Without TMEM106B, Lysosomal Traffic Jams Wreak Havoc in Neurons
Quick Links
TMEM106b, a gene carrying risk variants for frontotemporal dementia (FTD), coordinates the steady stream of lysosomal traffic along axons, according to a study published March 10 in Cell Reports. Researchers led by Markus Damme of Christian Albrechts University in Kiel, Germany, reported that, in two independent strains of TMEM106b knockout mice, motor neurons contained swollen vacuoles that bore markers of lysosomes. These defunct organelles piled up in axons, crowding their base at the soma. Mice missing TMEM106b had poor facial nerve responses and were wobbly on their feet, but it is unclear if these symptoms relate to risk for neurodegenerative disease. Prior reports had suggested that TMEM106b knockouts had milder phenotypes.
- Motor neurons in TMEM106b knockout mice had swollen vacuoles.
- The vacuoles piled up in axons, adjacent to the soma.
- Retrograde axonal trafficking of lysosomes ramped up in TMEM106b knockouts.
“This study is important as it may shed light on the function of TMEM106B, which has emerged as a key genetic risk locus not only for development of FTLD-TDP, but also for rate of progression of FTD and of Parkinson’s Disease,” Alice Chen-Plotkin of the University of Pennsylvania, Philadelphia, wrote to Alzforum (Tropea et al., 2019).
“It’s a beautiful study, which clearly shows a role of TMEM106B in lysosome trafficking along axons,” wrote Fenghua Hu of Cornell University in Ithaca, New York. “This study suggests that loss of TMEM106B potentiates the development of FTLD phenotypes.”
The bulging lysosomes spotted in these two mouse models contrast with results from two other TMEM106b knockout strains. They had subtler lysosomal changes and, in one case, even were reported to rescue deficits caused by mutations in progranulin, a cause of familial FTD (Jul 2017 news; Nicholson et al., 2018). The reason behind these discrepancies remains unclear, but Damme and co-authors raised the possibility that TMEM106b expression had not been completely extinguished in previous models.
TMEM106b was unknown until a decade ago, when polymorphisms near the gene emerged as modifiers of FTD risk in a genome-wide association study (Feb 2010 news; Van Deerlin et al., 2010). Researchers found that common variants boosted risk of FTD, while a rare variant offered protection from the disease, in people with or without a pathogenic mutation in progranulin. These variants were later found to augment risk in carriers of C9ORF72 expansions, another cause of FTD (Gallagher et al., 2014; van Blitterswijk et al., 2014). Subsequent studies suggested that risk variants upped expression of TMEM106b, and protective variants lowered it (Aug 2012 news; Oct 2017 news). Cell culture studies cast TMEM106B as a regulator of lysosomal trafficking and fusion, but the physiological function of the protein within the brain has remained elusive (Schwenk et al., 2014; Busch et al., 2016).
First authors Patrick Lüningschrör and Georg Werner set out to determine what TMEM106B really does in the brain. They used CRISPR to introduce into the TMEM106b gene a frame-shift mutation that corrupted its coding sequence and derailed transcription. Immunoblotting confirmed the complete absence of the TMEM106B protein in the brains and spinal cord lysates of homozygous knockouts.
While nothing appeared amiss in the visceral organs of the mice, the researchers spotted swollen vacuoles in specific brain regions by 10 weeks of age. These balloons—some larger than neuronal cell bodies—were most rampant in the facial motor nucleus. The FMN contains a bundle of neurons in the brainstem that innervate facial muscles. Vacuoles were also enlarged in other brainstem motor nuclei and in the frontal thalamus. Despite the burden of these vesicles, neuron numbers in these regions were unaffected, at least up to 22 weeks of age.
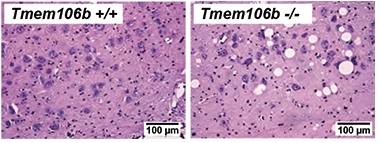
Bad Balloons. Giant vacuoles speckle the facial motor nucleus of TMEM106b knockout mice. [Courtesy of Lüningschrör et al., Cell Reports, 2020.]
A closer look at these vacuoles revealed that they expressed the lysosomal marker LAMP-1, and often the lysosomal protease cathepsin D. Compared with brain extracts from wild-type mice, those from TMEM106b knockouts had heightened lysosomal hydrolase activity, more cathepsin D, and more progranulin.
Shining an array of microscopy tools on FMN slices, the researchers found that the enlarged lysosomes primarily crowded the axon initial segment (AIS)—the axonal stretch closest to the soma. In many cases, a giant tubular lysosome filled out this entire segment, culminating in a bulging vacuole at the distal end of the AIS (see image below). The furthest reaches of the axons themselves appeared normal.

LAMP1 Bulge. In cultured motor neurons (red) from TMEM106b knockouts (bottom), LAMP1+ vacuoles bulged [inset] at the gateway from the axon into the soma. [Courtesy of Lüningschrör et al., Cell Reports, 2020.]
In cultured motor neurons from the knockout mice, retrograde trafficking of lysosomes—from the distal axons toward the soma—was significantly ramped up. More lysosomes moved toward the soma, but then appeared stuck there, unable to enter. This logjam prevented the organelles from fusing properly with autophagosomes, the double membrane-bound organelles that need lysosomal enzymes to degrade their contents. This failure to fuse was evident via a buildup in the spinal cord, hindbrain, and thalamus of autophagosomes and their cargo, which includes p62, ubiquitinated protein aggregates, and lipofuscin.
This axonal traffic jam had functional consequences that matched the location of the cellular pathology. The knockout mice had slight deficits in their eye-blink reflex and whisker movement, two responses that depend on signals from the facial motor nuclei. In addition, they wobbled on a balance beam and faltered while descending a vertical pole more than their wild-type counterparts did, even though their neuromuscular junctions were intact. This suggested that the motor problems stemmed from the vacuolar pile-up near the soma, where action potentials originate.
To confirm their findings, the researchers generated a second strain of knockout mice by way of a completely different method. They used a so-called gene-trap technique. Into intron four of the TMEM106b gene, they inserted a sequence containing an artificial splice acceptor site as well as a LacZ reporter gene. This derailed proper transcription of the TMEM106b gene, leading to expression of the LacZ reporter instead. However, because alternative splice acceptor sites can be “leaky,” the researchers took the additional step of crossing the mice to a “Cre deleter strain,” which targets the inserted sequence, nixing a section of the gene for good.
The resulting homozygous TMEM106b knockouts had the same major phenotypes as the CRISPR line did: enlarged vacuoles crowding axons near neuronal cell bodies, and a buildup of autophagosomes and their cargo in the FMN. They authors did not test balance or facial responses.
Together, the findings suggest that TMEM106B plays a key role in lysosomal transport from the axons to the soma, and may also be involved in lysosomal entry into the soma and/or fusion with autophagosomes.
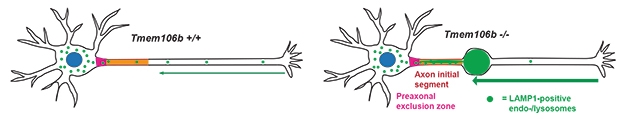
Hurry Up to Wait. In motor neurons lacking TMEM106b, lysosomal trafficking from the axons toward the soma revs up, causing a traffic jam near the soma. [Courtesy of Lüningschrör et al., Cell Reports, 2020.]
Curiously, the phenotype of these two new mouse strains stands in contrast to those previously described. One study reported that knocking out TMEM106b was beneficial, reducing lysosomal acidification and correcting lysosomal dysfunction caused by progranulin deficiency (Klein et al., 2017). Another found no rescue of C9ORF72 pathogenesis on TMEM106b knockout, but reported no bulging lysosomes (Nicholson et al., 2018).
Damme and colleagues pointed out that these other knockouts were generated using gene trap methodology without the additional step of permanent deletion, which may not have completely inactivated the gene. Alternatively, these previous studies may have missed the motor neuron pathology because they focused on FTD-related phenotypes in the cortex. Authors of those previous papers did not reply to a request for comment.
“These results differ from prior reports of TMEM106b knockout mice, where minimal phenotypes were seen,” noted Chen-Plotkin. “The fact that the authors got this vacuolization phenotype in two lines of knockouts, generated via different strategies, is reassuring,” she added, noting that the findings mesh with those of cell-culture studies implicating TMEM106B in lysosomal function.
Co-author Christian Haass of Ludwig Maximilians University in Munich told Alzforum that his lab has already crossed the TMEM106b knockouts with progranulin-deficient mice, and that preliminary data suggest that loss of TMEM106b exacerbates deficits, rather than rescuing them.
What do the new TMEM106b knockout mice say about the role of TMEM106B in FTD? Damme emphasized the knockouts were not made to model FTD, but to understand the physiological function of the gene. The FTD risk alleles do not wipe out expression of the gene, and it is possible that subtle changes in expression may have different effects than full deletion. More insight might come from crossing these knockouts with progranulin-deficient or C9ORF72 repeat expansion models.
Co-author Anja Capell of LMU noted that it is still unclear how disease risk variants near TMEM106b affect its expression and function. For example, though risk variants have been associated with higher levels of TMEM106B protein in the brain, it is possible that this is caused by a secondary build-up of TMEM106B protein in swollen vacuoles, rather than an increase in gene expression, she noted.—Jessica Shugart
References
News Citations
- TMEM106B and Progranulin Duke It Out at the Lysosome
- Genetics of FTD: New Gene, PGRN Variety, and a Bit of FUS
- FTD Risk Factor Confirmed, Alters Progranulin Pathways
- FTD Variant Alters Chromatin, Boosts Expression of TMEM106b
Paper Citations
- Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J, Siderowf A, Weintraub D, Grossman M, Irwin D, Wolk DA, Trojanowski JQ, Van Deerlin V, Chen-Plotkin AS. TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol. 2019 Jun;85(6):801-811. PubMed.
- Nicholson AM, Zhou X, Perkerson RB, Parsons TM, Chew J, Brooks M, DeJesus-Hernandez M, Finch NA, Matchett BJ, Kurti A, Jansen-West KR, Perkerson E, Daughrity L, Castanedes-Casey M, Rousseau L, Phillips V, Hu F, Gendron TF, Murray ME, Dickson DW, Fryer JD, Petrucelli L, Rademakers R. Loss of Tmem106b is unable to ameliorate frontotemporal dementia-like phenotypes in an AAV mouse model of C9ORF72-repeat induced toxicity. Acta Neuropathol Commun. 2018 May 31;6(1):42. PubMed.
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL, Ferrer I, Lladó A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IR, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, Decarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tuñón MT, Martínez MC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, Trojanowski JQ, Lee VM. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010 Mar;42(3):234-9. PubMed.
- Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, Rohrer JD, Halliday G, Van Broeckhoven C, Seilhean D, Shaw PJ, Frosch MP, Alafuzoff I, Antonell A, Bogdanovic N, Brooks W, Cairns NJ, Cooper-Knock J, Cotman C, Cras P, Cruts M, De Deyn PP, DeCarli C, Dobson-Stone C, Engelborghs S, Fox N, Galasko D, Gearing M, Gijselinck I, Grafman J, Hartikainen P, Hatanpaa KJ, Highley JR, Hodges J, Hulette C, Ince PG, Jin LW, Kirby J, Kofler J, Kril J, Kwok JB, Levey A, Lieberman A, Llado A, Martin JJ, Masliah E, McDermott CJ, McKee A, McLean C, Mead S, Miller CA, Miller J, Munoz DG, Murrell J, Paulson H, Piguet O, Rossor M, Sanchez-Valle R, Sano M, Schneider J, Silbert LC, Spina S, van der Zee J, Van Langenhove T, Warren J, Wharton SB, White CL 3rd, Woltjer RL, Trojanowski JQ, Lee VM, Van Deerlin V, Chen-Plotkin AS. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014 Mar;127(3):407-18. Epub 2014 Jan 19 PubMed.
- van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014 Mar;127(3):397-406. Epub 2014 Jan 3 PubMed.
- Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, Lichtenthaler SF, Hoogenraad CC, Capell A, Haass C, Edbauer D. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 2014 Mar 3;33(5):450-67. Epub 2013 Dec 19 PubMed.
- Busch JI, Unger TL, Jain N, Tyler Skrinak R, Charan RA, Chen-Plotkin AS. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet. 2016 Apr 28; PubMed.
- Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, Strittmatter SM. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron. 2017 Jul 19;95(2):281-296.e6. PubMed.
Further Reading
Papers
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M, Forloni G, Kertesz A, Knopman DS, Uitti R, White CL, Caselli R, Lippa C, Bigio EH, Wszolek ZK, Binetti G, Mackenzie IR, Miller BL, Boeve BF, Younkin SG, Dickson DW, Petersen RC, Graff-Radford NR, Geschwind DH, Rademakers R. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011 Feb 1;76(5):467-74. PubMed.
- Pottier C, Zhou X, Perkerson RB 3rd, Baker M, Jenkins GD, Serie DJ, Ghidoni R, Benussi L, Binetti G, López de Munain A, Zulaica M, Moreno F, Le Ber I, Pasquier F, Hannequin D, Sánchez-Valle R, Antonell A, Lladó A, Parsons TM, Finch NA, Finger EC, Lippa CF, Huey ED, Neumann M, Heutink P, Synofzik M, Wilke C, Rissman RA, Slawek J, Sitek E, Johannsen P, Nielsen JE, Ren Y, van Blitterswijk M, DeJesus-Hernandez M, Christopher E, Murray ME, Bieniek KF, Evers BM, Ferrari C, Rollinson S, Richardson A, Scarpini E, Fumagalli GG, Padovani A, Hardy J, Momeni P, Ferrari R, Frangipane F, Maletta R, Anfossi M, Gallo M, Petrucelli L, Suh E, Lopez OL, Wong TH, van Rooij JG, Seelaar H, Mead S, Caselli RJ, Reiman EM, Noel Sabbagh M, Kjolby M, Nykjaer A, Karydas AM, Boxer AL, Grinberg LT, Grafman J, Spina S, Oblak A, Mesulam MM, Weintraub S, Geula C, Hodges JR, Piguet O, Brooks WS, Irwin DJ, Trojanowski JQ, Lee EB, Josephs KA, Parisi JE, Ertekin-Taner N, Knopman DS, Nacmias B, Piaceri I, Bagnoli S, Sorbi S, Gearing M, Glass J, Beach TG, Black SE, Masellis M, Rogaeva E, Vonsattel JP, Honig LS, Kofler J, Bruni AC, Snowden J, Mann D, Pickering-Brown S, Diehl-Schmid J, Winkelmann J, Galimberti D, Graff C, Öijerstedt L, Troakes C, Al-Sarraj S, Cruchaga C, Cairns NJ, Rohrer JD, Halliday GM, Kwok JB, van Swieten JC, White CL 3rd, Ghetti B, Murell JR, Mackenzie IR, Hsiung GR, Borroni B, Rossi G, Tagliavini F, Wszolek ZK, Petersen RC, Bigio EH, Grossman M, Van Deerlin VM, Seeley WW, Miller BL, Graff-Radford NR, Boeve BF, Dickson DW, Biernacka JM, Rademakers R. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol. 2018 Jun;17(6):548-558. Epub 2018 Apr 30 PubMed.
- Li Z, Farias FH, Dube U, Del-Aguila JL, Mihindukulasuriya KA, Fernandez MV, Ibanez L, Budde JP, Wang F, Lake AM, Deming Y, Perez J, Yang C, Bahena JA, Qin W, Bradley JL, Davenport R, Bergmann K, Morris JC, Perrin RJ, Benitez BA, Dougherty JD, Harari O, Cruchaga C. The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol. 2020 Jan;139(1):45-61. Epub 2019 Aug 27 PubMed.
- Arrant AE, Nicholson AM, Zhou X, Rademakers R, Roberson ED. Partial Tmem106b reduction does not correct abnormalities due to progranulin haploinsufficiency. Mol Neurodegener. 2018 Jun 22;13(1):32. PubMed.
Primary Papers
- Lüningschrör P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D, Wanner R, Lüllmann-Rauch R, Wefers B, Wurst W, D'Hooge R, Uchiyama Y, Sendtner M, Haass C, Saftig P, Knöll B, Capell A, Damme M. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 2020 Mar 10;30(10):3506-3519.e6. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Penn Neurological Institute
I think this study is important, as it may shed light on the function of TMEM106B, which has emerged as a key genetic risk locus not only for development of FTLD-TDP, but also for rate of progression in clinical FTD and in Parkinson’s disease (Tropea et al., 2019). Specifically, two lines of mice lacking TMEM106B show vacuolization within neurons, and the data suggest that these vacuoles are a type of non-functional, or poorly functional, lysosome.
These results differ from prior reports of TMEM106B knockout mice, where minimal phenotypes were seen. However, variable phenotypes in what should be equivalent mouse models sometimes happen—the various C9ORF72 expansion mice are a key example. The fact that the authors got this vacuolization phenotype in two lines of knockouts, generated via different strategies, is reassuring. In addition, our work, and the work of other groups, using cells leads us to believe that TMEM106B may regulate aspects of lysosomal function and/or lysosome-autophagosome fusion (Chen-Plotkin et al., 2012; Busch et al., 2016). Their in vivo findings do agree with these expectations.
References:
Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J, Siderowf A, Weintraub D, Grossman M, Irwin D, Wolk DA, Trojanowski JQ, Van Deerlin V, Chen-Plotkin AS. TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol. 2019 Jun;85(6):801-811. PubMed.
Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, Busch JI, Akle S, Grossman M, Van Deerlin V, Trojanowski JQ, Lee VM. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci. 2012 Aug 15;32(33):11213-27. PubMed.
Busch JI, Unger TL, Jain N, Tyler Skrinak R, Charan RA, Chen-Plotkin AS. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet. 2016 Apr 28; PubMed.
Make a Comment
To make a comment you must login or register.