From a Million Samples, GWAS Squeezes Out Seven New Alzheimer's Spots
Quick Links
It took more than a million samples, but researchers have managed to extract seven fresh AD risk loci from a genome-wide association study. Published September 7 in Nature Genetics, this GWAS included 90,338 samples from people who were either diagnosed with AD or had a family history of the disease, as well as from 1,036,225 controls. It pegged 38 AD risk loci, 31 of which had been netted in previous GWAS. Seven new ones included two that had been previously tied to frontotemporal dementia, and five relative newcomers to neurodegeneration. In all, the findings build further support for the role of microglia, immune function, and protein homeostasis in AD.
- From more than 90,000 cases and a million controls, a GWAS pulled 38 AD risk loci.
- Among seven new ones, TMEM106B and GRN were previously tied to frontotemporal dementia.
- Variants implicate immunity and protein catabolism in Alzheimer's disease.
Despite eclipsing the million-person milestone, this latest GWAS, led by Danielle Posthuma of VU Amsterdam, The Netherlands, identified fewer risk loci than a recently posted study led by Jean-Charles Lambert at the Institut Pasteur de Lille in France, which identified 75 loci, including 42 new ones, from 111,326 cases and 677,663 controls (Feb 2021 news). First author Douglas Wightman attributed the bigger haul of Lambert’s GWAS both to the larger number of cases included in that study and to the authors’ generation of novel genotyping data.
For their study, Wightman and colleagues drew genotyping data from 13 cohorts, including the International Genomics of Alzheimer's Project (IGAP), deCODE, UK Biobank, 23andMe, BioVU, the Trøndelag Health Study, DemGene, TwinGene, STSA, GR@CE, Gothenburg, ANMerge, and Finngen. The 90,338 cases included 43,725 AD and 46,613 proxy cases, i.e., people with a family history of AD. Of the 1,036,225 controls, 318,246 were considered proxy controls, having no family history of AD. The study more than doubles the sample size of a previous GWAS led by many of the same authors, adding more than 18,000 cases and 650,000 controls (Mar 2019 news on Jansen et al., 2019).
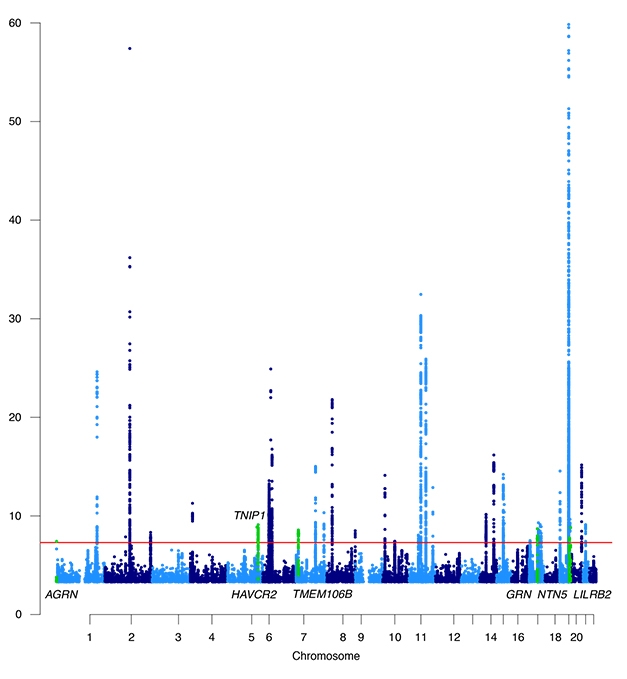
Growing AD Skyline. Manhattan plot of genome-wide significant AD risk loci. Newcomers are displayed in green. [Courtesy of Wightman et al., Nature Genetics, 2021.]
From this genotypic trove, the researchers identified 3,915 genome-wide significant variants across 38 independent loci. Of the seven new ones, five—AGRN, TNIP1, AVCR2, NTN5, LILRB2—had never been linked to a neurodegenerative disease in GWAS. Two—TMEM106B and GRN—are important in frontotemporal dementia.
The new analysis replicated most variants identified in Posthuma et al.'s previous GWAS, as well as another massive GWAS published around the same time (Kunkle et al., 2019 and Apr 2018 news).
The researchers used the genomic position of each variant, as well as co-localization with expression quantitative trait loci (eQTL) and previously published data, to estimate which gene might account for each of the 38 loci. These genes were involved in amyloid and tau aggregation, catabolism of plaques, immune cell recruitment, and glial cell function. Combing through single-cell RNA sequencing data, the scientists found the risk genes to likely be expressed in microglia.
“This study is a great example of what genetics can do,” said Carlos Cruchaga of Washington University in St. Louis. He was referring to the integration of tissue and cell type-specific expression data to narrow down the list of potential causal genes in each loci, adding that “These tools are helping us understand the biological context behind associations.”
What do scientists know about the newbies? TNIP1 previously popped up in an autoimmune GWAS; it is thought to fuel hyperinflammation (Shamilov and Aneskievich, 2018). TNIP1cropped up in a transcription module in inflamed, aging mouse microglia; there, it was regulated by Bcl3, a gene that ramps up in AD brain and has come up in AD biomarker studies (e.g., Cho et al., 2019; Marques-Coelho et al., 2021).
HAVCR2 has been spotted in aged microglia and appears to help them sense ligands and microbes (Olah et al., 2018; Hickman et al., 2013).
LILRB2 belongs to the leukocyte immunoglobulin-like receptor family. These transmembrane glycoproteins are MHC class 1 receptors, i.e., they influence immune activation (Zhang et al., 2017). A small literature going back eight years has shown LILRB2 is expressed in brain and tied it to AD by way of Aβ oligomer binding (Sep 2013 news; Oct 2018 news). Carla Shatz of Stanford University, who led this line of research, said she was gratified to see LilrB2 emerge as a potential risk gene in AD. “Their GWAS results show how important it is to take high-quality basic science studies seriously, and fund basic research even in the absence of support from GWAS results,” she wrote.
While the study's sheer size helped unearth more AD risk loci, it also came at the price of lower specificity, Cruchaga said. He noted that especially with the use of proxy cases, it is difficult to determine whether the genetic associations relate to AD specifically or perhaps other types of dementia.
John Hardy of University College London made a similar point. “We know the diagnostic accuracy even in the highly cited clinic-based GWAS is only about 80 percent, and so is undoubtedly less in these GWAS, which use reported (parental) cases,” he wrote. “As FTD genes start to show up, perhaps we should note this concern,” he added (full comment below).
Even with more than a million samples, the study only scratches the surface of genetic heritability underpinning AD, the authors noted. Anders Dale, University of California, San Diego, and colleagues estimate that 2.2 million samples would be required to detect 80 percent of genetic variance on chromosome 19, which houses the ApoE gene, while a whopping 7.8 million samples would be needed to detect 80 percent of the variance from the rest of the genome (Holland et al., 2021). Wightman estimated that their current GWAS was powered to explain about 6 percent of the variance outside of chromosome 19, and 59 percent of the variance within it. Besides continually growing GWAS samples, other approaches, including chasing rare and private variants, will be needed to dig up the remaining AD risk influencers.—Jessica Shugart
References
News Citations
- Massive GWAS Meta-Analysis Digs Up Trove of Alzheimer’s Genes
- Paper Alerts: Massive GWAS Studies Published
- GWAS, GWAX: bioRχiv Hosts Bonanza of Alzheimer’s Genetics
- Immune Receptor Binds Aβ Oligomers, Spurs Synaptic Loss
- Crystal Structure of Aβ and Proposed Receptor Solved
Paper Citations
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019 Mar;51(3):404-413. Epub 2019 Jan 7 PubMed.
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier JG, Harold D, Fitzpatrick AL, Valladares O, Moutet ML, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Del Zompo M, Fox NC, Choi SH, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MC, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, De Deyn PP, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujić-Čomić H, Frosch MP, Thonberg H, Maier W, Roshchupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernández I, Kamboh MI, Brundin R, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT Jr, Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Andre Uitterlinden AG, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu CK, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo Á, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frölich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JS, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kölsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hüll M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin LW, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jöckel KH, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann HE, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nöthen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O'Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossù P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, De Jager PL, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues JF, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz J, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Van Broeckhoven C, C O'Donovan M, DeStefano AL, Jones L, Haines JL, Deleuze JF, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang LS, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert JC, Pericak-Vance MA, Alzheimer Disease Genetics Consortium (ADGC),, European Alzheimer’s Disease Initiative (EADI),, Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE),, Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES),. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019 Mar;51(3):414-430. Epub 2019 Feb 28 PubMed. Correction.
- Shamilov R, Aneskievich BJ. TNIP1 in Autoimmune Diseases: Regulation of Toll-like Receptor Signaling. J Immunol Res. 2018;2018:3491269. Epub 2018 Oct 3 PubMed.
- Cho CE, Damle SS, Wancewicz EV, Mukhopadhyay S, Hart CE, Mazur C, Swayze EE, Kamme F. A modular analysis of microglia gene expression, insights into the aged phenotype. BMC Genomics. 2019 Feb 28;20(1):164. PubMed.
- Marques-Coelho D, Iohan LD, Melo de Farias AR, Flaig A, Brainbank Neuro–CEB Neuropathology Network, Lambert JC, Costa MR. Differential transcript usage unravels gene expression alterations in Alzheimer's disease human brains. NPJ Aging Mech Dis. 2021 Jan 4;7(1):2. PubMed.
- Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, Connor S, Yung CJ, Frangieh M, McHenry A, Elyaman W, Petyuk V, Schneider JA, Bennett DA, De Jager PL, Bradshaw EM. A transcriptomic atlas of aged human microglia. Nat Commun. 2018 Feb 7;9(1):539. PubMed.
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013 Dec;16(12):1896-905. Epub 2013 Oct 27 PubMed.
- Zhang J, Mai S, Chen HM, Kang K, Li XC, Chen SH, Pan PY. Leukocyte immunoglobulin-like receptors in human diseases: an overview of their distribution, function, and potential application for immunotherapies. J Leukoc Biol. 2017 Aug;102(2):351-360. Epub 2017 Mar 28 PubMed.
- Holland D, Frei O, Desikan R, Fan CC, Shadrin AA, Smeland OB, Andreassen OA, Dale AM. The genetic architecture of human complex phenotypes is modulated by linkage disequilibrium and heterozygosity. Genetics. 2021 Mar 31;217(3) PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, Rongve A, Børte S, Winsvold BS, Drange OK, Martinsen AE, Skogholt AH, Willer C, Bråthen G, Bosnes I, Nielsen JB, Fritsche LG, Thomas LF, Pedersen LM, Gabrielsen ME, Johnsen MB, Meisingset TW, Zhou W, Proitsi P, Hodges A, Dobson R, Velayudhan L, 23andMe Research Team, Sealock JM, Davis LK, Pedersen NL, Reynolds CA, Karlsson IK, Magnusson S, Stefansson H, Thordardottir S, Jonsson PV, Snaedal J, Zettergren A, Skoog I, Kern S, Waern M, Zetterberg H, Blennow K, Stordal E, Hveem K, Zwart JA, Athanasiu L, Selnes P, Saltvedt I, Sando SB, Ulstein I, Djurovic S, Fladby T, Aarsland D, Selbæk G, Ripke S, Stefansson K, Andreassen OA, Posthuma D. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021 Sep;53(9):1276-1282. Epub 2021 Sep 7 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
Institute of Neurology, UCL
My concern about this GWAS is that it is not a GWAS for Alzheimer’s disease but rather a GWAS for dementia. We know the diagnostic accuracy even in the highly cited clinic-based GWAS is only about 80 percent, and so is undoubtedly less in these GWAS which use reported (parental) cases. AS FTD genes start to show up, perhaps we should note this concern.
Another concern (not at all limited to this GWAS for dementia) is that everyone meta-analyses their data with previous datasets, so errors that include these diagnostic ones, but also other errors, get baked into the ever-increasing size and reach statistically significant but biologically misleading conclusions.
Institute Pasteur de Lille, INSERM
Douglas Wightman and colleagues report seven new loci associated with AD risk based on a large meta-analysis of GWAS. Even if the number of samples claimed by the authors is impressive, several points deserve comment and precision, some of them already fairly mentioned by the authors.
Why are the numbers of genes discovered in Wightman et al. and Bellenguez et al. so different, i.e., seven and 42, respectively? Below I briefly describe some of the characteristics and results of the main recently published GWAS in AD.
Click to Enlarge
First, it is important to keep in mind that, following our first IGAP publication in 2013, and the use of the U.K. biobank and proxy-AD cases in 2018, most of the GWAS meta-analyses shared the same main GWAS datasets, making these studies not independent of each other.…More
In addition, the number of controls grew, but not the number of cases. However, at the level of statistics, it becomes useless to have only more and more controls. Finally, methodologies are dissimilar between the studies: (i) with or without replication stage; (ii) using different panels of imputation, to name the most differentiating elements.
Taking into account these points, we can describe major differences between our GWAS:
(i) We analyzed almost 30,000 fully new, clinically diagnosed AD cases (discovery/replication), whereas Wightman et al. included mainly new controls through 23&Me and FinnGen.
(ii) We used the novel TopMed imputation panel, allowing us to double the number of SNPs analyzed with high imputation quality.
(iii) Wightman et al. included no replication stage in their study, unlike us (respectively, stage I = 90,338 cases, stage I+II = 85,934 + 25,392).
(iv) Wightman et al. included a new 23&Me dataset. This approach had been powerful in Parkinson's, helping to report dozens of new loci. However, this needs to be evaluated in Alzheimer's. The diagnosis is declarative, and no demographic information is reported in the paper, making it difficult to understand the main characteristics of this population and how this may impact the results.
It is important to note that the new loci described in the De Rojas, Schwartzentruber, and now the Wightman papers, are not in common (they share the main GWAS datasets), but a large part of them are detected in the Bellenguez’s paper. This likely indicates lack of statistical power and variability due to different designs.
Inversely, for those loci that are found only in one of these three studies, this may indicate that they are potentially false positives. This is the case with three of the loci described by Wightman et al., i.e., AGRN, HAVCR2, NTN5; they clearly require further investigation in independent datasets.
To conclude, it is more than likely that clinically diagnosed cases add power and, potentially, uncharacterized controls add noise. The difference in the number of novel cases defines the potential for novel discoveries in GWA studies.
More generally, we must carry out a GWAS gathering all the data, at least in populations of European origins, in order to present a landscape of the genetics of AD as clearly as possible to our community. As stated by John Hardy, this is important in order to avoid misleading the post-genomic studies that will follow. This also implies that GWAS of larger sizes relating to other neurodegenerative diseases, but also based on pathological diagnosis, have to be performed. Again as mentioned by John, it is indeed interesting but also disturbing to see genes involved in other neurodegenerative diseases being genetic determinants of AD. Does this represent a pathophysiological reality, or is it a bias associated with diagnostic uncertainty in GWASs?
It is important to answer these questions, because this can have a significant impact on future research strategies. In the Bellenguez paper, we also observed that some AD genes are linked to other neurodegenerative diseases, including Parkinson’s disease (the IDUA and CTSB loci), frontotemporal dementia (the GRN and TMEM106B loci) and amyotrophic lateral sclerosis (the TNIP1 locus). The presence of common causal variants in the same gene may indicate that these genetic factors have a shared pathological role downstream, whereas the presence of different causal variants may indicate specific mechanisms upstream. Importantly, these signals seemed to be independent of the U.K. biobank proxy-AD cases, and were still present and replicated in clinically diagnosed AD cases.
References:
de Rojas I, Moreno-Grau S, Tesi N, Grenier-Boley B, Andrade V, Jansen IE, Pedersen NL, Stringa N, Zettergren A, Hernández I, Montrreal L, Antúnez C, Antonell A, Tankard RM, Bis JC, Sims R, Bellenguez C, Quintela I, González-Perez A, Calero M, Franco-Macías E, Macías J, Blesa R, Cervera-Carles L, Menéndez-González M, Frank-García A, Royo JL, Moreno F, Huerto Vilas R, Baquero M, Diez-Fairen M, Lage C, García-Madrona S, García-González P, Alarcón-Martín E, Valero S, Sotolongo-Grau O, Ullgren A, Naj AC, Lemstra AW, Benaque A, Pérez-Cordón A, Benussi A, Rábano A, Padovani A, Squassina A, de Mendonça A, Arias Pastor A, Kok AA, Meggy A, Pastor AB, Espinosa A, Corma-Gómez A, Martín Montes A, Sanabria Á, DeStefano AL, Schneider A, Haapasalo A, Kinhult Ståhlbom A, Tybjærg-Hansen A, Hartmann AM, Spottke A, Corbatón-Anchuelo A, Rongve A, Borroni B, Arosio B, Nacmias B, Nordestgaard BG, Kunkle BW, Charbonnier C, Abdelnour C, Masullo C, Martínez Rodríguez C, Muñoz-Fernandez C, Dufouil C, Graff C, Ferreira CB, Chillotti C, Reynolds CA, Fenoglio C, Van Broeckhoven C, Clark C, Pisanu C, Satizabal CL, Holmes C, Buiza-Rueda D, Aarsland D, Rujescu D, Alcolea D, Galimberti D, Wallon D, Seripa D, Grünblatt E, Dardiotis E, Düzel E, Scarpini E, Conti E, Rubino E, Gelpi E, Rodriguez-Rodriguez E, Duron E, Boerwinkle E, Ferri E, Tagliavini F, Küçükali F, Pasquier F, Sanchez-Garcia F, Mangialasche F, Jessen F, Nicolas G, Selbæk G, Ortega G, Chêne G, Hadjigeorgiou G, Rossi G, Spalletta G, Giaccone G, Grande G, Binetti G, Papenberg G, Hampel H, Bailly H, Zetterberg H, Soininen H, Karlsson IK, Alvarez I, Appollonio I, Giegling I, Skoog I, Saltvedt I, Rainero I, Rosas Allende I, Hort J, Diehl-Schmid J, Van Dongen J, Vidal JS, Lehtisalo J, Wiltfang J, Thomassen JQ, Kornhuber J, Haines JL, Vogelgsang J, Pineda JA, Fortea J, Popp J, Deckert J, Buerger K, Morgan K, Fließbach K, Sleegers K, Molina-Porcel L, Kilander L, Weinhold L, Farrer LA, Wang LS, Kleineidam L, Farotti L, Parnetti L, Tremolizzo L, Hausner L, Benussi L, Froelich L, Ikram MA, Deniz-Naranjo MC, Tsolaki M, Rosende-Roca M, Löwenmark M, Hulsman M, Spallazzi M, Pericak-Vance MA, Esiri M, Bernal Sánchez-Arjona M, Dalmasso MC, Martínez-Larrad MT, Arcaro M, Nöthen MM, Fernández-Fuertes M, Dichgans M, Ingelsson M, Herrmann MJ, Scherer M, Vyhnalek M, Kosmidis MH, Yannakoulia M, Schmid M, Ewers M, Heneka MT, Wagner M, Scamosci M, Kivipelto M, Hiltunen M, Zulaica M, Alegret M, Fornage M, Roberto N, van Schoor NM, Seidu NM, Banaj N, Armstrong NJ, Scarmeas N, Scherbaum N, Goldhardt O, Hanon O, Peters O, Skrobot OA, Quenez O, Lerch O, Bossù P, Caffarra P, Dionigi Rossi P, Sakka P, Hoffmann P, Holmans PA, Fischer P, Riederer P, Yang Q, Marshall R, Kalaria RN, Mayeux R, Vandenberghe R, Cecchetti R, Ghidoni R, Frikke-Schmidt R, Sorbi S, Hägg S, Engelborghs S, Helisalmi S, Botne Sando S, Kern S, Archetti S, Boschi S, Fostinelli S, Gil S, Mendoza S, Mead S, Ciccone S, Djurovic S, Heilmann-Heimbach S, Riedel-Heller S, Kuulasmaa T, Del Ser T, Lebouvier T, Polak T, Ngandu T, Grimmer T, Bessi V, Escott-Price V, Giedraitis V, Deramecourt V, Maier W, Jian X, Pijnenburg YA, EADB contributors, GR@ACE study group, DEGESCO consortium, IGAP (ADGC, CHARGE, EADI, GERAD), PGC-ALZ consortia, Kehoe PG, Garcia-Ribas G, Sánchez-Juan P, Pastor P, Pérez-Tur J, Piñol-Ripoll G, Lopez de Munain A, García-Alberca JM, Bullido MJ, Álvarez V, Lleó A, Real LM, Mir P, Medina M, Scheltens P, Holstege H, Marquié M, Sáez ME, Carracedo Á, Amouyel P, Schellenberg GD, Williams J, Seshadri S, van Duijn CM, Mather KA, Sánchez-Valle R, Serrano-Ríos M, Orellana A, Tárraga L, Blennow K, Huisman M, Andreassen OA, Posthuma D, Clarimón J, Boada M, van der Flier WM, Ramirez A, Lambert JC, van der Lee SJ, Ruiz A. Common variants in Alzheimer's disease and risk stratification by polygenic risk scores. Nat Commun. 2021 Jun 7;12(1):3417. PubMed. Correction.
Schwartzentruber J, Cooper S, Liu JZ, Barrio-Hernandez I, Bello E, Kumasaka N, Young AM, Franklin RJ, Johnson T, Estrada K, Gaffney DJ, Beltrao P, Bassett A. Author Correction: Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer's disease risk genes. Nat Genet. 2021 Apr;53(4):585-586. PubMed.
Make a Comment
To make a comment you must login or register.