Stunning Detail: Single-Cell Studies Chart Genomic Architecture of AD
Quick Links
Why do some people descend into dementia as they age, while others remain sharp? How do genetic variants influence AD pathogenesis? Ditto for the myriad ephemeral states of microglia influence? These are burning questions in AD research these days, and many scientists are looking for answers at the level of single cells. A quartet of papers published September 28 in Cell represents the largest, most comprehensive exploration of transcriptomic and epigenomic landscape of single cells in the AD brain to date. Led by Manolis Kellis and Li-Huei Tsai of the Massachusetts Institute of Technology in Cambridge, the studies plumbed the genomic depths of more than 2 million cells inhabiting the brains of more than 400 people who died at various stages of the disease.
- Four studies examined transcriptomic and epigenomic data at single-cell level in hundreds of postmortem brain samples.
- As AD progresses, DNA damage response skyrockets in neurons.
- In advanced AD, cells lose their grip on epigenomic control, identity.
- AD variants sway gene expression and access to chromatin, mostly in microglia.
- Twelve microglial states tracked through disease progression.
To Kellis, single-cell approaches define a new era of AD research, akin to deploying a more powerful microscope to see molecular features at the heart of the disease.
The first of the four studies creates a transcriptomic atlas in which the authors found evidence that struggling neurons mount frantic responses to DNA damage as AD worsens, and that different types of inhibitory neurons reflect cognitive resilience in the face of AD pathology. Tracking shifts against disease progression in the structure of chromatin, i.e., the epigenome, the second study illustrated how some AD risk variants might operate, and laid bare widespread erosion of epigenomic regulation and cellular identity late in the disease. A third paper showed how double-strand breaks in DNA contribute to this crumbling genomic infrastructure. The fourth study focused on microglia, counting a dozen distinct cell states during disease. It proposes both master transcriptional regulators and therapeutic targets for different stages of disease.
“These extensive transcriptomic and epigenomic datasets illuminate our path forward as we continue our odyssey against AD,” wrote Li Gan, Wenjie Luo, and Wenhui Qu of Weill Cornell Medical School in New York, in an editorial in Cell. “Tremendous promise lies ahead for transformative discoveries that might lead to novel therapeutic strategies—enhancing cognitive resilience, reprogramming microglia, fortifying genomic integrity, and rejuvenating epigenomic landscapes.”
As described in one paper, co-first authors Hansruedi Mathys, Zhuyu Peng, Carles Boix, and colleagues used single-nucleus RNA sequencing to survey the transcriptomes of 2.3 million cells from the postmortem prefrontal cortices of 427 participants in the ROS-MAP longitudinal studies in Chicago (image below). The sample size tops the scientists’ previous snRNA-Seq atlas in this cohort by an order of magnitude (May 2019 news on Mathys et al., 2019). Samples came from 146 controls, 102 people with mild cognitive impairment, and 144 with AD dementia, who were grouped by Aβ plaque load, neuritic plaque burden, and Braak staging of neurofibrillary tangles.

AD Atlas. 427 ROS-MAP participants died at different stages of Alzheimer's pathology (blue) and cognitive impairment (orange). From their prefrontal cortices, researchers sequenced the transcriptomes of 2.3 million cells. [Courtesy of Mathys et al., Cell, 2023.]
What tales did these transcriptomes tell? First, the number of nuclei yielded statistical power to delineate 54 cell types: 14 excitatory and 25 inhibitory neurons, oligodendrocytes, oligodendrocyte precursor cells, three astrocyte subtypes, five immune cell types, including microglia, and several kinds of vascular cells.
Across cells, transcriptomes changed dramatically with increasing loads of AD pathology. For example, in several subtypes of excitatory neuron, transcripts of genes involved in synaptic signaling, chromatin reorganization, neuron projections, and mRNA metabolism increased as the burden of pathology rose. This may seem at odds with the known drop in synaptic function as AD progresses. The researchers surmised that more expression of synaptic genes reflects neurons’ attempts to compensate for synaptic degeneration.
Expression of genes needed for lipid metabolism, autophagy, and mitochondrial function also shifted across multiple cell types with worsening pathological burden. Notably, expression of genes encoding cohesin—a protein complex involved in DNA repair—cranked up in excitatory neurons and oligodendrocytes in late pathological stages, hinting the cells were overwhelmed with damaged DNA.
Mathys and colleagues used this dataset to investigate the cellular and molecular basis of cognitive resilience. They singled out subtypes of inhibitory interneurons that were abundant among those with the highest cognitive function. One, defined by expression of RELN and LAMP5, was overrepresented in people with the highest scores on cognitive tests, and also in people who retained cognitive sharpness despite harboring AD pathology. To Tsai, the findings are particularly interesting in light of the recent discovery of a variant in the RELN gene that protected an autosomal-dominant AD mutation carrier from the disease (May 2023 news).
An Eroding Epigenetic Landscape
Looking to how epigenomics change through the stages of AD, co-first authors Xushen Xiong, Benjamin James, Carles Boix, and colleagues surveyed nuclei from 48 ROS-MAP participants without AD, 29 with early AD, and 15 with late AD, as diagnosed by a combination of pathological and clinical factors. The authors used snRNA-Seq to measure the transcriptomes of 414,000 nuclei, and snATAC-Seq to map stretches of open chromatin in a separate set of 171,000 nuclei. So-called chromatin accessibility peaks reflect areas of active transcriptional regulation, where transcription factors and other machinery can bind. By correlating accessibility peaks with transcription of nearby genes, the researchers mapped cell-type specific epigenetic regulation (image below). On average, 21 peaks correlated with expression of a single gene, underscoring that a complex web of regulation toggles the abundance of a given transcript.
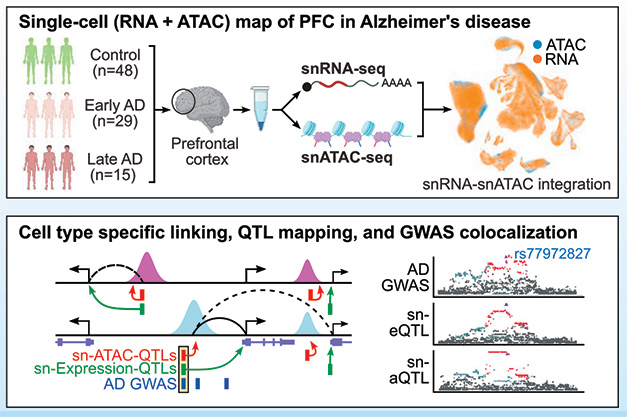
Tying Variants to Peaks to Genes. Integrating snRNA-Seq and snATAC-Seq yields maps of epigenetic regulation within specific cell types (top). Aligning those maps with GWAS variants reveals how the latter influences genomic regulation and AD risk. [Courtesy of Xiong et al., Cell, 2023.]
The researchers used this epigenomic map to probe how AD risk variants work. Because most variants lie in noncoding stretches of the genome, they are thought to modulate expression of nearby genes, but only in specific cell types. Aligning GWAS hits with accessibility maps revealed that many variants landed within stretches of chromatin that were open only in microglia. This supports the idea that microglia play a central role in exerting the effects of AD risk variants.
Interestingly, while many AD variants mapped to stretches of open chromatin, not all affected transcription of nearby genes. Kellis proposed that some genetic variants help open chromatin without immediately influencing gene expression. By “priming” chromatin in this way, these variants may heighten cells’ readiness to transition into new states, he believes. This mechanism may be particularly relevant in microglia, which are constantly “at the ready” as they surveil the brain.

Identity Crisis. In nuclei from late-stage AD samples, a fraction of each cell type becomes de-identified (deid), as its specific signature weakens. For some de-identified cells (grey), the original cell type is unclear. [Courtesy of Xiong et al., Cell, 2023.]
Apart from dissecting potential epigenetic underpinnings of AD heritability, the study unveiled profound cellular identity crises unfolding in late-stage AD. To evoke Auguste Deter's famous complaint to Alois Alzheimer, individual cells appeared to “have lost themselves,” in that the characteristic connections between their gene expression signatures and their chromatin accessibility peaks had loosened. This loss of identity occurred in all cell types; the researchers even spotted some cells whose original identity was untraceable (image at right).
This loss of cell-type specificity coincided with epigenomic erosion, defined as a progressive blurring of the lines between active, inactive, and repressed regions of the genome. In this scenario, typically active regions, such as promoters or enhancers, may shut down, while typically repressed regions may open up. For each cell, the scientists quantified this loss of epigenomic grip with an erosion score. De-identified cells scored highest.
“At the same time that a person is losing their identity due to AD, cells in the brain are doing the same thing,” Kellis told Alzforum.
Vivek Swarup of the University of California, Irvine, sees in this study a paradigm shift in our understanding of the regulatory genomics of AD. “It addresses critical gaps in knowledge—ranging from cell-type-specific regulatory elements to late-stage epigenomic aberrations—thus offering not just mechanistic insights but also revealing potential targets for therapeutic development,” he wrote.
When the Genome Comes Apart
In their study, first author Vishnu Dileep and colleagues zeroed in on structural changes to the genome in AD, including DNA breakage and mismatched repair.

Fusion Spells Trouble. Gene fusion events (top) resulting from inaccurate repair of DNA ultimately lead to fusion transcripts (middle). Such mRNAs turned up in neurons in late-stage AD, and tracked with loss of cellular identity and function (bottom). [Courtesy of Dileep et al., Cell, 2023.]
Among the snRNA-Seq data, the scientists hunted for hybrid transcripts—those with segments derived from two different genes. Such transcripts signify gene-fusion events that result from sloppy repair of double-strand DNA breaks (image below). The researchers found a plethora of such gene fusions in excitatory neurons from people in late-stage AD.
These fused transcripts were most abundant in cells that expressed high levels of the cohesin complex that repairs damaged DNA and was elevated in late-stage AD. Fusion occurred mostly in highly expressed and long genes, as well as in loop anchor regions. These are critical for supporting the three-dimensional architecture of the genome, where distal regulatory regions, such as enhancers, loop around to connect with their target genes. Notably, the number and integrity of these regulatory loops dropped dramatically as AD progressed.
Tsai thinks such genomic acrobatics not only up the odds of DNA breaks, but also render these regions vulnerable to failing DNA repair. The researchers proposed that accumulating DNA damage in AD ultimately overwhelms the repair machinery, leading to the loss of the three-dimensional architecture needed to maintain epigenomic regulation. This, in turn, strips away cellular identity, beckons senescence, and wipes out neuronal function.
Microglia: A Dozen States, Shifting by Stage
Equipped to respond rapidly to changes in the brain environment, microglia are shifty cells by nature. Their ephemeral states are thought to be intimately intertwined with AD risk and progression—but which ones are helpful, which are harmful, and how does that change as disease worsens? To get closer to addressing these questions, Kellis, Tsai, and colleagues focused squarely on microglia in the fourth study. Co-first authors Na Sun and Matheus Victor sequenced the transcriptomes of 194,000 microglial nuclei plucked from 443 postmortem brain samples that came from people who died across the spectrum of AD. The large number of microglia nuclei gave the researchers the power to delineate 12 transcriptional subsets. Based on the unique gene-expression signatures, the scientists identified homeostatic, neuronal surveillance, lipid processing, ribosome biogenesis, phagocytic, stress-related, glycolytic, anti-viral, cycling, and three inflammatory states of microglia (image below).

A Dozen Moods. Microglial transcriptomic signatures fell into 12 unique categories, each assigned with a functional proclivity (left). [Courtesy of Sun et al., Cell, 2023.]
Microglia in all 12 states were found in people across the spectrum of AD, and in multiple regions of the brain. However, the proportion of cells in each state differed by disease stage. Some of the highlights from the analysis? As AD progressed, microglia in lipid processing and inflammatory states increased proportionately, while homeostatic microglia, and those in neuronal surveillance mode, decreased. In a separate analysis, the researchers also identified more than 1,500 genes that changed expression across stages of AD. “Our data supports the concept that inflammation and lipid metabolism are tightly linked processes, and critically involved in the contribution of microglia to AD pathogenesis,” the authors wrote.
Sun and colleagues also gauged how AD risk variants influence expression of target genes in different microglial states and stages of disease. Enticingly, they found that expression of AD risk genes was highest in the microglial states overrepresented in AD brains, including lipid processing and inflammatory states. For example, TREM2 expression skyrocketed in the lipid processing state. In all, they found 35 AD risk genes were highly expressed in at least one microglial state.
Further, they identified transcriptional master regulators that dictate microglial transitions into different states, and investigated their function by tweaking their expression in human induced microglial (iMGL) cultures. For example, they identified a suite of transcription factors— FOXO3, HIF1A, and FOXP2—associated with transition from a weakly to a strongly inflammatory state. By turning down expression of this troika in iMGLs, they prevented this inflammatory leap. Conversely, overexpression of two other transcription factors—PPARG and RUNX—coaxed iMGLs into a more homeostatic state. With these studies, Kellis and Tsai aim to identify therapeutic targets that keep microglia in the most beneficial state for each stage of disease.

Open to Change. While 12 transcriptional states of microglia exist, only three distinct epigenomic states were found. Transcriptional state transitions were governed by master transcription factors, rather than by changes to chromatin accessibility (bottom). [Courtesy of Luo et al., Leading Edge, Cell, 2023.]
Finally, the researchers integrated epigenetic chromatin accessibility maps with transcriptomic data. Surprisingly, in contrast to the dazzling diversity of microglial transcriptomic states, they could delineate only three subsets of microglia based on chromatin accessibility: one homeostatic, and two activated (image above). To the authors, the lack of congruence between transcriptomic and epigenomic states in microglia could reflect the perpetual openness of the cells to respond to dynamic changes in their environment, and suggests that master regulator transcription factors, rather than epigenomic rearrangements, govern these rapid microglial transitions.
To Marta Olah of Columbia University in New York, this disconnect between the transcriptome and epigenome was the most intriguing finding of the study. “If confirmed, it could suggest that non-resting/activated microglia may maintain a permissive chromatin accessibility architecture that could allow dynamic state transitions to occur in response to changes in the microenvironment, and, indeed, in response to therapeutic targeting.”—Jessica Shugart
References
News Citations
- When It Comes to Alzheimer’s Disease, Do Human Microglia Even Give a DAM?
- Reelin Variant Wards Off Dementia in Colombian Kindred Siblings
Paper Citations
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019 Jun;570(7761):332-337. Epub 2019 May 1 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Mathys H, Peng Z, Boix CA, Victor MB, Leary N, Babu S, Abdelhady G, Jiang X, Ng AP, Ghafari K, Kunisky AK, Mantero J, Galani K, Lohia VN, Fortier GE, Lotfi Y, Ivey J, Brown HP, Patel PR, Chakraborty N, Beaudway JI, Imhoff EJ, Keeler CF, McChesney MM, Patel HH, Patel SP, Thai MT, Bennett DA, Kellis M, Tsai LH. Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer's disease pathology. Cell. 2023 Sep 28;186(20):4365-4385.e27. PubMed.
- Xiong X, James BT, Boix CA, Park YP, Galani K, Victor MB, Sun N, Hou L, Ho LL, Mantero J, Scannail AN, Dileep V, Dong W, Mathys H, Bennett DA, Tsai LH, Kellis M. Epigenomic dissection of Alzheimer's disease pinpoints causal variants and reveals epigenome erosion. Cell. 2023 Sep 28;186(20):4422-4437.e21. PubMed.
- Dileep V, Boix CA, Mathys H, Marco A, Welch GM, Meharena HS, Loon A, Jeloka R, Peng Z, Bennett DA, Kellis M, Tsai LH. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell. 2023 Sep 28;186(20):4404-4421.e20. PubMed.
- Sun N, Victor MB, Park YP, Xiong X, Scannail AN, Leary N, Prosper S, Viswanathan S, Luna X, Boix CA, James BT, Tanigawa Y, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, Tsai LH, Kellis M. Human microglial state dynamics in Alzheimer's disease progression. Cell. 2023 Sep 28;186(20):4386-4403.e29. PubMed.
Follow-On Reading
Papers
- Luo W, Qu W, Gan L. The AD odyssey 2023: Tales of single cell. Cell. 2023 Sep 28;186(20):4257-4259. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of California, Irvine
The paper by Xiong et al. represents a seminal contribution to our understanding of Alzheimer's disease from a functional genomics perspective, elucidating previously unrecognized aspects of the disease's molecular etiology. One of the most significant advances lies in the comprehensive mapping of the brain regulome, derived from the prefrontal cortexes of 92 individuals. By employing single-cell resolution for epigenomic and transcriptomic landscapes, the authors were able to resolve cell-type-specific regulatory modules and peak-to-gene links, thus overcoming the limitations posed by earlier bulk tissue analyses.
Of paramount interest is the identification of AD risk loci enrichment in microglial enhancers and in specific transcription factors, such as SPI1, ELF2, and RUNX1. This not only helps in understanding the transcriptional regulatory circuitry of AD but also paves the way for targeted therapeutic interventions. Furthermore, the discovery of 9,628 cell-type-specific ATAC-QTL loci, integrated alongside peak-to-gene links, provides an unprecedented depth to the study of AD variant regulatory circuits, thereby presenting avenues for identifying causal links between genetic variants and their regulatory roles in pathogenesis.
The paper is particularly illuminating when discussing the phenomenon of epigenomic erosion and loss of cell identity in late-stage AD. The use of advanced, single-cell technologies allowed the authors to distinguish “de-identified” cells with eroded cellular identities across various major brain cell types in late AD, demonstrating that this is a feature intrinsic to disease progression rather than a technical artifact. Strikingly, this epigenomic erosion was shown to manifest as a global dysregulation of the epigenome, corroborated by integrated erosion scores and changes in three-dimensional genome organization, suggesting a profound loss of cellular function and identity as AD progresses.
In summary, this paper provides a paradigm shift in our understanding of the regulatory genomics of Alzheimer's disease. It addresses critical gaps in knowledge—ranging from cell-type-specific regulatory elements to late-stage epigenomic aberrations—thus offering not just mechanistic insights but also revealing potential targets for therapeutic development. The study elevates our comprehension from isolated genetic loci to a more holistic view, focusing on the interplay between genetic, epigenetic, and transcriptomic landscapes in the context of Alzheimer's disease.
Columbia University Medical Center
Sun et al. provide a comprehensive assessment of human microglia phenotypes in the aged and Alzheimer’s disease brain. Utilizing the data-rich ROS/MAP cohort the authors established the presence of 12 different microglia transcriptional states, identifying gene sets that are differentially expressed within each microglia subset at different stages of AD, measuring changes in the relative abundance of microglia subsets with AD progression, and establishing relationships between microglia subset abundances and clinicopathological traits, all of which will serve as a great resource for the scientific community.
Importantly, the authors found good correlation between microglia phenotypes and ones described previously that were based on single cell RNA-seq, an approach that has been shown to be better suited to capturing microglial phenotypes than single nucleus approaches (Thrupp et al., 2020). Despite this, while the prior scRNA-seq study (Olah et al., 2020) identified one inflammatory state, the current study was able to identify three. These were governed by the same transcription factors, but had unique gene sets, and likely constituted phenotypes at different stages along a microglial inflammatory activation trajectory. The ability to detect transitioning microglia states was most likely enabled by the much larger sample size, justifying the generation of ever-larger datasets to increase resolving power.
Nevertheless, perhaps the most intriguing finding of this study is the observation that single-nucleus chromatic accessibility does not recapitulate the transcriptionally defined states very well, beyond the ability to discern a homeostatic state from one to two activated states. Studies usually find strong correlation between chromatin accessibility (as assayed by ATAC-Seq) and gene expression, so it is particularly interesting that this is not the case here. These findings will need to be further corroborated using orthogonal approaches (or better technology) to exclude the possibility that the lack of resolution comes from the sparseness of the snATAC-Seq data. Nonetheless, if confirmed, it could suggest, as the authors concluded, that non-resting/activated microglia may maintain a permissive chromatin accessibility architecture that could allow dynamic state transitions to occur in response to changes in the microenvironment, and, indeed, in response to therapeutic targeting. This retained microglial plasticity is the conceptual prerequisite and cornerstone of any future effort to devise therapeutic approaches that target individual microglia subsets to fine-tune the microglial population to maintain or regain tissue homeostasis.
References:
Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, de Strooper B, Fiers M. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020 Sep 29;32(13):108189. PubMed.
Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, Grün D, Kroshilina AA, Dionne D, Sarkis RA, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Prinz M, Vonsattel JP, Teich AF, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nat Commun. 2020 Nov 30;11(1):6129. PubMed.
Make a Comment
To make a comment you must login or register.