Nixing TMEM106b Fans the Flames of Progranulin Deficiency
Quick Links
For carriers of a pathogenic mutation in the progranulin gene, variants in another gene—TMEM106b—strongly influence when, and even if, they will develop frontotemporal dementia. A trio of recent papers, published in August and September in EMBO Reports, details how these two disease genes might together bring about neurodegeneration.
- Three labs generated TMEM106b, GRN double-knockout mice.
- Lysosomal dysfunction, gliosis, TDP-43 accumulation, and neurodegeneration were worse in the double knockouts than in single-KO mice.
- Double KOs developed motor deficits not seen in single KOs.
The papers also clear up discrepancies dogging previously published TMEM106b knockouts. The three studies reported that, compared to mice lacking either gene alone, double knockouts developed more severe lysosomal dysfunction, neuroinflammation, and TDP-43 deposits. Debilitating motor deficits only emerged in the double knockouts. Animals that expressed even a smidgen of TMEM106b due to an incomplete knockout of the gene fared better initially, but still succumbed to motor neuron degeneration as they got older. The findings suggest that a proper balance between the two lysosomal proteins is critical for neuronal health.
TMEM106b emerged as an FTD risk gene a decade ago, when two common variants in the gene were tied to increased risk of the disease (Feb 2010 news on Van Deerlin et al., 2010; Cruchaga et al., 2011). The risk variants were found to boost expression of TMEM106b while suppressing expression of progranulin, and this implied that the balance between the two lysosomal proteins tunes disease risk (Aug 2012 news; Oct 2017 news).
Subsequent studies found protective TMEM106b variants that lowered risk of disease in carriers of pathogenic mutations in progranulin or C9ORF72 (Gallagher et al., 2014; van Blitterswijk et al., 2014; Dec 2018 conference news). These variants resulted in lower TMEM106b protein levels and elevated progranulin (Nicholson et al., 2013; Finch et al., 2011).
Two initial reports of TMEM106b knockout mice described relatively mild phenotypes such as gliosis and subtle lysosomal changes (Nicholson et al., 2018; Jul 2017 news). Led by Rosa Rademakers of the Mayo Clinic in Jacksonville, Florida, and Stephen Strittmatter of Yale University in New Haven, Connecticut, both relied on a so-called gene-trap approach, in which a LacZ reporter gene, along with an artificial splice acceptor site, is inserted into intron four to disrupt the TMEM106b gene. This derails proper transcription, leading to expression of the LacZ reporter instead of TMEM106b. In a double knockout generated by the Strittmatter lab, loss of TMEM106b even appeared to ameliorate detrimental phenotypes in progranulin nulls.
Alas, controversy arose when researchers led by Markus Damme of Christian Albrechts University in Kiel, Germany, developed two additional TMEM106b knockout strains—one using a gene trap followed by Cre-mediated excision of the gene to remove it, and the other using CRISPR to edit out a section of the gene. In contrast to the earlier reports, these single knockouts had severely swollen lysosomes choking motor neuron axons (Mar 2020 news). The Damme lab knockouts also had slight deficits in eye-blink reflexes and whisker movement, and they were shaky on their feet.
At the time, Damme and colleagues suggested that the milder phenotypes reported by other groups may have stemmed from incomplete takedown of TMEM106b expression with the gene trap approach. Leaky expression of the full-length gene can occur when the splicing machinery occasionally skips over the artificial splice acceptor site.
Would a complete knockout of the TMEM106b gene protect GRN knockouts? Apparently not. The three new papers in EMBO Reports describe phenotypes of TMEM106b/GRN double knockouts. Rademakers and Mayo Clinic colleague Dennis Dickson headed one study, published August 5. They used the gene-trap method. Another, published August 10 and led by Fenghua Hu at Cornell University in Ithaca, New York, used CRISPR to delete a portion of the TMEM106b gene. The third study was led by Anja Capell at the German Center for Neurodegenerative Diseases in Munich, and published September 14. Damme is second author on the study, which knocked out TMEM106b by gene trap followed by Cre deletion. All crossed their respective TMEM106b knockouts with progranulin knockouts.
All three studies report that the double knockouts had motor deficits, which were either absent or much milder in mice missing either gene alone. Capell and Hu saw severe motor problems by 4 months of age. The mice were ataxic, weak on their hind limbs, and unstable. They shook, and clasped their hindlimbs when picked up by the tail—an abnormal reflex seen in mice with lesions in the spinal cords, cerebella, basal ganglia, and neocortices. These double knockouts had to be euthanized between 4 to 5.5 months of age, when their deficits became debilitating.
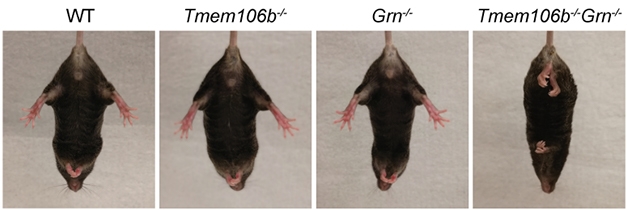
Double Clasp. Only Tmem106b/PGRN double-knockout mice abnormally clasped their hindlimbs when suspended upside down. [Courtesy of Feng et al., EMBO Reports, 2020.]
In Rademakers’ group, motor deficits emerged in double KOs at around 7 months, and the mice became paralyzed by 11 to 12 months. First author Xiaolai Zhou and colleagues found that these mice retained 5 to 10 percent residual expression of the full-length TMEM106b protein, and think this leaky expression explains the delay in disease phenotypes relative to the double knockouts described in the other two studies. Indeed, when Zhou and colleagues used CRISPR to make yet another TMEM106b knockout strain and crossed that one to progranulin knockouts, motor problems surfaced at 4.5 months, followed by paralysis and death at 7 months.
In a joint comment to Alzforum, Strittmatter and Hideyuki Takahashi, also from Yale, confirmed that they, too, had observed residual TMEM106b expression in their TMEM106b knockouts, which were also made using the gene-trap method. “The partial rescue phenotype we observed for TMEM106B reduction in Grn-/- mice at young-adult ages appears to depend on the allele being a hypomorph as opposed to a complete null,” they wrote. With the various complete null TMEM106B mice generated in the new studies, it is clear that complete loss of both genes becomes lethal by 5 months, they noted (see comment below). They added that subsequent experiments with their double knockouts revealed that, like Rademakers’ mice, motor deficits emerged months later.
All three studies reported severe neuropathology in the double knockouts. The Cornell and Munich groups found gliosis in the spinal cord and in multiple regions of the brain by 4 months of age. Levels of lysosomal enzymes soared, and autophagosomes and their cargo built up. What’s more, while TDP-43 inclusions were scarce in single knockouts, they were abundant in mice missing both TMEM106b and progranulin genes.
Gene expression analyses meshed with the neuropathology. Genes involved in microglial activation, autophagy, and lysosomal digestion were dramatically upregulated in brain and spinal-cord lysates from the double knockouts compared to single knockouts or wild-type mice, while genes involved in myelination were turned down.

Bad Bubbles. Autophagosomes (arrows) and lysosomes (arrowheads) accumulate in a spinal cord section of a TMEM106b/GRN double-knockout mouse. The “a” denotes a cross-sectioned axon. [Courtesy of Feng et al., EMBO Reports, 2020.]
Many of the same pathological characteristics, including lysosomal and autophagic dysfunction, gliosis, and TDP-43 inclusions, emerged in Rademakers’ double-knockout mice as well, albeit at 11 to 12 months. At this middle-aged stage, the researchers also detected myelin loss and profound motor neuron degeneration in the spinal cord.
The new studies explain discrepancies among the mouse models and suggest that a total loss of both TMEM106b and progranulin spells trouble for neurons. Even so, it remains unclear what the findings reveal about human disease. After all, the protective TMEM106b variants reduce its expression.
“It is critical to note that none of these TMEM106B alleles on the Grn-/- background accurately model human FTLD-GRN with TMEM10B variants,” noted Strittmatter and Takahashi. “In such clinical cases, there is only 50 percent reduction of PGRN expression, and the risk variants for the TMEM106B locus reportedly cause moderate changes in expression. Thus, continued investigation of GRN interaction with TMEM106B in neurodegeneration is required.”—Jessica Shugart
References
News Citations
- Genetics of FTD: New Gene, PGRN Variety, and a Bit of FUS
- FTD Risk Factor Confirmed, Alters Progranulin Pathways
- FTD Variant Alters Chromatin, Boosts Expression of TMEM106b
- 11th ICFTD Meeting in Sydney Sorts Out Clinical Subtypes
- TMEM106B and Progranulin Duke It Out at the Lysosome
- Without TMEM106B, Lysosomal Traffic Jams Wreak Havoc in Neurons
Paper Citations
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL, Ferrer I, Lladó A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IR, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, Decarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tuñón MT, Martínez MC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, Trojanowski JQ, Lee VM. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010 Mar;42(3):234-9. PubMed.
- Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, Goate A. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011 May;68(5):581-6. PubMed.
- Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, Rohrer JD, Halliday G, Van Broeckhoven C, Seilhean D, Shaw PJ, Frosch MP, Alafuzoff I, Antonell A, Bogdanovic N, Brooks W, Cairns NJ, Cooper-Knock J, Cotman C, Cras P, Cruts M, De Deyn PP, DeCarli C, Dobson-Stone C, Engelborghs S, Fox N, Galasko D, Gearing M, Gijselinck I, Grafman J, Hartikainen P, Hatanpaa KJ, Highley JR, Hodges J, Hulette C, Ince PG, Jin LW, Kirby J, Kofler J, Kril J, Kwok JB, Levey A, Lieberman A, Llado A, Martin JJ, Masliah E, McDermott CJ, McKee A, McLean C, Mead S, Miller CA, Miller J, Munoz DG, Murrell J, Paulson H, Piguet O, Rossor M, Sanchez-Valle R, Sano M, Schneider J, Silbert LC, Spina S, van der Zee J, Van Langenhove T, Warren J, Wharton SB, White CL 3rd, Woltjer RL, Trojanowski JQ, Lee VM, Van Deerlin V, Chen-Plotkin AS. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014 Mar;127(3):407-18. Epub 2014 Jan 19 PubMed.
- van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014 Mar;127(3):397-406. Epub 2014 Jan 3 PubMed.
- Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R, Hsiung GY, Mackenzie IR, Finger E, Boeve BF, Ertekin-Taner N, Graff-Radford NR, Dickson DW, Rademakers R. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem. 2013 Jun 6; PubMed.
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M, Forloni G, Kertesz A, Knopman DS, Uitti R, White CL, Caselli R, Lippa C, Bigio EH, Wszolek ZK, Binetti G, Mackenzie IR, Miller BL, Boeve BF, Younkin SG, Dickson DW, Petersen RC, Graff-Radford NR, Geschwind DH, Rademakers R. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011 Feb 1;76(5):467-74. PubMed.
- Nicholson AM, Zhou X, Perkerson RB, Parsons TM, Chew J, Brooks M, DeJesus-Hernandez M, Finch NA, Matchett BJ, Kurti A, Jansen-West KR, Perkerson E, Daughrity L, Castanedes-Casey M, Rousseau L, Phillips V, Hu F, Gendron TF, Murray ME, Dickson DW, Fryer JD, Petrucelli L, Rademakers R. Loss of Tmem106b is unable to ameliorate frontotemporal dementia-like phenotypes in an AAV mouse model of C9ORF72-repeat induced toxicity. Acta Neuropathol Commun. 2018 May 31;6(1):42. PubMed.
Further Reading
Primary Papers
- Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC, Parsons TM, Castanedes-Casey M, Phillips V, Librero AL, Kurti A, Fryer JD, Bu G, Lutz C, Dickson DW, Rademakers R. Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep. 2020 Oct 5;21(10):e50197. Epub 2020 Aug 5 PubMed.
- Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J, Katz II, Yu H, Xiong W, Hu F. Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep. 2020 Oct 5;21(10):e50219. Epub 2020 Aug 10 PubMed.
- Werner G, Damme M, Schludi M, Gnörich J, Wind K, Fellerer K, Wefers B, Wurst W, Edbauer D, Brendel M, Haass C, Capell A. Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep. 2020 Oct 5;21(10):e50241. Epub 2020 Sep 14 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Yale University School of Medicine
We are delighted that the three TMEM106B/PGRN double knockout studies from Zhou et al., Feng et al., and Werner et al. have further clarified the genetic interactions between TMEM106B and GRN. Full deletion of both genes clearly causes a synthetic lethal phenotype with motor dysfunction. The new results are distinctly different from our previous publication on this topic in 2017, which documented normalization of Grn-/- proteomic changes and reduced retinal degeneration in young TMEM106B/PGRN double gene-targeted mice.
Based on the report from the Rademakers laboratory of a TMEM106B gene trap line, we have examined the TMEM106B gene trap line that we used. Indeed, we confirm the Zhou et al. finding that TMEM106B gene trap mouse brain has about 5 percent residual full-length TMEM106B protein. This is presumably due to exon-skipping over the gene trap insertion during mRNA processing. In our case, this was observed by IP-western blot analysis, and confirmed by mass spectrometric peptide identification. We did not include a Cre recombinase cross to fully remove any TMEM106B coding exon (as was done in the Werner et al. paper), so the possibility of a spliced, full-length TMEM106B transcript always existed.
Thus, the partial rescue phenotype we observed for TMEM106B reduction in Grn-/- mice at young-adult ages appears to depend on the allele being a hypomorph as opposed to a complete null. With the various complete null TMEM106B mice generated in the new studies, there is clearly a synthetic lethal phenotype by 4-5 months.
Moreover, while the hypomorphic TMEM106B allele rescues some Grn-/- phenotypes at the 4- to 7-month time window, we have observed a severe, double-knockout motor phenotype beginning at 11 months that causes death. The latter is consistent with the description of gene trap hypomorphic mice in Zhou et al. (as opposed to early death in the CRISPR null line) and closely resembles the phenotype of 4- to 5-month-old, double-knockout mice with the complete null alleles.
It appears that certain Grn-/- phenotypes have a non-linear and biphasic dependence on reducing TMEM106B levels, and it is now clear that a full double knockout yields lethality.
It is critical to note that none of these TMEM106B alleles on the Grn-/- background accurately models human FTLD-GRN with TMEM10B variants. In such clinical cases, there is only 50 percent reduction of PGRN expression, and the risk variants for the TMEM106B locus reportedly cause moderate changes in expression. Thus, continued investigation of GRN interaction with TMEM106B in neurodegeneration is required.
—Hideyuki Takahashi is a co-author of this comment.
University of Kiel
As a minor addition to the comment: The cre-mediated Tmem106b full knockout used for generating the GRN/TMEM106B double knockout of the DZNE study was published before and was generated in the lab of Christian Haass. This strain was analyzed in parallel to the CRISPR-mediated Tmem106b knockout in my group in collaboration with Christian Haass and Anja Capell and, importantly, both showed a phenocopy and drastically enlarged vacuoles positive for lysosomal marker proteins.
References:
Lüningschrör P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D, Wanner R, Lüllmann-Rauch R, Wefers B, Wurst W, D'Hooge R, Uchiyama Y, Sendtner M, Haass C, Saftig P, Knöll B, Capell A, Damme M. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 2020 Mar 10;30(10):3506-3519.e6. PubMed.
Make a Comment
To make a comment you must login or register.