Do MicroRNAs Cause Mayhem Across Frontotemporal Dementia Spectrum?
Quick Links
RNA in all its guises was a central theme at the 9th International Conference on Frontotemporal Dementias, held October 23 to 25 in Vancouver, Canada. After all, several disease-related proteins are known to make a living comingling with this nucleic acid. But beneath the waves of mainstream RNA research presented at the conference, an undercurrent of findings revealed that tinier snippets called microRNAs could have an outsize impact on disease. Researchers contend that microRNAs, which dampen the expression of their target genes, are misregulated in cells derived from FTD patients and in animal models of FTD. Changes in the levels of these tiny off switches had big consequences for the function of neurons in the models, and researchers are trying to understand exactly how the microRNAs may act in human disease across the FTD spectrum.
MicroRNAs, or miRNAs for short, were first discovered in 1993, shortly before their larger cousins, called small interfering RNAs, made their splash. But it was not until the turn of the century that the regulatory role of miRNA was fully recognized. The little ones are 21- to 23-nucleotide sequences shaped like hairpins, which block the expression of their target genes. Transcribed from within introns and intergenic regions scattered across the genome, immature “pri-miRNAs” are processed into shorter fragments by the enzymes Drosha and DGCR8 in the nucleus. The resulting “pre-miRNAs” then cross into the cytoplasm, where the enzyme Dicer sculpts them into mature miRNAs. A portion of the miRNA molecule recognizes and binds to target sequences in 3’ untranslated regions of mRNA targets, while another portion of the miRNA brings along gene-silencing machinery to do its business. Typical miRNAs can potentially latch onto hundreds of different mRNA targets; their binding can trigger the destruction of mRNAs or block their translation.
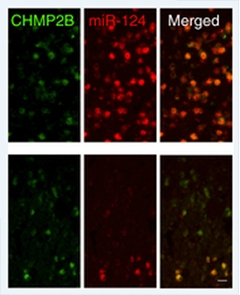
Missing Micro Manager.
Mice expressing a pathogenic mutant of CHMP2B (green, bottom panel) also express lower levels of miR-124 microRNA (red, bottom panel) than their wild-type counterparts. Proper regulation of some neurotransmitter receptors requires miR-124. [Image courtesy of Gascon et al., Nature Medicine 2014.]
A subset of miRNAs are expressed predominantly in the brain. There, their intricately choreographed expression patterns orchestrate neural development. However, recent studies have reported that some crucial jobs of miRNAs extend into adult life, making these curious fragments potentially important for neurodegenerative disease. Specific miRNA molecules seem to be misregulated in Alzheimer’s and Parkinson’s diseases (see Qiu et al., 2014), and growing evidence implicates them in FTD/ALS as well (see Gascon and Gao, 2014).
At ICFTD, Fen-Biao Gao of the University of Massachusetts Medical School in Worcester presented new findings underscoring the role of miRNAs in FTD/ALS. A recent study from Gao’s lab had reported that expression of miR-9 was turned down in induced pluripotent stem cell (iPSC)-derived neurons from FTLD-TDP patients, who develop neuronal inclusions containing the RNA-binding protein TDP-43. MicroR-9 is expressed primarily in the brain. The consequence of miR-9’s suppression in these patients is unknown (see Zhang et al., 2013). At the meeting, Gao presented data implicating yet another miRNA in FTD: miR-124. He and his colleagues looked at miRNA expression patterns in iPSC-derived neurons from people with behavioral variant FTD (bvFTD) including C9ORF72 expansion carriers. After two months in culture, these neurons drastically downregulated expression of miR-124. This microRNA was just reported independently by another group to target the GluA2 subunit of the AMPA receptor (see Ho et al., 2014), and miR-124 binding sites reside in the 3’ UTRs of three of AMPAR’s four subunit genes. Gao’s lab found that iPSC-derived neurons from people with bvFTD expressed more of these subunits, and mutating the miR-124 binding sites in these receptor genes returned receptor levels to normal. Postmortem brain samples from people with bvFTD also had lower miR-124 and higher GluA2 expression. Overexpression of the GluA2 subunit alters the ratio of calcium-permeable and -impermeable receptor subunits, altering the AMPA receptor function. This imbalance could spell trouble for neurons, Gao said.
This AMPAR/miR-124 relationship also revealed itself in the context of an entirely different FTD mutation. A completely penetrant CHMP2B mutation identified in a single Danish family causes neuronal inclusions containing p62, but not TDP-43. Gao presented findings from a new transgenic mouse in which researchers can switch on expression of the pathogenic CHMP2B mutant gene in the brain. These mice develop deficits in social interactive behavior at a relatively young age and groom themselves obsessively when they get older. These deficits make them useful models for bvFTD, Gao said. Like their human iPSC-derived counterparts, neurons in these mice expressed less miR-124 and more of certain AMPAR subunits than controls, but delivering miR-124 into their brain using adeno-associated virus restored normal AMPAR expression levels and partially rescued the animals’ behavioral phenotype. Gao’s study came out on November 17 (see Gascon et al., 2014).
Gao believes that some FTD/ALS causal mutations may somehow converge on similar changes in miRNA expression. “MiR-124 downregulation and downstream changes in AMPAR subunits might be common to other forms of FTD,” Gao told Alzforum. “Now we will try to figure out how different disease mutations trigger changes in miRNA expression.”
Previous studies implicated TDP-43 in processing miRNAs, suggesting that the protein’s aggregation in disease would prevent the production of some miRNAs (see Feb 2012 news story). However, the way CHMP2B mutations foul up miRNA processing could involve defects in autophagy, which also has been reported to affect miRNA activity by gobbling up Dicer and other miRNA co-conspirators (see Gibbings et al., 2013).
It appears that the entire spectrum of FTLD genes may be affected by miRNA. At ICFTD, Sébastien Hébert of Laval University in Quebec presented results on tau. Spurred by findings from his and other labs that the miR132/212 family of microRNAs were downregulated in the brains of FTLD-tau patients (see Hébert et al., 2013), Hébert’s lab generated miR132/212 knockout mice. Both the human and mouse tau genes contain a miR-132/212 binding site in their 3’ UTR. The mice expressed more tau protein, which became hyperphosphorylated by six months of age and formed insoluble aggregates by 12 months. The mice had memory problems and some behavioral deficits, Hébert said, but lived a normal lifespan without obvious neurodegeneration.
Their neurons had more and larger autophagic vacuoles. Hébert speculated that the upregulation of tau could have indirectly messed up autophagy. Alternatively, other genes regulated by miR-132 could be at play, but his group has already ruled out involvement of obvious autophagy-related genes.
Hébert told Alzforum that beyond affecting brain function in wild-type mice, knocking out miR-132 also exacerbated the phenotype of FTD or AD models. For example, deleting miR-132 in a mouse expressing human tau with a pathogenic P301L mutation accelerated tauopathy. The same held true for the 3xTg mouse, which expresses mutant tau, APP, and PS1 genes. Given these findings, it is possible that miR-132 may act both as a genetic risk factor in its own right, and as a disease modifier in the presence of other, causal mutations, he said.
This is not the first time miR-132 has been in the FTLD spotlight. A previous study led by Alice Chen-Plotkin and Virginia Lee at the University of Pennsylvania in Philadelphia reported that this microRNA family shuts down expression of TMEM106B, a known genetic modifier of FTLD. Patients expressed low levels of these microRNAs, which led to boosted TMEM106B expression. TMEM106B accumulated in endolysosomes, which disrupted the trafficking of proteins including progranulin (see Aug 2012 news story). Hébert said that unlike the human TMEM106B gene, mouse TMEM106B harbors no miR-132 binding site, so he was unable to study this relationship in his knockout mice. Coming full circle, Gao and colleagues re-analyzed Chen-Plotkin’s data and found that FTD patients with granulin mutations expressed lower levels of miR-124, as well.
Still other miRNAs have been implicated in FTLD/ALS. Rosa Rademakers at the Mayo Clinic in Jacksonville, Florida, found altered expression of five different miRNAs in the cerebellum of FTLD patients with progranulin mutations, and this correlated with dysregulation of 18 genes (see Kocerha et al., 2011). Rademakers first implicated miRNAs in FTD when in 2008 she reported variation of a miRNA binding site in the progranulin gene as a risk factor (see Nov 2008 conference coverage). Other studies reported upregulation of miR-155 in patients with ALS, and treated motor defects in a mouse model with antisense oligonucleotides targeting it (see Nov 2013 conference coverage).
Hébert believes that these growing findings support the notion that the genetic influence of miRNA spreads far and wide. Like transcription factors, miRNAs are likely to play broad, important roles in many disease processes, he said. Unfortunately, most large genetic studies pay them no mind. Exome sequencing discounts them entirely, and most GWAS have not looked at SNPs that fall into miRNA sequences or promoters, or within genes that regulate miRNAs or support their function. Hébert said he hopes emerging research implicating miRNAs in disease will motivate researchers to consider miRNAs in future genetic studies.—Jessica Shugart
References
News Citations
- Slicing and Dicing: TDP-43 Teams Up With Nucleases to Make MicroRNAs
- FTD Risk Factor Confirmed, Alters Progranulin Pathways
- DC: More MicroRNA Implicated in Dementia
- Blocking a MicroRNA Slows Motor Neuron Disease in Mice
Research Models Citations
Paper Citations
- Qiu L, Zhang W, Tan EK, Zeng L. Deciphering the function and regulation of microRNAs in Alzheimer's disease and Parkinson's disease. ACS Chem Neurosci. 2014 Oct 15;5(10):884-94. Epub 2014 Sep 25 PubMed.
- Gascon E, Gao FB. The emerging roles of microRNAs in the pathogenesis of frontotemporal dementia-amyotrophic lateral sclerosis (FTD-ALS) spectrum disorders. J Neurogenet. 2014 Mar-Jun;28(1-2):30-40. Epub 2014 Feb 10 PubMed.
- Zhang Z, Almeida S, Lu Y, Nishimura AL, Peng L, Sun D, Wu B, Karydas AM, Tartaglia MC, Fong JC, Miller BL, Farese RV, Moore MJ, Shaw CE, Gao FB. Downregulation of MicroRNA-9 in iPSC-Derived Neurons of FTD/ALS Patients with TDP-43 Mutations. PLoS One. 2013;8(10):e76055. PubMed.
- Ho VM, Dallalzadeh LO, Karathanasis N, Keles MF, Vangala S, Grogan T, Poirazi P, Martin KC. GluA2 mRNA distribution and regulation by miR-124 in hippocampal neurons. Mol Cell Neurosci. 2014 Jul;61:1-12. Epub 2014 Apr 28 PubMed.
- Gascon E, Lynch K, Ruan H, Almeida S, Verheyden JM, Seeley WW, Dickson DW, Petrucelli L, Sun D, Jiao J, Zhou H, Jakovcevski M, Akbarian S, Yao WD, Gao FB. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat Med. 2014 Dec;20(12):1444-51. Epub 2014 Nov 17 PubMed.
- Gibbings D, Mostowy S, Voinnet O. Autophagy selectively regulates miRNA homeostasis. Autophagy. 2013 May;9(5):781-3. Epub 2013 Feb 19 PubMed.
- Hébert SS, Wang WX, Zhu Q, Nelson PT. A Study of Small RNAs from Cerebral Neocortex of Pathology-Verified Alzheimer's Disease, Dementia with Lewy Bodies, Hippocampal Sclerosis, Frontotemporal Lobar Dementia, and Non-Demented Human Controls. J Alzheimers Dis. 2013 Jan 1;35(2):335-48. PubMed.
- Kocerha J, Kouri N, Baker M, Finch N, Dejesus-Hernandez M, Gonzalez J, Chidamparam K, Josephs KA, Boeve BF, Graff-Radford NR, Crook J, Dickson DW, Rademakers R. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genomics. 2011;12:527. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.