FTD Variant Alters Chromatin, Boosts Expression of TMEM106b
Quick Links
Like mysterious signals emanating from the vastness of genomic space, some variations rise above the din of genome-wide association studies (GWAS). For most of these statistical blips, scientists can only guess how they influence disease. Now, Alice Chen-Plotkin at the University of Pennsylvania in Philadelphia and colleagues may have a solid explanation for one them, a single-nucleotide polymorphism near the TMEM106b gene. Several SNPs near the gene correlate with higher odds of getting frontotemporal dementia, but which ones directly increase those odds, or just lie near other variants that do, has been unclear. Reporting in the American Journal of Human Genetics on October 19, the researchers narrowed the list of variants down to one—rs1990620. Lying near a well-documented FTD GWAS variant, rs1990620 boosts expression of TMEM106b, not of any other gene, in the brain and in immune cells.
- Scientists find a functional SNP near a TMEM106b GWAS variant.
- The SNP boosts TMEM106b expression by promoting binding of the chromatin remodeling factor CTCF.
- GWAS hits for other neurodegenerative diseases overlap with CTCF binding sites, suggesting a common mechanism.
The authors zeroed in on the mechanism, too, reporting that the SNP strengthens the binding of CTCF, a modulator of chromatin architecture. Strikingly, they also found an enrichment for CTCF binding sites abutting GWAS hits for other neurodegenerative diseases, hinting that the architectural mechanism could be a broad one.
Back in 2010, an SNP in a noncoding region of chromosome 7q21 was found to increase the risk of frontotemporal dementia 1.6-fold (Feb 2010 news on Van Deerlin et al., 2010). Multiple studies have since replicated the association, and found further associations between the SNP and other factors related to disease and aging, such as progranulin expression (Aug 2012 news). Chen-Plotkin and others pinned the association to TMEM106b, a gene involved in lysosomal degradation (Lang et al., 2012; Brady et al., 2013; Jul 2017 news). However, attempts to pinpoint which variants exert their influence on TMEM106b, and how they do so, have yielded conflicting results. Some researchers blamed a variant within exon 6 of the TMEM106b, while others failed to replicate that (Jun et al., 2015; Nicholson et al., 2013; Feb 2015 news).
First author Michael Gallagher and colleagues sought to nail down how FTD variations at chromosome 7q21 influenced TMEM106b expression. They started by tapping the Genotype-Tissue Expression (GTEx) project, which maps how genetic variations influence gene expression in 44 human tissues (Oct 2017 news). They found that the 7q21 SNP initially linked to FTD, the “sentinel” SNP, significantly associated with increased expression of TMEM106b in several tissues, including the hippocampus and nucleus accumbens in the brain, as well as the spleen and lymphoblast cell lines (LCLs). Notably, levels of no other transcript significantly associated with this variation.
If only this simple one-to-one association could explain everything. Instead, the researchers found that dozens of other SNPs in this 36kB locus also associated with TMEM106b expression. To tease out the causal variant(s) from those that were just along for the ride, the researchers turned to ENCODE, a project that maps regulatory elements within noncoding regions of the genome in different cell types, as well as to the NIH Roadmap EpiGenome Project, which similarly maps epigenetic modifications (Sep 2014 news; Feb 2015 news). They found that of 84 SNPs at the TMEM106b locus that could potentially regulate TMEM106b expression, only three landed in regions with potential regulatory activity. All three were binding sites for the mammalian chromatin organizing protein CTCF. According to chromatin immunoprecipitation sequencing (CHIP-Seq) maps from ENCODE, these three SNP sites bound CTCF in neuronal, glial, and lymphoblast cell lines.
The three SNPs, which include the sentinel, lay within perfect linkage disequilibrium with each other, meaning they are always co-inherited. This clue drove the researchers to the bench to tease out the functional culprit. Ultimately, they found that the minor, risk-associated A allele of rs1990620, located just downstream of the TMEM106b gene, bound to CTCF preferentially compared to the major, non-risk allele. Alleles of the two other SNPs, including the sentinel, bound CTCF equally well.
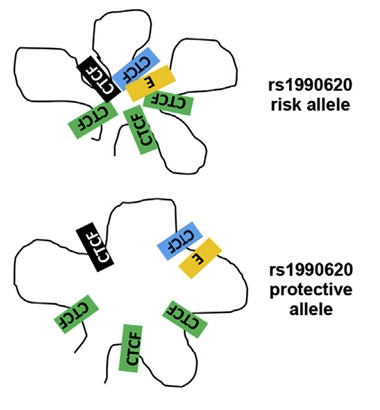
Looping You In.
Tucked into one of five CTCF binding sites, the rs1990620 risk allele (blue) brings an enhancer (yellow) close to the promoter (black) of the TMEM106b gene. [Courtesy of Gallagher et al., American Journal of Human Genetics, 2017.]
Clusters of CTCF binding sites are known to define neighborhoods of co-regulated genes. The researchers identified five such binding sites within the TMEM106b locus, including the one at rs1990620. CTCF binding at the SNP promoted a kind of looping of chromatin that juxtaposed TMEM106b promoter and enhancer regions, presumably ramping up expression of the gene (see image).
The researchers propose that enhanced binding of CTCF to the risk allele would only slightly enhance expression of TMEM106b. Could a small difference have harmful effects? Expressing TMEM106b at varying levels in HeLa cells, ranging from two- to 20-fold over baseline, the researchers found that each incremental increase in TMEM106b expression acutely worsened the cells’ condition, enlarging their lysosomes and promoting cell death. Doubling TMEM106b tripled the number of cells with overstuffed vacuoles, and decreased survival by 20 percent. Chen-Plotkin told Alzforum that this data suggests that even modest increases in TMEM106b expression over decades could gradually derail proteostasis, and thus hasten neurodegenerative diseases such as frontotemporal dementia.
“Increased TMEM106B levels are known to be a risk factor for FTLD patients who have progranulin mutations,” commented Fenghua Hu of Cornell University in Ithaca, New York. “This is a really nice study illustrating that the TMEM106B risk allele interacts with the chromatin-organizing protein CTCF with higher affinity, and this increased binding leads to higher TMEM106B transcription levels from the risk allele,” Hu said.
Might genetic variations associated with other neurodegenerative diseases also fall within CTCF binding sites? To investigate, the researchers cross-referenced 200 published risk SNPs for FTD, amyotrophic lateral sclerosis, Alzheimer’s, and Parkinson’s with mapped CTCF binding sites in the brain. They found a 1.6-fold enrichment of disease SNPs or their closely linked variants at CTCF binding sites. Interestingly, they found a similar enrichment when they only looked at CTCF binding sites relevant to leukocyte cell types, rather than those specific to the brain. This indicated that white blood cells could serve as proxies for CTCF-mediated gene regulation in the brain.
“The findings suggest that CTCF-mediated architectural changes to chromatin could play an important role in many neurodegenerative diseases,” Chen-Plotkin said. She added that these chromatin-looping interactions mediated by CTCF should be considered along with other epigenetic modifications to promotors or enhancers.
Chen-Plotkin told Alzforum that of the more than 200 loci associated with neurodegenerative disease so far, only a small fraction have functional studies to back them up. This type of in-depth study is needed to pin down the mechanisms that drive disease associations, she said. This will allow researchers to zero in on therapeutic targets.—Jessica Shugart
References
News Citations
- Genetics of FTD: New Gene, PGRN Variety, and a Bit of FUS
- FTD Risk Factor Confirmed, Alters Progranulin Pathways
- TMEM106B and Progranulin Duke It Out at the Lysosome
- FTLD Gene Bad Actor in Many TDP-43 Proteinopathies
- Gene Expression Map of Human Body Gives Value to Variants
- ENCODE Expands Analysis of DNA Regulation
- Consortium Debuts Biggest Set of Human Epigenomes To Date
Paper Citations
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL, Ferrer I, Lladó A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IR, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, Decarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tuñón MT, Martínez MC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, Trojanowski JQ, Lee VM. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010 Mar;42(3):234-9. PubMed.
- Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, Haass C. Membrane Orientation and Subcellular Localization of Transmembrane Protein 106B (TMEM106B), a Major Risk Factor for Frontotemporal Lobar Degeneration. J Biol Chem. 2012 Jun 1;287(23):19355-65. PubMed.
- Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet. 2013 Feb 15;22(4):685-95. PubMed.
- Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain. 2015 Dec 10;8:85. PubMed.
- Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R, Hsiung GY, Mackenzie IR, Finger E, Boeve BF, Ertekin-Taner N, Graff-Radford NR, Dickson DW, Rademakers R. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem. 2013 Jun 6; PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL, Chesi A, Manduchi E, Wells AD, Grant SF, Blobel GA, Brown CD, Chen-Plotkin AS. A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am J Hum Genet. 2017 Oct 16; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.