Held in the historic Austrian capital, the 13th AD/PD conference reflected a rapidly growing field. The meeting jammed science into five parallel sessions, with 545 talks running from early morning till late evening and some 1,200 posters vying for attention. There were no show-stopping announcements, but researchers noted biomarker advances on both the PET and CSF front, as well as a flourishing variety of basic science talks on tau, TREM2, epigenetics, and other topics. On the clinical side, one company touted a successful Phase 3 trial for transcranial magnetic stimulation to treat Alzheimer’s, but potential disease-modifying therapies for both AD and PD remain in development.
Next-Generation Tau PET Tracers Strut Their Stuff
The current crop of tau PET tracers have yielded new insights into the progression of Alzheimer’s disease, but they are plagued by problems. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Austria’s capital, Vienna, researchers debuted new tracers that appear at first glance to be able to overcome the limitations of the earlier compounds. In general, the newcomers boast higher brain uptake and more specific binding, yielding cleaner-looking scans with sharper distinction between positive and negative findings. While the older tracers work only in AD, some of the new ones appear to light up other tauopathies, as well. Researchers at Piramal Imaging wowed the crowd with scans showing a distinct, specific pattern of binding of their tracer in progressive supranuclear palsy (PSP) compared to AD. Earlier this year, researchers from Merck reported on their tau PET tracer, MK-6240, at the Human Amyloid Imaging (HAI) meeting held January 11-13 in Miami Beach, Florida. Like Piramal’s, Merck’s tracer is being readied for broad distribution. Researchers from Genentech and Roche reported longitudinal data for their in-house tracers in Phase 1 tests, while Janssen scientists at AD/PD showed massive brain uptake of their candidate in preclinical studies.
All five new tracers are at an early stage of development, but even as they undergo validation, clinicians across the field are urgently casting about for tau PET tracers to use in their observational and drug studies. They are in a pinch because the most validated tracer thus far, Lilly/Avid’s AV-1451 (aka T807, flortaucipir), is either unavailable or unaffordable for most groups. Another hopeful fizzled out thanks to off-target binding in key regions, and some of the new tracers are being developed by drug companies for internal and academic use. This situation leaves clinicians scrambling for a ligand to image tau pathology in their participants, with many now eyeing Merck’s and Piramal’s.
Researchers at AD/PD applauded the advent of more options. They called the preliminary data from the new batch promising, while cautioning that data from larger cohorts and in additional tauopathies are needed to verify how well these work in practice. “I am excited to see so many companies developing tau tracers. I hope to use one of these to develop new anti-tau treatments,” Philip Scheltens of VU University Medical Center, Amsterdam, wrote to Alzforum. At HAI, Bill Jagust of the University of California, Berkeley, quipped, “My favorite tracer is always the one I have not yet seen,” alluding to PET experts’ general experience that new tracers tend to start out looking great, until more scans in more people turn up their limitations.
Distinct Diseases. Piramal’s PI-2620 tracer has different uptake patterns in AD (top) and PSP (bottom). [Courtesy of Andrew Stephens.]
First Generation Tracers: A Mixed Track Record
Though imperfect, tau PET has already begun to transform the field. Studies to date, done mostly with AV-1451, have found that tau ligand binding matches Braak staging, advances with disease stage, and correlates with cognitive deficits (see Aug 2016 conference news; Aug 2016 news). Researchers are especially excited by emerging data hinting that tau tracer uptake increases rapidly as disease progresses, well within the detection time of a clinical trial, and that it appears to track cognitive decline early on in disease.
At the same time, researchers note problems, such as off-target binding in the choroid plexus and striatum, and low sensitivity for distinguishing AD cases from healthy controls, that stem from a relatively small dynamic range (see Feb 2016 conference news; Feb 2016 conference news). While AV-1451 binds to the paired helical fragments found in AD and in certain tau mutations causing frontotemporal dementia (FTD), this tracer does not recognize the three-repeat (3R) and four-repeat (4R) tau isoforms that predominate in tauopathies such as Pick’s disease, PSP, corticobasal degeneration (CBD), and most cases of frontotemporal dementia (see Sep 2016 conference news). AV-1451 also binds only weakly to tau in chronic traumatic encephalopathy (CTE).
Another existing tau tracer has been felled by nonspecific binding. This is THK5351, discovered at Tohoku University in Sendai, Japan, and licensed by GE Healthcare for commercial distribution. At this year’s HAI meeting, three groups reported THK5351 binding to the enzyme MAO-B, possibly on astrocytes. This clouds interpretation of the scans. Tohoku’s Ryuichi Harada reported that THK5351 bound to recombinant MAO-B in vitro and was displaced by an MAO-B inhibitor in basal ganglia of a human control. Qi Guo from AbbVie in Chicago showed that MAO-B accounted for up to 70 percent of the THK5351 signal in competitive binding and displacement studies in cynomolgus monkeys and homogenate of human AD entorhinal cortex. And in Pedro Rosa-Neto’s lab at McGill University in Montreal, the MAO-B inhibitor selegiline wiped out up to half of the prior THK5351 signal in four study participants with mild cognitive impairment or PSP.
“THK5351 has so much off-target binding that its value as a tau tracer is compromised,” Keith Johnson of Massachusetts General Hospital said at HAI. Other PET experts put it more bluntly: “Substantial off-target binding in a target region is the kiss of death,” said Chet Mathis of the University of Pittsburgh School of Medicine. Adding to the tracer’s woes, researchers at Janssen Pharmaceutica, Beerse, Belgium, reported on a poster at AD/PD that, in their hands, THK5351 also bound to aggregated Aβ in AD brain slices. Clinical researchers across the field who had been planning to use THK5351 held their horses once its MAO-B binding became apparent, and are seeking an alternative.
Goodbye GE, Hello … Piramal? Merck?
In Vienna, Andrew Stephens at Piramal Imaging, Berlin, Germany, described his group’s search for tau ligands. In collaboration with AC Immune, Lausanne, Switzerland, they screened compounds for binding to AD brain homogenate or to synthetic tau paired helical fragments. Their lead, PI-2620, bound to brain regions with high tau tangles but gave no signal from non-demented control brains. Neither did it bind Aβ fibrils. PI-2620 had but low affinity for MAO-B, or to MAO-A for that matter, the related enzyme that had temporarily mired the tracer AV-1451 in controversy until researchers agreed that AV-1451’s MAO-A binding in humans was of too low affinity to interfere with measuring tau. Importantly, PI-2620 bound strongly to 3R tau from Pick’s and 4R tau from PSP brains, Stephens said. The tracer also demonstrated suitable pharmacokinetic properties. In wild-type mice and nonhuman primates, it entered the brain well and washed out within an hour. PI-2620 is Piramal’s second tau tracer, replacing a weaker candidate called MNI-815.
Based on these findings, Piramal started a Phase 1 trial on four people with AD, three with PSP, and two healthy controls. It was run by John Seibyl of Molecular NeuroImaging, an imaging services company in New Haven, Connecticut, with PI-2620 receiving the designation MNI-960 for clinical testing. As in animal studies, the researchers saw robust brain uptake, fast washout, and little signal in the pertinent brain regions of controls. The signal plateaued 60 to 90 minutes after tracer injection, Seibyl noted in Vienna. Typical SUVRs for AD patients ran from 2.5 to 2.8.
Notably, AD and PSP scans revealed distinct patterns (see image above). In PSP, only a few discrete regions, mainly the pallidum and substantia nigra, lit up. In contrast, AD patients took up tracer in broader areas known to accumulate tau tangles, such as the lateral temporal lobe, hippocampus, entorhinal cortex, and precuneus.
Curiously, one of the AD patients had a negative tau scan. Stephens noted this patient had mild AD, with an MMSE of 26, and may not have accumulated much pathological tau yet. Incidentally, other PET experts, too, noted that as more research groups image both amyloid and tau pathology in the same cognitively impaired people, they are finding a few whose scans are amyloid-positive but tau-negative.
Importantly, the researchers saw no uptake of PI-2620 in choroid plexus, amygdala, or striatum, where other tracers have off-target binding. In particular, choroid plexus uptake can be nettlesome because this structure sits atop the hippocampus, hence uptake there could bleed into this important region and confound tau measurement. Overall, AD patients looked markedly different from controls in temporal regions, suggesting the tracer discriminates well between cases and controls, Stephens added.
Liana Apostolova of Indiana University, Indianapolis, noted that the absence of nonspecific binding in choroid plexus represents a significant advantage of this tracer over others. She was also impressed by the solid cortical binding in AD and specificity of the binding in PSP. “So far, PI-2620 is looking very good,” she wrote to Alzforum. Victor Villemagne of the University of Melbourne, Australia, who chaired the AD/PD session, said, “[PI-2620] truly represents a second generation of tracers.”
The data also raised questions. Some patients displayed an asymmetric pattern with more uptake on one side of the brain. “The heterogeneity of the tau distribution and load in patients who are β-amyloid positive is perhaps unexpected,” Stephens wrote to Alzforum. More research is needed to determine how this variable regional and temporal tangle accumulation relates to cognitive decline and amyloid, he noted.
At AD/PD, audience members observed that PI-2620 appears to bind to the eyes and to bone at the margins of the skull. Stephens said the eye signal might represent binding to melanin or other pigments. He speculated that tracer accumulating around the skull is not detecting bone or meninges, but might reflect a PI-2620 metabolite in blood that cannot enter brain. A meningeal signal is unwelcome if it is large enough to spill into the top of the cortex, a region of interest.
A healthy elderly volunteer (top) whose amyloid scan was negative showed low, homogeneous MK-6240 uptake across the brain, without a signal in hippocampus or medial temporal lobe. An older AD patient whose amyloid scan was positive showed high MK-6240 uptake in known tau pathology regions such as the medial temporal lobe and inferior temporal cortex. [Courtesy Jeffrey Evelhoch.]
Merck: Alternative for Wide Distribution?
If Piramal’s tracer holds up in further study, it would be sold to any drug developer or clinician, but it has competition. The pharma giant Merck has developed a tau tracer, called MK-6240, which has completed one Phase 1 trial and is recruiting for another. At HAI last January, Cyrille Sur, Jeffrey Evelhoch, and colleagues at Merck in West Point, Pennsylvania, presented data on the first use of 18F MK-6240 in people. By that point, Merck had studied the compound in about 10 middle-aged adult controls and 10 people with AD. They worked with researchers in Leuven, Belgium, and at the Boston-based imaging CRO InviCRO, in collaboration with Biogen, which has licensed this tracer for use in its aducanumab and other Biogen trials.
MK-6240 showed uptake in the brain regions consistent with Braak stage, including the parahippocampus, medial temporal cortex, and amygdala. Comparing the new tracer primarily to the standard bearer AV-1451, Cyr said that MK-6240 has a wider dynamic range. In addition, its higher affinity for neurofibrillary tangles means it requires lower doses, making it easier to run baseline and follow-up scans within a year, Cyr said. In Europe especially, radiation exposure laws can limit the repeat PET scans required for longitudinal studies or clinical trials.
Cyr further reported that in monkey studies and the initial human trial, MK-6240 showed none of the off-target binding in the choroid plexus and striatum seen with AV-1451, though other PET experts cautioned that additional research in older controls might yet turn up such binding. Thus far, the only overt off-target binding appears to be in a left leptomeningeal region, which could get in the way if it bleeds into the cortex there. Researchers who saw MK-6240 data at last year’s AAIC were cautiously optimistic (Aug 2016 conference news).
Both the preclinical characterization of this compound, as well as information on how it can be synthesized for widespread research use, are formally published (Hostetler et al., 2016; Collier et al., 2017).
Tracking Progression.
Genentech's GTP1 tau tracer reveals an increase in tau accumulation in a person with mild AD (right column) over six months. [Courtesy of Sandra Sanabria Bohorquez.]
Importantly for the field, Merck wants MK-6240 to become broadly available. “We want our tracer to get out there for the community to use in therapeutic trials,” Evelhoch told Alzforum. Merck routinely develops PET tracers for target engagement of its investigational drugs, but as a pharma company does not take on their scale-up, setup of distribution centers, and commercialization. Therefore, Merck in January 2017 licensed clinical development and sale of MK-6240 to Cerveau Technologies, a partnership between the Toronto-based company Enigma Biomedical Group and the Beijing-based Sinotau Pharmaceutical Group. In the months since, Cerveau has signed a manufacturing agreement with Siemens’ PETNET, and research and validation agreements with Rosa-Neta at McGill, Sterling Johnson at the Wisconsin Alzheimer’s Disease Research Center, Chris Rowe at Austin Health in Melbourne, with Biogen, and with Sinotau for development in China. Johnson told Alzforum that he was going to use THK5351 but switched to a combination of AV1451 and MK-6240 after the HAI meeting.
Genentech, Roche, Janssen: New In-House Tracers
Brain imaging researchers agree that the trouble obtaining AV1451 has spurred a welcome burst of international effort to come up with alternatives, and recent conferences featured updates on three more such programs. Researchers at Genentech, South San Francisco, are taking their next-generation tau tracer GTP1 through an 18-month longitudinal Phase 1 study. Last summer, Genentech’s Sandra Sanabria Bohorquez reported that the tracer signal intensified in the temporal lobes and hippocampi of AD patients over six to nine months, suggesting it is sensitive to small changes in tau load (see Aug 2016 conference news). At the time, the cohort consisted of six controls, six people with prodromal AD, and 10 mild to moderate AD patients. Adding new data, Sanabria Bohorquez in Vienna reported on 14 controls, 13 people with prodromal, and 25 with mild to moderate AD. The tracer signal matched the expected distribution of tau tangles at increasing Braak stages, and well distinguished cases from controls, and prodromal from mild to moderate, she reported. In general, people who have a higher amyloid plaque load also retain more GTP1.
As before, the GTP1 signal picked up changes over six to nine months in people with AD. This is important because it hints that GTP1 might track progression and possibly treatment response within the timeframe of a Phase 2 trial. At HAI, Sanabria reported that this tracer does not bind MAO-A or B. Similar to AV1451, in some subjects GTP1 does generate a signal in the basal ganglia that may be due to age-related changes other than tau. Genentech is building distribution centers to start using GTP-1 in therapeutic trials, including the crenezumab trials in sporadic disease (CREAD1 and 2) and Paisa mutation carriers in Colombia. Sanabria said Genentech will share its tracer with academic investigators, but currently has no plans to distribute it commercially on a larger scale.
At HAI, Mike Honer, Edilio Borroni, and colleagues at Roche presented the latest on their company’s tau tracer. RO6958948 is structurally similar to T807 (AV1451/flortaucipir), and it behaves similarly, too. In vitro binding studies on fresh-frozen tissue sections from people who had died with AD or a range of other tauopathies showed that RO6958948 binds tau aggregates in AD and in select tau mutation cases, as does AV1451. RO6958948 does not bind robustly in tissue from people who had Pick’s disease, PSP, or CBD. Also at HAI, Dean Wong of Johns Hopkins University in Baltimore, who collaborates with Roche in evaluating this tracer clinically, reported on the first four AD cases from a Phase 1 follow-up study of RO6958948. For all four people, MMSE score worsened between their baseline and follow-up scans some eight to 22 months later; for three of those four, tau PET SUVR values nudged up, as well. This trial is ongoing.
Borroni told Alzforum that Roche plans to use this tracer to help evaluate its investigational AD treatments, and would make it available to academic investigators and to international initiatives such as EPAD (see Aug 2015 conference news).
Last but not least, Janssen is getting into the action with a ligand that appears headed to Phase 1. In Vienna, Diederik Moechars showed that his team searched for new tau ligands by screening compounds for their ability to out-compete the original ones, T808 and T807. Janssen’s lead candidate, JNJ-067, bound tau tangles extracted from Braak stage 5 and 6 AD brains more strongly, and was more selective for tau over Aβ fibrils. It detected tau in AD brain slices. JNJ-067 had no affinity for MAO-A and low affinity for MAO-B, with binding starting only at concentrations of 1 μm or higher. It entered the brain readily and washed out quickly in a rhesus monkey. Notably, in this primate brain, uptake of JNJ-067 dwarfed uptake of T807, with a five times higher peak, while still washing out faster. If these properties hold up in humans, they could eventually shorten needed scan times with JNJ-067 compared to current tracers. Janssen is planning to take JNJ-067 into phase 1 this year but has not yet decided how to make it available, Moechars told Alzforum.
For the time being, however, Villemagne cautioned that other tau tracers have shown marked discrepancies between their preclinical profile and imaging in people. “Given this, it would be prudent to wait for the first human studies to see how it performs,” he wrote to Alzforum.—Madolyn Bowman Rogers and Gabrielle Strobel
This story was updated on 18 April 2017 to correct an error.
Cerebrospinal fluid biomarkers of Alzheimer’s disease have transformed clinical research, but transferring this technology to the clinic has not been easy. Even after intense efforts at quality control, biomarker readings using laboratory-grade assays swing widely between different labs, batches, and runs. Part of the problem can be solved by turning to automated systems, which tighten up biomarker readings from different runs within a few percent of each other. Even so, research labs still set their own biomarker cutoffs due to differences in the pre-analytical handling of samples. This made it impossible to agree on a single global cutoff for a biomarker level that indicates disease, as will be necessary to advance these markers into widespread diagnostic use.
The goal of a single cutoff has just gotten closer. Now, for the first time, researchers have validated a biomarker cutoff obtained in one cohort in a second, independent cohort. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, Austria, Oskar Hansson of Lund University, Sweden, described how cutoffs initially set in the Swedish BioFINDER (Biomarkers for Identifying Neurodegenerative Disorders Early and Reliably) study were transferred to ADNI.
The Swedish group first derived a correction factor based on differences in how the respective BioFINDER and ADNI protocols handle CSF before plunking the tubes into a machine for analysis. Then the researchers used an automated assay to determine the cutoffs that predicted amyloid positivity in the BioFINDER cohort, applied the correction factor, and found that the same cutoffs correctly identified ADNI participants with brain amyloid accumulation. This represents the first clinical validation of automated CSF assays, Hansson noted. “These assays can now be considered for routine clinical use,” he told Alzforum.
The talk was enthusiastically received by both academic and pharmaceutical researchers in attendance. “It’s a very impressive study,” said Michael Weiner of the University of California, San Francisco, who oversees ADNI. John Sims of Eli Lilly in Indianapolis noted that this work will facilitate the running of worldwide trials. Right now, trials have to ship all CSF samples to the same analytical site to reduce measurement variability, greatly increasing time and expense.
Robot Repeatability.
Use of automated systems like this one to analyze CSF samples has brought variation down to within standards of routine clinical chemistry. [Courtesy of Tobias Bittner, Roche.]
The development is the latest advance in an ongoing, years-long international effort to improve and standardize CSF measurements. Kaj Blennow of the University of Gothenburg, Sweden, leads the Global Biomarker Standardization Consortium. The consortium previously reported that despite its best efforts to standardize CSF assay protocols, identical samples run at different centers still vary by around 20 percent (see Mattsson et al., 2013). Using an automated system can bring this inter-lab variability down below 4 percent, meeting diagnostic standards (see Aug 2015 conference news). The machines do nothing to standardize the pre-analytical handling of samples, however.
To tackle this source of variation, Hansson, Blennow, and colleagues partnered with Roche Diagnostics, Basel, Switzerland. Roche makes the Elecsys immunoassays for Aβ42, total tau, and phosphotau. They run on the Cobas E601 machine that is used for routine clinical analyses across medicine, including cardiovascular care, diabetes, oncology, and infectious disease (see image above). Readings on this instrument have been shown to correlate closely with results obtained by a mass-spectrometry-based reference measurement protocol (see Bittner et al., 2016). Other companies, including Fujirebio Diagnostics, have developed similar systems.
Hansson and colleagues selected 277 BioFINDER participants with mild cognitive symptoms who had undergone both lumbar punctures and amyloid PET scans with the tracer flutemetamol. CSF samples were analyzed by Elecsys to find the cutoffs that separated amyloid-positive from amyloid-negative participants with 90 percent sensitivity. For Aβ42, this value turned out to be 1,100 pg/ml. While values lower than this reliably picked up people with brain amyloid, this cutoff also produced numerous false positives, with a diagnostic specificity of only 72 percent. By taking the ratio of either total tau or phosphotau to Aβ42 that achieved 90 percent sensitivity instead, the researchers improved the diagnostic specificity to 89 percent. The p-tau/Aβ42 cutoff was 0.022.
Notably, this 1,100 pg/ml Aβ42 cutoff is almost six times greater than the previously reported value of 192 pg/ml, obtained with the AlzBio3 assay, which distinguished people with autopsy-confirmed AD from healthy controls (see Aug 2010 news). Tobias Bittner of Roche attributes the different numbers to differences in standardization of the requisite assays. Elecsys was standardized to the LCMS-based reference method (Leinenbach et al., 2014), whereas the Alzbio3 assay was standardized based on weighted Aβ42 material. “Both cutpoints reflect the perfect clinical separation of amyloid-positive from amyloid-negative samples; just the numerical values are different,” Bittner wrote to Alzforum.
The Swedish researchers then compared CSF handling protocols between BioFINDER and ADNI. In Vienna, Hansson noted that because Aβ sticks to the sides of plastic tubes, a large amount of the protein can be lost just by aliquotting it into smaller tubes. A poster from Eline Willemse of VU University Medical Center, Amsterdam, in collaboration with the diagnostics company Euroimmun in Luebeck, Germany, corroborated this, reporting a 5-10 percent loss of Aβ every time CSF is transferred between tubes. Notably, the ADNI protocol involved many more transfer steps than BioFINDER’s. To quantify the loss, the researchers performed a small study on CSF obtained from 20 people undergoing lumbar punctures to treat normal-pressure hydrocephalus. The researchers split each sample in half and processed one half according to the BioFINDER protocol, the other ADNI’s. The ADNI protocol reliably produced peptide values about 20 percent lower than BioFINDER’s did. Thus, the researchers obtained a correction factor of 0.8 for converting Aβ42 cutoffs from BioFINDER to ADNI. Tau levels, on the other hand, did not significantly vary between the two cohorts.
Applying the correction factor, the researchers obtained cutoff values for the ADNI cohort of 880 pg/ml Aβ42, or a p-tau/Aβ42 ratio of 0.028. They used these cutoffs to stratify 646 ADNI participants who had either subjective memory complaints, mild cognitive impairment, or AD. All had undergone florbetapir amyloid PET scans. The corrected BioFINDER p-tau/Aβ42 cutoff separated ADNI amyloid-positive and amyloid-negative participants with a sensitivity of 88 percent and a specificity of 93 percent. The findings demonstrate that cutoffs from one cohort can be used diagnostically in another, Hansson said.
In both cohorts, the p-tau/Aβ42 and t-tau/Aβ42 ratios agreed with amyloid PET imaging 90 percent of the time. This is the maximum possible concordance with PET, since scans read by different radiologists agree only 90 percent of the time. The data establish automated CSF biomarker assays as equivalent to a PET visual read, Bittner told Alzforum. The Swedish study used visual reads rather than SUVRs because the former are approved for diagnostic use. However, SUVR data produce similar results, Hansson noted in Vienna.
While the use of a conversion factor allowed comparison of BioFINDER and ADNI data, researchers agree that the ultimate goal will be to have a standardized protocol for handling CSF samples. The Global Biomarker Standardization Consortium is developing such a unified sample handling protocol. Preferably, the protocol should involve as few steps as possible. Bittner suggested that, ideally, CSF would be collected in a single tube made of low-binding plastic, and then that tube would be plopped into the machine for analysis, with no transfers, freezing, centrifugation, or other manipulation of the sample. When a clinic needs to ship samples to a lab for analysis, they could be shipped fresh within 24 hours, Bittner added.
Benchtop Devices.
Companies such as Fujirebio (pictured) and Euroimmun make benchtop analyzers that run AD biomarker immunoassays. [Courtesy of Madolyn Rogers.]
This scheme would be practical because the automated systems are already in widespread use in clinics and labs worldwide. Bittner boasted that Roche has around 20,000 units in use globally, with no location more than 24 hours shipping time away from a unit.
Competing companies make similar systems. Fujirebio Diagnostics in Malvern, Pennsylvania, which acquired the Innogenetics CSF assays, runs them on a benchtop machine called Lumipulse G600II (see photo above). Hansson has worked with this system and reports similar precision to the Roche automated assays. Euroimmun also has a benchtop device, the EUROIMMUN RA Analyzer 15, that reads chemiluminescence immunoassays for Aβ42 and Aβ40.
All three companies are racing to get their AD assays to market. Roche expects to release its Aβ42 assay in June 2017, and its tau assays by the end of the year. Fujirebio reports that its assays for Aβ42 and total tau are already available in Europe, with phosphotau and Aβ40 in development. Euroimmun has finalized the technical validation for Aβ42 and Aβ40, and plans to have tau assays ready by year’s end as well. The Euroimmun assays use antibodies developed by its partner, ADx Neurosciences. All three companies will put a CE mark on their assays, indicating conformity with European standards, and are looking into requirements for approval by the U.S. Food and Drug Administration.
In addition, all three companies are validating the tests’ performance against patient samples. Manu Vandijck at Fujirebio said they have defined a normal range for Aβ42 in healthy subjects (see Wallin et al., 2015). Typical Aβ42 values in people with mild cognitive impairment or subjective cognitive decline were determined in the BioFINDER cohort, and that data has been submitted for publication, Vandijck added. Fujirebio is running studies on the other biomarkers, and also correlating CSF results with PET scanning. Britta Brix at Euroimmun noted that the company’s automated assays correlate almost perfectly (99 percent) with its manual ELISAs. Euroimmun is confirming that the biomarker cutoffs established for its ELISAs hold for the automated assays using clinical sample sets from Germany and elsewhere.
Tanja Schubert of Bioclinica Lab, a biomarker analysis company headquartered in Doylestown, Pennsylvania, performed some of the technical validation on the Euroimmun assays. To her mind, automated systems have an added advantage in that they can easily and cheaply run single patient samples. With ELISAs, each run requires a new kit, making it quite expensive to run small groups of samples, and clinical sites often wait with analysis until they have enough samples to fill a kit. The ability to run small samples quickly makes screening of participants for clinical trials more efficient, Schubert said.
One final piece to the puzzle of preparing CSF biomarkers in routine medical care will be the development of a standard CSF reference material against which all groups can calibrate their tests. That is in progress and expected to be ready later this year (see Oct 2015 news; Kuhlmann et al., 2016).—Madolyn Bowman Rogers
Kuhlmann J, Andreasson U, Pannee J, Bjerke M, Portelius E, Leinenbach A, Bittner T, Korecka M, Jenkins RG, Vanderstichele H, Stoops E, Lewczuk P, Shaw LM, Zegers I, Schimmel H, Zetterberg H, Blennow K, IFCC Working Group on Standardization of CSF proteins (WG-CSF).
CSF Aβ1-42 - an excellent but complicated Alzheimer's biomarker - a route to standardisation.
Clin Chim Acta. 2016 May 20;
PubMed.
ApoE and Tau: Unholy Alliance Spawns Neurodegeneration
ApoE4 doesn’t need to interact with Aβ to wreak havoc in the brain. Tau tangles can bring out the apolipoprotein’s bad side too, according to findings presented at the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29-April 2 in Vienna. David Holtzman of Washington University in St. Louis described a nexus of destruction in P301S-tau mice, reporting massive neurodegeneration in those that expressed the human ApoE4 allele. Without amyloid there, how would this work? The researchers found that ApoE4 jacked up the microglial response to tau pathology, which may have transformed astrocytes from neuronal supporters into neurotoxic entities. While the cause and effect relationships between these players are still fuzzy, it may soon be possible to define them in cell culture, as other researchers at the meeting reported they could generate bona fide astrocytes and microglia from human induced pluripotent stem cells (iPSCs).
Eloise Hudry of Massachusetts General Hospital in Boston commented that Holtzman’s work addressed a crucial missing link in the neurodegenerative disease field, i.e., the relationship between ApoE and tau. “This pioneering work stimulates people to ask more questions,” she told Alzforum. She added that the findings add fuel to growing excitement about new functions of ApoE, including its role as a transcription factor and in synaptic pruning (see Sep 2016 news; Jan 2017 news).
ApoE is known for influencing production and clearance of Aβ (see Jul 2011 webinar; Apr 2013 news; May 2014 news). However, several studies hint that the apolipoprotein may also affect neurodegenerative processes—to which tau pathology is closely linked—independently of Aβ. For example, AD patients carrying the ApoE4 allele generally have more tau and p-tau in the cerebrospinal fluid, even after correcting for their Aβ42 levels (see Apr 2013 news; Deming et al., 2017). ApoE4 also generally comes with greater brain atrophy in people with frontotemporal dementia, many of whom have tau but not Aβ pathology (see Agosta et al., 2009). Given the close relationship between tau pathology and neurodegeneration, Holtzman hypothesized that different ApoE isoforms may respond to or influence tau pathology differently.
To investigate this, graduate student Yang Shi crossed P301S mice, which express a tangle-forming FTD mutant of tau, to various ApoE knock-ins, as well as to ApoE knockout mice. To Shi and Holtzman’s great surprise, P301S mice expressing ApoE4 had profound neurodegeneration by nine months of age. Their brains were 25 percent smaller than those of the ApoE3 or ApoE2 mice. Animals without ApoE barely had neurodegeneration, suggesting that all ApoE alleles to some degree promote neuronal death in the presence of tauopathy.
Hypothesizing that ApoE4 may ramp up tau accumulation, Shi ran various biochemical and histopathological experiments to measure tau in the different mouse strains. However, she found only minor isoform-dependent differences between them, certainly not enough to explain the neurodegenerative effect, Holtzman said.
Next, Shi checked for a role of neuroinflammation in the degenerative cascade. She measured gene expression in microglia from nine-month-old P301S mice. Compared to microglia from P301S/ApoE knockout animals, microglia from P301S mice expressing any of the three human ApoE isoforms upregulated a suite of pro-inflammatory genes, and downregulated homeostatic ones. The skew toward inflammation was most dramatic in P301S/ApoE4 mice. Among the elevated proinflammatory genes were IL-1α, TNFα, and C1q—three microglial genes that a study led by Ben Barres at Stanford University recently implicated in turning normally neurotrophic astrocytes into killers (see Jan 2017 news). Holtzman therefore collaborated with Barres to measure gene expression of astrocytes in the various P301S mice. Ultimately, they found that the gene expression profile of astrocytes from P301S/ApoE4 mice fitted that of neurotoxic “A1” astrocytes previously identified by Barres.
Berislav Zlokovic at University of Southern California, Los Angeles, was excited by the convergence of tau, ApoE, and neuroinflammation in this work. That these factors conspire to cause neurodegeneration in the absence of Aβ is not surprising, he said, nor does it exclude a contribution of Aβ to the AD cascade. He noted that ApoE4 also compromises the integrity of the vascular system in the brain, which in turn can cause tau pathology (see May 2012 news). A compromised blood-brain barrier can activate microglia and astrocytes, placing vascular problems at the scene of the crime as well.
Holtzman proposed a model in which ApoE4 exacerbates the microglial response to tau aggregates, which in turn goads astrocytes into harming neurons. It is unclear how E4 inflames microglia more than do other ApoE alleles, Holtzman said. However, if ApoE4 triggers damaging neuroinflammatory responses to tau pathology, it could accelerate disease progression in AD as well as in primary tauopathies, a possibility Holtzman is investigating. Whether microglia in P301S mice are directly responsible for activating astrocytes is also unclear at the moment, but this is a question cell culture models may elucidate.
To that end, Julia TCW,a postdoc in Alison Goate’s lab at Mount Sinai School of Medicine in New York, presented her latest findings in astrocytes generated from human iPSCs. The researchers generated 42 astrocyte lines derived from 30 people, including men and women, as well as healthy controls and people with AD. The cells behaved similarly to primary astrocytes harvested directly from the human brain, gobbling up myelin in response to glutamate, TCW reported. They also appeared to exist in a quiescent, resting state unless given a stimulus. This quiescence was of utmost importance, TCW stressed, as hyper-reactivity is a common problem with primary astrocytes isolated from the human brain.
Bona Fide Astrocytes.
Star-shaped astrocytes differentiated from human iPSCs thrived in a quiescent state after differentiation. (Green: S100B; Red: GFAP; Blue: DAPI.)
[Image courtesy of Julia TCW.]
Interestingly, astrocytes generated from people carrying two copies of ApoE4 expressed higher resting-state levels of the inflammatory cytokines IL-6 and IL-8 than did cells from people with an E3/E3 genotype. Treatment of astrocytes with Aβ42 or tau oligomers boosted cytokine secretion regardless of ApoE genotype, however, E3/E3 astrocytes had a larger increase in cytokine secretion, perhaps owing to their lower baseline levels, TCW said.
While Holtzman proposed that ApoE may primarily modulate astrocyte activation indirectly via microglia, TCW’s work suggests that ApoE isoforms also directly affect astrocytes in both resting and activated states. TCW hypothesized that cross talk between microglia and astrocytes occurs within the complex cellular milieu of the brain, although cause and effect relationships are difficult to unravel. She also noted that ApoE’s lipidation state—which differs between mice and humans—influences its activity. This makes human iPSC-derived models more relevant to understanding how ApoE affects neuroinflammation, she added.
Man-made Microglia.
An iPSC-derived microglial cell fans out its processes following transplantation into mice. (Red: human cytoplasm marker SC121; green: resting microglial marker P2RY12; blue: Iba1.) [Image courtesy of Mathew Blurton-Jones.]
Completing the iPSC menagerie, Mathew Blurton-Jones of the University of California, Irvine, described the differentiation of microglia from the pluripotent cells. Graduate student Edsel Abud used a two-step differentiation procedure to mimic the conditions that the hemotopoietic cells first experience in the yolk sac, and then in the brain, as they develop into microglia. RNA sequencing revealed that the resulting cells expressed a similar homeostatic suite of genes as do human microglia at rest.
When treated with Aβ fibrils or tau oligomers derived from postmortem brain samples of AD patients, the microglia upregulated ApoE, as well as several genes associated with AD risk in genome-wide association studies. The cells also ramped up expression of neuroinflammatory genes, including the same troublesome triad—IL-1α, C1q, TNFα—that fire up A1 astrocytes. The researchers injected the microglia into neuronal organoids, and also into the brains of immunodeficient mice. Strikingly, the microglia migrated and took positions at regular intervals throughout the organoids and mouse brains, much as endogenous microglia tile throughout the brain.
Taking Position.
Microglia differentiated from human iPSCs tile across a mouse brain after transplantation. (Red: human cytoplasm marker SC121; green: microglial marker TMEM119.) [Image courtesy of Mathew Blurton-Jones.]
Blurton-Jones is collaborating with researchers in Goate’s lab to put iPSC-derived astrocytes into the organoid mix as well. He commented to Alzforum that it would be interesting not only to co-culture neurons, microglia, and astrocytes derived from the same person’s iPSCs, but also to mix and match cells with different ApoE genotypes. This could show which cells deliver the ApoE4 that activates microglia, and test whether the microglia truly turn astrocytes into killers. TCW and colleagues are now using CRISPR gene editing to generate isogenic lines in which only the ApoE genotype is changed. That way she can focus on the role of ApoE without having to deal with the influence of differing genetic background.—Jessica Shugart
Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, Carrell D, Cai Y, Fernandez MV, Budde J, Ma S, Saef B, Howells B, Huang KL, Bertelsen S, Fagan AM, Holtzman DM, Morris JC, Kim S, Saykin AJ, De Jager PL, Albert M, Moghekar A, O'Brien R, Riemenschneider M, Petersen RC, Blennow K, Zetterberg H, Minthon L, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Schellenberg G, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Peskind ER, Li G, Di Narzo AF, Alzheimer’s Disease Neuroimaging Initiative (ADNI), Alzheimer Disease Genetic Consortium (ADGC), Kauwe JS, Goate AM, Cruchaga C.
Genome-wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers.
Acta Neuropathol. 2017 May;133(5):839-856. Epub 2017 Feb 28
PubMed.
New Evidence Confirms TREM2 Binds Aβ, Drives Protective Response
Variants in the microglial receptor TREM2 heighten the risk for neurodegeneration, though exactly how they do that has remained unclear as initial research produced conflicting results. Now, however, researchers appear to be reaching a consensus that TREM2 protects the brain, and that the disease-causing variants all disrupt its function in some fashion. This weakens microglia through multiple pathways, including survival, migration, and phagocytosis.
“TREM2 acts as a signaling hub,” Christian Haass of the German Center for Neurodegenerative Diseases (DZNE), Munich, explained to Alzforum. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, researchers added more spokes to the paradigm. Speakers agreed that TREM2 activation triggers microglia to clean up messes in response to damage or disease. Some speakers said that both oligomeric Aβ and ApoE play a role in switching on this response, while others detailed how TREM2 variants linked to Alzheimer’s disease disturb it. However, it remains unclear at what point in disease TREM2 activity does the most good, and how it could be manipulated therapeutically.
“Great strides have been made in our understanding of TREM2 in the context of AD in just a short time since the initial description of AD-associated TREM2 variants,” wrote David Holtzman and colleagues at Washington University in St. Louis in an April 19 Neuron review article. So far, most of the evidence points toward TREM2 orchestrating the microglial response to amyloid pathology, they added.
Idling Microglia.
In year-old wild-type mice (left), microglia rev up their activity (red), but in TREM2 mutant knock-ins (right), the cells remain stuck in neutral. [Courtesy of Matthias Brendel and Christian Haass.]
Interest in TREM2 took off when geneticists pinned a tripled risk of AD on the R47H variant and linked other missense mutations such as T66M with frontotemporal dementia (see Oct 2012 news; Nov 2012 news). Researchers speculated that the receptor might aid phagocytosis, but initial studies disagreed on this, with some reporting no change in plaque load in TREM2 knockouts, while others did see changes in amyloid after TREM2 stimulation (see Jun 2014 news; Jul 2016 news; Dec 2016 conference news). Since then, scientists have developed a more nuanced view, agreeing that the effects of TREM2 depend on the stage of the disease. In several studies, AD mouse models with impaired TREM2 function accumulate fewer plaques than controls at four months of age, but a heavier load by eight months (see Jul 2016 conference news).
In Vienna, Peter St. George-Hyslop of the University of Toronto dug further into the relationship between plaques and phagocytosis. He wondered if instead of interacting with plaques themselves, TREM2 might sense something that leaches off plaques, such as Aβ oligomers. Supporting this idea, he found that synthetic Aβ co-immunoprecipitated with TREM2, an interaction that could be displaced by the presence of TREM2-blocking antibodies. The bound Aβ was oligomeric, as determined by two-color coincidence detection, a method that discriminates between monomers and oligomers. Binding of these oligomers to TREM2 triggered cleavage of the receptor in a dose-dependent fashion. This receptor cleavage released a soluble N-terminal fragment, sTREM2, whereas the C-terminal fragment was internalized. TREM2 processing seemed to stimulate phagocytosis, as incubating microglia with Aβ oligomers primed them to later scarf up debris from dead neurons. Microglia lacking TREM2 did not dial up phagocytosis. The results suggest TREM2 is needed to turn on phagocytosis in response to aggregated, toxic Aβ, St. George-Hyslop said.
Previous studies had found that TREM2 bound to Aβ complexed with LDL, but had not reported an interaction with Aβ alone (see Jun 2016 conference news). It is unclear if those studies examined oligomeric Aβ, however. In addition, prior studies focused on microglia engulfing Aβ through a TREM2-mediated process, but had not shown a role for Aβ itself in activating phagocytosis, Haass noted.
How would the AD variant R47H affect this process? Examining microglia from R47H knock-in mice, St. George-Hyslop found that they gobbled up fluorescent beads efficiently and signaled normally through TREM2 and its co-receptor DAP12. However, the R47H mutants only weakly bound Aβ oligomers. As a consequence, they released less sTREM2 and phosphorylated less DAP12 in response to the oligomers. Thus, the mutant microglia poorly activate phagocytosis in the presence of Aβ, St. George-Hyslop said. The results suggest that R47H is a hypomorph, meaning it causes a partial loss of function. This distinguishes it from variants that cause FTD, which never reach the cell surface and act as complete loss-of-function mutations (see Jul 2014 webinar; Apr 2015 conference news). The findings fit with previous work that found weak binding of the R47H variant, as well as other AD-causing variants such as R62H, to ApoE and cell-surface proteoglycans, too (see Kober et al., 2016).
The H157Y mutation, prevalent in Han Chinese, drives up AD risk 11-fold (see Jiang et al., 2016). In contrast to R47H, which attenuates sTREM2 shedding, this mutation heightens it, St. George-Hyslop said. Normally, the metalloprotease ADAM10 clips membrane-bound TREM2 right after histidine 157, the site of this mutation, with cleavage typically occurring within one hour. St. George-Hyslop found that microglia expressing H157Y shed sTREM2 sooner than this, and that the soluble and C-terminal fragments of the protein build up. Moreover, this cleavage was not inhibited by the ADAM10 inhibitor batimastat, suggesting a new metalloprotease might be responsible, although cleavage occurs at the same site. Haass said he has similar experimental results. His group also sees a rise in sTREM2 in H157Y microglia at the expense of cell-surface TREM2. The end result is a loss of microglial TREM2 signaling, just as with the R47H mutation, rendering H157Y effectively a hypomorph as well, Haass said.
“The ability of TREM2 variants to enhance or impair TREM2 signaling suggests that altered TREM2 homeostasis has serious consequences in regard to the development of AD,” Holtzman and colleagues wrote in their review.
While these hypomorphs provide clues to AD mechanisms, the complete loss-of-function variants draw a clearer picture of what TREM2 does. Haass shared data from studies in a T66M knock-in mouse, which expresses no cell-surface TREM2 receptor (see Sep 2016 conference news). He found the microglia to be incapable of activating properly, and prone to dying. Whereas wild-type mice continuously dial up microglial activation with age, the T66M microglia experienced but a blip at eight months, falling back to baseline by one year of age (see image above). These passive microglia were unable to perform many normal jobs. They put out fewer processes than their wild-type counterparts. The T66M knock-in mice also expressed fewer chemotactic proteins in the brain, which attract other microglia to damaged tissue. As a result, microglia neither moved toward apoptotic neurons in aging T66M brains, nor cleaned up damaged myelin. When these knock-ins were crossed with AD mouse models, the offspring had fewer microglia clustering around plaques. In essence, knocking out TREM2 locks microglia into a homeostatic torpor, such that they can no longer respond to external stimuli and activate normally to protect the brain, Haass concluded. The data are in press at EMBO Reports.
Oleg Butovsky at Brigham and Women’s Hospital, Boston, also believes that TREM2 helps rouse microglia out of homeostasis. When microglia chew up apoptotic cells, exposed phosphatidylserines in the damaged membranes bind and activate TREM2, he said. TREM2 signaling switches on a distinct pattern of gene expression, with ApoE being the most upregulated protein. Butovsky labeled this gene expression signature MGnD. It associates with disease, with MGnD microglia being the ones that cluster around neuritic plaques.
ApoE itself appears to counter the homeostatic phenotype, Butovsky found. Inducing ApoE jolted microglia out of homeostasis, while deleting the gene returned them to a quiescent state. Butovsky did not discuss whether the E4 allele affects this process differentially, though other researchers at AD/PD reported that ApoE4 sends microglia into overdrive in response to tau (see Apr 2017 conference news). Butovsky and colleagues were also able to return activated microglia to homeostasis in AD mice by ablating TREM2. As other groups have found, the lack of TREM2 led to fewer plaques at younger ages and to more plaque at late stages. On the other hand, in P301S tauopathy mice, ablating microglial ApoE lessened damage, implying that keeping microglia in a homeostatic state helped in this condition. Other work suggests that activated microglia exacerbate tau pathology (see Maphis et al., 2015). Whether or not microglial homeostasis is beneficial may depend on the stage and type of disease, Butovsky suggested.
Karel Otero of Biogen Idec, Boston, focused instead on TREM2’s role in promoting the survival of microglia. He noted that in healthy aging, fewer than 10 percent of microglia become dystrophic, whereas in AD patients, more than half degenerate. Likewise, TREM2 knockout mice display massive numbers of dying microglia. Loss of TREM2 also impairs microglial activation, proliferation, and migration in several other models of brain damage, such as stroke, toxin-induced demyelination, and prion infection.
How does TREM2 facilitate survival? Previous work suggested that this receptor cooperated with growth factor receptor colony stimulating factor 1 (CSF-1R), also located on microglia, to enhance its signaling (see Feb 2015 conference news). Otero therefore investigated whether TREM2 might act as a co-receptor for CSF-1R ligands. Although TREM2 did not bind to CSF-1R ligands by itself, TREM2-blocking antibodies did suppress CSF-1R signaling. In addition, once CSF-1R signaling was active, TREM2 and CSF-1R co-immunoprecipitated. The data suggest that TREM2 forms a complex with activated CSF-1R and amplifies the effects of its signaling to keep microglia alive, Otero said. Several groups have reported that CSF-1R’s ligands, CSF-1 and IL34, protect against amyloid pathology in mice (e.g., Boissonneault et al., 2009; Luo et al., 2013).
In Vienna, Otero reported that disease-causing TREM2 variants disrupt this process. For example, R47H does not bind CSF-1R. In mice carrying this variant, Otero found fewer dividing and more apoptotic microglia, suggesting harm to both proliferation and survival. Crossing Tg2576 mice with TREM2 knockouts, Otero found fewer microglia around plaques and worse performance in fear-conditioning tests in the offspring. His data, too, support a model where TREM2 is neuroprotective, and its loss leads to microglial dysfunction and neurodegenerative disease, Otero concluded.—Madolyn Bowman Rogers
Location, Conformation, Decoration: Tau Biology Dazzles at AD/PD
The well of tau is in no danger of running dry, if findings presented at the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, are any gauge. If anything, it was bubbling over as scientists put forth a steady stream of data about the microtubule binding protein linked to neurodegeneration. There was the massive impact of tau pathology on the epigenome of neurons, as well as distinctive adornments peppering the protein in each tauopathy. Tau’s intimate, toxic affair with synaptic vesicles was on display, as was a mouse model in which Aβ-laden neuritic plaques sparked a wildfire spread of bona fide tau tangles throughout the brain. Scientists even awed the audience with high-resolution images of tau fibrils from human brain.
The breadth of findings should help researchers understand the impact of tau pathology on the brain, and how to target the protein with greater precision in therapies for neurodegenerative disease.
Does Tau Crack Open Chromatin? Philip De Jager of Columbia University in New York described an impact of tau pathology on the epigenome, i.e., the collective series of DNA modifications that dictate which genes are expressed. Unlike the DNA sequence, which is static, the epigenome is subject to modification by its environment, and De Jager wanted to know how much sway Aβ and tau pathology held in that process. Working with David Bennett of Rush University Medical Center in Chicago, De Jager and colleagues extracted DNA from more than 600 postmortem dorsolateral prefrontal cortex samples from participants in the Religious Orders Study and the Memory and Aging Project. They mapped out more than 26,000 sites in the genome that were wrapped in acetylated histones—specifically H3K9Ac, a marker of open chromatin. The researchers then correlated a person’s acetylation pattern with the extent of his or her Aβ and tau pathology across five cortical regions. To De Jager’s surprise, tau’s impact on the epigenome topped that of Aβ’s by a full order of magnitude: Around 600 H3K9Ac marks correlated with Aβ, while nearly 6,000 correlated with tau. The intensity of these H3K9Ac peaks, which primarily landed in gene promoters or enhancers, correlated positively with transcription of associated genes.
Tau Pathology Opens DNA. A Manhattan plot of tau’s epigenetic influence across chromosome 1 reveals a pattern of “peaks of peaks,” in which tau influences large swaths of DNA. Each dot represents the size of tau’s effect on a single H3K9Ac peak. [Image courtesy of Philip De Jager.]
Was there a pattern to tau’s influence on the epigenome? Indeed, De Jager reported that while the epigenetic marks associated with Aβ were randomly scattered throughout the genome, those associated with tau clustered together in stretches containing hundreds of genes. The tau-associated H3K9Ac marks also coincided with binding sites for CTCF, a protein that regulates chromatin structure. The findings suggested that tau pathology somehow triggered large-scale alterations in chromatin structure—opening access to and enabling transcription of large swaths of the genome.
De Jager confirmed these findings in mice overexpressing tau and by forcing tau overexpression in human iPSC-derived neurons. On the flip side, the researchers blocked tau’s epigenetic influence by treating tau overexpressing neurons with the Hsp90 inhibitors alvespimycin or geldanamycin—two compounds predicted to reduce the effects of tau pathology. The impact of tau’s epigenomic effect on neurodegeneration is unclear, but De Jager proposed that elevated expression of so many genes could perhaps tie up the transcription machinery, leading to broad malfunction across the cell.
Even as tau pathology affects gene expression, the tau gene itself (aka MAPT) is not immune to regulation, either. According to Roberto Simone of University College London, a sequence in MAPT’s own 5‛ untranslated region keeps levels of the protein in check. Sequencing RNA from human brain samples and iPSC-derived neurons, Simone uncovered an antisense long non-coding RNA (lncRNA) called MAPT-AS1 that inhibited tau translation. LncRNAs, which are typically greater than 200 nucleotides in length, are emerging players in gene regulation, and their importance in neurodegenerative disease is only just beginning to surface (reviewed in Luo and Chen, 2016). Speaking at AD/PD, Simone suggested to the audience that MAPT-AS1 adheres to the tau mRNA’s internal ribosome entry site, where the lncRNA fends off approaching ribosomes via a mammalian-wide interspersed repeat (MIR) element. Silencing MAPT-AS1 boosted tau expression, while stable expression of the lncRNA in neuroblastoma cell lines repressed it, Simone reported. While this finding raises the idea that tau proteostasis could one day be influenced via lncRNAs for therapeutic purposes, basic research on these regulatory RNAs is in its early stages.
Tau: Form and Function
After tau translation is said and done, the resulting protein is subject to a slew of post-translational modifications, some of which contribute to its neurodegenerative power. While specific antibodies help researchers measure some of these adornments, such as phosphorylation of certain residues, these approaches do not draw a comprehensive map of modifications along the entire protein. At AD/PD, Judith Steen of Boston Children’s Hospital presented findings rendered from a mass spectroscopy-based technique called FLEXITau, which maps both the nature and quantity of tau modifications. Using an isotope-labeled version of tau as a standard, the method previously enabled Steen to map tau modifications in the Alzheimer’s brain (see Mair et al., 2016).
In Vienna, Steen described follow-up work using FLEXITau to compare modifications across different tauopathies. Analyzing 129 postmortem brain samples from people with AD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), or Pick’s disease (PiD), Steen found that tau in each disease had its own distinctive brand of phosphorylation, acetylation, ubiquitination, and fragmentation patterns. Based on these signatures, Steen developed an algorithm that correctly classified disease type in 90 percent of AD, CBD, PiD, and control samples, and around 80 percent of PSP samples. Steen is currently investigating whether FLEXITau done on CSF or plasma tau might work as a diagnostic tool. More importantly, she told Alzforum that understanding disease-related modifications should help researchers develop antibodies that specifically target pathological forms of tau in each disease.
Besides donning decorations, the tau protein also strikes a dizzying array of poses.
While neurodegeneration often accompanies its most infamous form—neurofibrillary tangles—researchers have long noted that synaptic defects, neuroinflammation, and even neuronal loss can occur in cells free of tangles, and before tangles are widespread (see Gomez-Isla et al., 1997; Feb 2007 news). Paralleling the Aβ story, researchers now think that soluble tau oligomers might be the silent culprit (see Berger et al., 2007; Brunden et al., 2008; Meraz-Rios et al., 2010; and Nov 2010 conference news).
In support of this idea, Eva and Eckhard Mandelkow of the German Center for Neurodegenerative Diseases in Bonn presented findings describing the structure and toxicity of small tau oligomers. They used bacterial cells as factories to pump out recombinant tauRDΔK, an aggregation-prone version of the full-length neurotoxic protein containing four repeat domains. They then tinkered with incubation buffers to coax the protein to oligomerize, and found that the resulting oligomers took on a globular shape, consisting primarily of dimers, trimers, and tetramers. When they purified these oligomers and added them to hippocampal neurons, the little tau blobs ramped up production of reactive oxygen species (ROS), boosted intracellular calcium, and reduced the density of dendritic spines in the neurons. However, the oligomers stopped short of killing the neurons. The Mandelkows proposed that soluble tau oligomers contribute to synaptic defects and inflammation as first steps on the road to neurodegeneration. In addition to globular oligomers, the Mandelkows observed more ordered filamentous tau aggregates in their protein preps.
Fibrillar forms of tau, including the paired helical filaments that dominate neurofibrillary tangles and the straight protofibrils that precede them, were the object of the first atomic-level structures of tau ever reported. At AD/PD, Anthony Fitzpatrick of the University of Cambridge, U.K., had a spellbound audience when he described 3.4 A resolution, cryo-EM structures of tau aggregates isolated from patient brain. He showed protofibrils, which were marked by β-solenoid structures previously described for prions. Alzforum will discuss these structures in detail when they are formally published. In a nutshell, Fitzpatrick in Vienna described a fibril structure held together by C-shaped protofilaments made up of tau’s R3 and R4 repeat domains. Tau’s first two repeats, as well as the protein’s N-terminus, formed a “fuzzy coat” around this R3/R4 core. Intriguingly, the structures included a putative binding site for the PET tracer AV1451/flortaucipir, and they will likely help inform structure-based drug design. These structures mark a new era in tau science, Eckhard Mandelkow told Alzforum.
Selina Wray of University College, London, called Fitzpatrick’s talk the highlight of the conference. She was particularly excited that the work may help explain how different structural polymorphisms of tau relate to the diversity of tau strains and pathologies.
Tau: Scene of the Crime
Tau’s effects on dendrites, and the postsynapses that sit on the dendrites, have been well-studied, partly because normal tau is scarce in those regions of the neuron and tau’s presence there struck researchers as a pathological target. However, some researchers, including the Mandelkows, have discovered that tau can also misbehave on its home turf. They previously reported that in model mice, tau aggregates stray from tau’s main site in axons into axon terminals, where they trigger synaptic dysfunction and loss from the presynaptic side (see Apr 2015 news).
In Vienna, Joseph McInnes of VIB KU Leuven, Belgium put forward a mechanism for this finding. Working with Patrik Verstreken and Bart De Strooper, McInnes examined the neuromuscular junctions of flies that express human wild-type or mutant tau, including P301L, V337M, and R406W. In flies with a mutant gene, tau aggregates accumulated at presynapses. The presence of tau there correlated with impaired vesicle fusion and neurotransmitter release. To find out why, McInnes turned to in vitro assays of purified tau and vesicles, finding that tau’s N-terminal domain bound the vesicles. In the fly synapses, tau aggregates in effect tied up vesicles, clustering them and preventing their release. Supporting this, a mutant tau transgene that lacked the N-terminal domain still localized to presynapses but did not harm function.
Vesicle Interloper?
Nanogold-labeled recombinant human Tau clings to synaptic vesicles (SV) isolated from the rat brain. [Image courtesy of Joseph McInnes.]
McInnes verified the finding in cultures of rat primary hippocampal and human neurons. When he added a peptide that competes with tau to bind vesicles, he prevented synaptic toxicity. He also detected high levels of phosphotau in synaptic vesicles isolated from postmortem AD hippocampal samples, suggesting the same mechanism could be at work in people. McInnes told Alzforum that he has zeroed in on a vesicular protein that latches onto tau in the presynapses. He aims to disentangle the relative contributions of pre- and postsynaptic tau to synaptoxicity by knocking it down.
Intrigued by McInnes’ finding, the Mandelkows pointed out that, similar to the role of tau in axons, the N-terminus of tau is relatively understudied compared to its C-terminus, which contains the repeat domains that bind microtubules and facilitate aggregation. “One known feature [of the N-terminus] is that it mediates the binding of tau to motor proteins and the axonal cytoskeleton, e.g. through dynactin, which would make sense in the context of axonal trafficking of synaptic vesicles,” they wrote to Alzforum (see Magnani et al., 2007).
A Better Mouse of Traveling Tau?
One hallmark of human tau neuropathology is its staged spread throughout the brain; alas, this has been difficult to recapitulate fully in mice because many mouse models of AD develop no neurofibrillary tangles, let alone tangles that propagate. This is true of models that express human mutant APP in a mouse tau background, such as APPPS1. More puzzling still, it even holds true in mice that express wild-type human tau in the presence of amyloid, or when human neurons are transplanted into AD mouse brain (see Dec 2016 conference news; Feb 2017 news). Some researchers have suggested that this may be because tangles require seeding to form.
Toxic Templating.
In APPPS1 mice expressing a fragment of four-repeat tau (Tau4R-AP), endogenous mouse tau by 18 months transformed into tangles that exacerbated at 25 months, both in cortex (CT) and hippocampus (HP). Single transgenic tau4R mice (right panel) developed no tangles. [Image courtesy of Li et al., Nature Communications 2016.]
At AD/PD, Philip Wong of Johns Hopkins University, Baltimore, presented evidence to back this idea. He generated mice that express an inducible fragment from the four-repeat isoform of human tau, which can be turned off by dietary tetracycline. The animals remained healthy, with no tau pathology, but when Wong crossed them to APPPS1 mice, the offspring developed neurofibrillary tangles throughout their hippocampi and cortices (see Li et al., 2016). Wong determined that these tangles, and their spread, started after Aβ-laden neuritic plaques had formed. Wong saw dramatic neuronal loss in the hippocampus and cortex at 15 months, along with extensive microgliosis and astrogliosis. He suggested that four-repeat tau may act as a template for misfolding wild-type tau, but that this happens only in the presence of neuritic amyloid plaques. Wong plans to switch off expression of the seed before or after wild-type mouse tau starts aggregating, to test if tau pathology can continue unabated once sparked. This mouse model could be useful for testing therapeutic strategies, he added.
Why did previous models using full-length human tau not convert tau into tangles and show spread of this process? Wong told Alzforum that perhaps tau fragmentation is necessary to create a proteopathic seed. This fragmentation may unfold over decades in humans, and mice expressing full-length human tau might not live long enough for it to happen, he said.—Jessica Shugart and Madolyn Bowman Rogers
Look in the MiR: MicroRNA Fans Neurogenesis in Old Alzheimer’s Mice
Tiny they may be, but don’t let their size fool you. At only 21-23 nucleotides in length, microRNAs orchestrate grand biological processes, and researchers are just starting to get a sense of their complex roles in neurodegenerative disease. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29-April 2 in Vienna, researchers presented new data on miR-132, a microRNA produced in the brain that wanes as AD sets in. miR-132, it turns out, stymies accumulation of Aβ and tau accumulation in young mice though, curiously, in older mice it appears to boost the birth of new neurons. Other researchers reported that this microRNA drops in people with vascular dementia as well. The findings cast microRNAs as dynamic genetic modulators that wear different hats throughout the aging and disease processes.
The emerging data should caution researchers against drawing simple conclusions about these small nucleic acid snippets, commented Sébastien Hébert of the University of Laval in Montreal.
Neuronal Nursery. With their cell bodies huddled in the subgranular layer (SGL) of the hippocampus, neural stem cells expressing Nestin and GFAP are the source of newborn neurons (GCL: granular cell layer, MCL: molecular cell layer). [Image courtesy of Evgenia Salta.]
Transcribed from the genomic backwaters of introns and intergenic regions, microRNAs block the translation of their mRNA targets, either by obstructing translation machinery or by recruiting gene-silencing equipment. The expression of microRNAs changes throughout development and aging, and researchers have also reported the under- or overexpression of particular microRNAs in the brains of people with neurodegenerative disease. MiR-132 expression is consistently downregulated in postmortem brain samples of people with AD (see Research Timeline 2010; Hébert et al., 2013; Pichler et al., 2017; and reviewed in Salta and De Strooper, 2017).
Two recent papers published in close succession—one led by Hébert, the other by his former mentor, Bart De Strooper at KU Leuven in Belgium—reported that deleting or downregulating miR-132 in young AD mice worsened Aβ and tau pathology, while (in De Strooper’s study) overexpressing miR-132 diminished pathology (see Hernandez-Rapp et al., 2016; Salta et al., 2016).
At AD/PD, Evgenia Salta, a postdoc in De Strooper’s lab, updated the crowd on a plot twist to this story. It came about when the researchers tested whether overexpressing miR-132 would slow cognitive decline in older, plaque-ridden mice. Indeed, Salta reported that 9-month-old APPPS1 mice given intracerebroventricular injections of miR-132 outperformed mice given a control microRNA on memory tests. Because this microRNA had vanquished Aβ and tau pathology in younger animals, Salta hypothesized that this is what would explain the cognitive benefit in the older ones. Not so. The researchers found no significant differences in Aβ pathology or phosphorylated tau in response to miR-132 overexpression, Salta reported. Hébert told Alzforum that, similar to what Salta observed in 9-month-old APPPS1 mice, he also found no consistent effect of miR-132 on Aβ and tau pathology in older 3xTg animals.
If not by lightening the pathology burden, then how might miR-132 boost memory in older AD mice? In search of hints, the researchers turned back to a previous paper, in which they had identified miR-132’s mRNA targets in the central nervous system of zebrafish (see Salta et al., 2014). In a nutshell, the researchers previously reported that miR-132 promotes the differentiation of new neurons from neural stem cells in the dentate gyrus, called radial glial progenitor cells. Given that neurogenesis takes a nosedive in multiple AD mouse models with age, might enhanced neurogenesis explain the memory boost in the 9-month-old APPPS1 animals treated with miR-132?
To test this, Salta and colleagues gave the animals running wheels. This exercise is a known neurogenesis booster. After a month, the researchers checked the radial glial progenitor cells for recent proliferation, the first step in neurogenesis. Among wild-type mice, both young and old had proliferating stem cells in response to running, but in APPPS1 mice, only youngsters did. Salta pointed out that both wild-type and AD mice ran similar distances, as measured by monitors fitted to the running wheels. Injection of miR-132 rescued this neurogenesis defect in the older AD mice. Conversely, knocking down miR-132 in old wild-type animals decreased proliferation, Salta reported.
Could boosting miR-132 expression slow cognitive decline in people with Alzheimer’s? Salta said existing data leave it unclear whether waning neurogenesis plays a role in cognitive decline in AD. She believes that even if flagging neurogenesis does not contribute to cognitive decline in AD, boosting it could still be beneficial. New neurons are fitter, more plastic, and better at absorbing new information than their older counterparts, she said. Salta and colleagues are working to confirm their preliminary findings, and to understand how miR-132 might drive neurogenesis.
Thickening the microRNA plot and expanding it into humans, at AD/PD Jose Gerardo-Aviles, a graduate student in Patrick Kehoe’s lab at the University of Bristol, U.K., described expression of seven microRNAs in the posterior cingulate gyrus of postmortem samples from people with AD, vascular dementia, or healthy controls. Gerardo-Aviles reported that the tissue concentration of four microRNAs—miR-16, miR-29a, miR-34a, and miR-125b—increased with higher Braak stage in AD patients, but not in people with vascular dementia, compared to controls. Together, these microRNAs reduced the expression of retinoic acid receptor-related orphan receptor-alpha (RORα), itself a transcriptional regulator involved in lipid metabolism, hypoxia, and the circadian clock.
An opposing pattern emerged for miR-132 and miR-212, both of which were downregulated in people with AD as the disease progressed, and in those with vascular dementia, Gerardo-Aviles reported. He proposed that the link to vascular dementia could be explained by the importance of pericyte-derived miR-132 in stimulating angiogenesis in response to hypoxia. “As highlighted by Salta’s talk, restoring miR-132 levels might represent a multi-hit therapeutic strategy in AD, and we consider this might be also helpful to vascular dementia,” he told Alzforum.
Hébert struck a more cautious tone. The changing role of miR-132 with age in his and Salta’s mice illustrates a key issue in the microRNA field, he said. Just as microRNA expression changes with age and in different disease states, so does the expression of their mRNA targets. Target expression differs between cell types, and neuroinflammatory or vascular problems might change target expression and render microRNA pathways less effective, he proposed. These fluid changes make it unlikely that a single microRNA will work as a therapeutic magic bullet, Hébert said. —Jessica Shugart
Transcranial Magnetic Stimulation for AD Boasts Success in Phase 3
In Alzheimer’s disease, synaptic activity goes haywire and brain networks gradually falter. This has made some researchers wonder if externally stimulating brain activity could help people with the disease function better for a while. The answer appears to be a tentative yes. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, Babak Tousi of the Cleveland Clinic Lou Ruvo Center for Brain Health in Lakewood, Ohio, reported positive results from a short Phase 3 trial of neuroAD, a therapy system developed by the Israeli medical technology company Neuronix. Classed as a medical device, neuroAD combines repetitive transcranial magnetic stimulation (rTMS) of several brain areas with cognitive training tailored to strengthen those same regions. Participants with mild AD who received the intervention for six weeks maintained stable cognitive abilities six weeks later, whereas cognition continued to decline in those who received sham stimulation, Tousi said. Though the effects were small, they met prespecified outcomes and were consistent with previous reports of efficacy from smaller studies. The system is already approved for use with AD in Europe, and Neuronix is now applying for marketing clearance from the U.S. Food and Drug Administration.
Marwan Sabbagh at Barrow Neurological Institute in Phoenix ran the trial along with Alvaro Pascual-Leone of Beth Israel Deaconess Medical Center in Boston. “The [beneficial] effect appears to be symptomatic, but very well tolerated. What needs to be determined from future studies is the duration and sustainability,” Sabbagh wrote to Alzforum
Other groups are investigating similar approaches. Lorenzo Pini works at the health care organization IRCCS Centro San Giovanni di Dio–Fatebenefratelli, Brescia, Italy. In Vienna, Pini presented preliminary results from a small ongoing trial of transcranial direct current stimulation (tDCS), which he said sharpened verbal abilities in participants. tDCS is similar to rTMS, but is delivered in a way that may be less bothersome to people with dementia. In addition, researchers are investigating whether brain stimulation may help other neurodegenerative disorders. Two poster presentations at AD/PD touted better motor control in Parkinson’s patients after TMS. This area of research has received far less attention in the neurodegenerative disease field than pharmacological approaches.
The neuroAD therapy system from Neuronix combines transcranial magnetic stimulation with computerized cognitive training. [Courtesy of Neuronix.]
In other disorders, rTMS, which has been around since 1984, has a history of success. rTMS has been FDA-approved since 2008 for the treatment of depression and since 2014 for migraine headaches. Many local clinics offer it, and consensus guidelines have been issued for its therapeutic use in various indications (e.g., Lefaucheur et al, 2014).
On the research front, scientists are investigating the benefits of the technology for neuropathic pain, schizophrenia, and AD. One recent review of 11 studies concluded that, in general, noninvasive brain stimulation improves cognition in AD patients (see Hsu et al., 2015). Some studies have found that rTMS of AD patients coordinates firing in the brain’s default mode network, and this correlates with better associative memory (see Dec 2011 news; Aug 2014 news).
To apply TMS, clinicians place a magnetic coil over particular spots on the head. The magnetic field induces electrical currents in the brain region below it, triggering neurons to fire. Patients may feel a slight knocking sensation on their heads as the magnetic pulses fire, and occasionally experience tingling, muscle twitches, or mild headaches or dizziness. Most applications use rTMS, rather than single-pulse, as it has more sustained effects.
Mixing Stimulation With Cognitive Training Boosts Efficacy
Neuronix combined rTMS with cognitive training based on pilot studies that found this combo worked better than either approach alone. In each treatment session, participants first take part in computerized training for language, memory, executive function, and spatial attention skills. The computer adjusts the level of difficulty to each participant’s ability based on his or her previous answers. Then participants undergo rTMS that targets six brain regions corresponding to these skills: Broca’s area, Wernicke’s area, the bilateral dorsolateral prefrontal cortex, and the bilateral parietal somatosensory cortex. These regions control, respectively, the production and understanding of speech, executive functions, and the integration of sensory inputs. They are all cortical regions that lie just under the skull. Three of the regions are stimulated in a single session, and the other three in the next.
In small pilot studies of AD patients, short-term use of the neuroAD system for five or six weeks reportedly improved scores on the ADAS-Cog by 3-4 points (see Bentwich et al., 2011; Rabey et al., 2012). Since then, small studies of the device in Korea and France likewise reported modest benefits to memory, language skills, and mood of AD patients undergoing the treatment (see Lee et al., 2016; Nguyen et al., 2017).
In Vienna, Tousi reported results of the pivotal Phase 3 study, which took place at nine U.S. sites and one in Israel. The trial enrolled 130 people between 60 and 90 years old who had mild to moderate AD according to NIA-AA criteria. About 85 percent of the cohort was classified as mild AD, based on having an ADAS-Cog equal to or less than 30. Participants taking acetylcholinesterase or memantine were allowed to continue on those drugs.
The cohort was randomized to either the active group or a sham condition. In the latter, participants interacted with a computer program and sat under a machine that made the same sounds as rTMS, but did not generate magnetic fields. Because some people feel sensations of TMS on the head, the research field has debated how well blinding works, and whether placebo effects might predominate in short studies. Sabbagh told Alzforum that participants were not told about the sensations they might experience from rTMS, and noted that all study staff except the TMS technician were blinded. He believes the sham condition was convincing. Supporting this, at the end of the trial participants were asked whether they thought they had received active or sham treatment, and people in both conditions were equally likely to guess sham.
Participants underwent rTMS or sham treatment for one hour five days per week for six weeks, or a total of 30 sessions. This trial length was pre-cleared with the FDA as sufficient to obtain marketing clearance, Tousi noted. Requirements for medical devices are much less stringent than for pharmaceuticals.
Adherence to this regimen was excellent, Tousi told the audience. More than 90 percent of participants did at least 90 percent of the sessions. Six people dropped out, most because they missed more than the allowed number of sessions. Overall, patients reported enjoying the treatment and asked to continue at the end of the study, Tousi noted. About 10 percent of participants had adverse effects related to rTMS, most commonly headache or skin irritation. Occasionally, participants experienced muscle twitches, which were alleviated by adjusting the settings on the device. All these effects went away within minutes to hours, and there were no serious adverse events, Tousi said.
Curiously, the cognitive benefits of treatment showed up only after the sessions stopped. At the end of the six weeks, the treatment and placebo groups looked similar on both the ADAS-Cog and the clinical global impression of change (CGIC), but six weeks later, the placebo group had worsened slightly on both measures, while the treatment group improved. Improvement occurred mainly in the 110 people with mild AD. In this group, ADAS-Cog scores at week 12 were 1.8 points better in the treatment group than in placebo, a statistically significant difference. For the CGIC, the difference was 0.45 in favor of treatment, missing significance (p=0.07).
It remains unclear how long the treatment benefit might last. No follow-up studies with the Phase 3 cohort are planned, but in some of the early pilot studies, researchers reported improved scores for as long as 4.5 months after treatment, Tousi told Alzforum. A Neuronix poster shown at AD/PD claims there can be long-lasting benefits. Jose Rabey of Tel Aviv University, Israel, and Evgenia Dobronevsky at Neuronix Medical Center in Ramat Gan treated a separate cohort of 30 AD patients with 30 sessions on the neuroAD system, following the protocol outlined above. In the treatment group, they saw improvement of 2.4 points on the ADAS-Cog and 1.7 points on the MMSE compared to control. This improvement lasted for at least nine months, the researchers reported (see Rabey and Dobronevsky, 2016).
How might rTMS be used in clinical practice? Tousi sees it as an adjunct to the standard of care and to potential future pharmacological interventions. He believes it may provide a symptomatic benefit that helps AD patients maintain their cognitive abilities and function better day-to-day. However, future studies are needed to determine how often a person might need to repeat the 30-session treatment for maximum benefit. Given that it worked better for people with mild AD, it is possible that those in prodromal stages might benefit more. That, too, remains to be investigated.
Direct Current Stimulation—Does Gentler Approach Hold Potential?
One drawback to rTMS is that the noise of the machinery and the sensations can be upsetting, particularly for dementia patients, making it less likely that patients will sit still through a session. A potential alternative might be transcranial direct current stimulation of the brain, which delivers electricity through electrodes placed on the scalp. Compared with rTMS systems, tDCS devices are portable, cheaper, and quieter, and sessions are shorter. However, tDCS effects are milder than those of rTMS, and tend to affect broader swaths of the brain, making it harder to target specific functions.
tDCS comes in two flavors, anodal (or positive) stimulation, and cathodal (or negative) stimulation. The former heightens the likelihood of neuron firing, while the latter dampens it. There has been relatively little research on the potential therapeutic effects of tDCS, although at least two small studies reported a cognitive benefit from anodal tDCS in AD patients (see Ferrucci et al., 2008; Boggio et al., 2011).
To gather data, researchers led by Michela Pievani at IRCCS started a small pilot study of both anodal and cathodal tDCS in AD patients. The researchers hope to enroll 20 participants; in Vienna, Pini reported on the first 16. They were between 55 and 85 years old and had mild AD, as judged by an MMSE of 18 or higher. Eight participants received 10 daily 25-minute sessions of anodal tDCS, eight cathodal. There was no placebo group. The treatment was targeted to the default mode network (DMN) by stimulating the right angular gyrus, and to the salience network by stimulating right dorsolateral prefrontal cortex (see Pievani et al., 2017). The researchers chose these networks based on evidence that, early in AD, connectivity wanes in the former while increasing in the latter (see Jul 2012 news; Aug 2014 news). They speculated that stimulation might normalize these networks and thus improve cognition. Participants underwent a battery of language, memory, and neuropsychological tests, as well as fMRI to measure changes in functional brain connectivity.
Anodal and cathodal stimulation had distinct effects, the researchers found. The cathodal tDCS group did not improve on any cognitive measure, but their global Neuropsychiatric Inventory (NPI) score dropped, suggesting behavioral improvement. Their functional connectivity showed no change.
The anodal group notched improvements on the Rey auditory verbal test, semantic fluency, clock drawing, and a language comprehension test, but not the other 11 tests in the battery. This group displayed fMRI changes, though surprisingly, connectivity in their DMN dropped, rather than increasing as might be expected given the cognitive improvement. The connectivity change did not correlate with test scores in this small sample, leaving it unclear whether it was related.
“We are still trying to understand this result,” Pievani told Alzforum. She noted that in healthy volunteers, high-frequency anodal stimulation has been reported to lower DMN connectivity, while low-frequency stimulation enhanced it (see Eldaief et al., 2011). In her study, as in most previous AD studies, the participants received high-frequency stimulation. “Future studies assessing different paradigms and collecting surrogate measures of network activity might help to identify the most appropriate intervention for AD,” she suggested.
So far, anodal tDCS appears to be more effective than cathodal for cognitive disorders, Pini said in Vienna. He noted that the treatment appeared safe, with few side effects, though two participants complained of headaches, and some noted burning sensations or itching skin.
The researchers are now looking for changes in structural connectivity using MPRAGE, a type of three-dimensional MRI, and diffusion tensor imaging. In addition, Pini will assess participants six months after treatment to see if the benefit persists. He believes these pilot results support running larger, placebo-controlled studies in AD and other diagnostic groups. Pievani stressed that future trials should collect biomarker data on amyloid and tau pathology as well as brain activity and connectivity. Animal studies hint that stimulating brain activity could increase amyloid deposition, though more recent work on inducing gamma waves in mouse brain by way of external stimulation had the opposite effect (see Dec 2005 news; Iaccarino et al., 2016).
Can rTMS help Parkinson’s patients?
Among other neurodegenerative diseases, Parkinson’s seems ripe for noninvasive brain stimulation, given that deep brain stimulation (DBS) is effective for many patients. Alas, most studies to date have found only mild benefits from rTMS (for review, see Benninger and Hallett, 2015), though a recent study of 50 PD patients by Pascual-Leone and colleagues did report motor improvement after targeting rTMS to the primary motor cortex (see Brys et al., 2016). Deep TMS, which uses several magnetic coils placed in a helmet worn by the patient to access deeper brain regions, may boost efficacy further (see Torres et al., 2015).
In Vienna, researchers from Japan and Korea added to this data. Tomoo Mano of Osaka University reported on a placebo-controlled crossover study of rTMS in 19 PD patients. Over the course of three days, participants received stimulation to their primary motor cortices, supplementary motor area (SMA), or dorsolateral prefrontal cortex. Stimulation of either the primary motor cortex or SMA correlated with better motor function on the UPDRS-III scale, Mano found. Targeting the dorsolateral prefrontal cortex, on the other hand, slightly improved mood but not motor abilities.
Similarly, Suk Yun Kang of Hallym University College of Medicine in Hwaseong-Si, Gyeonggi-do, Republic of Korea, described a study in 12 PD patients whose episodes of “freezing” during walking rendered them suddenly unable to move. After four brief rTMS sessions per day for two days, participants who received stimulation to their SMA had significantly fewer freezing episodes. The SMA helps plan and coordinate movements. Curiously, stimulation of motor cortex did not affect gait freezing.
Data from this and other studies of noninvasive brain stimulation emphasize that effects vary depending on which brain areas are stimulated, and how. Before conducting multicenter trials for neurodegenerative disorders, researchers will have to agree on the best regions to stimulate, as well as for how long, how often, and at what frequency or intensity, Pievani said.—Madolyn Bowman Rogers
Lefaucheur JP, André-Obadia N, Antal A, Ayache SS, Baeken C, Benninger DH, Cantello RM, Cincotta M, de Carvalho M, De Ridder D, Devanne H, Di Lazzaro V, Filipović SR, Hummel FC, Jääskeläinen SK, Kimiskidis VK, Koch G, Langguth B, Nyffeler T, Oliviero A, Padberg F, Poulet E, Rossi S, Rossini PM, Rothwell JC, Schönfeldt-Lecuona C, Siebner HR, Slotema CW, Stagg CJ, Valls-Sole J, Ziemann U, Paulus W, Garcia-Larrea L.
Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS).
Clin Neurophysiol. 2014 Nov;125(11):2150-206. Epub 2014 Jun 5
PubMed.
Treating Tau: Finally, Clinical Candidates Are Stepping into the Ring
While many of the more advanced Alzheimer’s therapies currently in trials target amyloid, tau-based approaches are stepping up to the plate. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, researchers shared updates on molecules wending their way through early stage trials. AbbVie reported Phase 1 data for its antibody, ABBV-8E12, against aggregated, extracellular tau. This antibody is in Phase 2. Genentech showed preclinical and Phase 1 findings of its pan-tau antibody, RO 7105705. Other therapies are headed to Phase 1. Biogen reported pharmacokinetic and dose-finding data from studies of its anti-tau antibody BIIB076 in non-human primates. Taking a different tack, Asceneuron presented preclinical data on its O-GlcNAcase inhibitor ASN120290 (previously ASN-561), which prevents tau tangles from forming and earlier this month was approved to enter human trials.
Dirk Beher of Asceneuron, Lausanne, Switzerland, applauded the field’s exploration of different ways to target tau, and suggested that approaches such as antibodies and O-GlcNAcylation could be synergistic. “The field had too much focus on β-amyloid in the past. Tau pathology drives clinical symptoms in AD—we need more therapies that target it,” Beher said in Vienna.
Preventing Tangles
Tau model mice treated for 3.5 months with an O-GlcNAcase inhibitor (right) develop few neurofibrillary tangles (black) compared to controls (left). [Courtesy of Dirk Beher.]
Researchers are keen to go after tau because imaging studies confirm that the progression of neurofibrillary tangles through the brain correlates with cognitive decline even at pre-dementia stages (see Nov 2013 conference news; Aug 2014 conference news; May 2016 news). This has raised hope that stopping tau pathology might arrest deterioration.
AbbVie developed ABBV-8E12 in collaboration with C2N Diagnostics, St. Louis. The antibody blocked tau seeding in cell cultures and prevented tangles in tau model mice (see Kfoury et al., 2012; Yanamandra et al., 2013; Yanamandra et al., 2015). In Vienna, Nuno Mendonca at AbbVie, Ludwigshafen, Germany, reported Phase 1 results of a study testing the antibody in 30 people with PSP at 12 clinical sites. Participants received a single IV dose of either 2.5, 7.5, 15, 25, or 50 mg/kg, with about one-fourth of the cohort receiving placebo. Twenty-seven people completed the study. The researchers monitored participants by blood sample and MRI scans for 84 days after dosing, and sampled cerebrospinal fluid at baseline and day 14.
The researchers reported a typical pharmacokinetic profile for an antibody. ABBV-8E12 had a half-life of about 30 days in the blood, with concentrations in CSF running 250 to 500 times lower than in plasma. Researchers are increasingly using a measure called CSF/plasma ratio for this purpose; for this antibody, it ranged from 0.2 to 0.4 percent. The maximum concentration in plasma increased linearly with dose. The treatment appeared safe. Some patients reported headaches, but these did not correlate with antibody dose. One person, who had a history of anxiety during medical procedures, withdrew from the trial due to agitation. The researchers were unable to detect anti-drug antibodies in the plasma of participants.
AbbVie is currently recruiting for two Phase 2 trials of this antibody. The first enrolls 180 people who have had progressive supranuclear palsy (PSP), a pure tauopathy, for less than five years. Most people with this disease survive no more than seven years. Sixty people will receive a low dose of the drug, 60 a high dose, and the remainder placebo. AbbVie did not disclose the doses it picked. After 52 weeks, the researchers will measure motor skills, cognition, and daily function via change on the PSP Rating Scale, and will follow participants for another 16 weeks. The other trial is in AD, and aims to test three different doses against placebo in 400 people with mild cognitive impairment who have a CDR score of 0.5 and a positive amyloid scan. The trial will run for 96 weeks, with the primary outcome being change on the CDR-sum of boxes.
Right behind ABBV-8E12 in terms of development is Genentech’s RO 7105705, now in Phase 1. In Vienna, Gai Ayalon of Genentech, South San Francisco, laid out preclinical evidence for this antibody. Developed in collaboration with AC Immune, Lausanne, Switzerland, RO 7105705 recognizes tau’s N-terminus and reacts with all six isoforms of human and primate tau, but not mouse tau. The antibody is indifferent to whether tau is monomeric or oligomeric, phosphorylated or not. Despite this promiscuity, the researchers believe it will act primarily on pathological tau because it targets extracellular forms of the protein. In P301L mice, which carry human mutant tau, 13 weeks of treatment with either 3, 10, or 30 mg/kg RO 7105705 curtailed brain pathology in a dose-dependent fashion, Ayalon reported.
At the same time, RO 7105705 treatment boosted tau levels in blood, Ayalon noted. This phenomenon has been seen with other anti-tau antibodies as well, and has been hypothesized to reflect stabilization of the protein in the periphery (see Apr 2017 news). To some researchers, this raises concerns that high plasma tau levels could interfere with brain clearance, given that a similar effect may have limited solanezumab’s ability to clear Aβ (see Jan 2017 news). Ayalon believes this is unlikely to be a problem for anti-tau therapy. “The proposed mechanisms of action for solanezumab and anti-tau antibodies are different,” he wrote to Alzforum. The latter attempt to stop the spread of misfolded tau through the brain, rather than clearing deposits.
To derive the RO 7105705 clinical candidate, Genentech researchers used an IgG4 backbone, which only weakly activates microglial Fcγ receptors. They did this because this type of microglial activation worsens inflammation in the brain, the researchers believe. In neuron-microglia co-cultures, the IgG4 version, as well as an “effectorless” version of the antibody lacking the Fcγ binding site, both protected neurons from toxicity better than the unmodified version did, suggesting that proinflammatory microglial activation in this case did harm neurons. In mouse brain, the effectorless antibody cleaned up tau aggregates as well as the normal antibody did (see Lee et al., 2016). Chronic dosing with RO 7105705 caused no adverse effects in mice or cynomolgus monkeys.
Genentech’s Geoff Kerchner reported Phase 1 data to date. The trial is fully enrolled but ongoing, comprising 55 healthy volunteers from 18 to 80 years old. They receive single IV doses of RO 7105705 ranging from 225 mg to a whopping 16.8 grams. Participants thus far have tolerated all doses well, Kerchner reported. Some complained of headaches or nausea, but as with the AbbVie antibody, it was unclear if this related to treatment. Some participants got a bruise at the injection site. The antibody’s half-life is 30 days, and it was detectable in CSF, with pharmacokinetic parameters as expected, Kerchner said. Genentech also tested subcutaneous administration of 1,200 mg of RO 7105705, and found that 70 percent of it became bioavailable.
The ongoing Phase 1 trial is now evaluating multiple once-weekly 8,400 mg doses of the antibody in healthy controls and AD patients. The latter meet NIA-AA criteria for AD, with CDR of 0.5-2 and a positive amyloid scan. In future trials, the researchers plan to use their in-house tau PET tracer, GTP1, to determine baseline tau pathology and any changes due to treatment, Kerchner said (see Apr 2017 conference news).
Meanwhile, Biogen in Cambridge, Massachusetts, now has two antibody candidates. Biogen recently licensed an antibody directed against fragmented extracellular tau, BMS-986168, from Bristol-Myers Squibb, and has announced plans to take it into Phase 2 trials in PSP and AD (see press release). BMS-986168 is currently completing a multiple ascending dose Phase 1 study in PSP.
Biogen has also developed its own antibody, BIIB076, which is still in preclincial testing. In Vienna, Danielle Graham reported that BIIB076 recognizes both monomeric and fibrillar human and primate tau, binding to the protein with subnanomolar affinity. In 3-year-old cynomolgus monkeys, a single 100 mg/kg dose of the antibody had a half-life of eight to 11 days in blood. It reached maximum CSF concentration within 24-48 hours. As with other antibodies, its concentration in CSF was 1,000 times less than in plasma. The researchers also measured tau concentrations in blood and CSF using ultrasensitive single-molecule array (Simoa) assays. In some animals, plasma total tau rose after antibody administration, as happens with other antibodies. In CSF, total tau did not change, but free tau (i.e., that not bound by the antibody) dropped by three-quarters after 24 hours, taking three weeks to return to baseline. This indicated target engagement in the central nervous system. The pharmacokinetic data will be used to select the doses and sampling times for human trials, Graham noted.
While antibody strategies currently hog the tau airwaves, some groups are trying to create buzz with fresh alternatives. Asceneuron’s Beher discussed preclinical data for ASN120290, a small molecule that inhibits O-GlcNAcase, the enzyme that strips sugars from tau. Tau decorated with sugar molecules is less likely to aggregate, though it is unclear why that is. Some believe that O-GlcNAcylation competes with phosphorylation for the same serine/threonine residues (see Liu et al., 2009; for review, Hart et al., 2011). Others think the sugar moieties simply prevent tau molecules from cozying up to one another. At any rate, in animal studies, O-GlcNAcase inhibitors suppress tau phosphorylation, prevent tangles, and boost neuronal survival (see Jul 2008 news; Mar 2012 news).
Asceneuron identified an inhibitor with favorable drug properties that enters the brain well. In JNPL3 tau mice, ASN120290 bumped up O-GlcNAcated tau 12-fold (see Aug 2014 conference news).
In Vienna, Beher presented new data on how ASN120290 affects tau. In young P301S mice fed ASN120290 for 3.5 months, 100 mg/kg of it boosted tau O-GlcNAcylation, whereas 30 mg/kg did not. At the higher dose, treated mice had less phosphorylated tau than controls, and developed 40 percent fewer paired helical filaments in cortex. This amount of reduction is similar to that seen with tau antibodies, Beher noted. Strikingly, 80 percent fewer neurofibrillary tangles formed.
“O-GlcNAcase inhibitors have a profound effect on tangle formation,” Beher said. Tangles never contain tau modified with sugars, he added. The data suggest that tau O-GlcNAcylation and PHF formation are mutually exclusive. “This is a druggable pathway,” Beher claims.
The first Phase 1 trial has gotten underway. It will include a placebo group and will test single and multiple doses of oral ASN120290 in healthy volunteers. Tau O-GlcNAcylation will be measured in peripheral blood mononuclear cells (PBMCs). In mice, this biomarker has been found to correlate well with levels of brain tau O-GlcNAcylation, as well as with levels of drug, Beher noted. Importantly, this biomarker translates well between rats and humans, and ASN120290 shows comparable potencies in the two species by this measure. In Phase 1, the researchers will not use any other measures of CNS target engagement, Beher said. The biomarker readings will help select the dose to take forward into a Phase 2 study in people with PSP, planned for 2018.
How about tau PET in later trials? Beher noted that, at present, most tracers have not been studied much in PSP, and the off-target binding in the midbrain seen with some current tracers could become a problem (see Sep 2016 conference news). Newer PET tracers may work better for PSP (see Apr 2017 conference news).—Madolyn Bowman Rogers
α-Synuclein Antibodies Enter Phase 2, Sans Biomarker
In the search for ways to slow down the progression of Parkinson’s disease, researchers currently have α-synuclein in their crosshairs. A handful of approaches are in development, and immunotherapy strategies are now advancing through clinical trials. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, speakers from Prothena and Biogen presented Phase 1 data on their respective α-synuclein antibodies. Prothena discussed plans for Phase 2, including smartphone monitoring to collect more detailed clinical data (see related AD/PD story). At the same time, researchers bemoaned the continued absence of good biomarkers to track disease progression and show efficacy of their drugs. What they really want is a PET tracer that detects pathological forms of the protein in living brain. Motivated by a large prize, scientists are working feverishly to find tracers, and in Vienna, Andreas Muhs of AC Immune, Lausanne, Switzerland, showed preclinical data on a candidate that selectively binds aggregated α-synuclein and appears to have suitable pharmacokinetics in rodents.
Academic and pharmaceutical researchers at AD/PD said they feel more hopeful than ever for meaningful progress on this disease. “The science has gotten more sophisticated, and we’re taking good bets in the clinical space,” noted Gene Kinney, who leads Prothena. At the same time, Kinney cautioned that many unknowns in these new types of trials make the way forward unclear.
α-Synuclein Specific.
A candidate α-synuclein PET tracer (blue) binds to aggregated α-synuclein (red; overlay appears pink), but not amyloid plaques (green) in PD brain sections. [Courtesy of AC Immune.]
One of these unknowns is whether lowering α-synuclein levels will actually slow disease progression, said Werner Poewe of Innsbruck Medical University, Austria, in a plenary talk. At the time PD is diagnosed, half of the neurons in the substantia nigra have already died, he noted. Eliezer Masliah, who heads the National Institute on Aging’s Division of Neuroscience, urged the PD field to learn from AD trials. “Don’t put antibodies into late-stage patients,” he advised.
There is broad consensus that preventative trials would be ideal. At the same time, many scientists believe that antibodies do hold potential to help symptomatic patients if they stop the transfer of pathological α-synuclein from cell to cell, thus halting its spread through the brain (see Jun 2014 news). Kinney noted that those antibodies that best prevent transfer in cell culture are the ones with the strongest protective effects in animal models. Even so, Prothena researchers have found that antibodies can mop up intracellular pathology as well, Kinney said. Serena Hung of Biogen, who leads her company’s PD program, said that she sees the same phenomenon in her studies, as well.
First α-Synuclein Antibodies Head to Phase 2
Both the Prothena and Biogen α-synuclein antibody programs are at a similar stage, completing safety and dose-finding studies. In Vienna, Joseph Jankovic of Baylor College of Medicine, Houston, described findings with Prothena’s antibody PRX002. It binds aggregated forms of α-synuclein 400 times more strongly than monomeric forms, Jankovic noted. In mice, it lowers pathology, protects synapses, and improves motor abilities. PRX002 previously completed a Phase 1 study in healthy volunteers, where it appeared safe and suppressed α-synuclein levels in the blood (Mar 2015 conference news; Schenk et al., 2017). The antibody is now being developed in collaboration with F. Hoffmann-La Roche in Basel, Switzerland.
In Vienna, Jankovic reported on the results of a Phase 1b trial in PD patients. He ran a site for this trial, which enrolled 80 people from 40 to 80 years old, predominantly white men, who had mild to moderate Parkinson’s. Each received antibody by IV once a month for three months at one of six doses: 0.3, 1, 3, 10, 30, or 60 mg/kg; a third of the cohort received placebo. After dosing, participants were followed for an additional 12 weeks.
The antibody appeared safe, Jankovic said. Participants did not make anti-drug antibodies. There were no serious adverse events, though some people reported skin reactions such as rash at the infusion site, and others had gastrointestinal complaints, headaches, or peripheral edema. Participants showed no improvement whatsoever on the Movement Disorder Society-Unified Parkinson's Disease Rating Scale (MDS-UPDRS), though that was not expected in this relatively short study, Jankovic said.
The pharmacokinetic profile was largely typical for an antibody, although the half-life was on the short side at 14 days. As with other antibodies, only 0.3 percent of the amount in blood made it into CSF. This CSF/serum ratio was the same for all dose groups at week nine. Unbound α-synuclein in serum dropped by as much as 97 percent at the highest antibody dose, indicating target engagement in the periphery. Antibody levels rose in CSF in accordance with dose, but the amount of monomeric α-synuclein in CSF did not change. This was expected, since PRX002 targets primarily aggregates, Jankovic noted. The scientists do not have an assay to measure the concentration of α-syn aggregates in CSF before and after treatment.
Based on these findings, Prothena and Roche will begin a Phase 2, year-long efficacy study in 300 PD patients this year, with the primary outcome being change on the MDS-UPDRS.
Meanwhile, Biogen’s antibody BIIB054 is not far behind. Like PRX002, this human monoclonal antibody binds pathological, aggregated α-synuclein while sparing physiological forms. It was not clear whether physiological means the monomer or a postulated tetramer, and Biogen researchers could not be reached for comment. In Vienna, Andreas Weihofen of Biogen, Cambridge, Massachusetts, presented preclinical findings. In cell culture, BIIB054 reduced spreading of aggregated α-synuclein between neurons, and in mice injected with α-synuclein fibrils, BIIB054 slowed down pathology and improved motor function, Weihofen said.
In 2015, Biogen started a Phase 1 single ascending dose study in 48 healthy people between age 40 and 65. At two U.S. sites, volunteers received infusions of either 1, 5, 15, 45, 90, or a whopping 135 mg/kg, reported Biogen’s Miroslaw Brys in Vienna. Participants underwent three MRI scans, at baseline, day three, and week four. They donated CSF samples at baseline, eight hours, 24 hours, and week three. Researchers followed participants for 16 weeks after dosing, doing clinical assessments and electrocardiograms in search of adverse effects.
Doses up to 90 mg/kg were well-tolerated, with similar adverse events on placebo and drug, Brys said. In the 135 mg/kg cohort—which translates to 9.4 grams in a person weighing 70 kg, or 154 pounds—one participant developed asymptomatic ischemia in the right parietal lobe; this dose will not be used further. Some participants complained of headache, dizziness, or pain related to the infusion, and one person developed a skin rash at the infusion site.
The pharmacokinetic profile was as expected, with a half-life of 28 days and a CSF/serum ratio of 0.2 percent at all doses. The maximum concentration in blood was proportional to the antibody dose given. The researchers are still analyzing what happens to plasma α-synuclein, Brys noted. This trial is ongoing, aiming to enroll 66 people, but based on the preliminary data, the researchers are already planning to take BIIB054 into Phase 2, Brys said.
Wanted, Oh So Badly: PET Tracer for α-Synuclein
One thing everyone in the field readily agrees on is the urgent need for a better biomarker of α-synuclein for trials. A PET tracer would allow researchers to track both accumulation of the aggregated protein in the brain at preclinical stages, as well as reduction due to anti-α-synuclein treatment. “A PET marker would be game-changing,” said Walter Koroshetz, who runs the National Institute of Neurological Disorders and Stroke (NINDS). Scientists have been working on this goal for years, with little success. To hurry things along, the Michael J. Fox Foundation has offered a $2 million prize to the first group to develop one.
In Vienna, Muhs debuted a candidate, saying that AC Immune has tested a chemical library to find compounds specific for aggregated α-synuclein. They found two compounds that fit the bill. They bind recombinant α-synuclein oligomers with 2-6 nanomolar affinity in vitro. In unfixed human postmortem brains from PD patients, both compounds lit up Lewy bodies and neurites. They also bound to α-synuclein aggregates in brain sections from people who had multiple system atrophy (MSA) type C.
One particular challenge for an α-synuclein tracer is that it be specific for the protein over aggregated Aβ, which also forms β-sheets and often occurs along with Lewy bodies in PD, dementia with Lewy bodies (DLB), and AD. At AD/PD, Muhs showed data to demonstrate that their candidate compounds do not bind Aβ. In late-stage AD and PD brains that contained Aβ plaques, the new compounds did decorate the fringes of plaques—but this was because α-synuclein also clustered around these structures. Triple staining confirmed that the compounds were binding only to bona fide α-synuclein structures, not Aβ, Muhs told the audience (see image above). The researchers also verified by way of postmortem tissue autoradiography studies that their compounds did not compete with amyloid tracers for binding to amyloid plaques in the amygdalas of AD patients.
In pharmacokinetic tests, one of the two compounds, dubbed “H,” had good brain uptake and fast washout, while the other had low brain uptake. Muhs said they will take H forward, with the next step being to radiolabel it. In answer to audience questions, he said it was not yet clear if other body tissues besides brain take up the compound, or how it might be cleared from brain. AC Immune is developing the compound in collaboration with Biogen.—Madolyn Bowman Rogers
Do Smartphones Collect Better Clinical Data Than Paper-and-Pencil Tests?
Potential disease-modifying therapies are entering trials for Parkinson’s disease even as researchers need better biomarkers to track clinical improvement. Some believe that smartphones might hold the key. At the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, scientists from Roche and Prothena discussed how they are using these devices to collect detailed clinical data from trial participants. The two companies collaborate on trials of Prothena’s anti-α-synuclein antibody PRX002 (see related conference story). In Vienna, Michael Lindemann of Roche Research & Early Development presented Phase 1 data from smartphones that suggested they more sensitively and accurately reflect small clinical changes than do traditional measures. Crucially, smartphone data closely correlated with the validated clinical measure for PD, the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS).
“This study shows you can use sensor data to correctly represent clinical severity,” Lindemann said. He believes these data are sensitive enough to track both disease progression and treatment effects. Potentially, the data could distinguish people who progress quickly from those whose disease advances slowly. Such information would help researchers stratify trials and better gauge whether treatments are working, Lindemann noted.
Currently, most PD trials rely on the MDS-UPDRS as the primary outcome measure. However, because the UPDRS is only assessed in the clinic, this scale cannot capture the day-to-day fluctuations in function that torment people with PD, and scientists have been searching for alternatives. Some groups are touting the benefits of technology such as smartphones to collect continuous data on neurodegenerative disease and paint a fuller picture of daily variability than an occasional clinic visit allows (see Dec 2012 news). This idea is starting to take hold in Parkinson’s disease, perhaps because, being a movement disorder, it is particularly amenable to the type of data easily collected by smartphone. Various projects have already begun to gather observational data from PD cohorts in this way (Mar 2016 news; PD Smartphone Data Challenge).
In Vienna, Lindemann described how the PRX002 Phase 1b trial used smartphones to monitor symptoms. These devices contain a gyroscope and accelerometer that enable them to detect movement of various types. Participants in the three highest dose cohorts were offered phones that they carried with them throughout the trial. This was optional, but all 44 participants in these cohorts decided to do it, Lindemann noted. The phones collected data for the full 24 weeks of the study. In addition to passive monitoring of gait and movement by phone, participants were asked to complete six specific tasks every day that measured their balance, walking, dexterity, postural tremor, resting tremor, and steadiness of voice.
Participants performed these tasks quite faithfully, Lindemann noted. At the beginning of the trial, the cohort as a whole collected 75 percent of the requested data points, and by the end of the six months, completed 50 percent of the requested tests. Notably, 90 percent of the participants performed the tasks at least once every four days, providing regular data points for analysis. This level of adherence is much higher than that seen in observational studies. Lindemann ascribes this partly to the personal relationship participants developed with the study center staff, and to the degree they trusted that their data would be used well and contribute to advancing PD treatments. He added that the research team also worked hard to make the apps easy to use and the daily tasks not unduly burdensome.
Others agree. Lara Mangravite of Sage Bionetworks, Seattle, ran a previous observational study. She said that trial participants typically feel a strong sense of commitment to provide as much data as possible to support development of new therapeutic agents for their disease. “Patients who provide mobile data collection within the context of a clinical trial consistently demonstrate greater adherence than those using mobile data collection as part of observational trials that are performed remotely,” she wrote to Alzforum.
Importantly, data collected from the smartphones correlated well with MDS-UPDRS scores, Lindemann said. However, smartphone data did not always match up with UPDRS categories of the same name. Balance detected by smartphone, for example, did not correlate that well with the “postural instability” category of the UPDRS, which is measured by the physician tugging patients backward and seeing how well they can regain their footing. Instead, the smartphone balance measure matched up almost perfectly with the “posture” category of the UPDRS, which assesses static balance. The smartphone dexterity test, which involves tapping spots on the screen, correlated with “dressing” in the UPDRS scale, because this task involves fine motor coordination as people manage buttons and zippers.
While scores from the UPDRS and smartphone tests matched, the latter included additional details that filled in the clinical picture, Lindemann said. Patients who scored a zero on the MDS-UPDRS scale for resting tremor, meaning the clinician did not see a tremor, in fact did have a tremor that their smartphone picked up. When patients self-assessed their own tremors, the results better matched their smartphone data than their UPDRS score, indicating that the phone more faithfully reflected the patient’s experience than did the clinician. Lindemann noted that in these cases, the patient’s tremor might not be present during the 15-minute office visit, but might come out at other times.
Moreover, smartphones were more sensitive at detecting small changes, Lindemann said. For example, one patient who started on a standard symptomatic therapy a month before the end of the trial showed distinct motor improvement on the high-frequency smartphone monitoring data, but the pattern was quite noisy. The effects were also subtle, amounting to about a 10 percent improvement in the dexterity measure. Sampling data from just two timepoints, as baseline and follow-up clinic visits would have done, likely would have missed this narrow improvement, Lindemann said.
The data indicate that smartphone technology is feasible to use in a trial, and can detect subtle and clinically meaningful motor impairments, Lindemann concluded. “Remote patient monitoring will transform clinical research,” he predicted, adding that smartphone monitoring will be included in the Phase 2 PRX002 trial.—Madolyn Bowman Rogers
Alzheimer’s researchers are pursuing treatments beyond amyloid and tau, and at the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, they shared news on drugs in Phase 1 and 2 trials that target inflammation, epigenetics, and regeneration. The candidates include two small molecules that soothe microglia and suppress inflammatory signaling, an attempt to tweak epigenetic regulation by way of lysine-specific demethylase, and the neurosteroid allopregnanolone.
Two Strategies to Calm Inflammation
Growing genetic and animal evidence pegs the immune system as a central influencer in both hastening and ameliorating Alzheimer’s progression. Because of this, many groups are looking at ways to tamp down harmful inflammatory signaling. Researchers led by D. Martin Watterson of Northwestern University Feinberg School of Medicine, Chicago, designed and synthesized compounds that specifically suppress pro-inflammatory cytokines, and turned up two related small molecules, MW151 and MW189. Both act on microglia to selectively turn down pro-inflammatory cytokines without affecting anti-inflammatory signaling, said Linda Van Eldik of the University of Kentucky, Lexington, who collaborates with Watterson to develop these candidates as drugs.
Targeting Inflammation.
The small molecule MW151 turns down harmful inflammatory signaling and is being developed in pill form for Alzheimer’s disease. [Courtesy of Linda Van Eldik.]
The researchers believe the compounds have potential to treat both acute brain injuries, which spark neuroinflammation, and Alzheimer’s and other neurodegenerative diseases. They developed MW189 as an intravenous formulation, and saw that it helped allay the destructive inflammatory effects of both amyloid pathology and brain injury in animal studies, Van Eldik reported in Vienna. To trigger amyloid aggregation, the researchers infused Aβ42 oligomers into mouse ventricles for four weeks. After the third week, half the mice received MW189 once daily for two weeks. MW189 treatment largely prevented the spike in brain astrogliosis and IL-1β seen in untreated control animals. Treated animals maintained normal synaptic density and performed like wild-type in the Y maze, in contrast to deficits on these measures in untreated mice.
Likewise, wild-type mice that received a brain injury followed by four days of twice-daily MW189 were able to cling to a rotarod longer than injured animals that did not receive treatment. They performed better in the Morris water maze than untreated animals, although not as well as wild-type. MW189 helped the animals when given as late as six hours after the injury, and at doses down to 0.1 mg/kg, Van Eldik said (see James et al., 2012).
In Phase 1a testing, 32 healthy volunteers were given a single IV dose of either placebo or 0.025, 0.05, 0.1, or 0.25 mg/kg MW189. Of eight people in each dose cohort, two received placebo. The researchers recorded no adverse events related to drug. In a separate Phase 1a study, an additional 18 healthy volunteers received 0.25 mg/kg MW189 or placebo, followed by 2 ng/kg lipopolysaccharide to induce an inflammatory reaction. Those who had MW189 in their systems made less pro-inflammatory TNFα and more anti-inflammatory IL-10 over the next 12 hours, supporting the compound’s ability to control inflammation. This protocol also appeared safe, van Eldik said.
MW189 entered a Phase 1b trial, in which 24 healthy participants receive either placebo or 0.075, 0.15, or 0.3 mg/kg by IV twice daily for five days, in the same proportions as the earlier study. If the compound is well-tolerated, the researchers will enroll people who have suffered a blunt-force traumatic brain injury in a Phase 2a trial. MW189 will be administered by IV within six hours of injury and given twice daily for five days, or until patients are discharged. The trial will enroll people whose brain injuries are severe enough to require external ventricular drains, Van Eldik noted. This will allow the researchers to collect CSF and measure changes in pro-inflammatory cytokines during treatment to determine target engagement. Follow-up visits with participants will assess whether the treatment made a difference in long-term outcomes.
The researchers are starting with brain injuries because these trials are shorter and have better-defined endpoints than AD trials, Van Eldik wrote to Alzforum. Ultimately, the researchers would like to apply the insights gained from these trials to the treatment of AD. “Future success with MW189 and TBI endpoints will inform us of the utility of targeting the pro-inflammatory cytokine aspects of neuroinflammation in more chronic CNS disorders, such as AD,” Van Eldik wrote to Alzforum. To that end, they have developed MW151 as an oral formulation. It suppressed inflammation and protected synapses in an APPPS1 knock-in mouse, and prevented a rise in harmful IL-1β and maintained learning and memory after brain injury in rodents (see Bachstetter et al., 2012; May 2012 conference news; Bachstetter et al., 2015).
Across the Atlantic, Ludwig Aigner of Paracelsus Medical University in Salzburg, Austria, is taking a different approach to modulating the immune system. He evaluates the approved asthma medication montelukast. This small molecule blocks leukotriene receptors that cause bronchial tubes to constrict. Leukotrienes are inflammatory molecules that white blood cells release during an asthma attack. Because neurons and microglia also express leukotriene receptors, Aigner decided to check if this type of signaling plays a role in brain inflammation. In aged, forgetful rats, six weeks of montelukast treatment shrank microglia, restored the blood-brain barrier, and boosted learning in the Morris water maze to that of young rats. Intriguingly, montelukast also boosted neurogenesis, suggesting some regenerative effects (see Oct 2015 news). Notably, leukotrienes rise during aging, and after stroke or brain damage.
The idea of repurposing montelukast for Alzheimer’s disease is buoyed by its excellent safety record; alas, the tablet formulation poorly enters the bloodstream, with only 63 percent of it becoming bioavailable. Because of this, Aigner and colleagues reformulated the drug in collaboration with IntelGenx Corp., Saint-Laurent, Canada. IntelGenx packaged the drug in a thin film that is placed in the mouth, either against the cheek or under the tongue. The film dissolves, delivering the drug to the bloodstream.
The researchers tested this delivery method in eight healthy volunteers, who took 10 mg montelukast, a common asthma dosage, both in the traditional tablet form and by film. The film resulted in higher absorption, reaching 95 percent in the bloodstream, an increase of 50 percent over the tablet’s bioavailability. This formulation will allow lower dosing, Aigner said in Vienna.
The drug crossed the blood-brain barrier, an essential feature for treating AD. After three hours, participants had 3.5 ng/ml in their CSF, rising to 4.25 ng/ml by seven hours. This is the pharmacologically active range, Aigner noted. The researchers next plan to do a Phase 2 study in AD patients in Canada, in collaboration with the research organization Consortium of Canadian Centres for Clinical Cognitive Research (C5R). Aigner noted that the oral film represents a new product that can be patented for commercial development.
Can the Epigenome Be Safely Targeted?
Many researchers have noted epigenetic changes in AD, but targeting these with a drug has proven difficult (see Nov 2012 conference news; Aug 2014 news; Nov 2014 news). Partly this is because histone deacetylase (HDAC) inhibitors, which derepress genes involved in learning and memory, have broad effects that can lead to toxicity with chronic dosing (see Dec 2008 conference news; Aug 2013 conference news).
Researchers at Oryzon Genomics in Barcelona, Spain, developed a different approach. They identified a small molecule, ORY-2001. It inhibits lysine-specific demethylase 1 (LSD1), an enzyme that forms part of a complex with HDAC1/2 to regulate gene expression. In Vienna, César Molinero of Oryzon noted that ORY-2001 has a second mechanism of action as well, inhibiting the mitochondrial membrane protein monoamine oxidase B (MAOB1). MAOB1 helps break down dopamine in the brain, so inhibiting it can help Parkinson’s patients, who suffer from a dearth of the neurotransmitter. The approved Parkinson’s drug rasagiline targets MAOB1.
ORY-2001 can be taken orally, has favorable pharmacokinetics, and crosses the blood-brain barrier, Molinero said. In senescence-accelerated (SAMP8) mice, a model of age-related cognitive decline, treatment for two to four months lowered the expression of pro-inflammatory genes in the hippocampus and restored learning and memory. The compound appeared to have no ill effects. Because LSD1 stimulates blood cell maturation by repressing stem cell genes, researchers looked for a drop in blood production, but treated mice had none (see Kerenyi et al., 2013).
The researchers are now testing single and multiple doses in Phase 1. Eighty-eight young, healthy volunteers took either 0.2, 0.6, 1, 1.5, 2.5, or 4 mg of ORY-2001. Of the eight people in each cohort, two got placebo, and multiple doses were given over five days. Molinero reported no serious adverse events so far, but with repeated dosing at 2.5 mg, platelet counts dropped by a third after eight days and took a week to rebound. The researchers added the 4 mg dose to explore these effects, and that testing is still ongoing, Molinero said.
After a single dose, the compound’s half-life in blood was 22 hours, but with repeated dosing, it accumulated, its clearance slowed, and its half-life stretched to four days. To check target engagement, the researchers measured the amount of LSD1 bound to ORY-2001 in peripheral blood mononuclear cells using chemiluminescent ELISA. They found a dose dependence, with the lowest single dose of ORY-2001 binding 20 percent of LSD1 while the highest dose bound 80 percent. After repeated dosing, the 0.2 mg dose resulted in 30 percent bound LSD1, while doses of 1 mg and higher topped out at 80 percent. The effects lasted for about one week, Molinero said in Vienna.
In a separate substudy, the researchers are measuring ORY-2001 in cerebrospinal fluid to find out if it crosses into the brain. They will also test the compound in healthy elderly participants. ORY-2001 will start Phase 2 later this year with support from the Alzheimer’s Drug Discovery Foundation (see news release).
Regeneration: Could it be Possible?
Neurons wither in the AD brain, and yet the age-old idea of sparking the birth of new neurons to replace them has been considered beyond the reach of reality. Enter the neurosteroid allopregnanolone. It revs up neurogenesis in AD mouse models, restoring learning and memory. All the while, it dials down inflammation and Aβ production. Based on these preclinical data, Roberta Brinton and Lon Schneider of the University of Southern California, Los Angeles, started a Phase 1 trial of the neurosteroid in 24 people diagnosed with mild cognitive impairment due to AD or early AD (see Aug 2013 conference news; Dec 2014 conference news).
Participants sit for a weekly infusion of intravenous allopregnanolone for three months. Each dose cohort consists of eight people, two of whom receive placebo. In Vienna, Brinton presented findings from the first two dose cohorts, who received 2 or 4 mg allopregnanolone. The third cohort, which tests doses from 6 to 18 mg, is still in progress. Administration of the first two doses appeared safe, with no adverse effects, Brinton reported. Toxicology studies in dogs and rats also found no ill effects after six and nine months of chronic dosing with allopregnanolone, she added.
Intriguingly, this short-term treatment revealed hints of efficacy, Brinton told the audience. Cognitive and physical abilities stabilized in people on the drug, whereas those on placebo continued to decline, she said. The treatment group reported being in better moods than the placebo group, and anecdotal reports from caretakers supported better function.
There were indications of possible structural benefits, Brinton added. In the 4 mg cohort, hippocampal volumes of treated participants shrank less than in the placebo group. In three treated participants, the researchers measured improved connectivity between the posterior cingulate cortex and the parietal and prefrontal cortices. These findings are preliminary, as the trial was not powered to detect efficacy. Allopregnanolone is slated to enter a 72-week Phase 2 study of 260 people with early AD, Brinton said.—Madolyn Bowman Roger
Anti-Amyloid Drug Pipeline Shows No Sign of Drying Up
Despite the litany of failed Alzheimer’s drugs, academic and industry researchers at the 13th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 29 to April 2 in Vienna, appeared confident that they are moving in the right direction. Asked why the field continues to go after the amyloid hypothesis even after billion-dollar losses, Samantha Budd Haeberlein of Biogen in Cambridge, Massachusetts, expressed the consensus view when she said, “The science for Aβ is very compelling.” Researchers stressed that they have learned from the past and believe the new trials stand a greater chance of success than previous ones, in part thanks to the recent emphasis on biomarkers.
At the same time, scientists conceded that therapeutic strategies must broaden beyond just targeting amyloid itself. “Instead of a linear amyloid cascade, it’s more of a swirling eddy,” quipped Brad Hyman of Massachusetts General Hospital, Boston. His point was that many other processes, such as inflammation and tau pathology, interact with amyloid to influence neurodegeneration. For an update on non-amyloid treatments, see May 2017 conference news. Roche’s Paulo Fontoura said the previous view—that amyloid is necessary and sufficient for the disease—has evolved into an acknowledgement that curbing amyloid may offer but a partial solution, and that combination therapy may be necessary for a truly effective treatment. In addition, the field still needs to identify the right form of Aβ to target. That goal is close, he believes. “We are on the verge of developing the first successful treatments,” Fontoura predicted. So what, in this big scheme of things, was the amyloid-related news at AD/PD?
Researchers in Vienna presented no breakthroughs. Instead, they offered updates on three drugs since Alzforum’s last coverage from the CTAD conference (see Dec 2016 conference news; Dec 2016 conference news). AstraZeneca reported Phase 1 data from the new anti-Aβ antibody MEDI1814; Eisai discussed Phase 2 pharmacodynamic data for a BACE inhibitor going on to Phase 3; and the biotech firm Alzheon in Lexington, Massachusetts, promoted its rationale for resurrecting the failed drug tramiprosate. Previously, Alzheon had noted a treatment benefit for the subset of participants who carried two copies of the APOE4 allele, and in Vienna, Martin Tolar said data from an open-label extension of a Phase 3 trial conducted in 2006 suggested a cognitive advantage over the original placebo group a year later. A version of the drug will enter Phase 3 this year.
Antibody Therapy: Sweeping Out Aβ
The strategy that has advanced the furthest through the development pipeline is that of clearing Aβ with antibodies. The 12 anti-Aβ antibodies currently in clinical trials target different forms of the peptide, employ distinct mechanisms of action, and produce varying side effects. Amyloid-related imaging abnormalities (ARIA) are of particular concern. These light spots on MRI scans reflect fluid buildup from leaky blood vessels (ARIA-E) or micro-hemorrhages (ARIA-H). Some researchers believe ARIA results from inflammation triggered by antibodies that bind amyloid in blood vessels.
In Vienna, Fabrizio Piazza of the University of Milano-Bicocca, Italy, noted that some people with cerebral amyloid angiopathy (CAA) spontaneously develop ARIA-like events characterized by edema in blood vessels. This phenomenon is known as CAA-related inflammation (CAA-ri), and Piazza has correlated its occurrence with the presence of endogenous autoantibodies against Aβ (see Piazza et al., 2013). These autoantibodies mobilize Aβ, removing it from plaques and leading to more deposition and damage in blood vessels. The condition models therapeutic-induced ARIA, Piazza said. “Aβ removal leads to a temporary overload at the vessels, particularly for people in advanced disease stages who have many plaques,” he explained.
One therapy option might be to pretreat participants at advanced stages of Alzheimer’s pathology with ponezumab, to lower vascular amyloid deposits before busting up parenchymal plaques, he suggested. Pfizer discontinued ponezumab for AD but is now developing it for CAA. Piazza leads the Inflammatory Cerebral Amyloid Angiopathy and Alzheimer’s Disease Biomarkers International Network. iCAβ is evaluating potential diagnostic and prognostic biomarkers of CAA and ARIA. Such biomarkers might help clinicians manage and treat ARIA, he believes.
In the meantime, some antibodies are being engineered to restrain microglial activation and thus lessen ARIA. One of these is MEDI1814, which recognizes monomeric Aβ42 and is being developed in collaboration with Eli Lilly. Built on an IgG1 backbone, MEDI1814 carries mutations that weaken its binding to microglial Fc receptors, potentially lowering the incidence of ARIA (see Nov 2015 news). In Vienna, Thor Ostenfeld of AstraZeneca Neuroscience Innovative Medicines & Early Development, Cambridge, U.K. reported results from the recently completed Phase 1 study. It enrolled 77 people between 55 and 85 years who were clinically diagnosed with mild to moderate AD and had low CSF Aβ42. In the first study phase, 45 participants received a single dose of drug or placebo in a 3:1 ratio. The tested doses were 25, 100, 300, 900, or 1,800 mg by IV, or 100 mg subcutaneously. In the second phase, the other 32 participants received three doses each over the course of two months. Dose levels for this group were 300, 900, or 1,800 mg by IV or 200 mg subcutaneously.
Participants were followed for four months. They had no serious adverse events, Ostenfeld reported. The most common complaints—dizziness, headache, gastrointestinal problems—seemed unrelated to dose. The researchers saw no irritation at the site of injection or reactions to the infusion. They found no evidence of ARIA-E or ARIA-H on MRI scans, supporting the idea that this antibody lowers the risk for this side effect.
The pharmacokinetic data resembled that for other antibodies. The antibody’s half-life in the blood was 17 to 20 days. There, it stabilized Aβ42, with plasma levels of the peptide rising after dosing. A small amount of MEDI1814 entered the brain, with CSF levels hovering around 0.1 to 0.6 percent of those in plasma, as is commonly seen with other therapeutic antibodies. About one-third of participants still had detectable CSF antibody a month after dosing stopped, Ostenfeld noted. MEDI1814 bound to Aβ42 in the central nervous system, as seen by the disappearance of free CSF peptide. A single IV dose of 300 mg cut the levels of unbound CSF Aβ42 in half, while multiple doses of 900 mg took it to near zero. For the subcutaneous formulation, about 33 percent of the dose became bioavailable, Ostenfeld said.
Crucially, the researchers saw a treatment effect on total CSF Aβ42. At the highest doses of drug, this biomarker tripled or quadrupled. Researchers have long debated how they would expect CSF Aβ42 to change if a drug worked. In Vienna, Ostenfeld said that because CSF Aβ42 levels fall in Alzheimer’s disease as the peptide becomes bound up in plaques, this rise may indicate mobilization of the peptide. A similar effect was seen in preclinical studies in rats and monkeys, Ostenfeld said. Aβ40 levels remained unchanged, demonstrating the antibody’s selectivity.
Other scientists stress the importance of determining a full dose-response curve for new therapeutics in order to pick the most effective dose. This was not always done for previous anti-amyloid therapeutics, including solanezumab, which fell short due to a puny effect size in Phase 3 trials of mild AD. Privately, researchers agree that more extensive dose ranging might have prompted a higher solanezumab dose in those trials (see Nov 2016 news; Dec 2016 news). Because AstraZeneca researchers found nearly complete engagement of CSF Aβ42 at high doses of MEDI1814, they believe they have the necessary dosage data, Ostenfeld said in Vienna. He told Alzforum that his team will take the lessons from solanezumab into account.
BACE Inhibitors: Squelching Production
BACE inhibitors represent a different anti-amyloid approach, seeking to turn off the spigot rather than pump up outflow. Several are in trials, with researchers encouraged by early signs that they appear safe enough for long-term use (see Oct 2016 conference news). In Vienna, Bruce Albala of Eisai Inc, Woodcliff Lake, New Jersey, presented safety and pharmacodynamic data from an ongoing Phase 2a trial of elenbecestat (formerly E2609). The study started in 2014 and enrolled 71 people who had prodromal or mild AD verified by a positive amyloid scan. Over the course of 18 months, they received either 5, 15, or 50 mg/day elenbecestat or placebo. A 12-week interim analysis determined that all doses were well-tolerated at that time, Albala said.
To model the pharmacodynamics, the researchers combined data from this trial with that from previous, smaller studies on healthy volunteers. They found that the drug dampened Aβ production to a similar degree in controls and patients (see Jul 2012 news). Specifically, with 17 ng/ml of elenbecestat in cerebrospinal fluid, CSF Aβ1-x levels fell by half. This concentration can be achieved by administering 20 mg/day of the drug, Albala said. At the 50 mg/day dose, modeling predicts that CSF Aβ1-x will fall by 70 percent, he added.
Eisai had initially planned to run a Phase 2b safety and efficacy study on another 630 participants. That trial would have used a Bayesian adaptive design, in which doses and other parameters could be adjusted as it went along based on interim data. The trial included other innovative features, such as using the new cognitive composite ADCOMS as an outcome measure. However, based on the Phase 2a data, Eisai decided instead to accelerate elenbecestat directly into two large pivotal Phase 3 trials with a more conventional design. These two-year studies, called MISSION AD 1 and 2, will enroll 1,330 people with early AD. The trials will test the 50 mg dose, with the primary endpoint being the Clinical Dementia Rating Sum of Boxes. Andrew Satlin at Eisai told Alzforum that this design was chosen after consultation with health authorities.
An audience member questioned whether BACE inhibitor trials are worth doing, given the recent failure of Merck’s verubecestat to budge endpoints in the Phase 3 EPOCH trial in mild to moderate AD. Albala said that Eisai is testing its inhibitor at an earlier stage of the disease, similar to the ongoing APECS trial of verubecestat in prodromal AD (see Feb 2017 news). The Eisai trials run longer, confirm amyloid positivity with PET or CSF, and use a different outcome measure than the EPOCH trial, Albala added.
Let’s Try This One Again: Preventing Aggregation
Tramiprosate has resurfaced to once again take a stab at amyloid. An analog of the amino acid taurine, tramiprosate was originally developed by Neurochem in Montreal. It failed to improve cognition in AD patients in an 18-month Phase 3 trial and was rebranded as a nutraceutical and directly sold to consumers (see Aug 2007 news; Nov 2007 conference news).
In 2013, Alzheon licensed the drug following a re-analysis of the original data that suggested a slowing of decline in the APOE4 homozygote subgroup (see Abushakra et al., 2016). APOE4 homozygotes make up about 2 percent of the general population and 10 percent of people with AD worldwide (see Ward et al., 2012).
In Vienna, Tolar presented data from an open-label extension of tramiprosate. The Phase 3 trial had comprised 1,053 participants taking either 100 mg or 150 mg twice/day, or placebo. Seven hundred and thirty-six participants continued past the blinded phase on the higher dose for another year. In this extension cohort, 104 people carried two APOE4 alleles. Among this group, those who had been on drug for the full 2.5 years maintained a cognitive benefit of 3.9 points on the ADAS-Cog compared to those who were switched from placebo, Tolar claimed. People with one copy of APOE4 had about half that benefit. ADAS-Cog scores stabilized for people who switched from placebo to active drug but never caught up to the active group; this suggests a disease-modifying effect, Tolar said. Among homozygotes, those with mild AD benefited more than those with moderate AD, again implying that early intervention may be more effective.
How might tramiprosate help AD patients? Previous studies claimed the drug blocked aggregation of Aβ42, but provided no mechanism. Alzheon researchers examined the drug’s interaction with Aβ in vitro using mass spectrometry and nuclear magnetic resonance spectroscopy, and in the April 24 CNS Drugs, reported that tramiprosate binds the Lys16, Lys28, and Asp23 amino acid side chains of Aβ42. Multiple molecules of the drug become tethered to each Aβ42 monomer, enveloping the peptides and preventing them from forming oligomers, the authors said. The same mechanism inhibited the elongation of existing oligomers. In vitro, a 1,000-fold excess of tramiprosate over Aβ42 reportedly prevented oligomer formation (see Kocis et al., 2017).
Tolar believes the evidence supports a new study. The researchers developed a tablet prodrug they call ALZ-801. This prodrug is converted to tramiprosate in vivo, but is more easily absorbed by the body and lasts longer in blood, the researchers claim (see Nov 2015 news).
In Phase 1 studies of about 160 healthy elderly participants, ALZ-801 appeared to cause less nausea than tramiprosate, and this side effect cleared up after one week on the drug, Tolar said in Vienna. The other most common complaints were headaches, dizziness, and falls, but these did not relate to dose.
The new formulation cut the person-to-person variability in drug levels in half, Tolar added. Because 150 mg twice daily of tramiprosate achieved a therapeutic effect in the Phase 3 trial, the researchers looked for the ALZ-801 dose that would produce an equivalent CSF exposure. This turned out to be 265 mg ALZ-801 twice daily. The researchers calculated that this dosage should result in a brain concentration of about 550 nM ALZ-801, or about 1,300 to 3,700 times the concentration of soluble CSF Aβ42 seen in AD patients, Tolar noted. This suggests the drug reaches the necessary concentrations to interfere with oligomer formation and have a therapeutic effect, he predicted. Alzheon plans to start a Phase 3 trial of 265 mg twice-daily ALZ-801 in APOE4 homozygotes this year.—Madolyn Bowman Rogers



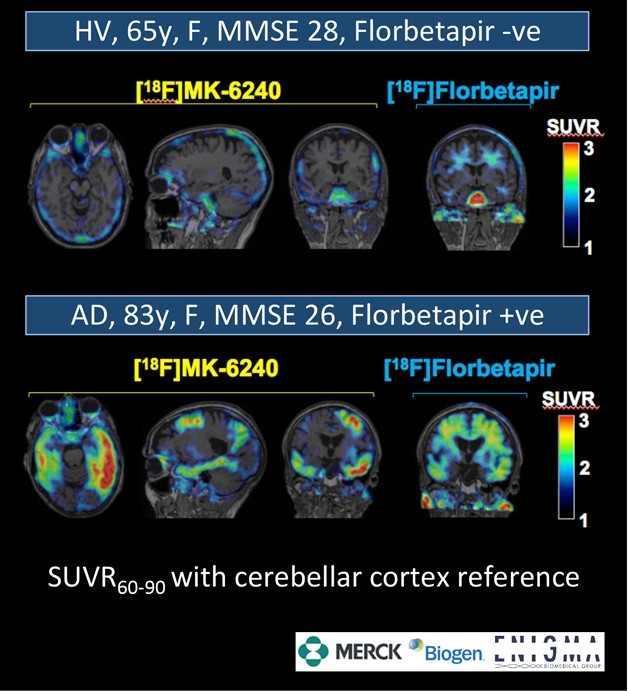



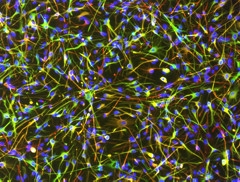

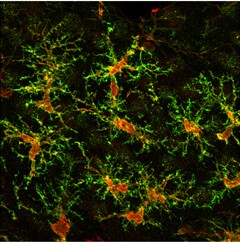



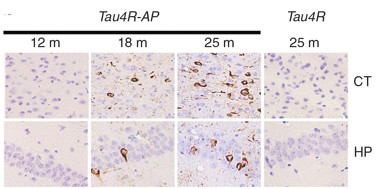




Comments
No Available Comments
Make a Comment
To make a comment you must login or register.