Massive Mouse Study Bolsters Rationale for Combination Therapy
Quick Links
Ron DeMattos and colleagues at Eli Lilly are challenging the field with extensive studies of combination therapies in mouse models of Alzheimer’s disease. At the Alzheimer's Association International Conference 2015, held July 18-23, DeMattos expanded a preclinical approach he had first described at AAIC in Copenhagen in 2014. To a packed room at the Walter E. Washington Convention Center in Washington, D.C., he reported the culmination of three years of work studying 700—yes, 700; that's no typo—mice given various combinations of Lilly's BACE inhibitor and anti-pyroglutamate Aβ antibody. "The take-away is that the work supports the rationale for combination therapy in the clinic," said DeMattos.
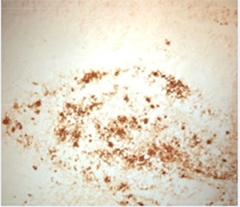
Pyrotechnics.
The anti pGluAβ antibody mE8 binds plaques in transgenic mice. [Image courtesy of Ronald DeMattos and Neuron.]
The wisdom of combination therapy has been hotly debated in recent years. Some, mostly in academia, argue that AD’s multiple pathologies necessitate a multipronged approach for a meaningful clinical benefit, and combination trials should start before the current batch of monotherapy trials read out (e.g., Apr 2015 conference news). Others counter that combination trials are a logistical nightmare and urge a go-slow approach (see Feb 2015 conference news; Mar 2014 conference news). Even starting combination trials of previously approved drugs is proving challenging (see Jun 2015 conference news).
Trying combinations of drugs in mice should be the first step, leaders in the field agree, but few have tried it. Among many possibilities for combination therapy, the Lilly researchers chose to focus on Aβ for now. They tested two drugs that could reduce amyloid deposits: the BACE inhibitor LY2811376 to turn off production of the peptide, and a humanized mouse monoclonal antibody (LY3002813) to the pyroglutamate form of N-terminal truncated Aβ to reduce existing plaques (see DeMattos et al., 2012). The pGluAβ antibody preferentially binds to dense-core plaques.
In Copenhagen last year, DeMattos reported that the combination of the two drugs worked better than either alone, knocking down plaques by 86 percent over four months when administered to PDAPP mice beginning when they were 18 months old. That study, in 180 mice, tested only one treatment duration and only one dose of each drug. In D.C., DeMattos shared data from two more recent studies, one that varied the doses of both drugs, and one that looked at the time course of combination therapy.
For the dose-response study, the researchers established 11 arms with around 28 PDAPP mice in each, covering a plethora of dose permutations. They included high (12.5 mg/Kg/week) or no antibody plus or minus high (30 mg/Kg/day), medium (3 mg/Kg/day), low (0.3 mg/Kg/day), or no BACE inhibitor. Medium- (4mg/K/week) and low-dose (1.5 mg/Kg/week) pGluAβ were combined with only the high dose of the BACE inhibitor. The study also had control arms for untreated and IgG2a-treated mice. The scientists treated the mice for four months beginning at 19 months of age, when plaques are well established in these animals. Total Aβ in guanidine HCl extracts measured by ELISA and immunohistochemistry served as end points.
For the high-dose combinations, the results matched those DeMattos reported last year. Antibody, BACE inhibitor, and the combination reduced total hippocampal Aβ by 30, 60, and 84 percent, respectively. For the BACE inhibitor, the dose response tightened when animals also received the antibody. That is, the 7 to 60 percent reductions in plaque load seen with increasing BACE inhibitor alone jumped to a 33 to 80 percent reduction in the presence of high-dose antibody as well.
Immunohistochemical analysis using the 3D6 anti-Aβ antibody paralleled the ELISA data. The high-dose BACE inhibitor strengthened the pGluAβ antibody dose response, such that 50, 63, and 84 percent of the Aβ was ablated by low, medium, and high doses of antibody, respectively. Low-dose antibody by itself removes about 30 percent of Aβ.
The longitudinal study was even larger. The researchers measured the effect of 4-, 8-, 12-, and 16-week treatments of high-dose BACE inhibitor and high-dose antibody either alone or in combination. Treatment began when the animals were 18 months old. Over the course of the 16 weeks, Aβ load as measured by ELISA slightly increased in controls, then reached a ceiling, which is very similar to what happens in AD patients, said DeMattos. In mice treated with the BACE inhibitor, the Aβ load decreased at four weeks, and continued to decrease slowly over the 16 weeks such that about half was removed. The antibody reduced Aβ by about 35 percent after four weeks and then it stabilized over the remaining 12 weeks. The important result, said DeMattos, was that on combination therapy, Aβ load fell faster over the 16 weeks than on either monotherapy alone, such that 80 percent was removed. The 16-week data in this longitudinal study lined up precisely with the four-month data from the previous two combination studies—all showing approximately 80 percent reduction in Aβ with the high doses of both drugs. "To repeat these experiments three times, over three years, with hundreds of animals, shows how robust this treatment and pharmacology are," said DeMattos.
What does might this mean for human trials? "Overall it fosters our confidence that combination therapy will result in more dramatic lowering of pre-existing plaque," said DeMattos. One upshot was that at medium and high doses of BACE inhibitor there was synergism with the antibody. "This tells us that when you have the two mechanisms engaged simultaneously, it promotes a feed-forward response resulting in more clearance of plaques," he said.
Others found this promising. "I have seen some of this data and find it very encouraging that combination therapies may have synergistic effects," wrote Reisa Sperling, Brigham and Women's Hospital, Boston. "I hope that other companies begin more animal studies and early safety work on combinations, including across mechanisms, such as anti-Aβ and anti-Tau." Other scientists noted that adding an antibody directed against plaques may allow researchers to go easy on the dose of the BACE inhibitor. Some BACE inhibitors on their own are able to reduce CSF Aβ levels down to about 80 percent, but because of BACE’s numerous reported substrates, some basic scientists have raised safety questions about doing so (see Nov 2014 news; Dec 2013 conference news).
DeMattos would not speculate on future mouse studies. He told Alzforum there is a lot more work to do to analyze the mice from the longitudinal study. The researchers have only processed the ELISA data, and still have to look at the histology, a large task with so many mouse brains to analyze.
Already the researchers know that plaque dynamics are complex. For example, plaques begin to dissolve with just the BACE inhibitor alone. DeMattos suggested that could be due to two ongoing processes, which are not mutually exclusive. First, as the inhibitor stops production of new Aβ, it may relieve overwhelmed clearance machinery, which can then work to remove existing Aβ deposits. Second, if free and plaque-bound Aβ are in equilibrium, then as the concentration of soluble Aβ falls in the brain, more may leech from the plaques. Add an antibody that reacts with a form of Aβ that mostly ends up in plaque cores, and the scenario becomes even more complicated. DeMattos said that it will be crucial to fully understand these dynamics. The longitudinal histology data will help with that, because it may reveal what happens to diffuse and dense-core plaques over time.
DeMattos said that besides providing proof of principle for combination therapy, the mouse study could inform the design of a human trial. The dose-response study will help clinicians establish rational choices for drug doses in humans, while data from the longitudinal study may predict when biomarker signals will change. "We will have an informed idea of when to do an amyloid PET scan, for example" said DeMattos. He cautioned, however, that all this will be contingent on what aspects of the animal model will hold true in people. Human plaques tend to be less soluble and the Aβ more truncated and modified. Paul Aisen, from the new Alzheimer's Therapeutic Research Institute, at University of Southern California, San Diego, co-chaired the session. He noted that it may be just as important to remove diffuse and dense-core plaques. "We have to take our insight and try to predict what will translate," said DeMattos.—Tom Fagan
References
News Citations
- As DIAN Plans Trial Number Two, the Goal Is to Go Big
- Alzheimer’s Disease Research Summit 2015: Three Years In
- Combination Trial Debate Energizes Keystone Symposium
- Advisory Panel Grapples with Combination Therapy
- At High Doses, BACE1 Inhibitors Hinder Synaptic Plasticity in Mice
- Blocking BACE—Do Adult Mouse Phenotypes Predict Side Effects?
Therapeutics Citations
Research Models Citations
Paper Citations
- Demattos RB, Lu J, Tang Y, Racke MM, DeLong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer's disease mice. Neuron. 2012 Dec 6;76(5):908-20. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Certara
This project is a major tour de force and likely an excellent example of the implementation of novel guidelines for making preclinical animal research more reproducible (Shineman et al., 2012). It will also provide a good database for preclinical drug metabolism/pharmacokinetisc modelers to derive the best treatment combination for achieving a specific reduction of the different Aβ peptides. This is important given the possible more neuroprotective effect of shorter Aβ isoforms vs. the neurotoxic effect of Aβ1-42 (Wang et al., 2013). Another advantage is possible dose-reduction of the antibodies and/or BACE inhibitors to expand the therapeutic window.
The big challenge, however, is the translation of these preclinical findings to functional outcomes in the clinical settings, where patients are on a diversity of co-medications that might affect the individual cognitive trajectory or where specific genotypes affect the resilience of synapse dysfunction. Those probably contribute to the lack of a direct correlation between amyloid load reduction and improvement on clinical cognitive scales in clinical trials, especially when the clinical effect size is small.
Simulations using our humanized ADAS-Cog-calibrated Quantitative Systems Pharmacology platform (Roberts et al., 2012; Nicholas et al., 2013) combined with a model for human Aβ synthesis from SILK data, biochemical data on the differential aggregation of Aβ peptides (1-40 and 1-42) and their differential impact on glutamatergic and α7 nAchR neurophysiology (Geerts 2014) suggest a clinical effect size in the range of 1-2 points for Aβ modification, assuming there is no neurotoxicity.
This same range of variations of 1-2 points on the ADAS-Cog scale along the cognitive trajectory can readily be observed with co-medications ranging from antidepressants to antipsychotics to benzodiazepines. In addition, this same variability can be reached when considering differential effects on synapse dysfunction (Perez-Nievas 2013).
Any imbalance in the different treatment arms could therefore substantially reduce the clinical effect size.
References:
Shineman DW, Basi GS, Bizon JL, Colton CA, Greenberg BD, Hollister BA, Lincecum J, Leblanc GG, Lee LB, Luo F, Morgan D, Morse I, Refolo LM, Riddell DR, Scearce-Levie K, Sweeney P, Yrjänheikki J, Fillit HM. Accelerating drug discovery for Alzheimer's disease: best practices for preclinical animal studies. Alzheimers Res Ther. 2011;3(5):28. PubMed.
Wang Y, Zhou TH, Zhi Z, Barakat A, Hlatky L, Querfurth H. Multiple effects of β-amyloid on single excitatory synaptic connections in the PFC. Front Cell Neurosci. 2013;7:129. Epub 2013 Sep 3 PubMed.
Roberts PD, Spiros A, Geerts H. Simulations of symptomatic treatments for Alzheimer's disease: computational analysis of pathology and mechanisms of drug action. Alzheimers Res Ther. 2012 Nov 26;4(6):50. PubMed.
Nicholas T, Duvvuri S, Leurent C, Raunig D, Rapp T, Iredale P, Rowinski C, Carr R, Roberts P, Spiros A, Geerts H. Systems Pharmacology Modeling in Neuroscience: Prediction and Outcome of PF-04995274, a 5HT4 Partial Agonist, in a Clinical Scopolamine Impairment Trial. Adv Alzheimer Dis. 2013 Sep; 2(3):83-98.
Geerts H. Using in silico mechanistic disease modeling to address the effect of amyloid beta manipulation on cognitive clinical readouts. Alzheimers Dement. 2011 Jul; 7(4 Suppl),S774–5.
Perez-Nievas BG, Stein TD, Tai HC, Dols-Icardo O, Scotton TC, Barroeta-Espar I, Fernandez-Carballo L, de Munain EL, Perez J, Marquie M, Serrano-Pozo A, Frosch MP, Lowe V, Parisi JE, Petersen RC, Ikonomovic MD, López OL, Klunk W, Hyman BT, Gómez-Isla T. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain. 2013 Aug;136(Pt 8):2510-26. PubMed.
Make a Comment
To make a comment you must login or register.