpened the techniques’ spatial resolution and scope, and they worked out how to simultaneously map transcriptomes and pathological proteins in the same tissue (Mallach et al., 2024; Aug 2022 conference news; Feb 2023 news).
Researchers at the Allen Institute for Brain Science in Seattle continue to improve these techniques, with the goal of mapping the mouse and, ultimately, the human brain, in stunning detail (Dec 2023 news). In Philadelphia, Allen’s Rebecca Hodge noted that while spatial omics technologies seem complex, they come in two main flavors.
Imaging-based approaches, such as MERFISH and Xenium, use nucleic acid probes that bind to specific RNA sequences within a tissue section. Using sequential imaging to quantify each target’s abundance, scientists can take stock of hundreds to thousands of transcripts at single-cell, even subcellular, resolution. This technique is limited by the number of transcripts it can detect.
On the other hand, sequencing-based techniques, such as Visium or Slide-Seq can survey entire transcriptomes but are limited in their cellular resolution. In these approaches, transcripts within tissue sections are transferred onto chips or slides that are dotted with barcoded spots that impart spatial information. Once the transferred transcripts are tagged with their location, they can be sequenced, yielding both spatial and transcriptomic information.
At AAIC, Hodge presented initial results from the Seattle Alzheimer’s Disease Cell Atlas. A partnership among the Allen Institute, the University of Washington in Seattle, and the Kaiser Permanente Washington Health Research Institute, also in Seattle, SEA-AD aims to understand how, and where, cells respond at different disease stages. To do this, scientists use neuropathology, single-cell transcriptomics, and spatial transcriptomics to analyze brain samples from people who died at different stages across the AD neuropathological spectrum. Findings are posted on bioRxiv (Gabitto et al., 2024).
A few highlights? For one, the researchers hunted for subsets of neurons that are vulnerable to AD pathology. With snRNA sequencing, they pinpointed transcriptional subtypes of cells in the middle temporal gyrus and the dorsolateral prefrontal cortex, which are affected by tau pathology early and late in AD, respectively. This identified 139 transcriptional subtypes of cells, including several that became scarcer as the pathological burden worsened. These included subsets of excitatory neurons as well as somatostatin-expressing interneurons. Because excitatory neurons express specific markers in different layers of the neocortex, the researchers inferred that the vulnerable subsets of these neurons resided in layers 2 and 3. However, because interneurons are distributed diffusely across different cortical layers, their locations are not closely tied to their transcriptomes.
Vulnerability Up Top. Most but not all subtypes of vulnerable neuron reside within the upper three layers of the neocortex (L1-L3). [Courtesy of Gabitto et al., bioRxiv, 2024.]
Enter spatial transcriptomics. Using MERFISH to map the expression of 140 transcripts with single-cell resolution across 76 sections of the MTG from 28 brains, the researchers spotted the most vulnerable subtypes of neurons dwelled in the outermost layers of the neocortex. They included somatostatin-positive interneurons, parvalbumin-expressing inhibitory neurons, and excitatory neurons. The lone exception was a subtype of excitatory neuron found deep within layer 5, which became vulnerable only in late stages of AD, i.e., among brain samples with tau tangles inundating the prefrontal cortex.
To Hodge, the findings exemplify that understanding where these cell types are within the context of intact tissue is important. “Now we can start to generate hypotheses about how the loss of these different neurons within the same layer, where they’re likely interacting, might impact the circuitry and function of the brain,” she said.
However, she added that this analysis lacks a critical piece of spatial information, namely, how being near pathological proteins influences a given cell’s gene expression. To address this question, Hodge and colleagues are adding immunostaining of Aβ and p-tau to spatial transcriptomics (image below).
Pathological Neighborhood. Different subtypes of neuron (A) and microglia (B) were mapped across neocortical layers with spatial transcriptomics, then Aβ and p-tau were stained (C). Overlaying this spatial data (D) could address how being near to pathology influences the states of nearby cells. [Courtesy of Rebecca Hodge, Allen Institute for Brain Science.]
Many challenges lie ahead for spatial transcriptomics, Hodge said. Chief among them: the sheer scale of the human brain. “Its size exceeds the imaging or capture size of every product on the market,” she said. To chip away at it, Allen Institute scientists are tiling across large anatomical structures to create series of tissue blocks that fit within their imaging machines. This technique is possible yet technically demanding, Hodge said, as it requires tracking many blocks followed by stitching images back together to recreate the anatomy of the brain.
Other scientists are approaching the problem by clearing sections of the brain with hydrogels. This allows them to image large, three-dimensional chunks. A recent study led by Kwanghun Chung at Massachusetts Institute of Technology used this method to visualize different morphological flavors of microglia congregating around Aβ plaques (Park et al., 2024). “It will be exciting to see how this technique might be used to look at both RNA and proteins in these large-scale tissues,” Hodge said.
De Strooper noted the pace of technological advancement in this field. “It’s fascinating to see how fast these things move,” he said after Hodge’s talk.
Yanling Wang of Rush University in Chicago investigates how Aβ plaques influence nearby neurons, and how glial cells recruited to the scene shake things up. In Philadelphia, Wang presented findings from her analysis of 21 postmortem brain samples from the ROS-MAP cohort, in which the researchers used Visium to map transcriptomes within sections of prefrontal cortex. Each finely sliced section was placed on a Visium slide dotted with nearly 260,000 spots, each big enough to cover the area of five to 10 cells. Once embedded with their spatial information, the transcripts were sequenced. For each section scrutinized in this way, two adjacent slices were subjected to immunohistochemistry to label Aβ, GFAP, and Iba1 to delineate plaques, astrocytes, and microglia.
Wang wanted to know how the amount of amyloid, and the maturity of plaques in each spot, influenced nearby neurons. Noting that plaques typically evolve from diffuse to compact to dense-core, Wang said that in these samples, spots with low levels of Aβ tended to host diffuse plaques, while those with more Aβ contained predominantly compact plaques and a few dense ones. Wang reported that neurons near diffuse, low-Aβ areas seemed to fare worse than neurons stationed near compacted plaques. Neurons adjacent to diffuse plaques had downregulated synaptic function genes and turned up apoptosis genes.
Once astrocytes and microglia were recruited to a plaque, neurons and other nearby cells appeared to be in trouble. Wang reported that in spots with high numbers of glia, neurons expressed a similar neurodegenerative profile. Spots with low Aβ and swarms of glia were the most hostile environs for neurons. Wang also tracked numerous other cell types, reporting, for example, that some subtypes of inhibitory neuron and oligodendrocyte precursor appeared most vulnerable to low-Aβ, high-glia conditions.
Wang zeroed in on the profiles of microglia near plaques, comparing them to previously reported subtypes. She found that plaque-adjacent microglia tended to be of the “ribosome biogenesis” persuasion, a subtype thought to closely resemble the disease-associated microglia (DAM) identified in mice (Oct 2023 news on Sun et al., 2023).
Wei-Ting Chen of Muna Therapeutics in Leuven has a related purpose in mind for spatial transcriptomics. She investigates how, despite their hefty burden of amyloid plaque, some people manage to fend off tau pathology and cognitive decline. Specifically, Chen wants to know which potentially protective cellular mechanisms might be at work around plaques among cognitively resilient people, or destructive ones in people with dementia. In collaboration with De Strooper’s lab at KU Leuven, Chen deployed Visium for spatial transcriptomics, single-nucleus RNA sequencing, and immunostaining for Aβ and p-tau on adjacent sections taken from cortical punches from the superior frontal gyri of 44 people. Donors came from two main autopsy cohorts (image below). One comprised octogenarians, including eight who died without amyloid pathology, eight who had amyloid plaques but no dementia, and eight who died with AD dementia. The second cohort comprised 20 cognitively resilient centenarians, who died with amyloid plaques but intact cognition.
Resilient or Not? To investigate why some people with amyloid plaques get dementia, while others stay sharp, scientists analyzed brain samples from octogenarians (left) who died with plaques but not dementia (AD-DEM) or with dementia (AD+DEM), or centenarians (right) who had plaques but not dementia. [Courtesy of Wei-Ting Chen, Muna Therapeutics.]
The researchers found only a slightly higher burden of amyloid plaques among people who died with dementia relative to resilient people. In contrast, p-tau was substantially higher among people who died with dementia relative to cognitively resilient octogenarians and centenarians (image below). Furthermore, resilient people had fewer neuritic plaques, which are known to beckon tau tangles and wreak havoc on nearby synapses.
Resilience, Spot by Spot. Tissue slices prepared for Visium spatial transcriptomics were first stained for Ab (red) and pTau (green). As seen across the entire array (left), as well as at the resolution of a single spot (right), people who died with dementia (bottom) had more pTau than those who remained dementia-free (top). [Courtesy of Wei-Ting Chen, Muna Therapeutics.]
What about cellular profiles near amyloid and tau pathology? Wang reported that regardless of whether there were tangles, amyloid plaques alone incited inflammatory gene expression in nearby cells, such as complement proteins and TREM2.
Once tangles entered the picture, transcriptional clusters of reactive astrocytes, as well as microglia expressing a suite of genes previously described as the ribosome biogenesis and HLA profiles, were dramatically more abundant near plaques, whereas a subset of excitatory neurons in cortical layer 5 became depleted. A cadre of inflammatory genes, including HLA-DRA, CD74, CD14, and SPP1, were rampant around plaques in samples with abundant tangle pathology relative to samples with only plaques. Dying with dementia had a similar effect on the cellular response to amyloid plaques as did tau pathology.
To Chen’s mind, this implies that these inflammatory responses might fuel amyloid-induced tau pathology and, ultimately, neurodegeneration and cognitive decline. Counteracting these responses early in AD pathogenesis might put a wrench in the cascade, she hopes.
Wang noted that the data produced thus far by spatial transcriptomics are but the tip of the iceberg. For example, most ROS-MAP participants harbor mixed pathologies, not just Aβ and tau. Moreover, spatial omics findings always must be functionally validated to move them beyond the realm of correlation, she added. De Strooper made a similar point. Indeed, how to draw meaning from the mountain of complex data spatial transcriptomics produces is the real challenge right now, he said.—Jessica Shugart
Gabitto MI, Travaglini KJ, Rachleff VM, Kaplan ES, Long B, Ariza J, Ding Y, Mahoney JT, Dee N, Goldy J, Melief EJ, Agrawal A, Kana O, Zhen X, Barlow ST, Brouner K, Campos J, Campos J, Carr AJ, Casper T, Chakrabarty R, Clark M, Cool J, Dalley R, Darvas M, Ding S-L, Dolbeare T, Egdorf T, Esposito L, Ferrer R, Fleckenstein LE, Gala R, Gary A, Gelfand E, Gloe J, Guilford N, Guzman J, Hirschstein D, Ho W, Hupp M, Jarksy T, Johansen N, Kalmbach BE, Keene LM, Khawand S, Kilgore M, Kirkla.
Integrated multimodal cell atlas of Alzheimer's disease.
2024 Feb 15 10.1101/2023.05.08.539485
(version 3)
bioRxiv.
Park J, Wang J, Guan W, Gjesteby LA, Pollack D, Kamentsky L, Evans NB, Stirman J, Gu X, Zhao C, Marx S, Kim ME, Choi SW, Snyder M, Chavez D, Su-Arcaro C, Tian Y, Park CS, Zhang Q, Yun DH, Moukheiber M, Feng G, Yang XW, Keene CD, Hof PR, Ghosh SS, Frosch MP, Brattain LJ, Chung K.
Integrated platform for multiscale molecular imaging and phenotyping of the human brain.
Science. 2024 Jun 14;384(6701):eadh9979.
PubMed.
Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N, Prosper S, Viswanathan S, Luna X, Boix CA, James BT, Tanigawa Y, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, Tsai LH, Kellis M.
Human microglial state dynamics in Alzheimer's disease progression.
Cell. 2023 Sep 28;186(20):4386-4403.e29.
PubMed.
Is the ‘Atrophy’ of Immunotherapy Just the Dismantling of Plaque ‘Suburbia’?
The brain shrinkage seen on amyloid immunotherapy remains an unsolved and concerning riddle. At the Alzheimer’s Association International Conference, held July 27 to August 1 in Philadelphia, Nick Fox of University College London offered a possible answer.
While the amount of Aβ removed by antibodies is too small to account for the lost volume, Fox pointed out that amyloid plaques are more than just Aβ. They include a built-up “suburbia” of other proteins, dystrophic neurites, and associated immune cells. All told, this occupies around 6 percent of the cortex. Indeed, some studies have found that the cortex thickens where plaques form. Removal of this material could in fact explain the cortical volume loss, Fox argued.
Greater cortical shrinkage was associated with slower cognitive decline in the Phase 3 lecanemab trial. Rather than calling this volume loss “atrophy,” which implies a degenerative process, Fox proposed calling it “amyloid-removal related pseudo-atrophy,” or ARPA. The data are in press at Lancet Neurology.
Cynthia Lemere of Brigham and Women’s Hospital, Boston, said the term ARPA makes sense, as does the theory behind it. Attendees in Philadelphia were receptive to the idea. Fox’s talk generated the most questions and subsequent buzz of its session.
Maybe a Shrinking Brain Really Can be Good for You
Accelerated volume loss on amyloid immunotherapy was first observed 20 years ago in the AN1792 vaccine trial. At the 2004 AAIC—also held in Philadelphia, and marked by the roving, cheerful, ever-present local “host” John Trojanowski—Fox first reported, to the audience’s consternation, that higher anti-Aβ antibody titers correlated with more brain shrinkage. Even in this discontinued first attempt at immunotherapy, drug response and brain shrinkage also correlated with lower cerebrospinal fluid tau and better cognitive outcomes. This raised the provocative question of whether there are circumstances when a bit of brain shrinkage can be a good thing (see Jul 2004 news coverage and associated comments).
Since then, a small amount of volume loss has become a consistent feature in many immunotherapy trials. This engendered debate over whether it indicates accelerated degeneration, or something good to do perhaps with dampened inflammation. In Philadelphia 2024, Fox argued for the latter. For one, brain shrinkage on immunotherapy is unlikely to represent neuron loss, because it does not correlate with neurodegenerative markers, he said. In the Phase 3 lecanemab trial, CSF total tau went down on drug, as did neurogranin, a marker of synaptic loss. Plasma NfL trended down as well (see related story).
Sign of Neuroprotection? In the lecanemab Phase 3 trial, people on drug (green) had preserved hippocampal volume relative to controls (black). [Courtesy of Bateman et al., CTAD 2022.]
Importantly, hippocampal volume is preserved in many immunotherapy trials. This fits with Fox’s explanation, because there are fewer plaques taking up space in AD hippocampus than cortex to begin with. In the Phase 3 donanemab trial, hippocampal volume loss was the same on drug or placebo (Jul 2023 conference news). In the Phase 3 lecanemab data, the hippocampus shrank less on drug than placebo, with the difference statistically significant (image above). Hippocampal shrinkage is a key indicator of AD, so its preservation could indicate neuroprotection, Fox said.
Further supporting the idea that the whole-brain volume loss on these antibodies is not harmful: It correlated with better cognitive outcomes. At the same degree of cortical shrinkage, people on lecanemab had less functional decline on the CDR-SB and ADCS MCI-ADL than did those on placebo, Fox calculated.
Sprawling Outward? Amyloid plaques and their associated cells and dystrophy occupy around 6 percent of the AD cortex (top) by volume; higher magnification at bottom. [Courtesy of Zane Jaunmuktane, Queen Square Brain Bank, UCL.]
Plaques Crowd the Cortex, Hog Space
Instead, Fox believes the shrinkage is due to clearance of plaque. This had been considered an unlikely explanation for volume loss, in part based on calculations that the AD brain contains a total of 6.5 mg of Aβ (Aug 2016 news; Roberts et al., 2017). Extrapolating from this, scientists have calculated that removing this tiny amount would account for only one-thousandth of the volume change. Fox decided to test this conclusion, noting that in the brain, these protein snarls and their associated cellular changes occupy extensive space.
Postmortem studies that directly measured plaques found they typically took up around 7 percent of the cortical volume (Bussière et al., 2002; Josephs et al., 2008; Clark et al., 2012). More recent papers reported similar numbers, with one multicenter study calculating that diffuse plaques occupy 5 to 8 percent of the cortex, and compact plaques 1 to 2 percent (Chen et al., 2021). Another found plaques accounting for almost 9 percent of frontal cortex, and 6.5 percent of temporal cortex (Abrahamson et al., 2022).
Seven percent of cortex would equate to about 3 percent of whole brain, Fox noted. Given that the excess brain shrinkage in the Phase 3 lecanemab trial was about 0.4 percent, plaque clearance could easily account for it, he said. He noted that the more plaque a person started with at baseline, and the more that was removed, the more their brain shrank compared with that of placebo controls.
The few autopsies of immunotherapy patients published to date show how extensive clearance can be. A woman on aducanumab for 2.5 years had almost no plaques left, while a man on lecanemab for 3.5 years had no diffuse plaques, and sparse neuritic plaques (Plowey et al., 2022; Aug 2022 conference news).
If plaque claims that much space in the brain, shouldn’t the brain get a little bigger as deposits form? Indeed, some studies have reported this. Gael Chételat of Cyceron in Caen, France, found that among cognitively healthy people, those with plaques have slightly bigger brains than those without (Chételat et al., 2010). Likewise, in presymptomatic people with a dominantly inherited AD mutation, the cortex thickens 15 to 20 years before symptom onset (Montal et al., 2021).
In his AAIC talk, Fox emphasized the role of inflammation as well. Astrocytes and microglia crowd around plaques, and the astrocytic marker GFAP rises as AD progresses. Removing plaque could get rid of these cells, slimming down the cortex. Anti-inflammatory treatments for multiple sclerosis initially shrink the brain, too, he noted.
A recent meta-analysis linked volume loss to ARIA. The more ARIA for a given immunotherapy, the more ventricles expanded on that drug. This implied a corresponding reduction in gray matter, the authors claimed (Apr 2023 news). Fox challenged the view that greater ventricular volume necessarily correlates with gray-matter loss. In the Phase 3 lecanemab trial, ARIA events were associated with 2.6 mL higher ventricular volume, but with no change in whole-brain shrinkage. Possibly, enlarged ventricles are caused by fluid shifts due to inflammatory changes, he suggested.
Slowing Shrinkage. In people who had cleared most plaque by 12 months on lecanemab (dotted green line), whole-brain volume loss slowed to about the rate seen on placebo (black), while in those who were still clearing amyloid (solid green), shrinkage remained accelerated. [Courtesy of Esai.]
Does Volume Loss Slow Over Time?
How to distinguish (good) shrinkage due to amyloid removal from (bad) shrinkage due to neurodegeneration? It’s a tricky problem. If amyloid immunotherapy protects the brain and slows neuron loss, then, as plaque gets cleared, accelerated shrinkage should level out. In time, volume loss would be expected to become less on drug than on placebo. Whether this happens is not yet clear, but there are some promising signs, Fox said.
There were hints of this even in the AN1792 trial. A subgroup of participants who responded well to the vaccine lost more brain volume than the placebo group for one year, but had bigger brains than placebo at year two. At AAIC, Fox reported there were hints of this in the 18-month Phase 3 lecanemab trial, too. People who had completely cleared plaque by 12 months lost less gray matter overall than did those who remained amyloid-positive at 12 months. After that point, their brains continued shrinking at nearly the same rate as in the placebo group.
Another finding in support of this hypothesis is the slower shrinkage of hippocampus on lecanemab than placebo. Because the hippocampus contains few plaques, it would be little affected by amyloid clearance, hence its size may more directly reflect neurodegeneration. Eventually, the cortex and whole brain would follow suit, with their rates of decline slowing relative to placebo, Fox said.
Because the lecanemab post-18-month extension is open-label, there are no longer-term comparative data. In addition, brain atrophy accelerates as disease advances, which would also make it hard to see any flattening of the curve (Chan et al., 2003). Fox stressed the importance of longer studies to track brain volume changes over several years on immunotherapy. He also recommended that drug companies make patient-level trial data available to help researchers parse out the relationships between regional plaque content, regional volume changes, and other markers of AD.—Madolyn Bowman Rogers
Now that lecanemab has been in clinical use in the U.S. for a little over a year, doctors are asking how long they should treat. This remains an open question. Drug maker Eisai has proposed to the FDA, in a supplemental biological license application, that patients stay on past 18 months and switch to half as frequent a dose once plaques and other, more sensitive biomarkers, have changed. Eisai is in discussions with the agency about that. What is the evidence for this proposal, and how would doctors know when to switch? These are the questions clinicians discussed at last month’s Alzheimer’s Association International Conference in Philadelphia.
Several talks showed how fluid biomarkers rebound after patients stop taking the drug. Likewise, cognitive decline returned to placebo levels, even in amyloid-negative patients. Scientists argued that this is due to the rebound of Aβ protofibrils in the brain. Lecanemab targets this species linked to synaptic damage and neurodegeneration. Eisai’s quantitative model predicted that 18 months of lecanemab will mop up most protofibrils in the brain. The same model projected that maintenance dosing would be sufficient to keep protofibrils low, biomarkers flat, and maintain cognitive benefits. These data stand in contrast to findings for donanemab, which targets fibrillar amyloid and can be discontinued once plaque is gone (see next story).
Other news from AAIC? Long-term lecanemab dosing in open-label extensions now goes out to three years, and continues to support the idea that the antibody slows disease progression. On cognitive measures, the treatment group maintains its edge over the former placebo group, as is expected for a disease-modifying drug, and people at the earliest disease stages reap the biggest cognitive benefits. Perhaps as Alzheimer’s disease progresses, non-amyloid processes come to dominate and immunotherapy is less helpful, Eisai’s Michael Irizarry told the audience.
Sustained Benefit? People on lecanemab for three years (green) maintain a cognitive edge over those who transitioned from placebo at 18 months (black to green); both do better than matched controls from ADNI (pink). [Courtesy of Eisai.]
Biomarkers Worsen Off Drug, Even in Absence of Plaque
Data from lecanemab trials offer a glimpse into what happens in the brain after dosing stops. After the Phase 2 trial ended, participants had to wait about two years before entering an open-label study. Most were amyloid-negative at the time. Scientists previously reported that plaque and plasma p-tau181 rose during the gap, creeping about a quarter back toward baseline levels, while plasma Aβ42/40 rebounded faster, halfway toward the baseline value (Mar 2022 conference news).
In Philadelphia, Eisai’s Larisa Reyderman showed more data. During the gap, the inflammatory marker plasma GFAP rose by nearly a third, similar to the rate of p-tau181. Plasma p-tau217, which reflects both plaques and tangles, rose more slowly, by 13 percent. These rates of worsening were similar to those seen in the placebo group. In effect, without lecanemab, disease progression resumed its usual pace.
How long before the treatment effect is lost? Half of it would be gone in six months for plasma Aβ42/40, and in less than two years for plasma p-tau181 and GFAP, Reyderman calculated. For plaque, it would take 12 years to move halfway back toward a patient’s load at trial baseline. Fluid biomarkers are more sensitive indicators of pathology than plaque, Reyderman concluded.
The picture changed once treatment resumed in the OLE, noted Charlotte Teunissen of Amsterdam University Medical Center. Biomarkers began improving again at the same rate as in the clinical trial, both in previously treated people and in the former placebo group.
In contrast to these Phase 2 data, in the Phase 3 Clarity study, participants transitioned directly into the OLE without a gap. For them, biomarkers responded continuously, with additional plaque clearance and further normalization of plasma Aβ42/40, p-tau181, p-tau217, and GFAP over six months of open-label treatment. “The biomarker data support continued treatment,” Teunissen said.
Off Treatment, Cognition Craters. During a dosing gap (middle), cognition declines at the same rate in the previous treatment (blue) and placebo (black) groups, but the numeric difference between them persists (red lines). The groups remain separated during open-label dosing (right). [Courtesy of Eisai.]
Cognition Tracks with Fluid Biomarkers
What about cognition? It followed suit. During the treatment gap after Phase 2, performance on the CDR-SB worsened at the same rate in the former treatment and placebo groups, Reyderman said. Nonetheless, the treatment group maintained its numerical advantage over placebo controls. The group difference persisted after 18 months of OLE treatment, as well (image above). That pattern is expected for a disease-modifying therapy, because people who were treated first remain at an earlier disease stage than those whose treatment started later.
This pattern occurred when there was no treatment gap, as well. Chris van Dyck of Yale School of Medicine in New Haven, Connecticut, showed 18-month OLE data from Clarity. These represent three years of continuous treatment with lecanemab. During the OLE, cognition on the CDR-SB slipped faster than during the trial, but the difference between the former treatment and placebo groups stayed constant, with the lines paralleling each other.
How does this compare with what would have happened off treatment? To approximate a placebo group, van Dyck compared these trajectories to those of matched controls from the ADNI observational study. For the 18-month core trial, ADNI trajectories overlaid those of the placebo group. During the OLE, however, the ADNI line steepened, diverging from the OLE curves. Cognitive decline is known to accelerate as disease progresses. At the end of three years, the difference in CDR-SB scores between the ADNI and original treatment groups was 0.95 points, twice as large as the 0.45 difference between treatment and control groups at 18 months (see image at top of story).
This is what would be expected for a disease-modifying therapy, where treated patients should continue to diverge from controls over time. Slicing the data another way, people on drug were 30 percent less likely to progress to a higher CDR-SB score than ADNI participants. External controls are less reliable than a true placebo group. Even so, van Dyck said, the data hint that lecanemab continues to slow cognitive decline over three years of treatment.
Benefits were bigger for people who entered the trial at very early disease stages, when their tau PET scans were below 1.06 SUVR. Previously, scientists had reported six-month OLE data from this “no-tau” subgroup (Nov 2023 conference news). In Philadelphia, van Dyck added 18-month OLE data. Again, the former treatment and placebo groups remained separated throughout the OLE. After three years of continuous treatment, 59 percent of these participants had lost no ground on the CDR-SB; 51 percent had improved their scores over baseline values. This compares with 76 and 60 percent, respectively, at the end of the placebo-controlled period, reinforcing again that AD does continue to progress even in these treated early stage patients.
Early Patients, Slow Decline. In a subgroup of Phase 3 participants with low amyloid burden, those who started on lecanemab (green) maintain separation at three years from those who switched to lecanemab (black to green). [Courtesy of Eisai.]
Because few people got tau PET scans in this trial, the groups were tiny, at 40 to 50 people each, and the data were noisy. To eke out a bit more statistical reliability, van Dyck selected early stage patients from the full cohort by using baseline amyloid loads of below 60 centiloids as a proxy for low tangle load. This resulted in about 150 people each from the former treatment and placebo groups. Again, their CDR-SB curves remained apart from and paralleling each other throughout the OLE, with less noise in the data and error bars separated (image above). After three years of continuous treatment, 46 percent of people in this larger group had stayed stable, 33 percent were above their baseline.
The apparent relative stabilization of cognition in this group, compared with the steepening decline in the overall cohort, highlights the importance of starting amyloid immunotherapy at an early disease stage, van Dyck said. Eisai’s Brian Willis agreed, noting that at later stages, pathological progression is driven by factors other than amyloid (see next story).
Dual Mechanism of Action—Plaques and Protofibrils
Why does lecanemab treatment matter after plaques are gone? Speakers evoked the antibody’s modus operandi, saying it both clears plaque and removes soluble protofibrils. Lecanemab has a higher affinity for protofibrils than do other antibodies (Nov 2021 conference news). Among plaque-clearing antibodies, aducanumab and gantenerumab bind soluble species more weakly than does lecanemab; donanemab only binds plaque.
Small species such as oligomers and protofibrils cause much synaptic toxicity, said Dennis Selkoe of Brigham and Women’s Hospital, Boston. His lab reported that oligomers isolated from AD brain inhibit long-term potentiation in hippocampal slices and blunt memory when infused into rat brain (May 2023 news). In cultured human neurons, they promote tau phosphorylation and trigger neurites to degenerate; neutralizing them delays tau pathology. Selkoe believes that this is why lecanemab continues to benefit the brain even in the absence of plaques. “Lecanemab’s dual mechanism of action supports continued dosing,” he said in Philadelphia.
For his part, Kenjiro Ono of Kanazawa University, Japan, linked Aβ protofibrils directly to neurodegeneration. To measure protofibrils in human cerebrospinal fluid, he first isolated them using lecanemab conjugated to magnetic beads, then quantified them via immunoassay. He found that protofibrils were significantly higher in CSF from 34 people with MCI-AD and 64 people with AD dementia than they were in 56 amyloid-negative people.
Curiously, however, protofibrils did not correlate with plaque load. Instead, they correlated with markers of neurodegeneration. Protofibrils were associated with CSF total tau, at r=0.6, and with neurogranin, at about r=0.45, but not with p-tau181, p-tau217, or Aβ42/40. The data suggest that protofibrils may directly promote neurodegeneration, Ono said. Eisai researchers collaborated on the study.
Staying Low. Modeling predicts that stopping lecanemab at 18 months would cause biomarkers to rebound (pink), and that maintaining therapeutic (aqua) or maintenance (blue) dosing would keep them flat. [Courtesy of Eisai.]
Modeling How Amyloid, Tau, and Cognition Interact
To try to predict what its antibody will do, Eisai has developed a quantitative systems pharmacology model (Mar 2022 conference news). In Philadelphia, Eisai’s Youfang Cao said the model integrates what is known about amyloid and tau pathology and their effect on cognition with the published literature on amyloid production, clearance, and tau kinetics and spread. Its 11 equations and 74 parameters attempt to simulate the AD pathological cascade, though without inflammatory components.
Next, the scientists plugged in data on lecanemab’s binding affinities for both protofibrils and plaque. Using data from ADNI and lecanemab’s Phase 2 and 3 studies on a total of 4,056 participants, they predicted how lecanemab would affect six outcomes ranging from amyloid and medial-temporal tau PET, plasma Aβ42/40, and p-tau181, to CDR-SB and ADAS-Cog scores. In every case, the model closely predicted actual trial results, Cao said.
Cao noted the model also predicts factors that cannot yet be easily measured, such as protofibril concentration, tau seeds, and neuronal loss. It estimated that 18 months of lecanemab treatment would clear 70 percent of a person’s starting plaque load, and 94 percent of protofibrils. The scientists will test this prediction by directly measuring protofibrils in stored CSF samples.
The scientists also tested the model on other antibodies, entering known data on the binding affinities of aducanumab, gantenerumab, and donanemab to predict final amyloid PET and CDR-SB values in those trials. Again, the model matched reported outcomes.
This model, then, helped Cao and colleagues determine a maintenance dose for lecanemab. If the drug were discontinued at 18 months, the model shows protofibrils rising 30 percent of the way back to control levels over the next 2.5 years, while medial-temporal tau PET would rise about a quarter back toward the level in the placebo group over the same time frame. If dosing continued at 10 mg/kg biweekly, protofibrils and tangles would remain flat. Ten mg/kg monthly would do the same (image above). Eisai requested marketing approval for that latter dose from the U.S. Food and Drug Administration (May 2024 news).
If approved, at what point should doctors transition their patients? In Philadelphia, Reyderman said their modeling suggests doing so at 18 or 24 months would be equally effective. Even for people who still have some amyloid at 18 months, monthly dosing would continue to clear plaque, and the clinical outcomes after four years would be nearly identical, she said. This means doctors could move to maintenance monthly dosing without needing to order an expensive amyloid PET scan to confirm amyloid negativity. Eisai’s Lynn Kramer said the company is in talks with the FDA about the best transition point. No final decision has been made.
Attendees in Philadelphia asked when to stop dosing. To that, Irizarry had a simple answer: when patients have progressed to moderate dementia stages. Accumulating evidence from multiple antibody programs indicates that amyloid immunotherapy does little good once cortical tau pathology takes off.—Madolyn Bowman Rogers
As amyloid immunotherapy is being rolled out, mostly in specialty care thus far, both treating physicians and researchers have many questions about it. Scientists at the Alzheimer’s Association International Conference, held last month in Philadelphia, offered a glimpse at some answers. For the FDA-approved donanemab, it appears that baseline amyloid load best predicts when to stop dosing, and that it may tell the doctor what the right time point might be to confirm with a second PET scan that the amyloid is gone.
Lilly also presented a few trial updates, including a new one in Down’s, details about its Trailblazer-Alz3 study, and that donanemab is going to be evaluated in the Alzheimer Prevention Initiative’s Colombian cohort.
For lecanemab, scientists at AAIC highlighted the promise of the fluid tau marker MTBR-243 to track tangle load, and they modeled how plaque clearance relates to cognitive benefit in an attempt to address a nagging question about this relationship.
See, You’re Negative. In donanemab trials, participants were amyloid-positive (gold) at baseline (top), and amyloid-negative by the end of the trial (middle). These latter scans were indistinguishable from those of people who never had amyloid (bottom), showing that visual reads could be used to determine a patient’s amyloid is gone. [Courtesy of Eli Lilly.]
New Donanemab Trials
First, the news. Robert Alexander of Banner Alzheimer’s Institute in Sun City, Arizona, reported that API has picked donanemab as the next drug to test in the large Colombian kindred who carry the Paisa presenilin 1 mutation that causes AD in mid-life. The first drug tested in this cohort, the oligomer-specific antibody crenezumab, nudged cognitive, imaging, and fluid biomarkers in the right direction but missed statistical significance (Jun 2022 news; Aug 2022 conference news). Ever since dosing in this study ended, a year and a half ago, participants have been waiting for the next opportunity (Dec 2022 conference news).
That will be donanemab. Donanemab targets plaque and mops it up fast. This may be an advantage in dominantly inherited AD, where fibrils also accumulate fast. Alexander said the study will enroll both mutation carriers and noncarriers, so that participants need not find out their mutation status. Noncarriers will be on placebo throughout the trial, taking both a pill and an infusion. For carriers, the trial will have two stages. In part one, all carriers will receive donanemab, plus a placebo pill. Amyloid PET scans will be done at baseline, nine, and 18 months. Once a person’s plaque load has fallen below 11 centiloids, they will transition into the second part of the study. All carriers will move to part two by 18 months. “Essentially, we’re trying to get everyone to the same baseline,” Alexander said. Below 11 centiloids baseline, that is.
In part two, scientists will swap the placebo pill for an active small molecule in carriers. They will use a factorial design with four treatment groups. One group will be randomized to the active molecule alone, one to donanemab alone, one to both, and one to placebo only. Placebo controls will be used for both drugs, so participants will not know their group. The idea is to learn which treatment best staves off plaque resumption, biomarker change, and cognitive decline in the years after donanemab has removed participants’ baseline amyloid. This trial may also help answer the niggling question of whether continued donanemab treatment after plaque clearance would be beneficial.
Alexander did not discuss what type of small molecule might be used. Roche is evaluating a γ-secretase modulator, RG6289, in Phase 2a (Aug 2024 conference news). Despite efforts to reactivate BACE inhibitors for low-dose treatment to keep amyloid formation at bay, none have re-entered clinical trials as yet.
In his talk, Alexander also offered small updates on Trailblazer-Alz3. This secondary prevention trial has been in the works for three years (Jul 2021 news). It is now fully enrolled, with 2,600 participants across the U.S. and Japan. All were cognitively healthy at baseline, posting a CDR of zero. Their amyloid positivity was confirmed via plasma p-tau217 rather than amyloid PET, simplifying enrollment and lowering cost.
All participants will receive nine monthly infusions of donanemab or placebo. The primary outcome is the time until they develop mild cognitive impairment as per a CDR 0.5 on two consecutive tests. The trial needs to record 350 such events to reach statistical significance. According to Alexander, the trial will genotype APOE, with disclosure optional, and include amyloid and tau PET substudies, using florbetapir and flortaucipir. The trial is expected to run until 2027.
Finally, Eli Lilly is now evaluating donanemab in Down’s. People with this syndrome inherit three copies of the amyloid precursor protein, and have a 95 percent lifetime risk of developing AD dementia. Because of this, the new clinical staging criteria for AD consider DS “stage 0,” along with familial ADAD mutations (Aug 2023 conference news). DS advocates have been demanding that immunotherapy be studied and made available to their loved ones. Michael Rafii of the University of Southern California will lead this Phase 4 trial to test safety and efficacy of escalating doses of donanemab versus placebo.
For these patients, safety is a particular worry in the design of an immunotherapy trial. A recent paper from scientists led by Lei Liu at Brigham and Women’s Hospital, Boston, examined binding of lecanemab to postmortem tissue sections from 15 DS brains. Donors were between 43 and 68 years old when they died. In each case, lecanemab detected parenchymal plaques, but also bound strongly to blood vessels, suggesting the presence of extensive cerebral amyloid angiopathy, the main risk factor for ARIA (Liu et al., 2024; Aug 2023 conference news).
In Philadelphia, Donna Wilcock of Indiana University, Indianapolis, a co-author on the paper, added that people with DS have an exacerbated inflammatory response and white-matter changes at baseline (Aug 2023 conference news). “They have a much greater risk of ARIA. I’m very concerned,” Wilcock said. One solution could be to start immunotherapy earlier in these patients, before CAA has formed, she suggested.
How Does the Doctor Determine When to Stop Donanemab?
Aside from describing new trials, talks in Philadelphia shed light on questions researchers want answered. Lilly’s Emily Collins addressed how to know when patients on donanemab should get a second amyloid PET scan to determine if their plaque is gone. Analyzing Phase 3 trial data, she found the best predictor for that was baseline plaque load. People who had completely cleared plaque by six months had started with an average of 85 centiloids; people who cleared it by one year, 104 centiloids; and by 18 months, 116. Thus, a patient’s baseline load could provide a rough guideline for clinicians on when to check for amyloid negativity. Collins noted that amyloid-negative scans from the Trailblazer trials resemble those from ADNI participants who never had amyloid, suggesting that visual reads would suffice to determine clearance (image above).
Will determining amyloid clearance always require expensive PET scans? Research has advanced plasma p-tau217 as an alternative indicator of brain amyloid. This blood-based biomarker predicts presence of plaques with high accuracy, and in trials it dropped as plaques were cleared (Dec 2023 conference news; Aug 2024 conference story). Alas, it may not be that straightforward. In donanemab trials, plasma p-tau217 did not predict amyloid negativity, Collins reported, its AUC of 0.55 being little better than chance. This could be because plasma p-tau217 also reflects tangle load, which plaque clearance barely budged, Collins said.
Tangle Trend? In the Clarity AD trial, MTBR-243 rose more slowly in the CSF of patients on lecanemab (green) than in those on placebo (black). [Courtesy of Kristin Wildsmith, Eisai Inc.]
Can a Fluid Biomarker Track Tangles?
Right as immunotherapies are entering clinical care, the toolbox of fluid-based tau markers is expanding, and scientists are intensely interested in using them to track what immunotherapy does to tangle load, which correlates more strongly with cognition. In her talk, Kristin Wildsmith of Eisai presented evidence that lecanemab slows accumulation of neurofibrillary tangles. Wildsmith and colleagues, including Kanta Horie at Washington University, St. Louis, used cerebrospinal fluid MTBR-243 as a proxy for tangle formation. Previously, Horie had found that levels of this tau fragment, which spans the microtubule binding region, correlate closely and linearly with tau PET, unlike other AD markers, such as p-tau217 (Dec 2020 news; Aug 2023 conference news). Wildsmith believes the fragment sheds from tangles as they form, leaking into the CSF.
In the Clarity AD Phase 3 clinical trial, tau PET signals trended higher in people on placebo, hinting that lecanemab slows tangle accumulation. Could CSF MTBR-243 detect this? Wildsmith showed data from a subset of 167 people in the 18-month trial who had CSF samples at baseline and follow-ups. At the trial’s start, CSF MTBR-243 correlated tightly with the tau PET signal across all six Braak stage regions. By 18 months, CSF MTBR-243 had inched up 23 percent in the 83 people on placebo and 14 percent in the 84 people on lecanemab (image above). The difference was not statistically significant, which Wildsmith attributed to the small sample size.
She noted that the trajectories resembled what was observed using tau PET. The data strengthen evidence that MTBR-243 might eventually become a fluid biomarker for tangles (see also Aug 2024 conference news).
It’s Not Just Amyloid. Modeling from Eisai shows that amyloid and non-amyloid pathways both affect cognition. Amyloid pathways act early and gradually, non-amyloid late and fast. [Courtesy of Eisai.]
How Does Plaque Removal Relate to Cognition?
Another burning question is how amyloid clearance correlates to cognitive benefit at the level of individual participants. Many scientists have asked to see spaghetti plots of the data. In Philadelphia, Eisai did not show such data. Brian Willis of Eisai told the audience that such plots are not very informative because the relationship between plaque clearance and cognition is complicated. As disease worsens, factors other than amyloid, such as tangles and synapse loss, exert an ever-greater effect on memory (image above). The more plaque a person starts with, the further along in disease they already are and the faster their cognition declines from there because more non-amyloid processes contribute. At the same time, the more plaque a person starts with, the more amyloid immunotherapy removes, confounding clearance and cognition data. As a result, the overall relationship is murky, with individual-level outcomes appearing as “data clouds” instead of linear relationships, noted Eisai’s Michael Irizarry.
Exposure-Response. Integrating pharmacokinetics of amyloid accumulation and clearance, with the effect of both amyloid- and non-amyloid-driven processes on cognition, Eisai’s model predicts amyloid-driven progression to be linear only between 0 and 105 centiloids. [Courtesy of Eisai.]
Eisai scientists created an exposure-response model that takes confounding factors into account. Willis said the model uses data from Phase 2 and 3 lecanemab studies and their open-label extensions, comprising 4,575 amyloid PET scans and 8,456 CDR-SB scores. It also incorporates what is known about the pharmacokinetics of plaque buildup and degradation, and includes a function for non-amyloid-driven cognitive decline. This type of decline kicks in at plaque loads above 105 centiloids, the scientists calculated. Between 0 and 105 centiloids, plaque accumulation correlates roughly linearly with amyloid-driven disease progression, allowing scientists to model the exposure-response relationship within this range (image above).
When tested against Phase 3 trial data, the model predicted a slowing of cognitive decline of 0.43 points on the CDR-SB at 18 months, close to the actual value of 0.45. Extrapolating forward, the model predicted that three years of treatment would slow decline by 1.45 points; four years, by 2.15. Because there are no placebo-controlled data past 18 months, these predictions cannot be verified.
Willis said the model could be used to predict a given person’s immunotherapy result. For someone who started with 40 centiloids of plaque, 18 months of lecanemab would affect their CDR-SB by less than a quarter of a point—a tiny effect. However, because the person is at an early disease stage with slow decline, this would represent a 40 percent slowing of progression. For someone who started with 80 centiloids, 18 months of treatment would slow decline by half a point on the CDR-SB, which would represent only 31 percent slowing of progression. As with modeling from other antibody programs, the data emphasize the importance of treating early.—Madolyn Bowman Rogers and Tom Fagan
Ever since variants in the gene for TMEM106b were tied to frontotemporal dementia, Alzheimer’s, and other neurodegenerative diseases, this endolysosomal protein has been a head-scratcher for scientists. Its ability to surprise is exemplified by the discovery of fibrils spun of the protein—but there’s much more. According to findings presented at the Alzheimer’s Association International Conference, held July 27 to August 1 in Philadelphia, an AluYb8 retrotransposon is situated within its 3ˈ untranslated region. Geneticists spotted it by scouring long-read sequences in search of structural variants around the gene. The twist? This retroviral relic was found sitting in the risk-boosting variant. Cognitively resilient centenarians tended to carry the protective, transposon-free version.
Importantly, this wee element represents the tip of an iceberg. The scientists found myriad other repeat sequences and expansions stationed near the gene. Moreover, different combinations of structural variants occur in people of African ancestry.
How this mounting pile of genomic variations affects TMEM106b’s expression, proper function, and dysregulation is a new area ripe for study. In Philadelphia, Henne Holstege of Amsterdam University Medical Center proposed a potential mechanism linking age-related demethylation, TDP-43 aggregation, and TMEM106b’s awakening transposon with neurodegenerative pathogenesis.
TMEM106b burst onto the neurodegeneration scene almost 15 years ago, when geneticists identified common variants in the gene that either increased or decreased a person’s risk of FTD and ALS (Feb 2010 news; Aug 2012 news). Later, TMEM106b’s sphere of influence expanded to AD and other neurodegenerative diseases (Sep 2021 news).
What might it do? TMEM106b knockout mice displayed endolysosomal dysfunction, which was worse in mice that also lacked progranulin (Sep 2020 news). Just when scientists thought they were getting a handle on TMEM106b’s modus operandi, structural biologists joined the fray and unveiled TMEM106b fibrils lurking in brain samples from people with neurodegenerative diseases and in many cognitively normal people older than 50 (Apr 2022 news). Subsequently it appeared that TMEM106b’s propensity to fibrillize goads TDP-43 pathology, which itself underlies many cases of FTD/ALS as well as limbic predominant TDP-43 encephalopathy (LATE) (Jan 2024 news). Exactly how genetic variation at the TMEM106b locus relates to its fibrillization and how that might tempt other proteopathic culprits to follow suit remain big puzzles in the field.
For Holstege’s team, protective variants in TMEM106b surfaced as top genetic discriminators of sharp centenarians. Holstege heads the 100-plus Study, which tracks comprehensive molecular, genetic, neuropathological, and health attributes of people who have managed to remain cognitively healthy into their 11th decade of life (Holstege et al., 2018). The study includes some 480 participants. They sit for annual testing, and many donate their brains after they pass on.
So far, Holstege has found that at autopsy, these centenarians had less brain Aβ, tau, and TDP-43 pathology than did people who died at a younger age with AD. Crucially, they also enjoyed remarkable cognitive resilience in the face of whatever proteopathy they did have until shortly before death (Zhang et al., 2022; Zhang et al., 2023).
Endolysosomal Endurance. Centenarians sported protective variants, or lacked risky ones, in three genes encoding proteins needed in endolysosomes: progranulin, sortilin, and TMEM106b. [Courtesy of Holler et al., eNeuro, 2017.]
How did they fend off these age-related scourges? In Philadelphia, Holstege presented results from a genetic analysis. First, postdoc Niccoló Tesi asked if the centenarians carried protective variants versus variants tied to increased AD risk. For 85 percent of the AD GWAS hits Tesi looked at, the cognitively healthy centenarians had a higher frequency of protective and lower frequency of risk alleles, relative to middle-aged people with AD or their age-matched controls. As such, the centenarians also had lower polygenic risk scores, and were likelier to carry the protective ApoE2 allele and unlikelier to carry ApoE4. Notably, protective variants in a trio of endolysosomal genes—SORT1, GRN, and, you guessed it, TMEM106b—were among those most strongly overrepresented in the centenarians (Tesi et al., 2024).
Next, Holstege noted that several risk TMEM106b SNPs had popped up in GWAS for AD, FTD, and ALS, including the coding variant T185S. They all corresponded to the same haplotype. Yet single-nucleotide polymorphisms are only one part of the picture of genetic variation. Also important are structural variants, such as short and long tandem repeats, transposable elements, and methylated CpGs, the latter of which regulate gene expression.
Only long-read sequencing can detect these structural changes. Postdoc Alex Salazar performed this new type of analysis on the genomes of 250 people with AD and 251 resilient centenarians. Lo and behold, he found a 317-bp AluYb8 retrotransposon lurking within the 3ˈUTR of people who carried the SNPs corresponding to the risk-raising allele, but not the protective one. The allele infiltrated by the transposon was enriched in people with AD; the protective one in centenarians (image below).
Retroviral Interloper. An AluYb8 retrotransposon (pink) was found lurking within the risk-boosting haplotype of TMEM106b (top), not within the protective variant (bottom). [Courtesy of Henne Holstege, Amsterdam University Medical Center.]
How might this mobile element affect TMEM106b’s expression or function? In Philadelphia, Holstege told the audience that Alu elements are retrotransposons that make up a whopping 11 percent of the human genome. They use a copy-and-paste strategy—i.e., reverse transcription and insertion, respectively—to propagate themselves across the genome. This frivolous behavior can disrupt expression and regulation of nearby genes, such as TMEM106b. In response, methylation of surrounding CpG sequences has evolved to ground the jumping genes. As methylation is known to wane with age, Holstege proposed that the Alu element might become increasingly active, potentially messing with TMEM106b expression among those who carry the risk allele.
As evidence that these mechanisms might be at play, Holstege reported at AAIC that 52 CpG sites in and around the TMEM106b gene were significantly likelier to be methylated in people carrying the risk haplotype than in people carrying the transposon-free one. Furthermore, the risk haplotype includes 19 unique CpG sites, hinting that the genome evolved to put the kibosh on the nearby Alu element, Holstege said.
Connecting the dots between TMEM106b and TDP-43, Holstege pointed out that TDP-43 has been reported to bind and suppress Alu elements (Morera et al., 2019). Moreover, TDP-43 itself becomes increasingly demethylated with age in the brain, hampering TDP-43’s normal autoregulation (Koike, 2024). The resulting glut of TDP-43 could then lead to its accumulation and aggregation, ultimately sequestering it from performing its functions—Alu suppression among them.
Demethylate, Degenerate? With advancing age, TMEM106b demethylation might stir up the AluYb8 retrotransposon. TARDBP demethylation and TDP-43 aggregation might also awaken the transposon, as physiological TDP-43 suppresses it. [Courtesy of Henne Holstege, Amsterdam University Medical Center.]
Holstege noted that TDP-43 pathology is seen in people with AD, FTD/ALS, and LATE, but rarely in centenarians. Therefore, she proposed that age-related demethylation may lift the lid on TMEM106b’s transposon in two ways: one, via demethylating TMEM106b itself, and two, by demethylating the Alu silencer, TDP-43 (image below). The findings are posted on medRxiv (Salazar et al., 2023).
Just how an activated AluYb8 element in TMEM106b’s 3ˈUTR affects the protein’s expression, much less fibrillization, remains unclear, Salazar told Alzforum. Two other groups found the same Alu element in the risk allele. One, led by Michael Koob of the University of Minnesota in Minneapolis, discovered the insertion when sequencing the TMEM106b region in preparation to make mouse models (Rodney et al., 2024). The other, led by Michael Greicius of Stanford University, reported that carriers of the transposon variant had more TMEM106b in their plasma and CSF (Chemparathy et al., 2023). “The mechanism by which the insertion may impact TMEM106B levels remains uncertain,” they wrote, proposing that changes in the 3ˈUTR could affect protein binding, translation efficiency, even subcellular localization of the mRNA. “Identifying a clear-cut mechanism linking the insertion to increased TMEM106B protein levels is still required to confirm that this is the causal variant at the locus,” they added.
Beyond Alu
But wait, even that is not all. In Philadelphia, Salazar reported that the Alu element was but one of many structural variants riddling the TMEM106b region. “There’s a lot more going on in this region than previously understood,” he told the audience. Case in point, just upstream of TMEM106b’s transcriptional start site, Salazar discovered a large, 4.6-kilobase stretch housing a slew of transposable elements. In a lone centenarian, this transposon stretch was missing. Salazar also unearthed a string of tandem AC repeats located 3ˈ of the Alu element. This sequence was multi-allelic, ranging from 35 to 80 bp in length. In all, Salazar spotted more than 60 structural variants located in or around the TMEM106b gene.
How do these genomic hijinks relate to the single-nucleotide variants that tracked with disease risk in GWAS? Salazar examined all of these variants at once. He generated more than 1,000 “haplotype fingerprints,” one for each copy of the locus carried by all participants in the 100-plus cohort. In this way, he identified a new protective haplotype. It lacks the Alu element but has different combinations of other structural variants relative to the original protective allele. This variant combo had double the protective power of the original, he reported.
This diversity of variants occurs in a homogenous population, i.e., people of European ancestry living in the Netherlands. How might this TMEM106b locus look in other ancestries? To find out, Salazar integrated whole genome sequencing data from the Human Pangenome Reference Consortium. Among people of African ancestry, he discovered two novel haplotypes. One included the SNPs corresponding to the risk-promoting allele but its 3ˈUTR matched the protective haplotype devoid of the Alu element. The other was the opposite, suggesting recombination events had produced the hybrid alleles. How these new combinations influence disease risk has yet to be investigated. In line with TMEM106b’s penchant for complexity, the researchers, even since AAIC, have unearthed even more unique haplotypes. They will describe them in an upcoming preprint.
Finally, in a separate AAIC talk, Tesi expanded the scope of the analysis beyond the tiny neighborhood of TMEM106b. Wondering if structural variants positioned near GWAS hits might be the actual drivers of disease, Tesi used long-read sequencing to explore the interplay between large such variants and AD risk SNPs in the 100-plus cohort. He identified 27,404 structural variants averaging 763bp in length, including more than 7,600 transposable elements and 17,500 tandem repeats. Among 85 GWAS hits, 37 were closely linked with a structural variant. Some, including ADAM10, SLC2A4RG, and CD2AP, were tied to more than one. For three genes—APOC1, SPI1, and ABCA7—the researchers found a significant association between the structural variant and AD risk within their cohort.
All told, the preliminary findings hint that structural variants might contribute to AD risk imparted by those genes, Tesi said. Pulling the most important pathways out of this genomic melee will take some time.—Jessica Shugart
Alzheimer’s is a multifaceted disease, particularly in its sporadic, late-onset form. Myriad factors—genetics, environment, cardiovascular health, metabolism, and inflammation—contribute to a decades-long process. Generating animal models that accurately reflect this neurodegenerative train wreck is a tall order, but scientists have long wanted to move beyond the flawed overexpression mice that they have relied upon for years. Funded by the National Institute on Aging in 2016, the MODEL-AD consortium rose to meet this challenge. Since then, scientists at Indiana University in Indianapolis, the University of Pittsburgh in Pennsylvania, The Jackson Laboratory in Bar Harbor, Maine, the biomedical research nonprofit Sage Bionetworks in Seattle, and University of California, Irvine, have used human datasets to guide development of dozens of mouse models of late-onset AD. Of the 80 strains made by consortium scientists so far, 74 are available to order, and colonies of six others are still being raised. Meanwhile, more than a dozen others are being developed.
Some express humanized versions of Aβ and tau, others express AD risk variants in ApoE, TREM2, and many other genes identified in GWAS and exome sequencing studies. Scientists can use these lines not only to study disease mechanisms, but also to test emerging therapeutics in preclinical pipelines.
Consortium scientists are eager for the field to start using models that more accurately capture the underlying biology of AD, Adrian Oblak of Indiana University told Alzforum. She co-leads that institution’s MODEL-AD core along with Bruce Lamb.
MODEL-AD aims to create mouse models that reflect different biological subtypes of AD that have been found in people, in hopes of improving drug discovery and tailoring treatments to different forms of AD (Jan 2021 news; Dec 2023 news). In 2022, Alzforum reported that some of the new models displayed subtler, potentially more translatable, phenotypes than mice overexpressing APP and PS1 genes saddled with familial AD mutations (Nov 2022 news). Since then, MODEL-AD has generated and characterized more models. Researchers presented the newest findings at AAIC in Philadelphia last July and in a coordinated special issue of Alzheimer’s & Dementia.
Just how well are these models catching on in the research community? Between 2018 and July of this year, 88 studies using MODEL-AD mice were logged into the Mouse Genome Informatics database, a number that is likely an underestimate, Jackson Lab’s Michael Sasner told Alzforum. Nevertheless, this pales in comparison to the number of studies using classic overexpression amyloidosis models introduced just after the turn of the century. PubMed identified 1,122 studies using the 5xFAD mouse strain alone since 2018, while 2,034 studies used APP/PS1 mice and 548 used 3xTg.
Still, Sasner expects more studies with MODEL-AD mice to come down the pike soon. Most of those published so far have used the ApoE and TREM2 models that were created in the early years of the consortium, but more recently, requests for new models, particularly those carrying a combination of AD risk variants, have risen substantially, Sasner said. While Jackson Labs, which is a nonprofit research institute, fielded orders for 500 MODEL-AD mice in 2018, that number had quadrupled by 2022, and more than 2,000 have already been ordered in 2024. Academic labs were the first to start using the mice, but now more than half go to biotech/pharma companies, he said.
Sasner pointed out that switching to a new mouse strain, let alone publishing the findings, is neither straightforward nor quick, since researchers have to phase out the strains they’re already working with, and rear new mice until they are old enough for AD-related experiments. Sasner said that older mice may become available for some of the strains.
Several of the new models—for example, LOAD1 and LOAD2 strains—are curated in Alzforum’s research model database, with more to be added soon.
LOADed with Variants
How well do these new models mimic late-onset AD? In Philadelphia, Oblak presented synaptic findings on LOAD2 mice. This triple mutant line carries two copies of humanized APOE4, the R47H AD risk variant in the endogenous mouse Trem2, and a humanized Aβ sequence within the mouse APP gene.
This strain is one of three late-onset AD “base models” generated by the IU MODEL-AD center. The others are LOAD1, which carries humanized ApoE4 and R47H-TREM2, and the newer LOAD3, which carries humanized APP and ApoE4 along with a humanized tau gene of the H1 haplotype. This strain is still being bred to sufficient numbers, and is not yet available .
Previously, Oblak had reported that by 18 months of age, LOAD2 mice had no amyloid plaques, mildly activated microglia and astrocytes, and barely any neuronal loss. However, LOAD2 mice fed high-fat chow that mimics the cardiovascular stress provoked by a typical Western diet did lose a small but significant number of neurons. This tracked with elevated plasma NfL, a marker of neuronal damage, and flagging performance on touch screen-based mouse memory tests (Jan 2024 news).
At AAIC, more synaptic snafus emerged. In collaboration with Nick Seyfried’s lab at Emory University in Atlanta, the Oblak lab compared the proteomic signatures of LOAD2 mouse brains with previously identified AD signatures (Feb 2022 news). Similar modules of dysregulated proteins correlated with disease in both. For example, among 95 post-synaptic proteins, the same ones were downregulated in LOAD2 mice and in people with AD. These disease-related correlations were strongest in mice on a high fat diet, but were still significant in those fed normal chow.
In further experiments with LOAD2 mice fed a regular diet, both the post-synaptic marker PSD95 and the pre-synaptic marker SV2A were dramatically reduced in hemi-brain extracts of 12-month-old LOAD2 mice relative to controls. However, when Oblak zoomed in on synapses, she was surprised to find that the LOAD2 terminals had almost twice as much SV2A as did control synapses. This could suggest a compensatory uptick in synaptic signaling, she told Alzforum. Possibly supporting this interpretation: the composition of receptor subunits within both NMDA and AMPA receptors was skewed in the LOAD2 mice. Their NMDA receptors contained more NR2B subunit, which is known to promote pro-apoptotic signaling, whereas their AMPA receptors contained a glut of GluA2 relative to GluA1 subunits, a shift that might lead to excitotoxicity, Oblak suggested.
Microglia seemed to have taken notice of these synaptic imbalances. In 18-month-old LOAD2 mice, Oblak found a dramatic uptick in microglia loaded up with synaptic material.
These synaptic changes appeared to have functional consequences. While control mice demonstrated a dip in long term potentiation—a measure of synaptic plasticity—with age, LOAD2 mice already had weak LTP at four months, which stayed low as they got older.
“Overall, these changes imply a reorganization of excitatory synaptic transmission in the absence of amyloid and tau pathologies,” Oblak said. She believes these mice model the inflammatory and synaptic problems that occur in the early stages of AD, before Aβ and tau pathology have inundated the brain, and invites labs across the field to use them.
Piling On More Risk
At AAIC, Jackson Lab’s Gregory Carter laid out current developments in using CRISPR to plant AD risk variants into LOAD1, and, more recently, the LOAD2 background. Previously, Carter and colleagues had reported 11 new mouse strains expressing coding variants in 11 different genes (image below and Sasner et al., 2024). The scientists used transcriptomics to compare how these variants affected biological pathways tied to AD in the AMP-AD cohort (Wan et al., 2020). They found, for example, that the A1527G variant in ABCA7 provoked expression changes in groups of genes involved in immune responses, cell cycle, myelination, and cellular stress responses, which corresponded to AD-related modules found in people. On the other hand, variants in SORL1 and PLCG2 genes skewed modules of neuron-related pathways.
Added Risk. Coding risk variants were introduced into LOAD1 mice, which already carry ApoE4 and TREM2-R47H risk alleles. [Courtesy of Sasner et al., 2024.]
Carter chose these variants based on five criteria: how well the wild-type allele is conserved between people and mice, predicted pathogenicity, well-replicated association findings, how common the variants are, and predicted biological function (image below). He included the last so the scientists would generate models for a variety of pathways involved in LOAD. Jackson Lab’s Sasner described this initial batch of coding variants as “low-hanging fruit,” in that there was little ambiguity about the identity of the causal risk gene, and the effect each variant likely had in mice and in people.
To Model, or Not? Starting with coding (C) and noncoding (NC) AD risk variants identified in human studies (top), scientists used five criteria (left) to predict if variants would behave in mice as in people. Those that met all five were CRISPRed into LOAD2 mice. [Courtesy of Gregory Carter, Jackson Lab.]
Carter used the same criteria to select which noncoding variants to model. Most GWAS hits reside in promoters, enhancers, and introns, from where they typically influence expression of nearby genes. Because human and mouse cells can regulate gene expression quite differently, it is trickier to model the effects of noncoding variants. What’s more, often it’s not even clear which gene is affected. Consequently few of these variants pass all of five criteria.
At AAIC, Carter showcased nine variant lines that made the cut (image below). Five have a single-nucleotide polymorphism in a promoter, enhancer, or intron of BIN1, CD2AP, EPHA1, PTK2B, SCIMP. One lacks a 2kb enhancer near the microglial ADAMts4 gene. Another has had the third exon in the IL1-RAP gene removed to mimic the effect of a SNP in a non-conserved intron in the human ortholog. This second batch also included two coding variants—the D57N missense mutation in PTPRB, and the Y213 stop-gain variant in IL-34. All these risk variants were introduced into the LOAD2 line and are available from Jackson Labs.
Beyond Coding. A new batch of mouse strains, on the LOAD2 background, carry noncoding AD risk variants introduced into promoters, enhancers, and introns. [Courtesy of Greg Carter, Jackson Lab.]
Once these mice were 1 year old, the researchers compared their transcriptomes with those of LOAD2 controls and human postmortem brain samples from the AMP-AD cohort. Focusing on 19 “biodomains”—biological processes such as immune response, synaptic function, lipid metabolism, mitochondrial function, APP processing, and apoptosis—they pinpointed pathways specifically influenced by each variant. For example, they found that the EphA1 variant dramatically altered genes involved in apoptosis, while the PTRB variant skewed expression of genes involved in APP metabolism—specifically, those related to Aβ clearance.
These new mouse strains move consortium scientists closer to their overall goal of creating a catalog of models that affect specific disease-related biological pathways, Carter said. “If you have a targeted therapeutic that you would like to test in vivo, we can then match the most suitable mouse models with that candidate,” he added.
Scientists from academia or biotech can purchase mice from Jackson Labs to run this type of testing on their own. Or they can have their candidate therapeutic tested in the NIA-funded preclinical testing core that is part of MODEL-AD. Led by Paul Territo at Indiana University and Stacey Rizzo at the University of Pittsburgh, the core evaluates selected drug candidates for pharmacokinetics and dynamics, in-vivo target engagement in relevant disease models, cognitive/behavioral effects, and potential disease-modification. As a proof of concept, they have applied their testing pipeline to drugs that have already gone through clinical trials, including the BACE1 inhibitor verubecestat and the anti-seizure drug levetiracetam (Oblak et al., 2022; Onos et al., 2022).
Interested? Investigators can apply via the STOP-AD Compound Submission Portal. A steering committee selects promising compounds, considering both the available mechanistic data in support of the drug, and how well it pairs with MODEL-AD mouse strains. At AAIC, Territo said they plan to put one or two compounds through this process per year. Sasner told Alzforum that about 10 drugs are already in the pipeline, though he would not disclose what they are. Once testing is complete, compounds, sponsors, and all data will be openly available, he said.
Modeling Protection
At the University of California, Irvine, another MODEL-AD center headed by Frank LaFerla, Andrea Tenner, and Kim Green, has produced its own batch of mice. Their menagerie includes human Aβ knock-ins, human tau knock-ins, and strains harboring both coding and non-coding AD risk variants (image below). In Philadelphia, Green presented findings from two of these. One carries a coding variant in the gene for the ABCA7 membrane transporter, while the other has the R154S Christchurch variant (aka R136S) in mouse ApoE.
More Risk Variants. UCI has “platform lines” that model Aβ and tau pathology, as well as lines incorporating AD risk variants. [Courtesy of Kim Green, UCI.]
The human ABCA7 variant is a V1599M mutation that came up in exome sequencing and is predicted to be deleterious. However, because it is so rare, it has been impossible thus far to definitively tie it to AD risk, Green told Alzforum. Using CRISPR-Cas9, the scientists introduced the corresponding mouse mutation, V1613M, into both copies of the mouse gene.
First author Claire Butler and colleagues wanted to know how this variant would affect amyloid plaques, so they initially crossed the V1613M strain to 5xFAD mice. The offspring reduced plaque load, dystrophic neurites, and plasma NfL. This was unexpected, Green said, because ABCA7 deficiency increases plaques. Rather than loss of function, the findings imply that this ABCA7 variant causes a protective gain of function. A recent paper tied this protection to reduced Aβ production brought about by altered APP trafficking (Butler et al., 2024).
Green said they are introducing the ABCA7-V1613M mutation into MODEL-AD humanized Aβ knock-in strains. This will teach them how it behaves under more physiological levels of pathology; 5xFAD mice overexpress mutant APP and produce copious amounts of Aβ.
While the protective effect of ABCA7-V1613M came as a surprise, the ApoE Christchurch mutation is well-known for fending off Aβ-induced tau pathology, and cognitive decline, in a carrier of the highly pathogenic Paisa mutation in presenilin-1 (Sep 2022 conference news). To try to understand this, Green used CRISPR to introduce the variant into the mouse ApoE gene, and crossed these ApoE-Ch mice to 5xFAD and to PS19 f tauopathy mice. Graduate student Kristine Tran and colleagues found that ApoE-Ch assuaged plaques in the former, but not tau pathology in the latter.
What is going on at the cellular level here? The scientists used single-cell and spatial transcriptomics to investigate the transcriptional state of microglia and astrocytes adjacent to Aβ plaques or tau-laden neurons. Essentially, they found that the Christchurch variant promoted microglia to adopt a disease-associated signature (DAM) around plaques (Jun 2017 news). In contrast, the variant put the kibosh on this DAM response in PS19 mice, and dampened a disease-associated astrocyte signature as well (Jan 2017 news; Habib et al., 2020).
The findings are reported in a preprint on BioRxiv (Tran et al., 2024). They jibe with previous studies implying DAM responses are beneficial in the context of amyloid, but harmful in the context of tau pathology. By promoting the former and inhibiting the latter, ApoE-Ch bolsters “good” microglial responses while scuppering the “bad” ones, Green suggested.
Situational Switch. The ApoE-Ch variant enhances (red arrows) disease-associated signatures around Aβ plaques (left), but restrains them (blue arrows) in the presence of tau aggregates (right).
Exactly how Christchurch tailors microglial responses depending on the pathological environment is the next question, Green told Alzforum. He thinks that more than any other discovery, this ApoE variant could point the way to therapeutics that promote beneficial glial responses to AD pathology, regardless of disease stage.
The findings dovetail with those reported from other ApoE-Ch mouse models of amyloidosis and tau pathology (Dec 2023 news). These ApoE-Ch mice are being crossed with humanized Aβ and tau knock-in models to study the effects of the variant in a more physiological context.—Jessica Shugart
Butler CA, Mendoza Arvilla A, Milinkeviciute G, Da Cunha C, Kawauchi S, Rezaie N, Liang HY, Javonillo D, Thach A, Wang S, Collins S, Walker A, Shi KX, Neumann J, Gomez-Arboledas A, Henningfield CM, Hohsfield LA, Mapstone M, Tenner AJ, LaFerla FM, Mortazavi A, MacGregor GR, Green KN.
The Abca7V1613M variant reduces Aβ generation, plaque load, and neuronal damage.
Alzheimers Dement. 2024 Jul;20(7):4914-4934. Epub 2024 Mar 20
PubMed.
Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A, Schwartz M.
Disease-associated astrocytes in Alzheimer's disease and aging.
Nat Neurosci. 2020 Jun;23(6):701-706. Epub 2020 Apr 27
PubMed.



















 Jérôme Braudeau
Jérôme Braudeau  Suzanne Schindler
Suzanne Schindler 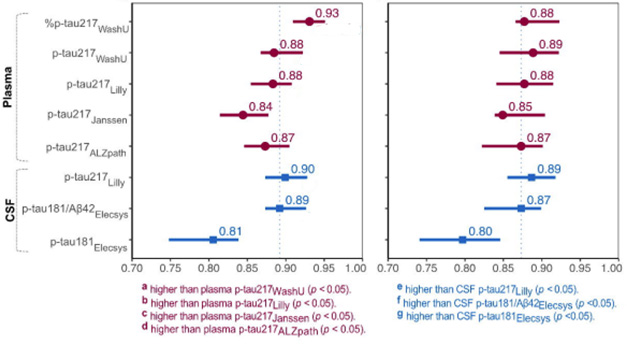




























Comments
University of California, San Diego
This study combines data from a group of primary care practices and from the BioFINDER study to analyze the value of plasma %p-tau217 (measured as part of the C2N Precivity2 assay and compared against the p-tau217 percentage occupancy alone) in improving diagnostic accuracy, with a prespecified single cutoff, or range of cutoffs, that optimized sensitivity and specificity at the expense of a “gray area” of indeterminate results. Plasma %p-tau217 showed great promise in both settings. The results are highly informative and promising for demonstrating feasibility and utility of plasma %p-tau217 as an aid to AD diagnosis.…More
The characteristics of patients who had clinical screening/assessment and then biomarker evaluation were remarkably similar in primary care and in the BioFINDER secondary care cohort, including age, sex and APOE e4 genotype. The last of these may have been an important aspect that contributed to the high diagnostic accuracy/predictive value in the primary care cohorts. There is a higher prevalence of APOE4 in Northern European countries than elsewhere, and it would be interesting to see comparable studies from countries or populations where APOE4 is less common. The primary care cohorts had higher rates of comorbidity than the secondary clinic cohort, which is more representative of a general population.
The accuracy of the primary care clinicians’ suspicion of AD was only 58% compared to %p-tau217, therefore the blood-based biomarker could make a difference to clinical judgment and have an impact on appropriateness of referrals. It would be interesting to know how (and whether) the primary care physicians diagnosed the categories of subjective cognitive decline, MCI or dementia, and what additional history or data may have been available besides the MMSE.
Also, the percentage of patients with SCD, MCI, and AD in primary care who were biomarker positive were not reported. Methods of initial history and cognitive screening could have an impact on how often a primary care physician might order a screening blood test, and the PPV will be lower if it is ordered in populations with lower prevalence APOE e4 or among people with low pretest likelihood of having AD. There may be room for improvement beyond the MMSE as a screening test in primary care, to increase the accuracy of pretest suspicion of AD.
It will be interesting to see further studies of how primary, and secondary, care clinics use and interpret blood-based biomarkers, especially in populations with diverse ethnic populations and among older patients, where multi-etiology dementia is common. How results are communicated to patients and whether blood tests can serve as stand-alone biomarkers that may rule a patient in or out as a candidate to receive therapy, will require further study. The present study makes important advances in addressing some of these questions.
View all comments by Douglas GalaskoMake a Comment
To make a comment you must login or register.