CONFERENCE COVERAGE SERIES
International Conference on Alzheimer's and Parkinson's Diseases 2023
Gothenberg, Sweden
28 March – 01 April 2023
![]()
CONFERENCE COVERAGE SERIES
Gothenberg, Sweden
28 March – 01 April 2023
![]()
Queen Silvia of Sweden opened the 17th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Sweden's southwestern city of Gothenburg. The queen, age 79, flew in from Stockholm through a snowstorm, toured the labs of Kaj Blennow and Henrik Zetterberg at Sahlgrenska University Hospital, and then addressed the 3,000 in-person and 1,000 virtual attendees at the city’s conference center. Her Majesty spoke movingly about losing her mother and, two years ago, her brother to Alzheimer's disease. She mentioned hope, and progress having been made on risk factors, immunotherapy, and fluid biomarkers. “Yet, there is still a lot to do, and that is why you are here,” she told the audience.

Queen Silvia
Indeed there is a lot to do, and Alzforum will do its part with a series of news summaries over the next two weeks. First up: progranulin as a therapeutic target.
In people with deleterious variants of the progranulin gene, the halving of its encoded protein that results from these mutations leads to frontotemporal dementia. This comparatively straightforward haploinsufficiency mechanism casts progranulin restoration as a leading therapeutic target, and a handful of progranulin-boosting therapies have already made their way into clinical trials. At AD/PD, scientists presented preclinical data on potential therapies coming down the pike. One was Arkuda’s ARKD-104, a small molecule that boosts progranulin levels as well as the activity of its cleavage products, the granulin peptides. Another, Denali’s DNL593, consists of a progranulin protein strapped to an antibody that whisks it across the blood-brain barrier; it relieved dramatic phenotypes in mice and human cells. Finally, scientists zeroed in on the lysosomal protease legumain as a link between progranulin deficiency and TDP-43 pathology. The findings could present opportunities to squelch the damage wrought by too little progranulin.
Though the concept of progranulin replacement sounds simple, the varied lifestyle of progranulin is anything but. In its full-length form, progranulin can be secreted, but also trafficked to the lysosomes, where proteases rapidly dice it up into multiple different granulin peptides. Considered the functional forms of progranulin, these peptides take part in processing various lipids within lysosomes, and play a critical part maintaining these organelles' acidification and function. What's more, progranulin rarely enters the lysosome alone. When outside the cell, it often buddies up with prosaposin, another protein that needs to gain access to the lysosome before being processed into its active forms, the saposin peptides. When bound together, the two full-length proteins can take advantage of different transport pathways to cooperatively traffic to the lysosome.
Multiple strategies attempt to boost progranulin levels. The furthest along is AL001, aka latozinemab. This antibody blocks the sortilin receptor, thought to internalize progranulin and whisk it to lysosomes for degradation (Aug 2021 conference news). It is in Phase 3, and had no presentations at AD/PD. Also underway are earlier-stage trials, for gene replacement using progranulin-expressing adeno associated viruses, including Prevail’s PR006 and Passage Bio’s PBFT02.
At AD/PD, scientists presented preclinical findings about the next drugs to enter the pipeline. Arkuda Pharmaceuticals, a small company in Watertown, Massachusetts, took a small-molecule approach to up progranulin levels. Raymond Hurst presented preclinical findings on the company’s current lead, ARKD-104, a brain-penetrant compound that stokes progranulin activity. In addition to raising the concentration of secreted progranulin, the compound also ratcheted up levels of granulins and saposins within the lysosome.
In GRN knockout mice and in iPSC-derived neurons from people with FTD-GRN, these elevated progranulin cleavage products appeared to be doing their job. Hurst reported a rise in bis(monoacylglycero)phosphates (BMPs), a type of lipid that is known to be abnormally low in both progranulin mutation carriers and in mouse models of progranulin deficiency. BMPs are required for the catabolism of glycosphingolipids within the lysosome, so BMP deficiency is thought to promote lysosomal overflow (Boland et al., 2022).
The treatment also restored flagging activity of acid lysosomal hydrolases such as GBA1. The exact mechanism of ARKD-104 remains unknown, but experiments thus far suggest that it enhances progranulin trafficking through the vesicular system, leading to its secretion, Hurst said.
In cynomolgus monkeys that received the drug orally, Hurst’s team measured a near tripling of the progranulin concentration in the cerebrospinal fluid within eight hours of the first dose. Levels held steady with daily dosing, but fell quickly back to baseline after dosing stopped. Hurst predicts that AKRD-104 could similarly normalize progranulin in humans. Arkuda plans to start a Phase 1a trial in healthy volunteers in early 2024.
Some in the audience asked whether such a bump in progranulin might be toxic, noting that elevated progranulin has been tied to cancer and inflammation. Hurst said that the goal of the treatment is to restore progranulin to physiological levels in people who are deficient. If anything, Hurst said, preclinical work suggests that restoring progranulin levels should reduce inflammation.
Others asked about off-target effects. Could the drug interfere with expression of other proteins? Hurst said that other than a small number of other lysosomal proteins that rose along with progranulin itself, no other proteins have been found to budge in response to the drug so far.
How does this small-molecule approach measure up to other ongoing strategies to boost progranulin? Hurst told Alzforum that, relative to biological drugs such as antibodies or gene therapy vectors, ease of use is the obvious benefit of ARKD-104. This is particularly true in the case of progranulin deficiency, which would need to be continually corrected throughout life. Hurst noted that while treating people with FTD-GRN is the first and foremost goal for ARKD-104, it is conceivable that using small molecules to slightly elevate progranulin could benefit people with other neurodegenerative conditions in which flagging endolysosomal dysfunction plays a part.
Another way to boost progranulin in the brain is to strap it to a shuttle. Denali’s DNL593 makes use of so-called “brain shuttle” technology. Also called a protein transfer vehicle, it comprises a progranulin protein fused to an antibody fragment that binds the transferrin receptor. This facilitates passage of the pair across the blood-brain barrier via transcytosis (Logan et al., 2021).
This drug is in Phase 1/2 and had no presentations at AD/PD; however, Christian Haass of Ludwig Maximilians University in Munich presented preclinical data exploring a related approach. Haass' team infected mice in the liver with an adeno-associated virus expressing the genes encoding this progranulin protein transport vehicle. The scientists treated mice deficient in both progranulin and TMEM106b, a gene involved in lysosomal function that modifies progranulin disease risk (Sep 2020 news on Werner et al., 2020).
Without treatment, these mice display drastic motor deficits. They quickly fall off a spinning rod, and can't get back up after being pushed over. Treatment with the progranulin shuttle not only gave them staying power on the rod, but also made them resistant to being pushed over in the first place, Haass said. The brain-shuttle-expressing virus also corrected a barrage of other disease phenotypes. It reduced soaring CSF NfL, calmed inflamed microglia, boosted autophagy, and lessened the buildup of ubiquitinated proteins. Importantly, it also reduced the accumulation of insoluble phosphorylated TDP-43 in neurons.
Haass’ group, in collaboration with Dominik Paquet and Anja Capell’s groups at LMU, also tried out the approach in human cells. Graduate students Marvin Reich and Sophie Robinson generated iPSC-derived neurons and microglia, with or without expression of progranulin and TMEM106b. Surprisingly, they found that when cultured alone, double-knockout neurons showed no signs of TDP-43 pathology. However, when either wild-type or double-knockout neurons were co-cultured with double-knockout microglia, the neurons not only developed TDP-43 aggregates, they also died. Adding Denali’s brain shuttle construct to the culture medium restored progranulin expression in the microglia, quelled their lysosomal dysfunction, and saved the neurons. Haass said the findings imply a role for neuron-microglial cross talk in the development of neuronal TDP-43 pathology and neurodegeneration caused by progranulin deficiency.

Deadly Conversation? When microglia missing both progranulin and TMEM106b were co-cultured with wild type neurons (left), TDP-43 aggregates (yellow, right) accumulated in neurons (MAP2, white). Microglia, Iba1, pink. [Courtesy of Christian Haass, Ludwig Maximilians University, Munich.]
Legumain: The Missing Link?
How are progranulin-deficient microglia triggering TDP-43 aggregation in neurons? Anja Capell, also at LMU, presented data pointing to one possible explanation. She reported that in microglia lacking progranulin, the lysosomal protease legumain became much more active. Using a series of biochemical experiments, Capell unraveled a striking chain of events.She found that progranulin somehow slows the cleavage of pro-legumain into its active form. Therefore, without progranulin around, legumain activity shoots up. Legumain, it turns out, also contributes to the cleavage of progranulin into granulins, and in the activation of cathepsin proteases. What’s more, progranulin-deficient microglia churn out pro-legumain, which is taken up by nearby neurons. Once inside, legumain becomes activated, and then cleaves—none other than TDP-43. This, in turn, sets off pathological accumulation of TDP-43 in the neurons.
Using a co-culture of iPSC-derived microglia and neurons similar to those that Haass used, Capell showed that legumain released from progranulin-deficient microglia played a pivotal role in instigating TDP-43 pathology in co-cultured neurons.

Legumain Link. Progranulin deficiency leads to excess legumain in microglia, which secrete a precursor of this lysosomal protease. Neurons absorb and process it into its active form, which subsequently cleaves TDP-43, inciting aggregation. [Courtesy of Anja Capell, Ludwig Maximilians University, Munich.]
In postmortem brain samples from people with FTD-GRN, Capell detected an excess of activated legumain, suggesting this pathway is active in disease, even in real-life circumstances milder than a full, experimental progranulin deletion.
That latter finding is important. One audience member noted that, in contrast to hyperactivated microglia seen in progranulin knockout mice, microglia with but one defunct copy of progranulin remain relatively "calm," hence progranulin knockouts do not truly model FTD-GRN. Capell readily agreed. She is continuing her work with human cells missing one copy of functional progranulin.
The detailed biochemical mechanisms linking progranulin, legumain, and TDP-43 remain to be ironed out. Even so, scientists at AD/PD suspect that this pathway likely will present even more opportunities for therapeutic targeting.—Jessica Shugart
No Available Further Reading
With amyloid-lowering treatments in hand, the field's next wish is for a way to clean up tau tangles, the pathological hallmark that correlates more closely with cognitive decline. So far, tau antibodies and aggregation inhibitors have struck out in clinical trials. At the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, Biogen researchers debuted the first data showing clearance of tau tangles in the brains of people with AD.
The drug, BIIB080, is an antisense oligonucleotide that suppresses production of all forms of tau. In a small Phase 1 study, a year of treatment lowered the tau PET signal below baseline in all six cortical brain regions examined. “Reducing tau ameliorates tau aggregates,” Dominic Walsh, who leads neurodegeneration research at Biogen, told Alzforum. “That’s good news for tauopathies.”
The findings traveled quickly through AD/PD’s hallway and bar conversations. The word “phenomenal” was uttered. “Some people might have doubted that reducing soluble tau would reduce the aggregated tau seen on PET,” said Adam Fleisher at Eli Lilly, Indianapolis, adding, “These are groundbreaking findings.” At the same time, researchers cautioned that the number of participants was small, and pointed to questions about effect size and long-term consequences. As was the case with anti-Aβ drugs, whether a slowing of neurodegeneration or cognitive decline will follow this biomarker result remains to be shown.

Bye Bye Tangles. In two people with mild AD (left), tangles (red) worsened over six months on placebo (middle), but cleared up during a year of tau ASO treatment (right). [Courtesy of Dominic Walsh, Biogen.]
This research started at Washington University in St. Louis, where scientists led by Timothy Miller, David Holtzman, and John Cirrito found that in tau transgenic mice, the ASO suppressed seizures, tangles, and neurodegeneration and, in monkeys, lowered tau protein in the hippocampus (Aug 2013 news; Jan 2017 news).
Biogen is trialing the therapy in partnership with Ionis Pharmaceuticals, Carlsbad, California, which makes ASOs for a range of neurodegenerative diseases. Biogen began its Phase 1b trial in 2017, and previously reported that BIIB080 lowered tau in cerebrospinal fluid (Aug 2021 conference news).

Four Regimens. The Phase 1 trial tested four doses, with the lower two featuring a longer gap before the beginning of the open-label extension (top row), and the highest dose testing quarterly administration (bottom row). [Courtesy of Dominic Walsh, Biogen.]
In Gothenburg, Biogen’s Jessica Collins discussed these biomarker data, adding results from the long-term extension and tau PET. The trial enrolled 46 people with mild AD from 12 sites in Canada and Europe. Participants were split into four dose cohorts, receiving either 10, 30, 60, or 115 mg BIIB080 or placebo via injection into the spinal cord. One-quarter of each cohort received placebo. Collins said the participants tolerated treatment well, with all completing dosing and no serious adverse events in treated participants during the placebo-controlled portion of the trial.
In the lower-dose cohorts, of eight people each, participants had their first dose at baseline and then monthly thereafter for three months, i.e., four injections total. After about a six-month gap period, they entered an open-label long-term extension, where all participants received 60 mg quarterly for a year. In these cohorts, CSF p-tau181 and total tau dropped rapidly, and rebounded during the gap. In the 10 mg group, tau biomarkers fell about 30 percent, and rapidly returned to the levels seen in placebo controls. On 30 mg, tau markers dropped further, about 40 percent, and rose only halfway back to control levels during the gap, suggesting a dose-dependent suppression of tau production. These data had been reported before; new at AD/PD were the long-term extension findings. During that year, tau markers fell again, bottoming out at six months of treatment around 60 percent suppressed. These two cohorts did not include tau PET data.
The higher-dose cohorts received either four doses of 60 mg, or two of 115 mg, over three months. In them, effects were slightly stronger. On either regimen, CSF p-tau181 and t-tau fell by 60 percent, and remained flat during a three-month gap period. During the LTE, the cohorts received either 60 or 115 mg quarterly for a year, but their tau markers fell no further, suggesting this represents maximal suppression with this treatment.
In Gothenburg, Collins offered a first peek at tau PET data from the two higher-dose cohorts. Imaging with the MK6240 tracer was done at baseline and six months; it comprised six composite regions that together encompassed the whole cerebral cortex. For participants on placebo, during the initial three-month treatment period, the tau PET signal rose in medial temporal, parietal, cingulate, frontal, and occipital cortices, and was unchanged in temporal cortex. For people who received two 115 mg doses of BIIB080, tangle accumulation slowed in the parietal, cingulate, frontal, and occipital cortices, and dropped slightly in the medial temporal lobe. For those who received four 60 mg doses, the tau PET signal dropped from baseline in all six regions. The effect was small, amounting to 0.2 SUVR in the medial temporal lobe, the region with the greatest effect.

Across The Cortex. A year of open-label treatment cleaned up tangles in most cortical regions examined, regardless of the dose a person had received initially. [Courtesy of Dominic Walsh, Biogen.]
During the year-long LTE, effects were more dramatic, though the cohort was tiny. Only 12 people in the LTE underwent PET. Two who had been on placebo caught up to those in the treatment groups, with similar results across the dozen. The PET signal dropped about 0.6 SUVR in medial temporal and temporal lobes, 0.4 in parietal and occipital lobes, 0.3 in cingulate, and 0.2 in frontal. Importantly, the change in tau PET was associated with total drug exposure in CSF, with the correlation around 0.60 in parietal, medial temporal, and cingulate cortex, 0.50 in frontal, and 0.40 or lower in temporal and occipital.
The findings suggest that tau monomers and aggregates are in equilibrium, such that suppressing monomer production triggers dissociation of aggregates, Walsh said. He added that as aggregates dissolve, tau may be disposed of by normal cellular clearance mechanisms such as the proteasome. A similar dynamic was seen in tau mice in the WashU study (DeVos et al., 2017).
Adam Boxer at the University of California, San Francisco, agreed this is plausible. He noted that the largest changes in tau PET signal occurred in earlier Braak stage regions, where there would be a heavier tangle load. “That makes the data more believable,” he told Alzforum. In future data, he suggested showing baseline tau PET values, as well, to give an idea of the relative reduction in tangles.
But does clearing tangles slow neurodegeneration? This is unknown. Walsh noted that the trial included CSF markers of inflammation and degeneration such as neurogranin, YKL40, and NfL. During the placebo-controlled portion, no dose-responsive differences from baseline were seen, and LTE data are still being analyzed. Likewise with the clinical measures, CDR and CDR-SB, no consistent trends were observed in the initial study, and LTE analysis is ongoing.
The researchers also included MRI to monitor safety issues such as enlarged ventricles, which were seen in a trial of the Huntington’s ASO tominersen; on this measure, too, there were no robust differences. Data from the placebo-controlled portion of the Phase 1 trial is in press at Nature Medicine, and a second paper detailing the LTE CSF and PET tau findings will follow shortly thereafter, Walsh said.
These questions will be better addressed in the Phase 2 trial enrolling now in the United States and Canada. It aims to enroll 735 participants with MCI or mild AD, and will compare two doses of BIIB080 over 72 weeks against a placebo control. The lower dose will be given biannually, the higher both biannually and quarterly. Researchers are testing biannual dosing because that would be less burdensome for clinical use, Collins said in Gothenburg. The primary outcome measure is the dose response on the CDR-SB, but the trial will include numerous other cognitive and biomarker outcomes.
Boxer believes that drugs lowering tau might be most efficacious in pure tauopathies, such as progressive supranuclear palsy or some forms of frontotemporal dementia, where there are no other pathologies to complicate the clinical picture. “We’re interested in testing drugs like this in these populations, where there is arguably an even greater unmet medical need,” he told Alzforum.—Madolyn Bowman Rogers
No Available Comments
For almost 10 years, scientists around the world have been using cell-based fluorescent sensors to study the protofibrils that spawn neurofibrillary tangles in tauopathies. Some have claimed the tau chimeras seeded in these cells cannot twist into the same type of fibrils found in the brain. Not so, said Sarah Shahmoradian, University of Texas Southwestern Medical Center, Dallas, at the International Conference on Alzheimer's and Parkinson's Diseases 2023, held March 28 to April 1 in Gothenburg, Sweden. Her high-resolution two-dimensional and three-dimensional cryo-electron microscopy and tomography images showed that these chimeras fold and stack into a typical amyloid structure, with their fluorescent peptide moieties evenly spaced along the outside of the fibrils. Her findings support the contention that these cells make good models for studying tau fibril formation in the brain.
Indeed, in a separate AD/PD presentation, Ulrich Hartl, Max Planck Institute of Biochemistry, Martinsried, Germany, reported that the fibrils in these same cells are pulled apart by valosin-containing protein. This chaperone then tosses the tau fragments into the proteasome for degradation. While that sounds like a blessing, VCP can also release new seeds that might fuel the spread of tangles in tauopathies, Hartl discovered.
Together, these studies elucidate the dynamic nature of tau fibrillization in cells, perhaps with broad consequences for tau pathology in the brain. Still, whether the structures formed in these cell lines are identical to those found in the tauopathies remains to be seen.
Marc Diamond, now at UT Southwestern, had developed the biosensor cell lines in question when he was at Washington University, St. Louis. The HEK293 cells express two tau chimeras. Each comprises an aggregation-prone repeat domain of tau coupled to cyan (CFP) or yellow fluorescent protein (YFP). When the chimeras are close together—as in a fibril—fluorescence from the CFP excites YFP in a process called fluorescence resonance energy transfer. FRET is visible under the microscope, and indeed, Diamond and colleagues used it to identify morphologically distinct polymorphs of fluorescent tau in sensor cells seeded by extracts taken from different tauopathies (May 2014 news). The cells even detected tau seeds in brains that had no overt signs of tau pathology (Oct 2014 news).
Scientists in other labs began using the cells. Then, three years ago, Eckhard Mandelkow’s lab at the German Center for Neurodegenerative Diseases, Bonn, threw cold water on them, claiming that the bulky fluorescent proteins prevented templated misfolding and fibrilization of tau as it happens in the human brain (May 2020 news).
Shahmoradian, a biophysicist, deployed some high-resolution cryo-electron microscopy techniques to take a closer look. In Gothenburg, she showed cryo-EM and cryo-electron tomography images of fibrils isolated from HEK sensor cells that had been seeded with brain extract from the tauopathy corticobasal syndrome. After placing fibrils from the sensor cells on an EM grid, Shahmoradian used their fluorescence to pinpoint them for cryo-EM, then tilted the grid through multiple angles to obtain three-dimensional cryo-ET images. This vastly increases the amount of information one can get from one fibril, she said.
From individual fibrils, Shahmoradian found hints of teeny blobs dotting their periphery. By summing analysis of 1.3 million of these fibril particles, she found that these moieties densely decorated the tau fibrils and that they had the same dimensions as—YFP. In short, her lab’s data indicated that brain extracts had seeded fibrils in the sensor cells, and that those fibrils were formed by the tau-YFP chimeras (image below).

Tau Twist. When computationally derived fibril structures are aligned (top), a fibril twist becomes recognizable, whereas the YFP moieties appear dim. When the alignment is coarse (bottom), bright dots matching YFP densely decorate the fibril and can be distinguished from each other. [Courtesy of Sarah Shahmoradian.]
But did these in vitro fibrils resemble those found in human brain? Fast Fourier transform analysis was able to resolve repeated features in the fibrils. It predicted that protein chains were stacked perpendicularly to the axis of the fibril at 4.7Å apart, and that they formed cross-β sheets in the fibril core. These are typical features and dimensions of amyloid fibrils, including the paired-helical fragments (PHFs) of tau that aggregate into neurofibrillary tangles.
Mandelkow’s group had calculated that tau-YFP chimeras couldn’t form PHFs, because YFP would not fit into the 4.7Å gap. “We cannot explain why the Mandelkow group could not produce recombinant tau-YFP fibrils,” Shahmoradian wrote to Alzforum. “Their interpretation that their negative data indicates biosensor cells cannot create tau fibrils, however, is incorrect,” she added. She concluded that steric hindrance does not keep these chimeras from templating fibrils.
Shahmoradian found similar fibrils within iPSC-derived human neurons. When she seeded such neurons expressing the FRET twins P301L-tau clover and P301L-tau ruby, fibrils formed that were unlike any natural filaments, such as microtubules or neurofilaments. These were detected by plunge-freezing the neurons, homing in on the FRET fluorescence, and using cryo-focus ion beam milling—a cellular equivalent of sand blasting—to thin the material sufficiently for cryo-TEM. The findings hint that, in human neurons, the fluorescent reporters form the same type of amyloid Shahmoradian had found in the sensor cells. She has no high-resolution cryo-EM tomography on those fibrils yet.
Breaking Up Tau Fibrils
Hartl collaborates with Diamond and uses the same sensory lines. He also believes that the fibrils in these cells are amyloids. Evidence came from a collaboration with Mark Hipp and Wolfgang Baumeister, also at Max Planck in Martinsried. Qiang Guo and Rubén Fernández-Busnadiego in Baumeister’s lab noticed that the fibrils looked quite different than microtubules and other cytoskeletal polymers, such as actin filaments, and that they bound the amyloid dye Amylo-Glo.
Cryo-ET suggested that the tau fibrils associate with organelles such as mitochondria, the Golgi network, and the endoplasmic reticulum. The latter is famous for its ability to disassemble protein complexes as part of the ER-associated proteasome pathway for degradation. Could that system dismantle these tau fibrils, too?
To test if the fibrils come apart, Itika Saha and colleagues in Hartl’s lab stopped tau-YFP production in sensor lines that had been seeded, then watched to see what happened to tau. The number of inclusions in the cells dropped 10-fold within a day and they shrank to half their size.
For tau fibrils to vanish so quickly, there would have to be some sort of machinery in the cells tearing them asunder. To find out what that might be, Saha compared the proteomes of cells with and without tau inclusions. The former had upregulated components of the proteasome—and VCP.
The chaperone piqued Hartl’s interest, because mutations in its gene have been associated with TDP-43 proteopathies, and even cause a rare form of frontotemporal dementia (Neuman et al., 2007; Oct 2020 news).

Beautiful Beast. The hexameric chaperone VCP yanks polypeptide chains through its central pore. Pathogenic mutations are shown in yellow. [Courtesy of Tang and Xia, 2016.]
The hexameric VCP complex forms a ring structure with a central pore (image at right). Powered by two ATPase subunits, it grabs the ends of proteins and pulls polypeptide chains through the pore, unravelling the whole thing in the process. Dubbed a protein extractor, it is highly conserved and essential for proteostasis from yeast to people.
Is VCP the dis-aggregase that pulls tau fibrils apart? Saha added an inhibitor of VCP's ATPases to tau sensor cells after they had formed tau inclusions. This time when she shut down tau production, the inclusions hung around (see image below). When she added the inhibitor to cells that were making fibrils, inclusions grew larger. “We see a net reaction between formation of aggregates and constant disaggregation,” said Hartl. Blocking the proteasome also prevented disassembly, suggesting a coupling between VCP's and the proteasome's actions.

Fibril Dynamics. Add tau seeds to tau reporter cells, and fluorescent tau aggregates form (top left). Add doxycycline to stop tau production, and the aggregates disassemble (top right). Block VCP (bottom left) or the proteasome (bottom right), and the disassembly slows. [Courtesy of Saha et al., 2023 Nature Communications.]
Disassembly of tau inclusions is good, right? Not always. Patricia Yuste-Checa in Hartl’s lab wondered if blocking VCP would affect the spread of toxic forms of tau from cell to cell. She added it to sensor cells as they were actively fibrillizing tau, extracted their aggregates, then used the aggregates to seed a new set of sensor cells. Surprisingly, the VCP inhibitor halved the seeds' potency, suggesting that the chaperone helped these toxic protofibrils to form. Blocking the chaperone Hsp70, or the proteasome, had no effect.
Hartl proposes that when VCP unravels the end of a fibril, it might release monomers of tau that get degraded by the proteasome, but if VCP starts pulling fibrils apart in the middle, this might release fragments that then act as seeds for the growth of more fibrils (see model below). These seeds might be involved in transcellular seeding of tau fibrils in the brain. If so, VCP would increase the danger of this phenomenon. The work appeared on February 2 in Nature Communications.

Dicey Disassembly. In pulling apart tau fibrils, VCP might release toxic seeds that accelerate aggregation of normal tau. [Courtesy of Saha et al., Nature Communications, 2023.]
Does this dynamic regulation happen in the human brain? “Although I would be surprised if VCP did not function in disaggregation in the brain, this remains to be addressed,” Hartl wrote to Alzforum. One hint that it might play out that way came from mouse primary neurons expressing fluorescent reporter tau chimeras. Blocking VCP dramatically increased the number of tau inclusions.
All that said, a major question remains. Are the amyloid fibrils formed in these sensor lines, and in induced human neurons, truly the same as those that form in the human brain? “At this point, we cannot be sure that the fibrils of tau that are disaggregated in our various cellular models are the same as those in patient brain,” Hartl wrote.
In Gothenburg, Sjors Scheres, MRC Laboratory of Molecular Biology, Cambridge, England, cautioned that the structures that form in seeding experiments may be different than those in the seed. His and Michel Goedert's labs had previously shown that α-synuclein fibrils formed in vitro bore little resemblance to the structure of the α-synuclein used to seed them (Lövestam et al., 2021). In the case of tau, more than 70 different fibril structures formed in response to varying fibrillization conditions, and only two were identical to tau structures found in disease (Lövestam et al., 2022).
Still, Scheres believes that experimental systems, be they in vitro, in cells, or in animals, will be crucial for studying how these protein fibrils form in the brain. “Are the structures that are formed the same as those in disease? We should answer this question. If so, then one could hope that some of the molecular mechanisms in the models are relevant for disease,” he said at AD/PD.
Shahmoradian is on it. “Our lab is now working on solving the core structure of these cell-extracted fibrils so that we can compare them to human brain tissue-extracted fibrils,” she wrote to Alzforum.—Tom Fagan
No Available Comments
Here’s a radical idea. Perivascular macrophages—those innate immune cells sitting oh-so-innocently on the small blood vessels in our brains—might be why ApoE4 carriers are more susceptible to cerebrovascular disease. So said Costantino Iadecola, Weill Cornell Medicine, New York, in his presentation at AD/PD 2023, held March 28 to April 1 in Gothenburg, Sweden. Iadecola reported that in mice, these macrophages both produce ApoE4 and react to it, all the while unleashing reactive oxygen species, aka free radicals, that curb cerebral blood flow. Iadecola believes that, in people who carry an ApoE4 gene, these cells might exacerbate cerebral amyloid angiopathy and ARIA, an inflammatory condition caused by Alzheimer's disease and anti-Aβ immunotherapy.
Previously, Iadecola's group had reported that perivascular macrophages pump out reactive oxygen species in response to Aβ, putting the squeeze on endothelial cells and narrowing blood vessels (May 2017 news). What's more, they found that cerebral blood flow, endothelial function, and neurovascular coupling were restricted in ApoE4 targeted replacement mice (Koizumi et al., 2018).
Could perivascular macrophages explain these deficits, as well, even without any toxic Aβ present? “These cells are very much plastered on the surface of the arterioles of the brain, and they are loaded with ApoE receptors and with free radical-producing enzymes, so they are the perfect place to make radicals that then affect the blood vessels,” Iadecola told the audience.
To explore this idea, Antoine Anfray, Laibaik Park, and colleagues in the lab first studied ApoE in wild-type mice. They applied some of it directly to the brain surface through a hole in the skull, and then, through a glass window, observed how blood vessels in the whisker-barrel cortex responded. This part of the cortex is an established system for studying cortical responses to sensory stimulation. Blood flow and electrical field potentials increase there when individual whiskers of the mice are tickled.
Indeed, just as in the targeted replacement mice, ApoE4 reduced endothelial function and neurovascular coupling when applied to wild-type mouse cortex. ApoE3 did not. Lipidated ApoE4, a more potent form of this lipid-packing protein, had a slightly stronger effect, while RAP, an inhibitor of ApoE receptors, completely suppressed it.

Militant Macrophage. In the brain of a living mouse, a perivascular macrophage sitting on a blood vessel wall takes up blue dextran (left). Adding ApoE4 to the brain boosts the cell's production of reactive oxygen species, which are detected with the red fluorescent reporter dihydroethidine (DHE). ApoE3 has no effect (right). [Courtesy of Costantino Iadecola.]
Was it the perivascular macrophages? A series of in vivo and in vitro experiments suggested as much. Two-photon microscopy revealed PVMs as a major source of free radicals in response to ApoE4, while the NADPH oxidase inhibitor gp91ds prevented the vascular dysfunction. PVMs are loaded with NADPH oxidase, which is a major source of reactive oxygen species (ROS) in these cells.
Further hints of PVM involvement came from examining cells isolated from the brain. PVMs from ApoE4 targeted replacement mice produced more ROS than PVMs from wild-type. Anfray and Park saw no such spike in other cell types, including microglia, another major source of free radicals. More direct evidence came when the scientists ablated PVMs by injecting clodronate into the brain ventricles. This bisphosphonate drug was developed for osteoporosis. It tempers osteoclasts; however, when macrophages take it up, it induces apoptosis. In ApoE4 targeted replacement mice, clodronate slashed macrophages on the ipsilateral side of the brain to a 20th of their normal number; this completely rescued endothelial function and neurovascular coupling in response to whisker stimulation.
If PVMs are reacting to ApoE4, then where does it come from? Iadecola suspects the macrophages themselves. Single-cell transcriptomic analysis of myeloid cells from the mouse brain suggested that PVMs make six times more ApoE than do microglia, endothelial cells, or blood vessel mural cells. Still, that does not prove the cells are responding to “home-made” ApoE in a cell-autonomous fashion. To test this directly, scientists in his lab developed conditional knockouts by crossing macrophage-driven Cre recombinase mice with “floxed” ApoE3 or ApoE4 mice. Cre, which can be induced by adding tamoxifen in this system, removes genes flanked by flox sequences.
The crosses behaved exactly like TR mice until the recombinase was turned on. In other words, neurovascular coupling was suppressed in the ApoE4 mice, but not once the PVM ApoE got spliced out. Then the mice behaved like wild-type, indicating that the PVMs indeed were the source of the ApoE that had been causing their vascular trouble.
The icing on the cake? When the scientists added back ApoE4 to the brains of these knockouts, blood flow was once again suppressed, as was neurovascular coupling. “Everything else required for this dysfunction was present, except there was no ApoE4 to activate the necessary receptors,” said Iadecola.
As in Alzheimer's disease, there was an allele difference. Replacing ApoE3 PVMs with ApoE4 ones caused vascular dysfunction, while replacing ApoE4 PVMs with ApoE3 ones ameliorated it. This was done by irradiating mice to kill off endogenous macrophages and then giving them fresh bone marrow of the alternative genotype.
Other scientists at the meeting called the presentation impressive. Christian Haass, Ludwig Maximilians University in Munich, wondered if the PVMs upregulate ApoE4 in response to some sort of challenge, such as presence of Aβ in cerebral amyloid angiopathy, just as disease-associated microglia do in the Alzheimer’s disease brain parenchyma. “Absolutely, this would be our assumption,” said Iadecola. “Now that we have the knockout mice, we can cross them with other mice to test these hypotheses."—Tom Fagan
No Available Comments
Epidemiological evidence has linked infection with viruses, such as influenza and herpes, to higher odds of AD and other neurodegenerative diseases (Feb 2023 news). How might these microbes contribute to neurodegeneration? Perhaps by stealthily slipping tau aggregates into healthy cells, proposes Ina Vorberg of the German Center for Neurodegenerative Diseases in Bonn. At the International Conference on Alzheimer’s and Parkinson’s Diseases held from March 28 to April 1 in Gothenburg, Sweden, she reported that envelope proteins from endogenous retroviruses hidden within mouse and human genomes enabled tau seeds to spread in cell cultures. Inhibiting expression of these envelope proteins may be a target for tauopathies, Vorberg believes.
Vorberg previously reported that viral envelope proteins, namely a glycoprotein from the vesicular stomatitis virus (VSV) and the SARS-COV-2 spike protein, drive the spread of tau aggregates by promoting the release of extracellular vesicles (EVs) containing the toxic cargo (Oct 2021 news). The viral proteins decorate EVs containing tau aggregates, enabling their fusion onto nearby cell membranes and release of the aggregates into the cell.

Tau Goes Viral. Tau aggregates (brown) spread from one cell to another by direct cell contact or through exocytosis of vesicles (top). This mirrors how viruses infect new cells (middle), leading researchers to suspect that extracellular vesicles decorated with viral glycoproteins (blue knobs) may take tau aggregates along for the ride (bottom). [Courtesy of Heumüller and Vorberg, Biospektrum, 2022.]
In Gothenburg, Vorberg reported that proteins from endogenous retroviruses also speed the spread of tau seeds in mouse and human cells. While studying yeast prion aggregates in N2a mouse neuroblastoma cells, she and colleagues noticed that the longer they grew the cells, the more cells were infected with prion aggregates. Compared to cultures passaged seven times, those passaged 16 times contained fivefold more aggregate-filled cells, and they made EVs that spread aggregates more efficiently. Why? Proteomics showed that the highly passaged cells expressed more murine leukemia virus (MLV) proteins than their counterparts.
MLVs are endogenous retroviruses that are usually epigenetically silenced but can be activated to make leukemia virus particles (Kozak, 2014). When Vorberg separated the N2a culture medium via centrifugation, she found MLV envelope proteins enriched in the EV fractions, and the reverse transcriptase enzyme made by active viruses in other fractions. Only the EV fractions facilitated prion spreading.
Is the MLV envelope protein, not the active virus, responsible for prion seeding? Indeed, when Vorberg added small interfering RNA against the envelope protein to the N2a cells, neither EVs shuttling yeast prion nor prion-infected cells transmitted their aggregates to healthy cells.
Could the same be true for tau aggregates in human cells? Vorberg's team turned to HEK kidney cells expressing fluorescently labeled soluble tau, which aggregates upon adding AD brain tissue homogenate. While the aggregates barely spread between cells, transfecting these donor cells with plasmids encoding the MLV envelope protein enabled a bit more tau spread.
Seeding jumped fourfold upon expression of both the envelope and gag/pol genes. Gag encodes structural proteins; pol encodes viral enzymes, such as the reverse transcriptase, integrase, and protease. Vorberg attributed the better tau seeding to the structural proteins’ ability to increase virus-like particle secretion and the protease’s ability to activate the envelope protein to induce membrane fusion following receptor engagement. Adding EVs isolated from MLV protein-expressing cells similarly boosted tau seeding.
Intriguingly, cell-to-cell tau aggregate spread doubled in cells expressing all three MLV genes and a viral transfer vector, which enabled the assembly of active viral particles that were able to integrate into the genome but not replicate. Vorberg was not sure why this was, but speculated that perhaps the formation of active viral particles resulted in the formation of EVs with increased aggregate-inducing activity, or that virus production altered the ratio of viral proteins or their arrangement at cell membranes.
MLV is an animal virus. However, other retroviruses do hide in the genome of people. Human endogenous retroviruses (HERVs) are usually silent. They can become derepressed during aging, producing viral proteins though not active viruses. Proteins from one such virus, HERV-K, have been detected in postmortem brain tissue from people who had amyotrophic lateral sclerosis or frontotemporal dementia but not in healthy adults (Dec 2010 news; Phan et al., 2021).

Retroviruses Spread Tau. Compared to human kidney cells containing tau aggregates (red, left), those expressing the HEVR-W envelope protein spread aggregates to more cells (right). [Courtesy of Liu et al., 2022, bioRxiv.]
Vorberg expressed HERV-K and HERV-W envelope proteins in human kidney cells containing tau aggregates. These cells spread tau aggregates among themselves 1.5-fold better than did cells without the viral proteins (see image above). “You don't need an active virus [to increase tau seed spreading]; it's sufficient to express the HERV protein,” Vorberg said.
Vorberg found that certain antiviral drugs stop aggregate spreading in these cells. The HIV protease inhibitor amprenavir also inhibits the MLV protease and prevents cleavage of the envelope protein into its mature form. Adding amprenavir to N2a cells expressing the yeast prion prevented aggregates from spreading. However, Vorberg noted that HIV inhibitors may not work for HERVs because their envelope proteins do not need to be cleaved to be fully functional. Still, another type of HIV drug—the reverse transcriptase inhibitor TPN-101 —is being tested in Phase 2 trials in people with ALS, FTD, and progressive supranuclear palsy.
Vorberg and colleagues are now focusing on finding drugs that prevent HERV protein expression in hopes they might treat tauopathies. The scientists are partnering with clinicians to isolate and clone antibodies against HERV proteins from people with tauopathies, as they likely have higher levels of these antibodies. Another approach might be resilencing HERVs, though it's still unknown why they become derepressed in the first place.
“We know very little, so researchers should be encouraged to look at these HERVs much more closely to understand what they really do and how they contribute to disease,” Vorberg told Alzforum.—Chelsea Weidman Burke
Even though there were no big revelations from immunotherapy trials at this year’s International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, new data from several programs deepened the field’s understanding of what happens when amyloid plaque is cleared from a person's brain. Different sets of trial data painted a remarkably consistent picture, showing that mopping up large amounts of plaque blunts downstream biomarkers of tau pathology, inflammation, and neurodegeneration. Clinical differences only become detectable once Aβ is mostly banished, i.e., below the threshold for brain-wide positivity.
Not everyone benefits equally from amyloid immunotherapy. Several presentations reported that people with a high tangle load clear less plaque, and their downstream biomarkers respond less robustly. Women tend to have more tangles than men at a given stage of disease, producing a sex difference in outcome in some studies. Some speakers offered a glimpse at long-term data that hint removing plaque can stabilize biomarkers and flatten disease trajectories, though the number of people remaining in studies after several years is tiny, hence subject to bias (see next story, Part 7 of this AD/PD series).
Throughout the conference, speakers extolled the historic nature of the meeting, the first AD/PD to be held since the field witnessed unambiguous Phase 3 results that removing amyloid slows disease progression (Dec 2022 conference news). Appropriately enough, that molecule, lecanemab, was developed in Sweden, originally by Lars Lannfelt and colleagues at Uppsala University.
“This meeting marks a milestone—117 years after the discovery of β-amyloid, and 39 years after identifying the polypeptide sequence, we have succeeded in removing it from the brain,” Roger Nitsch of Neurimmune, Switzerland, said in the meeting's opening address. He noted that this now allows scientists to investigate whether the aging brain can restore itself toward healthier function in the absence of plaque, and if so, by how much.
“We are witnessing the validation of the first disease-modifying target for Alzheimer’s disease,” Wagner Zago of Prothena said, adding, “The success of anti-amyloid immunotherapy increases the odds that other therapies will work. We now know that Alzheimer’s disease can be slowed.”
Clinicians and researchers have been desperate for a win, after decades of trying and failing. This conference reverberated with a sense of relief about finally having made what feels like a real start toward disease modification. This story will cover how plaque clearance affects the brain, and what factors determine whether a given person responds.

Threshold Effect? Read from right to left, this diagram shows that positive immunotherapy trials pushed plaque load to below 25 centiloids. Trials falling just short of that were negative, implying that something crucial happens in the brain when plaque is substantially eliminated. [Courtesy of Roger Nitsch.]
Plaque Gone? Downstream Markers Edge Toward Normal
No matter the antibody, removing large amounts of plaque seems to be key for nudging other biomarkers back toward healthier values. Consider data from Lilly’s Trailblazer-Alz4 trial, which compared donanemab and aducanumab treatment head-to-head, without a placebo group. The company had earlier reported that during the first six months, donanemab mopped up 62 centiloids of plaque, four times as much as aducanumab, partly due to donanemab’s faster titration to the effective dose. In addition, plasma p-tau217 fell by a quarter on donanemab (Dec 2022 conference news).
In Gothenburg, Hong Wang of Lilly added six-month data showing that other biomarkers of inflammation and neurodegeneration moved, as well. Plasma p-tau181, which reflects the presence of both plaques and tangles, fell 16 percent in the 66 people on donanemab, while rising 5 percent in the 64 on aducanumab. GFAP, a marker of gliosis, fell 8 percent on donanemab, while rising 9 percent on aducanumab. NfL, believed to be a marker for damaged neurons, rose in both groups, by 11 percent on donanemab and 17 percent on aducanumab. All these group differences were statistically significant. Importantly, all indicated normalization of biomarkers with large plaque removal, but continued progression with only modest removal. About 40 percent of people on donanemab became amyloid-negative by six months, versus a single person on aducanumab.
Wang linked these biomarker changes directly to clearance, showing that across the cohort, people who cleared more plaque lowered their plasma p-tau217, p-tau181, and GFAP by more. Plasma NfL was an exception. It did not correlate with amyloid removal; however, it did weakly correlate with the other plasma markers.
Data from the negative Phase 3 Graduate 1 and 2 studies of gantenerumab paint a similar picture. Roche had previously reported insufficient amyloid clearance in this trial, with participants on drug losing about 53 centiloids over more than two years, short of the expected clearance of 70. Only a quarter of participants became amyloid-negative. Cognitive decline on the CDR-SB and ADAS-Cog 13 nudged down an average of 11 percent, below statistical significance (Dec 2022 conference news). In Gothenburg, Roche’s Janice Smith added MMSE data to this, reporting a similar non-significant slowing of 9 percent on drug.

Treatment Effect. On gantenerumab (blue), CSF biomarkers of amyloid, tau, and neurodegeneration consistently normalized compared to untreated controls (gray). [Courtesy of Tobias Bittner, Roche.]
As with donanemab, plaque removal tracked with downstream markers. Tobias Bittner presented data from Roche’s NeuroToolkit suite of fluid biomarkers. In the two Graduate studies, a total of 293 people on gantenerumab and 269 on placebo donated cerebrospinal fluid. In this pooled CSF subgroup, Aβ42 rose 26 percent with treatment, while p-tau181 and total tau fell 23 and 16 percent, respectively. These changes normalized biomarkers, were statistically significant, and seemed to be a response to plaque removal. Surprisingly, CSF Aβ40, which fell 7 percent on placebo, fell further, by 17 percent, in those on gantenerumab. The difference was nominally significant, though it is unclear why it happened, Bittner noted.
Likewise, the inflammatory markers GFAP and S100B nudged downward by 5 and 14 percent on drug, while the synaptic markers neurogranin and α-synuclein fell 21 and 15 percent. Both inflammatory and synaptic markers rise in AD, the latter perhaps due to damaged synapses leaking these proteins into interstitial fluid. Meanwhile, the synaptic marker NPTX2 gave puzzling results, falling 19 percent on placebo and 26 percent on gantenerumab, a nominally significant difference. Bitter noted that NPTX2 is a newer marker in the toolkit and is not yet fully understood (see also Mar 2023 news). Finally, the neurodegeneration marker NfL, which rose 20 percent in the placebo group, rose 11 percent on drug.
The scientists also found effects in plasma, which was collected from all participants. As expected, Aβ42, Aβ40, and the Aβ42/40 ratio all rose on gantenerumab. This was likely not an effect of plaque removal, but rather due to antibody binding Aβ in the blood and prolonging its half-life, Bittner said. More telling was plasma p-tau, which does reflect brain pathology. P-tau181 and p-tau217 rose 9 and 13 percent, respectively, on placebo, but fell 16 and 32 percent on drug. Likewise, plasma GFAP rose 13 percent on placebo, and fell 10 percent on drug. There was no treatment effect on plasma NfL, perhaps due to the confounding effects of peripheral production, Bittner noted.
Notably, plasma markers were sensitive to the degree of plaque removal. Participants in Graduate 2 cleared about 10 centiloids less plaque on average than those in Graduate 1. This small difference showed up in blood, with the Graduate 2 cohort displaying a slightly smaller therapeutic effect on all biomarkers, even Aβ. The difference between studies did not show up in CSF because those data were pooled, due to the smaller number of participants. In plasma, the amount of plaque clearance correlated with the change in Aβ42 at r=-0.43, p-tau181 at 0.34, and GFAP at 0.16, again linking cleanup to widespread effects on pathology.
“These are quite robust treatment effects,” Bittner said, adding that this was surprising, given the clinically negative findings in these studies. Future analyses should compare participants’ final biomarker levels with values in healthy age-matched controls, to determine how close to normal they came, Bittner suggested.
Scientists at AD/PD said that even small differences in plaque removal around the clearance threshold appear to matter. Nitsch noted that all positive immunotherapy trials have brought plaque burden below 25 centiloids, whereas negative trials, such as Engage, Graduate 1, and Graduate 2, ended only slightly higher, with average amyloid loads of 35 centiloids or more (see image above). This implies that something important might happen around this threshold, and that near-complete plaque clearance is crucial.
To Christian Haass of Ludwig Maximilians University in Munich, the data validate the connection between amyloid, neuroinflammation, and tau. “The amyloid cascade is no longer a hypothesis,” he said in Gothenburg.
How to Find Likely Responders? Tangles Might Help
Some people clear amyloid faster than others—and have better outcomes. Are there characteristics, such as disease stage or APOE, that might help determine how well a person will respond to amyloid immunotherapy? In Trailblazer-Alz4, APOE genotype made no difference, Wang reported. However, a person’s tangle load at baseline did. People who started out with a lot of tau pathology, defined as a baseline tau PET SUVR of above 1.46, cleared less plaque over six months on donanemab, about 50 centiloids, compared to nearly 70 in the lower-tau subgroups.
This relatively small difference led to a stark effect on plasma biomarkers, with the high-tau subgroup gaining no benefit from donanemab. Their plasma p-tau181 stayed flat, while p-tau217, GFAP, and NfL rose at the same rate as in the aducanumab treatment group. These findings add to the evidence that amyloid removal may help people the most early in disease, before tau pathology takes off.
Wang noted different demographics in the high-tau group than the rest of the cohort. As might be expected, they had more clinically advanced disease and higher baseline biomarkers but, perhaps surprisingly, they also tended to be younger. Curiously high tau PET loads relative to other markers in younger patients have been cropping up in various studies ever since tau tracers came onto the scene (see Apr 2018 conference news) . These high-tangle trial participants also included more women. This could be due to women’s greater propensity to accumulate tangles (Aug 2018 conference news; Feb 2019 news; Nov 2019 news).
A similar effect popped up in subgroup analyses from the Graduate gantenerumab trials. Neither age nor APOE genotype affected the primary outcome, Smith reported in Gothenburg. Clinical stage did, with the mild cognitive impairment subgroup reaping slightly more benefit than the mild dementia group. However, that effect was weak, as slicing disease stage a different way, by baseline CDR score, did not show this difference.

A Girl Can’t Catch a Break. In the Phase 3 gantenerumab trials, women gained no clinical benefit from treatment (left), while men did (right). Findings were similar across all four clinical measures. [Courtesy of Janice Smith, Roche.]
Sexist Antibodies?
In contrast, the one characteristic that robustly affected outcomes was sex. Women gained no benefit from gantenerumab removal on any cognitive or functional measure, with their progression lines nearly identical to the placebo group’s. But men taking gantenerumab declined more slowly than did men on placebo—by 16 percent on the CDR-SB, 22 percent on the ADAS-Cog 13, 19 percent on the ADCS-ADL, and 16 percent on the FAQ. All four measures were statistically significant.
What is going on? Smith found no demographic differences between the male and female groups at baseline. They had the same clinical status, with around 55 percent MCI and the rest mild AD. They received the same number of gantenerumab doses and cleared about the same amount of amyloid. However, women in the study started with about 10 centiloids more plaque than men, 100 versus 90. This means they also ended the study further above the amyloid-negative threshold than did men.
Tellingly, perhaps, women also had more tau pathology at baseline in all four brain regions examined. In the two regions with the highest tau PET signal, the medial and lateral temporal lobes, women had SUVRs of 1.54 and 1.46, respectively, to men’s 1.35 and 1.28. Both these values would fall into the “high tau” category in the donanemab Trailblazer-Alz4 study. In their parietal and frontal lobes, women averaged 1.23 and 1.15, below this threshold but still higher than men, who had 1.11 and 1.05, respectively.
An audience member wondered if matching men and women by tangle load would eliminate the sex difference. Smith said the numbers in this substudy were too low to parse the data that way, but agreed it would be worth looking at. She also suggested exploring whether co-morbidities in women might contribute to their lack of response to immunotherapy. If the sex difference is solely due to tangle load, it would imply that anti-amyloid treatment might have to start sooner in women than men, placing even more importance on early detection of AD pathology in the clinic.
Frederik Barkhof of University College London reported the overall tau PET findings from the Graduate studies, not broken down by sex. Only 180 participants entered this substudy, and more than half dropped out, with 48 people on gantenerumab and 29 on placebo reaching the two-year mark. In the whole tau PET cohort, tangle load at baseline was 1.47 in the medial temporal lobe and 1.46 in the lateral temporal load, both above the “high-tau” threshold identified in the donanemab program, and there was no treatment effect. Barkhof did not discuss if this high baseline tangle load contributed to gantenerumab’s weak clinical effect in Phase 3.
A larger analysis from Mark Mintun of Eli Lilly jibed with his findings, though. Mintun examined data from positive trials of aducanumab, lecanemab, and donanemab to learn which participant characteristics predicted who would respond best. He found no consistent difference by APOE genotype or disease stage. However, the degree of amyloid clearance mattered; it determined the strength of the brake on tau pathology. In the donanemab Phase 2 trial, people who became amyloid-negative had less tangle accumulation during the trial than did those with only partial plaque clearance. In addition, amyloid removal in general correlated roughly linearly with a drop in p-tau.
Baseline tangle pathology also seemed to matter, Mintun added, referencing Wang’s data on downstream biomarkers stalling in the high-tau subgroup. This effect of baseline tangle load was independent of the amount of plaque clearance. In other words, even if plaques were cleared to below threshold in a person who started with a high tangle load, their downstream biomarkers barely budged. Mintun did not break down data by sex. The field urgently needs more data on how baseline tangles affect outcomes, he concluded.
Others have speculated that after tau pathology takes off in the cortex, Alzheimer’s disease will progress independently of amyloid. Thus, plaque clearance at that stage would do little good. The idea of a tangle threshold was proposed by Eric Karran, Marc Mercken, and Bart De Strooper as one possible model for Alzheimer’s disease, with the other being that accumulating tangles had a continuous, incremental effect (Karran et al., 2011; Alzforum webinar). Data now favor the threshold hypothesis.
More recently, the inflection point where tangles take over has been dubbed the “ca-tau-strophe” by Keith Johnson at Massachusetts General Hospital, Boston.
For more on the long-term effects of amyloid immunotherapy in early AD, see next story.—Madolyn Bowman Rogers
No Available Comments
With two plaque-clearing anti-amyloid antibodies now approved for clinical use, Alzheimer’s researchers are digging deeper into how banishing amyloid affects the brain. At AD/PD 2023, held March 28 to April 1 in Gothenburg, Sweden, scientists at Eli Lilly and Roche reported numerous effects of donanemab and gantenerumab on downstream biomarkers of tau, neuroinflammation, and neurodegeneration, as well as flagging a lack of benefit in people who started with too high a tangle load (see Part 1 of this series). Researchers also looked ahead. A burning issue for amyloid immunotherapy is what will happen over several years on drug, or a year or two—or five—after plaques have been cleared completely and a person has come off therapy. No society wants, and can afford, immunotherapy forever. Will plaques come back? More importantly, will the small clinical benefit stay stable, disappear, or grow?
“With disease-modifying therapies, the delay in progression should become greater over time,” Jeffrey Cummings of the University of Nevada, Las Vegas, said in Gothenburg. Others have projected this as well, but so far, real-world data have been lacking (Aug 2022 conference news).
Presentations in Gothenburg offered some early, tiny glimpses into such a future. Lilly’s Cynthia Evans presented long-term extension data from the donanemab Phase 2 Trailblazer study. The original study enrolled 257 people, of whom 131 took donanemab and 126 placebo for 18 months. Treatment was stopped at the end of the study, with a long gap period of more than a year before participants were invited back for a follow-up assessment. As a result, only 17 people from the donanemab cohort and 43 from the placebo group returned to take part. Those who did tended to be healthier and have less advanced disease than those who did not, Evans noted, acknowledging survivor bias. That said, these data offer a hint of how disease progresses in some after amyloid removal.

Less Plaque, Sharper Mind? Data from the few remaining participants in the Phase 2 Prime study of aducanumab link the degree of plaque clearance at four years (left) with better cognitive performance (right), with people on the effective dose of 10 mg/kg (dark blue, orange) scoring 4.5 points better on the CDR-SB than those on the lowest doses. [Courtesy of Roger Nitsch.]
Among the 17 treated participants, amyloid plaque fell from 109 to 26 centiloids during the trial, approximately the threshold for amyloid-negativity. During the gap, it reaccumulated at a rate of about 4 centiloids per year; this is similar to the rate of plaque buildup in an early AD population. The clinical benefit, as measured by iADRS and CDR-SB, was sustained during the gap, with lines continuing to slightly diverge compared to the 43 people who received placebo.
Meanwhile, the rate of tangle buildup had slowed during donanemab treatment by as much as 60 percent in frontal cortex, and somewhat less in neocortex. Eighteen months after the end of the trial, tangles continued to accumulate at this slower rate in people who had received donanemab. The findings hint at sustained benefit, though there was no tau PET data from the previous placebo group as a comparator.
Curiously, plasma p-tau217 showed a different profile from tangles. It fell about 20 percent during treatment and stabilized at that level for the next 18 months. This may be because p-tau217 reflects plaques more than tangles (Mar 2020 news; Aug 2022 conference news; Dec 2022 conference news).
Evans noted that the ongoing donanemab Phase 3 extension study will offer a deeper look at long-term effects, as participants will be rolled into that long-term extension with no gap, no unblinding of treatment groups, and more tau PET data. Top-line results from that study are expected to read out in June, hence LTE data are still several years away.

Time to Heal? A model of amyloid immunotherapy suggests that plaque clearance phase is followed by recovery and improving brain health (blue), resulting in a flattening of the trajectory of clinical decline (green). [Courtesy of Roger Nitsch.]
Complementing these data, Roger Nitsch of Neurimmune, Switzerland, presented four-year findings from the Phase 1b Prime extension study of aducanumab. He noted this is the first four-year immunotherapy data to be discussed. Biogen showed three-year data from this trial five years ago, and the few remaining Prime participants have since been folded into Biogen’s ongoing Embark extension trial, with some of them now approaching a decade of treatment (May 2018 news; Nov 2021 conference news). In the Prime extension study, the placebo group was shifted to a low dose of 3 mg/kg aducanumab, as was the initial 1 mg/kg treatment group. The other treatment groups, of 3, 6, and 10 mg/kg remained on those doses throughout the extension. The numbers of participants remaining in the study at four years was about a dozen per treatment group.
Small as they are, the data separated cleanly by dose, with those on the lower doses continuing to steeply decline on the CDR-SB. By contrast, the 18 people taking 10 mg/kg cleared more amyloid than the other groups, and their CDR-SB scores leveled out into a shallower trajectory, seeming to stabilize at year four. At the end of the study, the highest-dose group scored 4.5 points better on the CDR-SB than did the lowest-dose groups. Clinical outcomes improve with higher dosing and treatment duration, Nitsch concluded.
To Nitsch’s mind, the data evoke a model of amyloid immunotherapy that might progress in three stages: 12 to 18 months of amyloid clearance, followed by a phase of lessening neurotoxicity where biomarkers worsen more slowly compared to untreated patients and, finally, a stabilization phase where cognitive decline might level out. In this model, interrupting the amyloid cascade would allow gliosis to subside, resilience mechanisms to kick in, and the brain to—dare we say—heal.
Future data will help determine whether the aging brain has such capacity. Nitsch noted that more than 6,000 people have taken part in trials of aducanumab, donanemab, and lecanemab to date. Many of them are now in long-term extension studies. In addition, secondary prevention studies such as AHEAD 3-45, DIAN Tau NexGen, and Trailblazer-Alz3 enroll thousands more (Jul 2021 news; Nov 2021 conference news). The field should soon have more concrete answers to the question of long-term benefits.—Madolyn Bowman Rogers
No Available Comments
Since the discovery of rare, yet potent risk variants in the TREM2 gene 11 years ago, the microglial receptor has emerged as a pivot point in the pathogenesis of Alzheimer’s disease. Findings presented at the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, fleshed out TREM2 signaling mechanisms that could be amenable for therapeutic targeting.
More AD risk genes were implicated in the TREM2 signaling pathway. The protein encoded by the risk gene rhomboid family member 2 apparently stabilizes a protease that snips TREM2 off the microglial surface. Levels of this soluble piece (sTREM2) in the cerebrospinal fluid served as the basis for an entire GWAS, from which more genes, including one encoding TGFβ receptor 2, were found to tweak TREM2 cleavage. Some scientists showcased preclinical findings on up-and-coming small-molecule TREM2 agonists, while others identified chemokines as potential biomarkers of TREM2 activation for future trials.
From its perch as a transmembrane receptor on the microglial cell surface, TREM2 senses an ever-growing cast of ligands, including various lipids as well as Aβ (which, in a separate talk, was proposed to be itself lipidated most of the time). TREM2 signaling can switch microglia from homeostatic to an ever-growing number of responsive states. Besides the internal signaling cascade set off by the full-length receptor, TREM2 can also be cleaved from the cell surface by metalloproteases. The resulting soluble fragment rises in CSF in the early stages of AD, and has been found to aid in the clearance of Aβ plaques, among other functions (Jan 2016 news; Jan 2018 news; Apr 2019 news). The relative roles of the full-length versus soluble forms of TREM2 in various disease states are under intense investigation.
At AD/PD, Stefan Lichtenthaler of Ludwig Maximilians University in Munich emphasized that while metalloproteases such as ADAM10 and ADAM17 are known to shed TREM2 from the surface of myeloid cells, little is known about how that is regulated, particularly in microglia. To investigate one piece of this cleavage puzzle, Lichtenthaler dug into a previously reported genetic finding, which had tied elevated expression of the RHBDF2 gene to increased risk of AD (De Jager et al., 2014). RHBDF2 encodes inactivated Rhom2 protein. As its name suggests, iRhom2 has no catalytic activity. Instead, the protein reportedly ushers the ADAM17 protease from the endoplasmic reticulum to the trans-Golgi network, where the protease becomes activated before moving out to the plasma membrane and cleaving its substrates (McIlwain et al., 2012; Adrain et al., 2012). TNFα is one of ADAM17’s infamous substrates and, in myeloid cells lacking iRhom2, the marooning of ADAM17 thwarts the release of this potent inflammatory cytokine. Might iRhom2 have a similar effect on another ADAM17 substrate … TREM2?
Sure enough, Georg Jocher and colleagues from Lichtenthaler’s lab found that, in microglial cell lines and in primary microglia, knockout of iRhom2 drastically reduced the secretion of sTREM2, while bolstering expression of the full-length receptor. The resulting uptick in TREM2 signaling promoted a transcriptional shift into the disease-associated microglia (DAM) state. Notably, when Jocher unleashed microglial cells onto plaque-laden brain slices from APP/PS1 mice, he found that iRhom2 deficiency boosted their phagocytosis of plaques by 50 percent. However, removal of iRhom2 also doubled the proportion of microglia harboring lipid droplets, suggesting alterations in lipid metabolism. At the moment, it’s unclear whether these lipid droplets signify stepped-up internalization, and/or a slowdown in endolysosomal processing in the iRhom2-deficient microglia. Lichtenthaler later noted that in contrast to the consequences of iRhom2 deficiency, treatment with TREM2 agonist antibodies reduces these droplets.

iRhom2, the Handler. The iRhom2 proteins ushers ADAM17 from the ER up to the plasma membrane, where it can cleave TREM2. iRhom2 deficiency inhibits sTREM2 cleavage, promoting signaling through the full-length receptor.
The findings cast iRhom2 as a new genetic modifier of sTREM2 release, and confirmed ADAM17 as a major TREM2 protease in microglia, Lichtenthaler concluded. He proposed that in addition to agonistic TREM2 antibodies, perhaps blocking iRhom2 could be a way to enhance signaling through the full-length receptor.
Inhibiting TREM2's cleavage may enhance signaling through the full-length receptor, but it also halts production of soluble TREM2, which itself could also affect microglial function, noted Beth Stevens of Boston's Children’s Hospital. How do the new data address this dichotomy? Lichtenthaler acknowledged the issue. He said that in addition to reducing soluble TREM2, iRhom2 deficiency also puts the kibosh on release of TNF-α and CSF-1R. “A goal is to understand whether blocking iRhom2 is purely beneficial in the context of AD, or may also have unexpected detrimental consequences, potentially due to the reduced cleavage of other ADAM17 substrates,” Lichtenthaler wrote to Alzforum.
Bart De Strooper of UK Dementia Research Institute in London saw a silver lining in these potentially multipronged effects, noting that iRhom2 inhibition could theoretically hit two birds with one stone—strengthening TREM2 signaling while also quelling neuroinflammation wrought by TNF-α. Lichtenthaler agreed, noting that SciRhom, a Munich-based biotech company he is unaffiliated with, is developing small-molecule iRhom2 inhibitors for the treatment of inflammatory disorders involving TNF-α. He added that because iRhom2 is a multipass transmembrane protein that travels to the plasma membrane, it is particularly amenable to targeting with antibodies. However, further investigation is needed to fully grasp its function within the brain. To that end, Lichtenthaler’s group is using iRhom2-deficient mouse models of amyloidosis. Thus far, these mice suggest that halving iRhom2 assuages amyloidosis.
Approaching the investigation of TREM2 mechanisms from another angle, Carlos Cruchaga of Washington University in St. Louis and colleagues paired genomics and proteomics to hunt for factors involved in TREM2 signaling and shedding. To do this, they compiled CSF samples from more than 3,000 participants across eight AD research cohorts, measured thousands of proteins—soluble TREM2 among them—and ran a GWAS to find variants associated with its concentration. Four loci reached genome-wide significance.

Ties to sTREM2. Across the genome, four loci significantly associated with the CSF concentration of sTREM2. [Courtesy of Carlos Cruchaga, Washington University.]
One was TREM2 itself. Specifically, carriers of the R47H AD risk variant tended to have less sTREM2 in their CSF than noncarriers. Another signal came from MS4A. This AD risk locus previously has been implicated in tweaking TREM2 cleavage and, more recently, in microglial lipid metabolism (Aug 2019 news; Mar 2023 news). In the CSF GWAS, two variants within this multigene locus—one in the MS4a4a gene and the other in the MS4a6a gene—associated with higher sTREM2 levels.
A third GWAS signal popped up in the ApoE locus. Surprisingly to Cruchaga, the association was wholly independent of ApoE2, E3, or E4 genotype. What could it be? In other datasets, this polymorphism associated with high mRNA expression of a nearby gene, Nectin-2, and its encoded protein, poliovirus receptor-related 2 (PVRL2), Cruchaga told Alzforum. A transmembrane protein with Ig-like domains, PVRL2 stands accused of rolling out the welcome mat for herpesviruses, which themselves have been implicated in AD. Nectin-2 variants have also been tied to AD risk, but the gene’s close proximity to ApoE has complicated efforts to study its independent relationship with disease (Zhou et al., 2019). The findings hint that PVRL2 could play a bona fide role in in AD risk by influencing TREM2 homeostasis.
The fourth hit came from a locus harboring two genes—RBMS3 and TGFBR2. Further experiments in cultured macrophages suggest that the latter, which encodes the TGFβ2 receptor, was the one influencing CSF sTREM2, Cruchaga reported. While he does not know how this variant shifts sTREM2 levels, he did note that signaling through the TGFβ2 receptor is known to influence microglial responses, and the findings cast this receptor as yet another therapeutic target. Dovetailing with that idea, Oleg Butovsky of Brigham and Women’s Hospital, Boston, reported at AD/PD that ApoE4 promotes TGF-β signaling in microglia, locking the cells in a nonresponsive, homeostatic state. Butovsky, too, suggested TGF-β signaling as a therapeutic target.
Finally, Cruchaga told the audience that among the more than 3,000 participants included in the analysis, higher CSF sTREM2 correlated with a reduced risk of AD. This jibes with previous data from the DIAN cohort, where mutation carriers with the strongest uptick in CSF sTREM2 had a slower worsening of disease (Mar 2022 news).
In toto, what do the findings say about full-length versus soluble TREM2 signaling in AD? Cruchaga hesitated to suggest that one is more important than the other. “Obviously full-length TREM2 is a signal transduction protein, but at the same time, we think soluble TREM2 is doing something that may be protective,” he said. “We need more studies about the human biology of TREM2.” How to target Trem2, then? Cruchaga favors the idea of promoting TREM2 signaling with agonists, rather than solely blocking its cleavage.
A handful of TREM2 agonists are in clinical development. Antibodies that tickle TREM2 signaling, including Denali’s DNL919 and Alector’s AL002 are being evaluated in Phase 1 and 2 trials, respectively, though neither reported results at AD/PD. Vigil Neuroscience, a company in Watertown, Massachusetts, is evaluating its TREM2 agonist antibody, VGL101, in people with ALSP, a rare genetic disorder caused by a mutation in the CSF-1R gene. However, for AD—a much bigger indication that will require chronic dosing—Vigil is taking a small-molecule approach. The company originally bought its TREM2 agonist antibody and a suite of TREM2 small-molecule agonists from Amgen in 2020, after that company pulled the plug on its neuroscience programs.
At AD/PD, Vigil’s Christian Mirescu showed preclinical data on the company’s lead TREM2 agonists, which have been selected and polished for brain permeability, oral availability, and potency. Mirescu claimed they work like “molecular glue,” i.e., by promoting the clustering of TREM2 receptors on the microglial cell surface. In cultured human microglia, this huddling boosted TREM2 signaling and reduced shedding. The agonists worked with the known TREM2 AD risk variants and in conjunction with endogenous TREM2 ligands. For example, one agonist upped the maximal response to sulfatide, a damage-associated TREM2 ligand, sixfold.
In humanized TREM2 mice fed with a Vigil TREM2 agonist, the molecule readily crossed into the brain. There, it incited microglia to shift into a disease-associated state, and stoked brain levels of IFN-γ inducible protein (IP-10), a chemokine that recruits other immune cells. These responses resembled those triggered by injection with a TREM2 agonist antibody, Mirescu reported. Feeding the small molecule agonist to cynomolgus macaques triggered a dramatic drop in levels of soluble TREM2 in their CSF over the first 24 hours, followed by a return to baseline levels three days later.

Glue That TREM2. Vigil’s small-molecule TREM2 agonists promote clustering and signaling through the full-length receptor (left). They prevent TREM2 cleavage and shedding, as seen by a drop in CSF sTREM2 in monkeys fed a single dose (right). [Courtesy of Christian Mirescu, Vigil Neuroscience.]
Vigil aims to file an IND application for its lead TREM2 agonist in the second half of 2023. Before testing the agonists in the broader AD population, the company will evaluate them in people who carry loss-of-function mutations in TREM2 or other microglial genes, Mirescu told Alzforum.
Even as the field gears up for trials, it still lacks specific ways to track TREM2 signaling. Sure, soluble TREM2 levels may dip in the CSF following engagement of some agonists, but how will scientists know that their agonists are enhancing signaling through the receptor? To that end, Choya Yoon of Merck shared results from her lab's hunt for such TREM2 activation biomarkers.
Yoon surveyed a panel of cytokines and chemokines in brain homogenates from the TgCRND8 mouse model of amyloidosis, on a wild-type or TREM2-deficient background. She found that two chemokines—Ccl4 and IP-10—ramped up in response to amyloidosis, but only when mice expressed TREM2. Yoon also found Ccl4 and IP-10 to be elevated in the mouse CSF, but not plasma, in response to amyloidosis. Both cytokines were detectable in the CSF of cognitively normal people, suggesting they could be feasibly tracked in trials, Yoon said.
How might these chemokines respond to TREM2 agonists? Yoon tested this in different mouse models. In TgCRND8 mice, both chemokines spiked in the CSF 24 hours after treatment with an agonistic, anti-mouse TREM2 antibody. This also happened in humanized TREM2 mice treated with an anti-human TREM2 antibody developed by Merck.
Curiously, neither IP-10 nor Ccl4 budged after repeated dosing, perhaps reflecting a desensitization of the chemokine response after chronic TREM2 stimulation, Yoon said. In ongoing experiments, she is trying to define this refractory period, in hopes of devising a proper dosing interval for clinical trials. The Merck scientists are also measuring these chemokines in CSF from people across the AD spectrum, and are correlating them with the other known AD biomarkers. Yoon's biomarker talk at AD/PD reflected part of a larger effort at Merck to understand and target Trem2.—Jessica Shugart
No Available Comments
No Available Further Reading
Signaling through the CD33 receptor counteracts the known beneficial microglial responses to plaques, while clusterin is thought to promote Aβ aggregation. Up until now, these two AD risk factors were thought to be going about their nefarious ways independently. Not anymore. According to data presented at the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28-April 1 in Gothenburg, Sweden, it appears the two have been working together all along. Peter St. George-Hyslop of Columbia University, New York City, reported that full-length CD33, the isoform favored by risk variants in the gene, forms dimers on the microglial surface—to which none other than clusterin tightly binds. This instigates inhibitory signaling through CD33, which ultimately spoils the cell's appetite for Aβ plaques.
What’s more, this cascade is maximally riled if clusterin is complexed with Aβ oligomers while binding CD33. The findings offer an intuitive explanation for how both genes drive AD risk, and support the therapeutic strategy of blocking CD33.
The data complement news on TREM2 signaling and therapeutic strategies presented at AD/PD (see Part 8 of this series). The two receptors share a yin-yang-like relationship, with TREM2 promoting beneficial microglial responses, and CD33 thwarting them (Chan et al., 2015; Jul 2019 news). As such, efforts are underway to beef up TREM2 signaling with agonistic antibodies and small molecules, while blocking CD33 might be a complementary, arguably more straightforward tactic (Apr 2019 conference news).

Longer Means Riskier. Alternative splicing produces two isoforms of CD33; the long isoform, aka CD33M, contains a ligand binding site. The short version, aka CD33m, lacks that site and is favored by the protective variant. [Courtesy of Peter St. George-Hyslop.]
CD33 was identified as an AD risk gene 15 years ago (Oct 2008 news). Also known as Siglec-3, the transmembrane protein binds sialic acid and regulates innate immunity through its inhibitory ITIM motifs. Alternative splicing yields two isoforms: a long, “CD33M” isoform containing an extracellular ligand binding domain; and a short, “CD33m” isoform sans this domain. Importantly, AD risk variants were found to favor production of the longer form, while protective variants skewed toward the short version. AD risk variants in CD33 reportedly inhibit helpful microglial functions including Aβ phagocytosis, while the protective variants counteract those effects (Griciuc et al., 2013; Aug 2013 news).
How might one inhibit CD33 signaling, and what are the ligands responsible for imparting AD risk through the receptor? St. George-Hyslop aimed to answer these questions. To understand how the receptor works, his group took a structural approach, crystallizing the extracellular domain of the receptor and resolving its structure at 2.4 angstrom resolution. At AD/PD, St. George-Hyslop showed that the extracellular portion of the receptor folds into two domains, each consisting of two β-sheet sandwiches separated by a flexible linker region. The ligand binding site resided in one of these sandwiches, with an arginine residue positioned to bind sialic acid.

CD33 Club. The extracellular domain of CD33 folds into two β-sheet sandwiches, separated by a linker domain that acts as a hinge. The ligand binding domain resides in one of these sandwiches. [Courtesy of Peter St. George-Hyslop.]
St. George-Hyslop was surprised to realize that CD33 formed dimers—a coupling that was later found to be required for CD33 to travel to the membrane. In these dimers, the ligand binding sites from each molecule of CD33 were slightly offset from one another. “This arrangement, where you have two geometrically offset ligand binding sites with a flexible stalk, raises the possibility that CD33 dimers could potentially bind much larger polysialylated ligands,” he proposed.
What polysialylated ligands might those be? After testing out sialylated sugars, which only loosely bound CD33, as well as soluble sialylated ApoE, which bound CD33 not at all, the scientists hit paydirt with another sialylated protein: clusterin. Also known as ApoJ, clusterin joins ApoE as one of the two most abundant apolipoproteins in the brain.
Before it was pegged as an AD risk factor, clusterin was found to bind Aβ and bend its aggregation toward more toxic forms (Jul 2002 news). Now considered a top AD risk gene, clusterin has been implicated in all manner of mischief, ranging from Aβ aggregation to bungling communication between different regions of the brain (Jul 2010 news; Dec 2011 news). Elevated levels of clusterin in the blood or CSF associate with worse AD pathology and neurodegeneration (Apr 2011 news; Jan 2014 news).
So what are clusterin and CD33 doing together? First of all, St. George-Hyslop ascertained the rules of engagement: Only the long isoform of CD33 bound clusterin, and clusterin needed to be sialylated for binding to occur. In human AD brain samples, his team spotted the two proteins buddied up within microglia. Their partnership was most intense in microglia near plaques; in microglia distant from plaques, or microglia in control brains, the two proteins mostly kept to themselves.
In cultured human cell lines, and in peripheral blood mononuclear cells (PBMCs), clusterin binding set off signaling through CD33’s ITIM domains. It evoked an even stronger signaling cascade when complexed with Aβ oligomers, St. George-Hyslop reported. The functional consequences of clusterin binding to CD33 were borne out in subpar microglial phagocytosis of Aβ plaques. When the scientists let loose human PBMCs onto brain slices containing Aβ plaques, they found that PBMCs from CD33 risk variant carriers had a harder time mopping up plaques than did PBMCs from carriers of the protective CD33 variant. Adding clusterin to these cultures exacerbated the poor phagocytic performance of cells harboring the risk variant.
In toto, the findings unveiled a synergism between two Alzheimer's genes. Because the CD33 risk variant contains the ligand binding domain, it is open for stimulation by clusterin and other ligands. Conversely, the protective variant of CD33 lacks the ligand binding domain, making it less likely to nix beneficial microglial responses.
To what extent the interaction between clusterin and CD33 explains how these proteins contribute to AD risk remains unclear. “It is certainly possible that clusterin binding to CD33 is part of the reason that both genes/proteins contribute to risk,” commented David Holtzman of Washington University, St. Louis. “Clusterin also directly can interact with Aβ to influence its fibrillogenesis, and this may be another way that clusterin contributes to AD risk that is distinct from mechanisms relating to its interaction with CD33.”—Jessica Shugart
No Available Comments
No Available Further Reading
Retroviruses lurking in the human genome aren’t the only potential troublemakers in neurodegenerative diseases (see Part 5 of this series). Herpes simplex virus type 1 (HSV-1) integrates into the genome of trigeminal ganglia nerves, can lie dormant for decades, and flare up as cold sores throughout a person’s life. Infection with this virus has been epidemiologically tied to a higher risk of developing Alzheimer’s disease. As more laboratories around the world are investigating the herpes virus hypothesis in search of mechanistic explanations for AD, one lab is now implicating TREM2 signaling.
The microglial receptor helps the cells fend off HSV-1 infection, while the virus quiets the antiviral signaling triggered by TREM2 post-infection, according to Stefanie Fruhwürth and Søren Paludan, University of Gothenburg, Sweden. At the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Fruhwürth reported that TREM2 deficiency made mice and cultured human microglia more susceptible to herpes infection. In turn, herpes-infected microglia downregulated genes in the TREM2 pathway and in a downstream antiviral sensing pathway; this reduced the cells’ ability to detect and prune infected neurons.
Other AD/PD presentations associated genetic susceptibility to developing cold sores with increased AD risk, and ApoE4 with newly formed viral particle budding on cell membranes.
“These presentations provided important new insights into the mechanistic interplay between HSV1 and TREM2 and APOE, respectively, and thereby contributed to our understanding of the mechanisms by which HSV1 could drive AD processes,” wrote Hugo Lövheim of Umeå University, Sweden. “Direct mechanistic links to known AD risk-associated genes/proteins strengthen the HSV1-AD hypothesis.”
People infected with HSV1 have been reported to have a 1.5- to 2.7-fold higher risk of developing dementia, yet an 11 percent lower risk if they have taken an HSV antiviral drug (Feb 2021 news; Apr 2021 news). At AD/PD, Erika Vestin in Bodil Weidung's lab at Sweden's Uppsala University added to this data. She showed that, among 1,002 cognitively normal septuagenarians from Uppsala's PIVUS longitudinal population-based cohort, those who were seropositive for HSV1 were twice as likely to develop dementia over 15 years, but those who took anti-herpes medication were not at greater risk. Separately, a pilot trial in 33 people with early AD who took valacyclovir for a month saw their Mini-Mental State Exam scores rise one point (Weidung et al., 2022).
But what are the mechanisms by which HSV can set the stage for dementia? Researchers have seen virus trigger amyloidosis, tauopathy, and neuron death in three-dimensional human neuron cultures and mouse models (May 2020 news; Jun 2018 news). Some think Aβ may be antiviral, forming sticky nets to ensnare HSV particles.
At AD/PD, Fruhwürth added TREM2 modulation to HSV’s hit list. To study the role of human microglia during HSV1 infection, she analyzed RNA sequencing data from infected iPSC-derived microglia. The most upregulated genes were antiviral and proinflammatory, including IL-6, IFN-β, and TNF-α.
Among the most downregulated genes were APOE, TREM2, DAP12, and SYK. They caught Fruhwürth’s eye because loss-of-function mutations in TREM2 increase AD risk. DAP12 and SYK are part of the receptor's signaling pathway. Fruhwürth then discovered that, once infected with HSV, microglia expressed almost no Trem2 protein, and that stimulating the receptor with the agonistic antibody AF1828 boosted the microglia’s antiviral response by ramping up their expression of IL-6, IFN-β, and TNF-α. In contrast, blocking SYK, a protein kinase downstream of TREM2, with the selective inhibitor ER 27319 decreased expression of those antiviral genes.

HSV Halts Antiviral Cascade. When HSV infects neurons (left), microglia sense the virus and spew interferons and cytokines. Within microglia (right), the DNA sensor cGAS detects HSV DNA, activating the antiviral STING pathway to increase transcription of interferon-β, interleukin-6, and tumor necrosis factor-α. But HSV turns down TREM2 signaling, hobbling both the STING cascade and phagocytosis of infected neurons. [Courtesy of Fruhwürth et al., bioRxiv, 2022.]
SYK also functions in the cGAS-STING innate antiviral signaling pathway. Within microglia, the DNA sensor cyclic GMP-AMP synthase (cGAS) lies in wait of viral DNA, churning out cGAMP upon binding HSV DNA. This activates the stimulator of interferon genes (STING) pathway that, as its name suggests, ups transcription of antiviral interferons and inflammatory cytokines, such as IFN-β, IL-6, and TNF-α (see image above). Therefore, tamping down TREM2 signaling stunts both SYK activation and antiviral sensing.
Indeed, knocking down TREM2 by RNA interference rendered the microglia more prone to HSV1 infection. These microglia had abnormally low expression of the antiviral IL-6, IFN-β, and TNF-α genes and double the HSV titers as infected wild-type cells. What's more, TREM2 knockdown microglia barely nibbled at infected neurons, engulfing 60 percent fewer fluorescently labeled human iPSC-derived neurons infected with HSV1 than did wild-type microglia (see image below).

Less TREM2, Less Appetite. Compared to wild-type human microglia (top), TREM2 knockdown cells (bottom) phagocytosed fewer HSV1-infected neurons (red). [Courtesy of Fruhwürth et al., bioRxiv, 2022.]
What about in vivo? Fruhwürth infected the brainstems of wild-type mice by injecting HSV1 into their eyes; four days later, TREM2 expression was halved. When she infected TREM2 knockout mice, they had twice as much virus in their brainstems as did infected wild-type mice.
Fruhwürth believes TREM2 loss-of-function variants that increase AD risk, such as R47H, may predispose a person to HSV1 infection. This could explain the higher prevalence of HSV1 seropositivity in people with AD. She is now studying how HSV1 infection affects iPSC-derived microglia engineered to carry R47H TREM2.
To learn if TREM2 downregulation is a broader phenomenon, Fruhwürth is investigating if other neurotropic viruses, such as varicella-zoster and cytomegalovirus, interfere with its expression.
It's Not All About TREM2
Could susceptibility to HSV infection via other gene variants also fuel AD pathogeneis? People who carry a certain variant of C21ORF91 frequently get cold sores (Kriesel et al., 2011). Karin Lopatko Lindman in Lövheim’s lab analyzed blood samples and medical records from 1,244 cognitively normal adults and 331 with AD diagnosed at an average age of 71. She showed on an AD/PD poster that C21ORF91 variant carriers were twice as likely to get AD over a decade as were people without this variant. Curiously, she only saw this for APOE4 noncarriers. Lindman wasn’t sure what to make of this. “Our findings may reflect the complex genetics of AD,” she wrote to Alzforum. Lövheim agreed that this could be due to APOE4 being such a strong risk allele that minor variants show no effect in this type of study among E4 carriers.
APOE genotype has been linked to HSV susceptibility. For example, APOE4 transgenic mice have higher brain viral burdens than E3 mice (Jan 2021 news). At AD/PD, Lifeng Liu in Marta Bally’s group, also at Umeå U, found much the same in green monkey kidney (GMK) cells, commonly used to study HSV (Liu et al., 2023). Adding human ApoE3 or E4, but not E2, proteins to HSV1-infected GMK cells boosted HSV1 titers and accelerated how quickly titers rose. Liu thinks this is because ApoE3 and E4 bind to, and become incorporated into, virus particles budding on the outer membrane of GMK cells, enabling their release. This could explain the higher viral burden seen in the APOE4 mice, and could be explored in human cells, as well.
Even as the circle of scientists who are studying common neurotropic viruses in Alzheimer's is widening (e.g., Wennberg et al., 2023), the idea that the viruses play an important role in this disease is far from generally accepted. Some research groups find little to no evidence of higher AD risk, of elevated viral loads in the brains of people who had AD, or, indeed, of the proposed antiviral effect proposed for Aβ (Jan 2021 news; Jan 2020 news; Bocharova et al., 2023).—Chelsea Weidman Burke
No Available Comments
No Available Further Reading
Seed amplification assays, those PCR-like reactions for toxic misfolded proteins, are starting to look pretty good. In the May Lancet Neurology, scientists led by Andrew Siderowf, University of Pennsylvania, Philadelphia, and Luis Concha-Marambio, Amprion, San Diego, reported the largest SAA CSF study to date, of more than 1,100 samples from the Parkinson’s Progression Markers Initiative. Their α-synuclein seeding assay detected sporadic PD with high sensitivity and specificity.
But there were some twists, and not necessarily of the molecular kind. The best detection rate, 99 percent, came from people with a poor sense of smell, a common though not universal symptom of PD. Among carriers of mutations in the LRRK2 gene, only 67 percent tested positive.
“The test is very specific, but the other interesting thing is that it reveals heterogeneity in the disease,” Siderowf told Alzforum. That heterogeneity could be important when planning, or interpreting, clinical trials. For example, 86 percent of people who had symptoms of prodromal PD tested positive in the seeding amplification assay (SAA), while only 8 percent of asymptomatic LRRK2 and GBA mutation carriers did. “These findings suggest a crucial role for the α-synuclein SAA in therapeutic development, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined, at-risk cohorts,” wrote the authors.
“The good news is that we have entered a new era of biomarker and treatment development for Parkinson’s disease,” wrote Daniela Berg, Christian Albrechts-University, Kiel, and Christine Klein, University of Lübeck, both in Germany, in a comment to Lancet Neurology. “The possibility of detecting a misfolded α-synuclein, the pathological hallmark of Parkinson’s disease, by employing a seed amplification assay (SAA), is a seminal development,” they added.
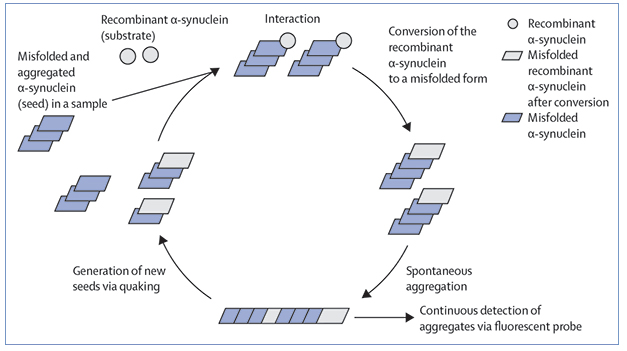
Synuclein SAA. In Real Time Quaking-Induced Conversion (RT-QuIC) assays, fibrils of α-synuclein coax recombinant forms to misfold and extend the fibril. Shaking fragments the fibrils, setting the whole process in motion again. [Courtesy of Inga Zerr, Lancet Neurology, 2021].
As was evident at AD/PD 2023 in Gothenburg, Sweden, scientists are turning to SAAs for other neurodegenerative diseases, as well. Scientists led by Oskar Hansson, Lund University, Sweden, are looking to SAAs to identify α-synuclein pathology in people with AD. Lewy bodies are the most common comorbidity in people with AD under age 85, and Hansson thinks it is important to understand how these inclusions contribute to clinical symptoms, especially since they are not targeted by approved anti-amyloid immunotherapies or by promising anti-tau therapies (Apr 2023 conference news).
Others are trying to optimize assays to detect tau seeds in the CSF, which has proven a tough nut to crack. Bryan Frey from AbbVie, Ludwigshafen am Rhein, Germany, reported that he sees higher signals in samples from amyloid-positive donors than from amyloid-negative ones, although the signal overlaps and does not correlate with cognition or other tau markers. In short, while researchers are making progress on these tests, questions remain.
Seed amplification assays are based on the principle of templated misfolding. A protein fibril binds a native monomer and stabilizes it in a toxic conformation, whereby it joins the end of the growing fibril. In practice, the tests sound simple enough—spike a solution of protein monomers with a biological sample containing fibrils and wait for the seeds to do their thing (see image above). In practice, it is tricky, requiring the growing fibrils be split apart to create new seeds to amplify the process. Success depends on the number of such cycles and the quality of the monomers used, since many of the proteins of interest tend to self-aggregate, yielding false positives.
Fragmentation can be done by physically shaking or sonicating the reaction mixture, as in Real Time Quaking-Induced Conversion, or RT-QuIC, developed by Byron Caughey’s lab at the Rocky Mountain Laboratories, National Institutes of Health, Hamilton, Montana, or the Protein-Misfolding Cyclic-Amplification (PMCA) method developed by Claudio Soto at the University of Texas Medical School, Houston (Mar 2014 news). Soto is a senior author on the Lancet Neurology paper. Those tests were initially developed to detect prion proteins; they are used to diagnose Creutzfeldt-Jakob disease in people and prion diseases in animals.
SAAs are now widely used by manly labs, and those for α-synuclein, at least, seem to work quite well. A prior analysis of 80 PPMI samples by Soto, Caughey, and Un Jung Kang’s group at New York University Grossman School of Medicine found that three different versions performed equally, identifying clinically confirmed PD cases with high sensitivity and specificity (Russo et al., 2021). Siderowf and colleagues expanded that analysis to 1,123 PPMI samples. “It’s a nice extension of what a number of other studies have found with smaller numbers of patients in multiple cohorts,” Caughey told Alzforum.
Previously, Caughey and Hansson had reported that the RT-QuIC SAA of CSF samples from the BioFinder cohort in Sweden identified people with Lewy body disorders with 95 percent sensitivity, while in collaboration with Kang, Caughey reported that the assay detected α-synuclein seeds in 102 PPMI CSF samples with high sensitivity (Hall et al., 2022; Orrù et al., 2021). Caughey, Piero Parchi at the University of Bologna, Italy, and colleagues found that RT-QuIC accurately identified synucleinopathy among 439 clinically characterized volunteers who had donated samples to the Institute of Neurological Sciences there (Rossi et al., 2020). That cohort included people with isolated REM sleep behavior disorder (IRBD) and pure autonomic failure, two clinical symptoms that commonly precede PD. Likewise, scientists led by Alison Green at the University of Edinburgh and Alex Iranzo at the Hospital Clínic de Barcelona found that among 52 people with IRBD, 32 developed PD or dementia with Lewy bodies within seven years, and CSF from 31 of those had tested positive for α-synuclein seeds at baseline, suggesting that the assay detects prodromal disease (Iranzo et al., 2021).

Better Than a Smell Test? People with sporadic or familial PD who have a dopaminergic deficit (y axis) and test positive on the α-synuclein SAA (closed circles) don’t always have a poor sense of smell (x axis). Horizontal and vertical bars represent cutoffs for dopamine transporter SPECT imaging and the UPSIT smell test, respectively. [Courtesy of Andrew Siderowf, Lancet Neurology, 2023.]
Siderowf and colleagues came to a similar conclusion. Among the PPMI volunteers they tested, 545 had been diagnosed with PD. Of them, 373 had sporadic disease, 123 carried a LRRK2 Gly2019Ser mutation, and 49 an Asn409Ser mutation in their GBA gene. Another 54 volunteers had parkinsonism, but appeared normal on brain imaging scans for dopaminergic deficit, while 51 had evidence of prodromal PD, be that IRBD or hyposmia, i.e., loss of smell. They also tested CSF from 310 asymptomatic carriers of LRRK2 or GBA mutations and from 163 healthy controls.
The SAA detected sporadic PD with a sensitivity of 88 percent, while the specificity for ruling out normal controls was 96 percent. Among sporadic cases who performed below the 15th percentile on the University of Pennsylvania Smell Identification Test, the sensitivity was even higher, at 99 percent (see image above). About 90 percent of people with PD have hyposmia. Among PD patients without hyposmia, 78 percent tested positive.
The sensitivity was lower in other subgroups, also. In LRRK2 PD, only 67 percent tested positive, falling to 35 percent in those who passed the scratch and sniff test. Why these numbers are low remains to be investigated. One possibility is that LRRK2 carriers who don’t test positive may have a different type of Parkinsonism, said Siderowf. Indeed, some LRRK2 PD cases have been reported to have no Lewy body pathology (Kalia et al., 2015). In the PPMI cohort, 15 of the PD volunteers had died and their brains were autopsied. One had no Lewy bodies or Lewy neurites, but had lost neurons in the substantia nigra. This person turned out to have the LRRK2 mutation and normal sense of smell.
Why the SAA test does better in those with hyposmia needs investigation, too. “In prodromal participants, we looked hard at all the clinical features—cognitive, autonomic, affective disorder, neuropsychology, etc., but none correlated with a positive SAA, bar olfactory deficit,” said Siderowf. It has been hypothesized that PD starts in the olfactory bulb, and Siderowf believes the SAA test presents an opportunity to test that further (Braak et al., 2003; Jan 2010 news; Dec 2015 conference news).
Can the SAA predict who will develop a Lewy body disorder? Of the 310 asymptomatic carriers, 25 tested positive, suggesting these might be in the early stages of Parkinson’s. Future analysis may tell, since the PPMI is a 10-year study, and only the baseline SAA data are reported in this paper. In the meantime, the current data hint that the test is prognostic. Among 51 volunteers who had hyposmia or IRBD, 44 tested positive on the SAA test but only 13 of those had SPECT-DAT imaging suggesting dopaminergic deficit. This suggests that the SAA picks up synuclein pathology first. “DAT scans typically detect deficits three years before symptoms, but the SAA test could push that back another three to five years,” suggested Siderowf. “Among GBA mutation carriers, about 7 to 8 percent were positive on SAA but not on DAT scans, so it’s possible that in these cases the SAA turns positive many years before the patient develops PD,” he said.
Hansson thought this was very exciting. “I found this extremely encouraging, especially for preclinical trials,” he told Alzforum. Still, Hansson would like to see more studies of normal people to see how many have Lewy bodies according to this assay. Indeed, Iranzo and colleagues had reported that four of 40 healthy controls in the Barcelona cohort tested SAA-positive, and 10 years later three were still healthy, while the fourth, who had been admitted to the hospital with an infection, was unable to complete the study. “The probability of some participants having a subclinical α-synucleinopathy that did not evolve into Parkinson’s disease or dementia with Lewy bodies within the observation period cannot be excluded and requires further investigation,” wrote Inga Zerr, University Medical Center, Georg-August University, Göttingen, Germany, in a Lancet Neurology editorial at the time.
Siderowf acknowledged the possibility. Indeed, in PPMI, about 4 percent of healthy controls also tested positive. “Is this random variation, false positives, or is there a number of people in the general population who do have misfolded seeds of α-synuclein in their CSF who do not, or will never, get PD?” he asked. Such resilience might hold the key to novel therapeutics, he suggested.
SAAs for Other Diseases
These assays have potential as diagnostics for other disorders, as well. They would be particularly helpful when no imaging or fluid marker is available, such as in TDP-43-based diseases, or mixed pathologies. On the latter, Hansson showed some preliminary data from almost 2,000 volunteers in the Swedish BioFinder cohort. It suggested that the α-synuclein seeding assay, together with AD biomarker analysis, detects comorbidities.
He reported that among cognitively unimpaired volunteers, about 12 percent had AD, 6 percent had Lewy body disease, while about 2 percent had both. In cognitively impaired people, the numbers for AD, LBD, and AD/LBD jumped to 42, 12, and 11 percent, respectively. Hansson promised to show more at the AAIC meeting next July. He believes these numbers will have important ramifications for clinical trials because both the Lewy bodies and tau pathology correlated equally well with memory loss in cognitively unimpaired volunteers. “I was quite surprised by this,” Hansson told the audience. He said longitudinal data suggests that having Lewy body pathology in AD is detrimental. “It will be important to better understand variability in response to therapies in trials to see what happens to these individuals,” he told Alzforum.
Frey’s talk in Gothenburg emphasized that it's still difficult to optimize SAAs for other proteins. Indeed, researchers in Marc Diamond’s lab, UT Southwestern Medical Center, Dallas, found no tau seeds in CSF from AD patients using biosensor cell lines (see Part 3 of this series) , but scientists in Caughey’s lab were able to detect seeds in frontotemporal dementia CSF using an RT-QuIC assay (Hitt et al., 2021; Saijo et al., 2020). Notably, the tau fibrils that form in AD and FTD are different. The former comprise tau with both 3- and 4-repeat units, whereas the latter is 4R tau only. Frey and colleagues at AbbVie tried to optimize an SAA test for the 3R/4R forms of tau found in AD.
By changing the substrate in the assay to a small fragment that includes the repeat domains and an elongated C-terminal tail, Frey struck pay dirt. In this system, AD CSF boosted fluorescence, an indication of fibrillization, by 5,000-fold over that seen with CSF from healthy controls. In a pilot study, the assay detected 40 percent more fluorescence from AD samples than from Aβ-negative, age-matched control CSF. However, there was considerable overlap between the 10 AD and five control samples. A larger study of 48 samples indicated a bigger group difference of 65 percent, but again, the overlap was substantial. Frey is not sure why. It could be that the assay needs to be optimized further, or it might be that controls also have tau seeds. Some scientists believe that fibrils of tau can begin to form in the brain with age, even without any amyloid present (Nov 2014 news).
Caughey noted that, in his hands, tau SAAs are less robust than α-synuclein assays. “We get more chatter from healthy controls, so while a lot of people have tau seeds in their CSF, the data are not quite as consistent,” he told Alzforum. “The presence of different types of pathological and non-pathological aggregates and co-pathologies is part of the landscape,” he said. Still, he says it’s early days and he thinks tau RT-QuIC assays will become helpful diagnostically. As for TDP-43, one recent paper reported that RT-QuIC detects seeds in the CSF of people who had ALS/FTD (Scialò et al., 2020).—Tom Fagan
No Available Comments
Anti-amyloid immunotherapy has reached a milestone with the approval of two antibodies for clinical use; alas, the race is far from over. At the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, researchers enumerated many challenges that remain before immunotherapy can become standard treatment for AD. The field needs to improve safety, figure out which types of patients are at higher risk of side effects, and make treatments more convenient, Dennis Selkoe of Brigham and Women’s Hospital, Boston, said in an opening talk. In addition, researchers must continue gathering data on whether these treatments offer substantial benefits to people with early AD.
Various presentations at AD/PD dealt with these issues. Subjective quality-of-life assessments from the Phase 3 Clarity trial of lecanemab offered a fresh look at clinical meaningfulness. Calculations of “time saved” by various antibodies were used to make a case for slowed disease progression. On risk, Clarity data showed that people taking anticoagulants have greater odds of suffering the most serious side effect of immunotherapy, large brain bleeds. New Appropriate Use Recommendations suggest not prescribing the drug to this group.
Eli Lilly researchers shared data about two troublesome side effects of donanemab, infusion reactions and antidrug antibodies, and debuted a successor, remternetug, that appears to be free of these drawbacks. Finally, researchers at Prothena described their new Phase 1 antibody, PRX012, which may require less frequent administration than previous molecules, potentially easing the burden of treatment for patients.
“Cognitive decline is the challenge of our age,” Nick Fox of University College London said in Gothenburg. He reminded the audience that one in three people alive today are projected to develop dementia, and half will look after someone with the disease. He believes these new therapies will brighten this outlook. “We have the opportunity to preserve function longer, provide hope, and change the mindset about dementia. No more nihilism,” Fox said.

Vanishing Plaques. Remternetug mopped up amyloid with a speed that varied by dose (left); spaghetti plots show how many people on each dose became amyloid-negative within six months (right). [Courtesy of Yan Jin, Eli Lilly.]
How Much Does Amyloid Immunotherapy Really Help?
The question of whether the slight slowing of cognitive decline quantified in immunotherapy trials matters to patients has engendered fierce debate among clinicians. This is because the treatment effect is not large and obvious to everyone, and the meaningfulness of small effects is difficult to measure (Feb 2023 news).
In Gothenburg, Sharon Cohen of the Toronto Memory Program made the case that the cognitive benefit achieved thus far does matter to patients. Cohen, a site investigator for the Clarity trial, believes in asking patients themselves. She based her conclusion on quality-of-life assessments completed by trial participants and their caregivers. Clarity included three such measures: the European Quality of Life scale, the Quality of Life in AD, and the Zarit Burden Inventory. The 859 participants on lecanemab and 875 on placebo completed these assessments at baseline, and repeated them every six months over the 18 months of the trial.
The European Quality of Life scale measures five aspects of health: activities, self-care, mood, mobility, and pain; it goes from 0 to 100, high numbers being better. Clarity participants started with an average score of 82, as expected for an early AD population. Over the course of the trial, people on placebo lowered their rating of their own quality of life by four points, while those on drug lowered it two, for a 50 percent slowing of decline. The benefit showed up in the scores for activities, self-care, and mood, not mobility or pain, as might be expected for a drug that sharpens cognition.
On the Quality of Life in AD scale, which ranges from 13 to 52 and is scored by both patient and caregiver, average scores started at 39. The placebo group dropped by more than a point during the trial, the treatment group half a point, again halving the rate of decline. Caregivers perceived less treatment benefit, rating their loved one’s decline as nearly 2.5 points on placebo and a bit under 2 on lecanemab. Caregiver ratings correlated with those of patients across the cohort.
Finally, on the Zarit Burden Inventory, caregivers scored their own stress level from 0 to 88, where high numbers mean more stress. Their starting value was 17, and their stress worsened by 6 points in the placebo group and 3.5 in the treatment group, for a 38 percent slowing of decline. Unlike the other measures, which only became statistically significant at 18 months, on this scale the difference between groups became apparent at six months, and grew over time, Cohen showed.
On all three measures, benefits were consistent across APOE genotypes and other personal characteristics such as age and sex. Because these differences were self-reported, the data suggest that treatment gains were meaningful and detectable to patients and their loved ones, Cohen said.
Suzanne Hendrix of Pentara Corporation, Salt Lake City, assessed meaningfulness another way. She calculated how much sooner people on placebo reached the same level of cognitive decline that people on a given antibody had reached at the end of the study. In the donanemab Phase 2 Trailblazer trial, averaging data from the iADRS and CDR-SB, this calculation came to 5.5 months. In the lecanemab Clarity trial, combining data from the ADAS-Cog, CDR-SB, and ADCS-ADL, it was five months. Both trials were 18 months long, hence those values jibe with the one-quarter to one-third slowing seen with these antibodies. For aducanumab, averaging data from the Emerge and Engage trials on the same three measures as in Clarity, progression slowed by 2.5 months. This is less than for the other antibodies because Engage was negative.
For all three antibodies, the amount of “time saved” grew throughout the trials, as would be expected for disease-modifying therapies that bend the slope of progression. Hendrix believes time is an inherently meaningful measure that enables a comparison between different disease-modifying therapies.
Anticoagulants and Amyloid Immunotherapy—A Bad Combination?
How to balance the benefits of amyloid immunotherapy against its risks, chiefly the brain swelling and bleeding known as ARIA? In Gothenburg, Marwan Sabbagh of the Barrow Neurological Institute in Phoenix presented more data on how this varies in people taking anticoagulant or antiplatelet drugs, which can increase bleeding. Anticoagulant or acute thrombolytic use has been associated with three deaths in the open-label extension of the Clarity trial (Jan 2023 news).
Sabbagh first went over general data from this OLE. The trial enrolled 1,795 people, of whom 90 percent, or 1,612, rolled over into the extension. As in the placebo-controlled portion of the trial, in the combined core and OLE data, ARIA-E rates were lower than those of other antibodies. They averaged 13.6 percent, compared to around one-quarter for people on donanemab, and one-third on aducanumab (Mar 2021 news; Dec 2021 news). As expected, the risk varied by APOE4 genotype, with noncarriers at 7, heterozygotes at 12, and homozygotes at 35 percent. All of these numbers were similar to those in the trial itself. Likewise, the amount of symptomatic ARIA-E stayed steady in the OLE, at about 3 percent.
The Clarity trial differed from other antibody programs in that it allowed people on anticoagulant or antiplatelet therapy to enter. In the core trial plus OLE data, the 156 people on anticoagulants had somewhat lower incidence of ARIA-E and microhemorrhages than the 1,456 people not on these drugs. However, their risk of macrohemorrhage was six times higher, going from 0.4 to 2.6 percent. This amounted to four cases in this group.
Though macrohemorrhages are rare, they have much more severe effects than other forms of ARIA. Ominously, the risk of dying with an intracerebral hemorrhage was much higher for people on anticoagulants, rising from 0.1 to 1.4 percent. Sabbagh offered no details on these patient deaths. By contrast, the use of antiplatelet drugs such as aspirin or clopidogrel did not increase the risk of brain bleeds. Unlike with ARIA-E, there was no apparent effect of APOE genotype on macrohemorrhages.
The higher risks of brain bleeds for people on anticoagulants, and of ARIA-E in APOE4 homozygotes, worry many clinicians, leading to efforts to restrict antibody use for these groups until more is known. The lecanemab Appropriate Use Recommendations were published March 27, coinciding with the start of AD/PD (Cummings et al., 2023). In Gothenburg, Jeffrey Cummings of the University of Nevada, Las Vegas, gave an overview. Like the aducanumab AUR, the new publication suggests that lecanemab not be prescribed for people taking anticoagulants, or anyone with a clotting disorder, strokes, or seizures. People taking lecanemab should not be treated with acute thrombolytics such as tPA, though common antiplatelet medications are allowed.
Like the revised aducanumab AUR, the lecanemab AUR recommend APOE genotyping before starting therapy, so clinicians can discuss potential risks with patients. The AUR do not restrict prescription to APOE4 homozygotes, but other groups may. Cummings noted that the Department of Veterans Affairs has decided to provide lecanemab for its beneficiaries, though not if they carry two copies of APOE4.
Per the AUR, MRIs should be done at baseline, and before the fifth, seventh, and 14th biweekly infusions to monitor for ARIA. The AUR also suggest scans at one year for APOE4 carriers and people with any ARIA during the first year. The schedule differs from that recommended for aducanumab, in which scans are done before the fifth, seventh, ninth, and 12th monthly doses (Aug 2022 conference news).
Alireza Atri of the Banner Sun Health Research Institute in Sun City, Arizona, emphasized the need for clinicians to stay in regular contact with patients and track their progress and symptoms. “It’s not ‘diagnose, infuse, and adios,’” he said in Gothenburg. Selkoe spoke of an internal system among neurologists at his hospitals, whereby one of them will be on call especially for the hospitals’ amyloid immunotherapy patients at all times.

Blunted Efficacy. Anti-drug antibodies against donanemab slow plaque clearance, with people above the median amount of ADAs (gray) clearing about 10 centiloids less than those below the median (tan) over 18 months. [Courtesy of Garrett Mullins, Eli Lilly.]
Anti-Drug Antibodies to Donanemab: a Problem. Remternetug: a Solution
For donanemab, ARIA is not the only concern. From its beginning, this antibody has been plagued by the generation of anti-drug antibodies in most patients, as well as occasional severe infusion reactions (Dec 2022 conference news). In Gothenburg, Lilly researchers offered data on these side effects.
Lilly’s Paul Ardayfio discussed infusion reactions. In Phase 2, these occurred in 10 out of 131 people on drug, or 8 percent. Reactions affected the whole body, causing flushing, chills, headaches, nausea, cramping, or chest tightness. Two of the reactions were characterized as severe, though Ardayfio gave no details. Most resolved within one day. The reactions typically happened after the second, fourth, or fifth infusions. They were more likely in people who made more antidrug antibodies, with none occurring in the lowest tertile of ADAs.
Garrett Mullins of Lilly elaborated on these ADAs. In Phase 2, 92 percent of participants developed ADAs that neutralized donanemab. The amount ranged widely, with the median titer being 1:10,000, meaning the antibodies could still be detected after diluting blood samples 10,000-fold. These ADAs hastened the clearance of donanemab from blood, with the highest titers mopping up the antibody about a third faster than the lowest titers. The maximum blood concentration of donanemab was not affected, but at the low point, donanemab concentration dipped to half the level seen in people without ADAs. As a result, high ADAs lessened donanemab’s efficacy by about a quarter, and attenuated plaque clearance, Mullins said. In answer to an audience question, Mullins said the Phase 2 cohort was too small to tell if the effect of ADAs increased over time. Lilly is investigating this in Phase 3, and also whether the cognitive benefit is less in people with high ADAs.
The data hint at problems for long-term use of donanemab. To solve this, Lilly made a new antibody, remternetug, with similar binding characteristics to donanemab, but fewer safety issues. Lilly’s Yan Jin presented interim Phase 1 data in Gothenburg. The trial enrolled 41 people who had MCI or mild AD and at least 37 centiloids of plaque. It compared 250, 700, 1,400, and 2,800 mg monthly to placebo, as well as a titration cohort going from 700 to 1,400 mg remternetug. Most groups comprised five people on drug and one on placebo, except for the 700 and 1,400 mg cohorts, which had twice as many participants. Each group took remternetug monthly for six months, i.e. seven doses, followed by a one-year extension. As the trial is still running, Jin presented only six-month data on plaque reduction and safety.
In this brief period, plaque dropped by as much as 100 centiloids with treatment. Effects were dose-dependent. At 250 mg, no one dropped below the amyloid-positivity threshold, defined as 24 centiloids. In the 700 mg group, four out of 10 people did, and in higher dose groups, everyone. On 2,800 mg, everyone dropped below 24 centiloids within three months. This is faster clearance than with donanemab, which slashes plaque about 60 centiloids in six months, bringing 40 percent of people below the threshold.
Safety data are still blinded, so treatment and placebo groups cannot be compared. However, Jin noted that there were a total of 10 ARIA-E and seven ARIA-H cases. These were scattered among treatment arms, with no obvious dose correlation. All the ARIA-E cases occurred in APOE4 carriers. One was symptomatic, with the participant developing balance, visual, and language problems that went away after dosing was stopped.
Crucially, no ADAs were detected in any participant. There were also no systemic infusion reactions, although two people developed local reactions at the injection site. Remternetug is now in Phase 3.

Better Binding? In vitro data from new anti-amyloid antibody PRX012 (gold) shows it hangs onto Aβ protofibrils better than lecanemab (gray, left) and clears plaque more potently than donanemab (gray, right). [Courtesy of Prothena.]
Will a More Potent Antibody Make This Less Burdensome?
At the moment, aducanumab, lecanemab, and donanemab require monthly or biweekly infusions. This will limit the number of people who can take these drugs. All antibody programs are testing subcutaneous dosing, but even then, the need for monthly injections may discourage some patients.
Prothena scientists set out to develop a more powerful treatment that could be taken less often. In Gothenburg, Brian Campbell of Prothena explained why they believe their antibody PRX012 fits the bill. PRX012 was designed to bind all aggregated forms of Aβ, including oligomers, protofibrils, and fibrils, with high affinity. Like other anti-amyloid antibodies, it targets the N-terminus, but unlike them, once it binds, it does not let go. Its very slow off-rate allows it to bind with picomolar affinity, Campbell said. In head-to-head comparisons of lecanemab and PRX012, the former had a dissociation constant of 2 nM from protofibrils, the latter 0.1 nM, giving it 20 times higher affinity.
What about plaques? When the researchers added PRX012 and microglia to postmortem AD brain tissue, almost half the fibrillar Aβ was gone three days later—including pyroglutamated Aβ, even though PRX012 does not bind this form. This is because once plaques are opsonized by antibody, microglia gobble all plaque material indiscriminately, Campbell said (for a new hypothesis of how a little microglia eats a huge plaque, see next story in this series).
In a direct comparison with donanemab, PRX012 cleared the equivalent amount of pyroAβ at a concentration three to eight times lower, he claimed. Prothena had earlier showed that PRX012 bested aducanumab in a direct comparison of plaque clearing, as well (Aug 2021 conference news).
Because of these features, Prothena researchers believe PRX012 could be given at lower doses than other antibodies, the company’s Chad Swanson said in Gothenburg. PRX012 has completed Phase 1 single-ascending-dose testing, and is now in a multiple-ascending-dose trial. The trial tests infrequent, low doses, Swanson said, but provided no further details. Swanson noted that PRX012 antibody was designed to be given under the skin, making administration easier.
In answer to audience questions, Swanson said PRX012 enters the brain at about the same rate as other antibodies, at 0.1 times the concentration in blood, and has a similar half-life. Results from the trial are expected in 2024. Meanwhile, Prothena has teamed up with Walgreens to accelerate and diversify enrollment in the MAD, as well as future studies of the antibody (see press release).—Madolyn Bowman Rogers
No Available Comments
The scientific diet at this year’s International Conference on Alzheimer’s and Parkinson’s diseases, March 28 to April 1 in Gothenburg, was especially rich in news about the ins and outs of the microglial “gastrointestinal tract,” aka their endolysosomal system. From binging to indigestion to purging, research on microglial peristalsis, essentially, moved scientists' understanding on cellular and molecular mechanisms in neurodegenerative disease.
Results converged largely on the lysosome. Sensing Aβ aggregates, microglia reportedly send these digestive sacs to the cell surface, where they dump their enzyme juices onto plaques. But Aβ aggregates gave some microglia a sour stomach. They festered in their lysosomes, growing larger over time, ready to contribute to the spread of amyloidogenic seeds when spewed out later. TMEM106b, a lysosomal protein with genetic ties to several neurodegenerative diseases, connected the dots between AD risk and floundering microglial phagocytosis (see Part 14 of this series). In all, the findings appear to put malfunctions in microglial lysosomes at the heart of the early, cellular phase of neurodegenerative disease.
Microglia are intertwined with every stage of amyloid plaque development, from construction to containment to clearance. It is clear that the cells engulf Aβ aggregates via phagocytosis, but how do they manage to consume plaques that are several times their size? Unlike a snake that unhinges its jaw bones when eating animals larger than itself, microglia might well digest plaques without eating them at all. At AD/PD, Santiago Sole-Domenech, a research associate in Frederick Maxfield’s lab at Weill Cornell Medical College in New York, showed how that could work.
Graduate student Rudy Jacquet and Sole-Domenech took a cue from the way macrophages digest large LDL particles, or osteoclasts reabsorb bone. Maxfield’s group previously showed that, using a process called digestive exophagy, these other myeloid cells direct their lysosomes to the cell surface, where these vesicular troops fuse with the plasma membrane, hurling the brew of enzymes within them onto their extracellular prey (Singh et al., 2016). Could microglia use the same technique to degrade Aβ aggregates?
Jacquet found that when exposed to fluorescently labeled aggregates of Aβ, primary microglia quickly rearranged their plasma membranes to form an invagination surrounding each aggregate.

Digestive Hug. Exposure to Aβ aggregates (red) triggered microglia to rearrange their cytoskeleton (green) and wrap the aggregates in pockets of plasma membrane. [Courtesy of Santiago Sole-Domenech, Weill Cornell Medical College.]
Using pH-sensitive dyes, the scientists found that microglia released acidic, lysosomal contents into these aggregate-clutching pockets, a phenomenon Maxfield had christened as “lysosomal synapses.” They only formed in regions of the membrane in direct contact with Aβ aggregates. The same phenomenon appeared to occur in the brain. Using electron microscopy of brain slices from 5xFAD mice, the researchers spotted microglia forming membrane pockets right where they touched plaques. These extracellular compartments brimmed with acid phosphatase, an abundant lysosomal enzyme.

Lysosomal Synapses. A microglial cell (gray) sits underneath an Aβ aggregate (red). At the point of contact, it released acidic contents of its lysosomes, as detected by pH-sensitive dye (green, arrowhead). [Courtesy of Santiago Sole-Domenech, Weill Cornell Medical College.]
While this external digestion may initially shrink plaques, it could potentially backfire, Sole-Domenech reported. He found that when microglia were first allowed to gorge themselves on small Aβ fibrils before encountering large aggregates, the cells filled up their lysosomes with fibrils, then later spewed them out onto large Aβ aggregates via lysosomal synapses.
Akin to the way the process is regulated in LDL-digesting macrophages, Sole-Domenech and Maxfield found that signaling via MyD88, downstream of toll-like receptor 4 and CD14 receptors, instigated digestive exophagy in microglia.
How might signaling through TREM2, a microglial receptor that promotes phagocytosis of Aβ aggregates, influence this external digestion? Strikingly, Jacquet and Sole-Domenech found that microglia deficient in TREM2 doubled down on digestive exophagy, unleashing even more lysosomal contents onto Aβ plaques. He hypothesized that TREM2-deficient microglia might ramp up these external digestive mechanisms to compensate for their deficits in phagocytosis. In particular, TREM2-deficient microglia poorly polymerize actin, and phagocytosis requires more actin polymerization than does digestive exophagy, Sole-Domenech told Alzforum. “It is therefore ‘easier’ for the cells to exocytose lysosomal contents during digestive exophagy,” he added. In support of this idea, he said wild-type and TREM2-deficient microglia form lysosomal synapses with equal measure when they are treated with GM-CSF, which boosts actin polymerization.
In all, Sole-Domenech cast digestive exophagy as a double-edged sword: It allows microglia to cut Aβ plaques down to size without internalizing them, but if microglia also fill themselves with smaller Aβ aggregates via phagocytosis, they run the risk of regurgitating—and potentially spreading—undigested toxic amyloidogenic peptides.
This exact flavor of microglial indigestion was the topic of Anja Schneider’s talk at AD/PD. Schneider, of the German Center for Neurodegenerative Diseases in Bonn, used an isotope-labeling technique to track the evolution of aggregates within the mouse brain. When 5xFAD mice reached 6 months of age, she swapped out their regular chow with food containing 13C-labeled lysine. This heavy isotope can be distinguished from the lighter 12C variety using nanoSIMS, a form of mass spectrometry that can track isotopes with nanoscale resolution. Schneider was then able to distinguish between relatively younger and older proteins within the mouse brain.
Schneider found that roughly 30 percent of Aβ aggregates resided inside of cells. Microglia stomached most of that intracellular pool within their lysosomes. Isotope scanning of the lysosomal aggregates revealed that Aβ peptides in the core were substantially older than those in the outer shell, suggesting that fresh Aβ peptides were continually joining up with older aggregates that were sitting within the lysosome.
The results took Schneider by surprise. “We had assumed that Aβ aggregates are being degraded in the lysosome, not that they grow there.”

Old at Heart. In Aβ aggregates within microglial lysosomes, the oldest peptides (red) accumulate in the core, with progressively younger peptides forming the shell. [Courtesy of Anja Schneider, DZNE, Bonn]
Schneider came to similar conclusions about tau. This protein accumulated within microglial lysosomes of P301L mice and, once again, younger tau proteins surrounded a comparatively ancient aggregate core. Schneider proposed that instead of clearing amyloid or tau aggregates, microglial phagocytosis might sometimes lead to the formation of amyloidogenic seeds. If the contents of these lysosomes were to be secreted, say, by a mechanism like digestive exophagy, this could lead to propagation rather than containment. Schneider and Sole-Domenech agreed that their findings complement each other, and are consistent with the hypothesis that buildup of aggregates within lysosomes could ultimately contribute to amyloid dissemination. In light of these findings, Schneider believes the field should approach therapies aiming to cautiously increase microglial phagocytosis.
Marc Diamond of UT Southwestern Medical Center questioned the conclusion that aggregates were actually growing within the lysosomes. He posed an alternative explanation, that aggregates were fully formed—with old proteins in the core and young ones in the shell—prior to their phagocytosis. Schneider acknowledged that possibility, adding that one would need to label proteins at different time points to determine exactly when and where the aggregates formed. Whether the aggregates grew in the lysosomes or not, the findings do suggest that microglia fail to degrade them.
What prevents microglia from fully digesting Aβ aggregates within their lysosomes? Schneider and Sole-Domenech noted that the acidic pH of late endosomes and lysosomes provide the optimal environment for Aβ aggregation. “We think that low pH and high local intralysosomal concentrations may favor aggregate formation,” Schneider said.
The AD/PD meeting was not the first time microglia have been painted as disseminators of amyloid. Previous studies have implicated the cells in the propagation of tau pathology, reporting that microglia readily consume tau aggregates but fail to destroy them, leading to their eventual release (Jul 2018 conference news; Wang et al., 2022; Hopp et al., 2018). In a variation on this theme, studies from Seiko and Tsuneya Ikezu’s lab, then at Boston University School of Medicine and now at Mayo Clinic Jacksonville, Florida, reported that microglia release tau aggregates encased in extracellular vesicles (Clayton et al., 2021).
At AD/PD, Seiko Ikezu expanded on these findings. She described what happened when microglia were deprived of TSG101, a member of the ESCRT-I complex that forms extracellular vesicles. In conditional knockout mice, removing TSG101 from microglia stymied the propagation of tau tangles from the entorhinal cortex into the hippocampus, a propagation route the researchers had previously described (Sep 2021 news). In a P301S mouse model of tauopathy, nixing TSG101 from microglia roughly halved the burden of tau tangles in the hippocampus, and rescued memory deficits.
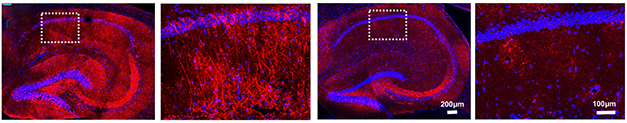
No Transport for Tau. Misfolded tau (red) accumulated in the hippocampi of PS19 mice (left panels). Deletion of TSG101 in microglia cut the tau burden in half (right panels). [Courtesy of Seiko Ikezu, Mayo Clinic.]
Curiously, without TSG101, microglia were less likely to shift into a disease-associated transcriptional state. They expressed less of the complement receptor C3aR1, and munched on fewer synapses than did their TSG101-replete counterparts. In culture, primary microglia sans TSG101 had little appetite for synaptosomes, while microglia from wild-type mice readily consumed them.
The Ikezus’ data indicate that the microglial production of extracellular vesicles exacerbates tau pathology. Exactly how cutting off EV secretion might also counteract microglial activation needs further investigation, Ikezu said. Much as constipation reduces appetite, blocking the release of EVs apparently blocks microglial phagocytosis. Ikezu believes that phagocytosis of apoptotic neurons, damaged synapses, or aggregated proteins is an essential part of the microglial transition into the DAM state, so this could explain how TSG101 deletion ultimately douses microglial activation, she told Alzforum. It is also possible that the EVs contain inflammatory ligands, such as nucleic acids or mitochondrial proteins, that push microglia into the DAM state, she added.
In a startling example of microglia gone terribly wrong, Charles Arber, working in the lab of Selina Wray at University College London reported that the cells are the sole purveyors of the amyloidogenic peptide that causes familial British dementia. This rare genetic disorder is marked by amyloid plaques and tau tangles, but its plaques contain no Aβ. They are made of a peptide cleaved from the C-terminus of the transmembrane protein ITM2B. Mutations that cause FBD disrupt a stop codon in the ITM2B gene, resulting in production of the aggregation-prone, 34-amino acid “A-Bri” peptide.
Together with Sarah Wiethoff, Arber acquired fibroblasts from two people with FBD and two controls, and differentiated them into iPSCs. Try as she might, Emma Augustin from Arber’s lab found no trace of ITM2B, the A-Bri peptide, or any phenotype whatsoever, in iPSC-derived neurons from the two people with FBD. Taking a hint from other gene-expression datasets that suggested ITM2B was expressed in microglia, the scientists differentiated the iPSCs into microglia. Lo and behold, microglia from both control and patient samples expressed ITM2B, and A-Bri was found only in FBD. Microglia were also spotted manufacturing A-Bri in brain samples from a person with FBD, as well as from a person with familial Danish dementia, a related disorder caused by mutations in the same gene.
A dive into single-cell transcriptomic datasets revealed that ITM2B is a disease-associated microglia (DAM) signature gene, rising in step with TREM2, Tyrobp, and other DAM genes. What does this mean? Arber proposed that FBD could be sparked when an inflammatory event provokes microglia to shift into the DAM-like state. In support of this idea, Arber said that many people develop FBD symptoms after experiencing such events, such as a flu infection or a stroke. In other words, in people carrying a causative ITM2B mutation, an otherwise beneficial response to, say, a viral infection or a brain trauma, causes microglia, of all cells, to produce an amyloidogenic protein that leads to dementia.
David Holtzman of Washington University in St. Louis called the production of A-Bri by microglia “an amazingly cool finding. I don’t know of any other extracellular amyloid that is mostly produced by microglia.” Regarding the idea that the disorder could be sparked by neuroinflammation, Holtzman wondered if familial British dementia, like AD, starts with a long preclinical phase where plaques steadily grow. If so, how would one reconcile that long asymptomatic period with the idea that symptoms of disease surface soon after an inflammatory event? Arber said those remain open questions, given the dearth of preclinical samples or biomarkers for this disease. “Still, it is exciting to think that inflammation is part of the onset and progression,” Arber said.
This sampling of microglial behavior on display at AD/PD offers but a glimpse of the myriad ways the cells react to different stimuli. How do these reactions—and the transcriptional states that control them—influence neurodegenerative disease? In the next part of this series, read how scientists grapple with this question by marrying cutting edge omics and human microglial cell culture techniques.—Jessica Shugart
No Available Comments
No Available Further Reading
New data presented at the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, further reinforced the field's growing recognition that microglia respond in myriad ways to different environmental stimuli (for a selection, see Part 13 of this series). How best to study brain cells of such astounding reactivity, at the levels of both transcription and function? This question was the focus of Beth Stevens’ talk at the conference (spoiler alert: lysosomes end up stealing the show, again).
In their previous efforts to document microglial behavior across neurodegenerative diseases, Stevens, who is at the Broad Institute of MIT and Harvard, and others have collected massive genomic datasets detailing how expression of thousands of microglial genes change in response to various insults. Stevens wondered whether this diversity could be recapitulated in vitro, to better manipulate and study it. To find out, scientists in her lab generated iPSC-derived microglia, and exposed them to different triggers commonly encountered in the brain. Think aggregated Aβ, α-synuclein, tau, apoptotic neurons, myelin debris, and synapses.
Alzforum covered some findings from this study, which was completed in collaboration with the group of Evan Macosko, also at the Broad, when they appeared on bioRxiv (Oct 2022 news on Dolan et al., 2022). In essence, the scientists found that, before exposure, the iMGLs in these monocultures appeared identical in every way, but when confronted with a challenge, their diversity sprang to life. The cells transformed into a striking array of transcriptional states, including homeostatic, DAM-like, interferon-responsive, antigen-presenting, and proliferating clusters. The proportion of cells in these respective states changed depending on the specific exposure. For example, the researchers detected two DAM-like transcriptional clusters, one of which was provoked by all substrates except amyloid, the other only by amyloid fibrils and apoptotic neurons.
With this culture system in place, the scientists deployed omics techniques to decipher the master transcriptional regulators of these different states. One outcome was the discovery that the transcription factor MITF controls the shift into DAM-like states. Microglia in the AD brain were recently reported to upregulate this transcription factor in response to pathology (Smith et al., 2022). Then, wielding a lentiviral infection technique honed by postdoc Saša Jereb, the scientists understood a functional consequence of this transcriptional state. Overexpression of MITF in iMGLs not only switched them into the DAM state, it also quintupled their appetite for apoptotic neurons. The researchers are continuing to apply this method to connect multiple transcriptional regulators to different functional states, Stevens said.
At the same time, they started a new project that aims to decode the functional, disease-related consequences of genetic variation in microglia. “Imagine that instead of looking at one cell line at a time, we could compare 100 or more different lines from different patients, with high versus low disease risk, or resilience,” Stevens said at AD/PD. She described how her lab is doing just that.
She introduced the concept of a so-called “village culture system,” whereby iMGLs differentiated from multiple donors are pooled and grown together. Scientists can then expose these microglial “villages” to an array of conditions and probe their functional responses, such as changes in viability, proliferation, mitochondrial or lysosomal function, and phagocytosis, to name a few. Using single-cell barcode tags to track each microglial cell back to its original donor, the scientists are then able to connect the dots from genotype to phenotype, Stevens said.
The method, called Census-Seq, was initially developed by Broad/MIT’s Steve McCarroll and Kevin Eggan, who applied it in human neuronal cells (Mitchell et al., 2020; Wells et al., 2023).
In Gothenburg, Stevens shared some early learnings from the application of this analysis technique in iMGL cultures. Postdoc Martine Therrien started with a pool of 27 donor lines, which came from people with a range of genetic risk for AD. She “fed” the cells fluorescently labeled, apoptotic neurons, and ranked the phagocytic prowess of each cell based on how much shiny debris it consumed. Using Census-seq, the researchers discovered that microglia that came from donors with a high polygenic risk score for AD—meaning they carried a heavy burden of risk variants—tended to be lackluster eaters of dying neurons. This suggested that AD risk variants may provoke disease by hobbling microglial phagocytosis, Stevens said.
Did any specific gene account for this effect more than others? By assessing which variants had the strongest association with phagocytosis, the researchers found TMEM106b—an endolysosomal protein with genetic ties to AD, ALS/FTD, and PD—sitting at the top of the list.
Dovetailing with other findings presented at AD/PD (see Part 13), the data underscore the importance of the microglial endolysosomal system in AD. By connecting genetics to function, the results go a step further, Stevens emphasized, suggesting that microglial digestive troubles are upstream drivers, rather than merely downstream consequences, of AD.
The scientists are starting to explore a next frontier of this line of work by taking cells from their in vitro system back in vivo. They are injecting such microglial “villages” into the brains of disease mice, using chimeric models as developed in the labs of Mathew Blurton-Jones and Bart De Strooper (Aug 2019 news and Oct 2022 news on Mancuso et al., 2022). After a while, they will remove the cells again and compare how the genetics of each microglial donor dictated their cells' differential response to the disease environment they encountered in the host brain.
The ultimate goal of these analyses is to tie genetics to transcriptional states and to function, Stevens said. In so doing, the scientists hope to illuminate essential disease mechanisms that could point to new therapeutic targets.—Jessica Shugart
No Available Comments
No Available Further Reading
Springing a leak is rarely good news, but when microglial mitochondria start oozing, it can be particularly bad in situations of tauopathy. So conclude scientists led by Li Gan and Sadaf Amin, Weill Cornell Medicine, New York, in the April 24 Nature Neuroscience.
The authors report that when mitochondrial DNA drips into the cytosol, it sets off a series of reactions that unleashes an interferon response. That, in turn, douses expression of neuronal genes that protect against tau pathology. Neutralizing the mtDNA threat rescued learning and memory in mouse models of tauopathy. The findings are the latest in a string of reports linking neurodegeneration to wayward nucleic acids, be they double-stranded RNA or DNA. Gan first reported on this work at AD/PD 2022 in Barcelona, and expanded on the data at the 2023 meeting, held in Gothenburg, Sweden, March 28 to April 1.
“There is a lot to be excited about here,” wrote Russell Swerdlow, Kansas University Medical Center (comment below). “If tau indeed messes with mitochondria, and induces a mitochondria DNA leak into the cytoplasm that triggers microglial activation via the cGAS-Sting pathway, that could have important implications for the basis of AD brain inflammation.”

A “Gascade?” Foreign DNA that enters the cytosol activates cGAS, setting off inflammation via activation of STING and phosphorylation of interferon regulatory factor 3. IRF3 drives transcription of interferons in the nucleus. By letting DNA out of mitochondria, pathogenic tau drives the same “gascade.” [Image courtesy Ding et al., 2022, Frontiers in Molecular Neuroscience.]
In a prior study of tau protein interaction networks, Gan and her team were surprised to discover that the wild-type microtubule binding protein cozies up to mitochondrial partners, and that tau carrying a pathogenic mutation rendered the mini power plants much less efficient (Jan 2022 news).
As Gan described in Barcelona last year, Amin then extended the work to microglia. She found that fibrils of tau spark an interferon response cascade in these innate immune cells that is typically driven by viral RNA or DNA (see model above). Amin found that the fibrils of tau somehow damaged the microglial mitochondria, causing them to leak their DNA into the cytosol, activating cyclic GMP-AMP synthase (cGAS), which in turn sets off STING, aka stimulator of interferon genes. By knocking out cGAS or dosing mice with the cGAS inhibitor TDI-6570, Amin was able to dampen the interferon response and preserve synapses and memory loss in P301S tau mice (see image below and Apr 2022 conference news).
However, cGAS suppression did not affect tau pathology. How, then, did it protect neurons?

With, or Without, cGAS? In wild-type hippocampal fields (left panels), knocking out cGAS (bottom) has no effect on levels of the postsynaptic marker PSD-95 (red). In P301S mice (right panel), PSD95 drops (top), but cGAS knockout restores it (bottom). [Courtesy of Udeochu et al., Nature Neuroscience 2023.]
Gan addressed this in Gothenburg. Co-first authors Amin, Joe Udeochu, and Yige Huang found that if they deleted cGAS, neurons up- and downregulated a plethora of genes. Among them, myocyte enhancer factor 2c stood out as being highly expressed among inhibitory and excitatory neurons. Mef2 is a family of transcription factors that are induced by neuronal activity, and they modulate synaptic density (Flavell et al., 2006). Genetic variants near the Mef2c locus have been linked to Alzheimer’s disease, and previously, researchers led by Li Huei Tsai at MIT reported that the gene confers resilience to neurons in mouse models of tauopathy (Mar 2019 news; Barker et al., 2021). Gan’s findings fit with that work, indicating that the cGAS pathway suppresses Mef2c function. Tsai is a co-author on the paper.
To test this idea more directly, the authors injected mice five times over 12 days with 5,6 dimethylxanthenone-4-acetic acid, a STING agonist, to chronically induce IFN-I responses. In wild-type mice, but not in mice lacking IFN receptors, the treatment suppressed Mef2c.

Beefing up Mef2c. In the CA1 hippocampal layer in P301S tau mice (top), neurons (green) express little Mef2c (red). When cGAS is knocked out (bottom), Mef2c expression jumps about 40 percent. [Courtesy of Udeochu et al., Nature Neuroscience, 2023.]
What about genes downstream of Mef2c? The transcription factor regulates 400+ genes. Gloria Huang, a graduate student in the Gan lab, found that expression of more than half of those also change in P301S mice when cGAS is knocked out. Many of these are resilience genes identified by the prior work from the Tsai lab. Those same changes could be evoked by treating mice with the cGAS inhibitor TDI-6570.
All told, Gan believes that by preserving the expression of resilience genes, cGAS-STING inhibition could be protective not only in tauopathies, but in other neurodegenerative diseases as well. At AD/PD, Valina Dawson, Johns Hopkins University, Baltimore, noted in her talk that knocking out STING protects against dopaminergic neuron loss and motor deficits when α-synuclein fibrils are injected into mouse brain. She showed data suggesting that levels of the STING protein double in the substantia nigra in people with Parkinson’s compared to healthy controls (Hinkle et al., 2022). Familial PD mutations damage mitochondria and activate the cGAS/STING pathway, and TDP-43 does the same in ALS (Sliter et al., 2018; Yu et al., 2020).
Furthermore, tau has been implicated in the production of double-stranded RNA in astrocytes, and to cause inflammation in this way, while knocking out cGAS appears to reduce plaque accumulation in mouse models of AD (Jan 2023 news). The cGAS/STING pathway might also help explain links between viral infections and a person’s risk for AD (see Part 10 of this AD/PD series).
cGAS knockout mice seem healthy. To Gan's mind, this implies that targeting this enzyme in multiple neurodegenerative diseases might be safe. “We would propose to continue to understand how cGAS/STING drives neurodegeneration as a common mechanism, most likely triggered, partially at least, by mitochondrial dysfunction,” said Gan.—Tom Fagan
After years of languishing in obscurity, the topic of disruptions in the brain's lipid metabolism is moving center stage. The number of publications in this area is growing, and at the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, lipid dysfunction debuted as a session topic. At the session, six presenters showed off a tool kit of newfangled imaging and analysis methods that promise to transform this famously complex field of research. Their data converged on changes in phospholipid, fatty acid, and cholesterol metabolism in AD, PD, and Lewy body dementia. It appears that these perturbations occur early in the disease trajectory, perhaps even preceding amyloid plaques.
“I think the session was excellent and really shows the coming of age of how we understand and measure lipid changes in Alzheimer’s disease (and Parkinson’s disease),” Rik van der Kant, Vrije Universiteit, Amsterdam, who did not speak at this session, wrote to Alzforum.
Ole Isacson of Harvard Medical School, Boston, co-chaired the session. “This [work] provides fundamental understanding for how these diseases can be caused by lipid dyshomeostasis and metabolic disturbances that drive downstream amyloidogenic pathways. Lipid dyshomeostasis is more of a driver than perhaps we have had time to investigate,” he told Alzforum. Laura Beth McIntire of Weill Cornell Medical College, New York City, welcomed the new focus on advances in lipidomics in neurodegenerative disease. “Lipid metabolism is a major pathway in AD,” she said.
Already in 1906, Alois Alzheimer saw glial cells that appeared bloated with lipid droplets under the microscope. Much later, human genetics pointed to lipid metabolism in neurodegenerative diseases. Variants of the lipid binding proteins ApoE and PICALM, the lipid transporter ABCA7, the genes MS4A4A and PLCG2, and others have been linked to higher odds of AD, as well as variants in the glycolipid hydrolase GBA1 to LBD and PD (Feb 2021 news; Apr 2018 news; Mar 2021 news). Up to 40 percent of AD genome-wide association study hits are involved in lipid metabolism, making it the major contributor to disease risk, McIntire noted.
To understand how lipid metabolism goes awry in AD, McIntire analyzed gene expression in cortical postmortem tissue samples from 639 participants from the Religious Orders Study and the Rush Memory and Aging Project (ROSMAP) cohort. After accounting for amyloid plaque and tau tangle loads, she found that people whose cognition slipped fastest had upregulated phospholipase A2 (PLA2) and downregulated lysophosphatidylcholine acyltransferase 2 (LPCAT2) genes. The former converts the lipid phosphatidylcholine (PC) to lysoPC; the latter recycles lysoPC back to PC in what is called the Lands cycle or acyl chain remodeling, a core constituent of the making and breaking of the phospholipids that form cell membranes (see image below). These and other gene-expression changes suggest that an abundance of lysoPC might reflect lipid metabolism dysregulation in AD, McIntire concluded at AD/PD.

Lipid Flux. To recycle and replenish phospholipids within cell membranes, phospholipase A2 (PLA2) cleaves a fatty acid tail from the lipid to make a lysophospholipid. A lysophospholipid acyltransferase, such as LPCAT2, tacks on a new tail to make a new phospholipid. The headgroup (X) distinguishes lipid subtypes, with phosphatidylcholine sporting a choline molecule. [Courtesy of Bankaitis, 2009, Journal of Cell Biology.]
Next, the scientists performed lipidomics on samples from 99 ROSMAP participants. Limitations in their lipid panel precluded them from defining which species were up- or downregulated in AD, but even so, they were able to analyze patterns in lipid abundance based on participants' clinical diagnosis and APOE4 genotype. Two profiles emerged. Lipidomes were similar in cognitively normal APOE4 carriers and all people with mild cognitive impairment, yet were significantly different from cognitively normal E4 noncarriers and all people who had AD. “If we’re only looking at advanced AD compared to cognitively normal people, we may be missing important lipid changes,” McIntire concluded.
To McIntire, this implies lipid metabolism changes so early in AD that they would precede amyloid plaques and neurofibrillary tangle deposition. Indeed, in ApoE4 carriers, PET imaging with a tracer binding the fatty acid DHA reflects a DHA deficiency in gray matter as early as age 35 (Yassine et al., 2017).
DHA levels change in young amyloidosis mice, too. Artur Lazarian, who works in McIntire’s lab, analyzed brain slices from wild-type and Tg2576 mice using imaging mass spectrometry. This technique ionizes molecules from a specific area within a brain tissue slice, measures the mass and abundance of each ion there, and creates maps of how each lipid species populates the brain. Incidentally, Lazarian mentioned that his lab has a new imaging mass spectrometer and invited collaboration inquiries from scientists interested in studying lipidomics.
Compared to wild-type mice, the transgenics accumulated more polyunsaturated fatty acids, including DHA, in the brain by 4 months of age, before plaques appeared. These lipids remained high and unchanged while amyloid accumulated. How would DHA, or other fatty acids and lipids, influence subsequent amyloid accumulation? “The role of lipids in amyloid pathology is potentially important, though still enigmatic,” Jörg Hanrieder, University of Gothenburg and University College London, wrote to Alzforum.
McIntire offered one explanation. It appears that certain types of fatty acid stick to Aβ and influence its accumulation. Most of the lipids at different abundances in the ROSMAP profiles contained polyunsaturated fatty acids. In an in vitro assay, McIntire found that the polyunsaturated DHA, but not the saturated stearic acid, bound fluorescently labeled Aβ42. Nuclear magnetic resonance and molecular modeling detected DHA docking into a pocket on Aβ42 created by residues 12 through 20. Thioflavin T aggregation assays showed that DHA prevented Aβ42 aggregation while stearic acid did not, likely because the latter did not bind to Aβ42.
This finding implies a caution for mechanistic studies using Aβ. “Aβ is probably characteristically lipidated, and it is unlikely that studies using the unlipidated peptide give us changes that are biologically relevant,” McIntire said. Van der Kant agreed. “I don’t think it would be farfetched to hypothesize that adding unlipidated synthetic Aβ to cells or mouse brains would lead to toxicity simply because this ‘empty’ Aβ starts searching for lipids to bind and extract,” he wrote to Alzforum.
To get a better look at how lipids and plaques interact in the brain, Hanrieder’s group has been developing a sophisticated method that uses imaging mass spectrometry to detect simultaneously which lipids are found in amyloid plaques and where they are within a given plaque. Hanrieder has studied brain tissue slices from Tg-ArcSwe mice (Kaya et al., 2017; Kaya et al., 2017; Michno et al., 2018). The scientists sharpened the 10-micron spatial resolution of their imaging MS down to 300 nanometers by combining it with hyperspectral confocal microscopy, which uses two different structurally sensitive fluorescent amyloid dyes to label diffuse or cored plaques, respectively (Wehrli et al., 2023). In essence, this multimodal analysis renders specific chemical information from mass spec at the high spatial resolution of amyloid microscopy, Hanrieder explained.
Hanrieder is using it on the human cortex. He started with postmortem samples from five people with familial AD and a presenilin 1 mutation, and is now analyzing sporadic AD tissue, from UCL's Queen Square Brain Bank. He was able to “see” phospholipids, ceramides, cerebrosides, and gangliosides—importantly, all of them localized to amyloid plaques.
One ganglioside, GM1, was the main lipid in cored plaques. In contrast, sulfatide, a major lipid component of myelin sheaths, was relatively absent from plaques and the surrounding areas (see image below). Hanrieder thinks that this suggests demyelination of nearby neurons.

Gangliosides, No Sulfatides. In presenilin 1 ADAD mutation carriers, amyloid plaques (green, left) include the ganglioside GM1 (green, middle) but lack a sulfatide species (blue, right). [Courtesy of Jörg Hanrieder, University of Gothenburg.]
At AD/PD, Li-Huei Tsai of Massachusetts Institute of Technology, Boston, also presented on disrupted myelination in APOE4 carriers with AD. Tsai's lab recently published results of single-nucleus RNA sequencing on cortical tissue from 32 ROSMAP participants, comparing gene expression between APOE genotypes. Relative to E3/3 carriers, most of the differentially expressed genes in E4/4 carriers mapped to lipid metabolism pathways, with myelination down- and cholesterol synthesis upregulated in oligodendrocytes. Cortical tissue from APOE3/4 carriers was reported to be less myelinated than in E3/3 carriers, and cultured iPSC-derived human APOE4/4 oligodendrocytes to contain more lipid droplets than their E3/3 counterparts. Tsai concluded that ApoE4’s impaired ability to transport the lipid cholesterol might hobble oligodendrocytes' ability to properly myelinate axons (Blanchard et al., 2022). Van der Kant noted that, somehow, AD risk variants seem to cause a broken feedback loop, where cholesterol accumulates intracellularly, yet cells keep on producing more cholesterol as if they were starved of the lipid.
Another AD GWAS hit, the lipid efflux pump ABCA7, reared its head here, too. Much like APOE4 carriers, 12 ROSMAP participants with a loss-of-function mutation in ABCA7 had upregulated cholesterol synthesis genes in their oligodendrocytes, according to Djuna Von Maydell from the Tsai lab. Oddly, their lipid metabolism was downregulated in other cells. Excitatory neurons expressed the most ABCA7, yet those harboring the AD risk variant downregulated genes involved in lipid metabolism. Lipidomics analysis showed that ABCA7 loss-of-function brains contained less of almost every lipid species, including phosphatidylcholine, than did noncarriers. Van der Kant wondered if cholesterol metabolism was abnormal in people with mutations in other AD-linked genes. “[I believe it] will likely be a key aspect of more AD risk variants,” he wrote.
What About Dementia with Lewy Bodies and Parkinson's?
Scientists have been implicating lipid dysregulation in LBD and PD for longer than in AD. Besides driving up risk of AD, APOE4 also puts a person at greater risk of developing Lewy body dementia. Mutations in the glucocerebrosidase GBA1 increase risk for LBD and Parkinson’s. GBA1 hydrolyzes glucocerebroside, an intermediate metabolite needed to make the glycolipids that are a component of cell membranes. At AD/PD, Isacson showed that blocking GBA1 in wild-type mice increased the amount of ApoE protein in their brain cortices and hippocampi, suggesting that ApoE adapts to the presence of more lipids when GBA1 no longer hydrolyzes them.
To explore how GBA1 and APOE influence brain lipid homeostasis, Isacson and colleagues treated wild-type and APOE knockout mice with the GBA1 inhibitor conduritol b epoxide for 18 days. In hippocampal tissue from both mouse lines, lots of lipids—cholesterol, glycolipids, triglycerides—formed large clusters between brain cells (see image below). Microglia multiplied, and levels of the complement activator C1q and the intracellular cholesterol trafficking protein NPC1 rose.
APOE knockouts had more severe phenotypes than did wild-types, likely because ApoE was not there to compensate for the lipid influx. “GBA1 inhibition causing glycolipid accumulation is functionally connected to the loss of APOE function causing cholesterol and lipid accumulation,” Isacson told Alzforum.
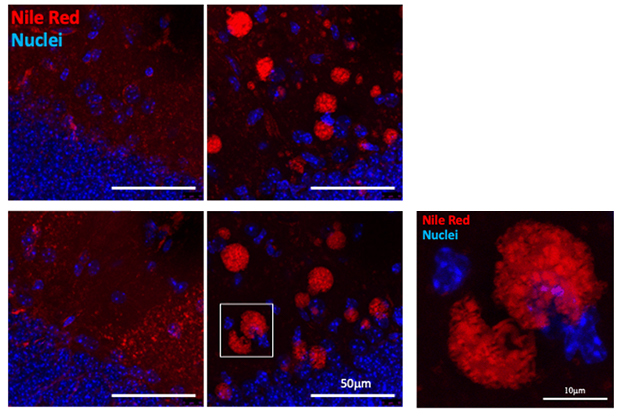
Little Balls of Fat. In the hippocampi of wild-type (top) or APOE knockout mice (bottom), lipids (red) formed blobs between brain cells when GBA1 was inhibited (right; nuclei in blue). [Courtesy of Ole Isacson, Harvard Medical School.]
Moreover, ApoE helped clear cholesterol from within cells, but how well it did so depended on the allele. ApoE binds to the outer cell surface, becomes lipidated, then carts the fats away. ApoE4 carries a lighter lipid load than E3 and E2, hence is worse at clearing intracellular lipids. In human astrocyte cultures, blocking NPC1 spurred cholesterol and triglyceride accumulations, and those co-localized with APP. Adding ApoE2 to the culture medium brought intracellular cholesterol and APP down to almost normal. Adding ApoE3 did so a little bit, while ApoE4 barely removed excess cholesterol and APP. “ApoE2 and ApoE3 can shuttle cholesterol out of the cell more effectively than ApoE4,” Isacson concluded. “This is very important for showing that functional differences in cholesterol lipid transport by ApoE isoforms correspond to the risk for developing LBD and AD.”
Kimberly Paul of the University of California, Los Angeles, took a different approach to learning about lipids in PD. She used mass spectrometry data of the metabolomes of serum samples from 642 people with early stage PD and 277 controls from UCLA’s Parkinson’s Environment and Genes study (Paul et al., 2022). Analyzing the data through a metabolome-wide association study, she found 104 of 4,762 identified metabolites to be specific to PD. Lo and behold, quite a few fell into lipid metabolism pathways.
Among the metabolites most associated with PD were various lysoPC species from the Lands cycle. People with PD had up to four times more of these lipids in their blood than did controls. LysoPCs also turned up at higher levels in the substantia nigra in a rat model of PD (Farmer et al., 2015). Functionally, these lipid metabolites are a “find me” signal on apoptotic cells to cue macrophages and microglia for phagocytosis. They have been linked to neuroinflammation and demyelination. "We are still teasing out the chain of events and mechanisms for lipid dyshomeostasis in PD, but lysoPCs seem to play a role,” Paul noted.
Could Choline Help Fix Lipid Dysfunction?
While phosphatidylcholine and lysoPC metabolism were disturbed in AD and PD, other signs point to perturbations in the lipid’s precursor molecule, choline. Carrying a mutation in the choline synthesis enzyme PEMT, and consuming insufficient choline in one's diet, are both linked to AD (Bi et al., 2011; Yuan et al., 2022). In amyloidosis mice, withholding dietary choline altered synaptic transmission and upped amyloid plaques and phospho-tau species, whereas APP/PS1 mice fed a high choline diet had fewer plaques and better spatial memory (Dave et al., 2023; Velazquez et al., 2019).
At AD/PD, Tsai reported similar results in APOE4 knock-in 5xFAD mice. A diet rich in choline cut amyloid load and lowered levels of the lipid droplet-associated protein perilipin1 in brain tissue. These findings mirror a reduction in lipid droplets Tsai saw within iPSC-derived human APOE4/4 astrocytes after adding choline to the culture medium (Mar 2021 news).
Taking choline supplements to treat or prevent AD has not been shown to work in clinical studies, though some are ongoing. It is unclear if dietary supplementation could overcome a deficit in choline transport or utilization, and, if so, how much choline would be required for a treatment benefit, McIntire wrote to Alzforum.—Chelsea Weidman Burke
Do microglia thwart neurodegenerative disease, or help it along? Do they keep amyloid in check with one hand, while goading tau entanglement with the other? Do they protect neurons early on in disease, but sour into synaptic slayers later? As the field gears up to target these reactive immune cells in clinical trials (see Part 8 of this series), these fundamental paradoxes remain unsettled. At the International Conference on Alzheimer’s and Parkinson’s diseases, held March 28 to April 1 in Gothenburg, Sweden, scientists leveraged longitudinal data from human cohorts to get at these questions.
A load of findings came from the Montreal-based Translational Biomarkers of Aging and Dementia cohort. TRIAD deploys a trio of PET scans to map amyloid plaques, tau tangles, and microglial activation within a person's brain. Its data indicate that ApoE4 goads microglial activation, which, in turn, appears to worsen tau pathology and neurodegeneration. In TRIAD, fluid markers of phosphorylated tau tracked with heightened microglial activation in brain regions where tangles accumulate early in AD. Curiously, people with revved-up microglia tended to be more irritable than those with calmer microglia.
While these TRIAD results cast microglia as bad actors, data shown at AD/PD from a Swedish cohort implied the opposite conclusion. Based in Lund, BioFinder used CSF markers to gauge microglial activity, and those results associated microglial responses to reduced tau accumulation and slower cognitive decline.
“These clinical findings are really important. We have a clear divide in the microglial field between whether the microglia responses seen in the AD brain protect against aspects of the pathology, or contribute toward them,” commented Kim Green of the University of California, Irvine. “In reality, it is likely disease stage- and brain region-specific.” To study this, Green said the field sorely needs animal models that develop the spectrum of AD pathologies.
Pedro Rosa-Neto of McGill University in Montreal heads TRIAD. This cohort is unique in that participants periodically undergo three scans with second-generation PET tracers: [18F]AZD4694 for amyloid, [18F]MK6240 for tau, and [11C]PBR28 for TSPO. TSPO is an outer mitochondrial membrane protein that cranks up as microglia become activated.
Including this PET scan comes at a cost. A polymorphism in the gene encoding TSPO influences how tightly the protein binds the PET tracer. Therefore, TRIAD participants are screened for these polymorphisms, and only carriers of the high TSPO-binding version—about 60 percent of participants in this predominantly non-Hispanic white population—undergo TSPO scans, Rosa-Neto told Alzforum. Rosa-Neto believes that the extra screening is worth the trouble, as zeroing in on how microglia, Aβ, and tau interact at the regional level is critical for understanding how they influence AD pathogenesis. In an example of this, Rosa-Neto and Tharick Pascoal, now at the University of Pittsburgh, previously reported that microglial activation arises in step with Braak stage regions, just before tau tangles inundate a given region (Sep 2021 news).
At AD/PD, the scientists added ApoE4 to this heady mix. Setting microglia aside for a moment, João Pedro Ferrari-Souza’s talk focused on the role of ApoE4 in potentiating the effect of Aβ plaques on the subsequent spread of tau tangles. A graduate student in Pascoal’s lab, Ferrari-Souza studied amyloid- and tau-PET scans from 104 TRIAD participants, including 72 cognitively unimpaired people, 25 with MCI, and seven with AD dementia. Ferrari-Souza found that relative to noncarriers with or without amyloid at baseline, ApoE4 carriers with amyloid had dramatically higher levels of tau deposition in medial temporal lobe regions over the following two years. A mediation analysis then suggested that ApoE4 exacerbated the connection between Aβ pathology and tau entanglement. Notably, serial measurements of plasma p-tau217 in the same participants indicated that the synergistic effect of Aβ and ApoE4 on tau tangles happened via phosphorylated tau.
“Indeed, the ApoE4 potentiated effects of Aβ on tau tangle accumulation occurs through tau phosphorylation,” Ferrari-Souza told the audience.

E4 Sparks Tau, but How? In ApoE4 noncarriers (blue), baseline Aβ burden did not correlate with tau tangle deposition two years later. In carriers (pink), baseline Aβ burden predicted a rise in plasma p-tau217, which, in turn, predicted tau tangle accumulation. [Courtesy of Ferrari-Souza, University of Pittsburgh.]
How does ApoE4 influence tau tangle accumulation? Ferrari-Souza and colleagues wove microglial activation into the equation. The study included 118 participants across the AD spectrum, including 79 who were cognitively normal, 23 with MCI, and 16 with AD dementia. At baseline, ApoE4 carriers had significantly higher microglial activation, according to TSPO-PET, than noncarriers. This E4 effect cropped up predominantly in areas corresponding to Braak stage regions, and it was strongest in early Braak regions. People with the “hottest” TSPO-PET signals in Braak regions at baseline tended to decline more steeply on cognitive tests, and their hippocampi shrank more over the following year. This suggested that ApoE4-triggered microglial responses speed up neurodegeneration.

E4 Riles Microglia. TSPO-PET scans (left) reveal that ApoE4 carriers have more activated microglia than noncarriers in medial temporal regions. The difference is greatest in early Braak stage regions (right, percent area showing E4-related TSPO-PET boost). [Courtesy of Ferrari-Souza et al., Science Advances, 2023.]
Why would ApoE4 prod microglia more in some regions than others? The researchers got a hint from the Allen Human Brain Atlas. Its spatial RNA sequencing data revealed that ApoE mRNA expression tracked with Braak stage regions, with expression being highest in the earliest-stage regions, including the transentorhinal cortex, entorhinal cortex, and hippocampus. Importantly, these ApoE expression patterns predicted the strength of the TSPO-PET signal across brain regions among TRIAD participants, Ferrari-Souza reported.
It remains unclear what factors dictate the distribution of ApoE expression, or even which cells are responsible. Production of the apolipoprotein by astrocytes, activated microglia, and even stressed neurons can reportedly jolt microglia out of their homeostatic state (Oct 2019 news; Apr 2021 news; Feb 2023 news). Still, the findings suggest that microglial activation depends on levels of ApoE expression.
Notably, ApoE4's microglia-stoking effect in the medial temporal lobe was observed regardless of the person's global Aβ burden, local tau tangle burden, or clinical diagnosis. The tie between ApoE4 and microglial activation in these early Braak regions was strongest among people with a higher burden of tangles in those same regions, suggesting that E4-triggered microglia promote tau pathology.
Ferrari-Souza used a statistical method called structural equation modeling to tease out the causal relationships among all these contributors to AD. He reported that heightened TSPO-PET signals in the medial temporal lobe were partially responsible for ApoE4’s promotion of tau pathology. In a separate, parallel pathway, amyloid burden also contributed to a boost in tau tangles. Both the Aβ-dependent and independent contributors to tau pathology were tied to hippocampal shrinkage and steeper cognitive decline. The findings were published in Science Advances on April 6 (Ferrari-Souza et al., 2023).

Parallel Paths to AD? Statistical modeling calculates relative causal relationships among different pathological contributors. Larger numbers (β-estimates) signify stronger causal ties. Black arrows represent significant effects; grey arrows, nonsignificant. [Courtesy of Ferrari-Souza et al., Science Advances, 2023.]
At AD/PD, Nesrine Rahmouni, a graduate student in Rosa-Neto’s lab and coordinator of the TRIAD cohort, presented fluid biomarker findings from the TRIAD cohort. They dovetailed with Ferrari-Souza’s work. Essentially, Rahmouni found that CSF markers of phosphorylated tau, including p-tau181, p-tau217, and p-tau231, all correlated with TSPO-PET signals within the medial temporal lobe.
In cognitively normal people, this association was independent of Aβ or tau burden, as gauged by PET scans. However, among those with MCI or AD dementia, the relationship between CSF p-tau and TSPO-PET was largely dependent on tangles, Rahmouni reported. In statistical models, a combination of CSF p-tau and tau-PET best predicted microglial activation as measured by TSPO-PET. Her interpretation? CSF p-tau biomarkers—which reflect the infancy of tau pathology—have an independent relationship with brain inflammation early in disease, but at later stages, tau tangles take precedence in mediating this effect.
Marc Diamond of UT Southwestern Medical Center in Dallas broached the chicken-and-egg conundrum: “Is neuroinflammation causing tau pathology, or the other way around?”
“That’s the million-dollar question,” Rahmouni responded. “We think it’s both.” She said that inflammation may help at first, only to become chronic, and therefore deleterious, later on. A deeper knowledge about inflammatory pathways at work in the brain will be needed to understand how and when that switch occurs, she said.
Rosa-Neto believes the findings are consistent with several parallel pathological pathways contributing to AD, as opposed to a sequential cascade of single events. “Perhaps AD is a tale of two proteinopathies that start to cause disease when they converge,” Rosa-Neto said. Microglia represent one point of this convergence, he said, as "well-meaning" cells eventually transform into harbingers of degeneration. In ApoE4 carriers, the path to this damage-prone state might be shortened both by microglial reactivity as well as by higher vulnerability among neurons.
Pascoal agrees that multiple players are likely involved in the overall progression of AD. He thinks future studies will become more granular at every level, and in that way zero in on cell type- and disease-stage specific contributors that converge on neurodegeneration.
The TRIAD results jibe with mouse studies from David Holtzman’s lab, in which ApoE4 dramatically worsened tau pathology in a way that depended upon microglia (Sep 2017 news; Oct 2019 news). “This new human data would argue that microglial reactivity may be useful in the future in predicting outcome, and be useful to measure in clinical trials,” Holtzman commented to Alzforum. “It would be very helpful going forward to have even more specific markers that we can use in humans to determine the particular microglial state that is present, whether via imaging or fluids, to better get at the details of how the microglial reactive state is changing.”
As microglia take blame for promoting tau propagation (see Part 13 of this series), how can scientists square this heinous act with the fact that loss-of-function variants in TREM2—the receptor that controls myriad microglial responses—boost a person's risk of AD? This contradiction has sparked confusion in the field, Joana Pereira of the Karolinska Institute in Stockholm said at AD/PD.
To investigate how TREM2-mediated signaling in microglia influences tau pathology and other aspects of AD, Pereira looked in CSF for indicators that microglia had transitioned to the disease associated microglia-2 (DAM2) state. This transcriptional state relies on TREM2 signaling, and is characterized by enhanced phagocytosis, mediated at least in part through TAM receptors Axl and MerTK (Jun 2017 news; Apr 2021 news). Pereira hunted for proteins churned out by DAM2 cells, including soluble TREM2 (sTREM2), Axl, and MerTK along with their ligand, Gas6, as well as LPL, CST7, SPP1, and CSF1.
Her analysis included 344 cognitively unimpaired participants from the BioFinder cohort, split into three groups based on results of their baseline amyloid and tau-PET scans: 121 had evidence of amyloid plaques but not tau tangles, 64 had both plaques and tangles, and 159 had neither. Participants underwent repeated scanning, CSF sampling, and cognitive testing two and four years later.
What did they find? Starting with soluble TREM2, Pereira reported that among all participants with amyloid at baseline, higher levels of sTREM2 predicted a slower rise in amyloid over the following four years. Among those who started the study with some evidence of tau pathology, Pereira found the same beneficial effect of higher sTREM2 on tau accumulation.
For the other DAM2 markers, Pereira found no association with future amyloid accumulation. Alas, she did find that higher CSF concentrations of three—Gas6, Cst7, and CSF1—predicted a slower progression of tau pathology. This was true in Braak III/IV and Braak V/VI regions, which correspond to neocortical tangles. These associations were independent of amyloid. Meshing with her tau findings, Pereira also reported that higher CSF DAM2 markers predicted slower cognitive decline as measured by MMSE scores. Curiously, this salubrious effect on cognition was stronger in women than in men.
SPP1 was a notable exception. High levels of this cytokine foreshadowed a faster spread of tau, and a steeper drop in cognition. To Pereira's mind, SPP1’s outlier status makes sense in light of the finding that SPP1 incites perivascular macrophages to nosh on synapses (Feb 2023 news). “Not all myeloid responses in the brain are beneficial,” Pereira said. The findings were published in Nature Aging (Pereira et al., 2022).

TREM2 Transition. When microglia stay in a homeostatic state (top), Aβ plaques and tau tangles can more quickly spread across the brain. When the cells transition into the DAM2 state, they keep AD pathology in check. [Courtesy of Pereira et al., Nature Aging, 2022.]
How to reconcile the helpful responses Pereira saw with the TRIAD results, which cast microglia as accomplices to tau propagation? Potential explanations abound. They include differences in the number and disease stage of participants in the cohorts, and the modalities used to gauge the microglial mood. Pereira hypothesized that, similar to the nefarious SPP1 cytokine in her study, perhaps TSPO-PET illuminates destructive microglia or macrophages. “I think TSPO-PET is probably detecting a harmful microglial phenotype that is associated with synaptic phagocytosis and degeneration, whereas DAM2 are more associated with protective effects,” she wrote to Alzforum.
A way to answer this question is to track both TSPO-PET and DAM2 CSF biomarkers in the same cohort, said Henrik Zetterberg of the University of Gothenburg in Sweden, who co-authored both papers.
To Pascoal’s mind, the TRIAD and BioFinder results don’t necessarily conflict. Instead, they offer a glimpse at the dramatic heterogeneity of microglial states within the human brain. Alluding to single-cell RNA sequencing studies, Pascoal said that not only are the cells in different states at any given time, but these states themselves change with time, age, and disease stage. Cracking open this heterogeneity is the focus of intense research (see Part 14 of this series). Just like the false dichotomy of M1 versus M2 phenotypes of the past, labeling microglia as “good” or “bad” is a gross oversimplification.—Jessica Shugart
No Available Comments
Tau pathology is proving a tough nut to crack, with most antibodies directed against it posting negative results in clinical trials thus far. At the recent International Conference on Alzheimer’s and Parkinson’s Diseases in Gothenburg, Sweden, researchers discussed how they might hone antibodies and vaccination strategies to become effective.
Acknowledging that the bulk of tau stays inside cells, some speakers touted single-domain antibodies, small fragments that can more easily enter neurons. Others introduced vaccine strategies designed to provoke a stronger immune response or hit multiple types of fibril. For example, the vaccine PRX123 elicits antibodies against both Aβ and tau. Likewise, scientists at a pan-amyloid company debuted a peptide-antibody fusion, or “peptibody,” that binds Aβ, tau, and α-synuclein, three pathologies that often co-occur in Alzheimer’s disease.
These strategies are still preclinical, so it remains to be seen how well they perform in people. Nonetheless, the presentations showed a field branching out as it learns from early failure.

One Domain Curbs Tau. Tauopathy flies (solid red) die much faster than do wild-types (solid black), but treatment with anti-tau single-domain antibodies sdAbs (dotted red) extends (left) or restores (right) their lifespan. [Courtesy of Einar Sigurdsson.]
Get Small, Sneak In
Initially, most tau antibodies were intended to stop the spread of toxic aggregates from cell to cell. However, more recent research suggests that replication of tau seeds within neurons does more to propagate tangles than extracellular spreading (Nov 2021 news). In addition, aggregated tau may pass between cells via exosomes or nanotubes that shield it from antibodies (e.g., Leroux et al., 2022). These findings have heightened interest in targeting intracellular tau.
In Gothenburg, Einar Sigurdsson of New York University School of Medicine stressed the importance of this, noting that intracellular tau is 10,000 to 100,000 times more abundant than extracellular species (Han et al., 2017; Congdon et al., 2022). While antibodies can be taken up by cells to reach intracellular tau, cells do that mostly through receptor-mediated endocytosis. Antibody fragments, on the other hand, can be engulfed by bulk endocytosis and thus may enter cells more readily, Sigurdsson said.
To shrink antibodies, one option is to use single-domain versions. sdAbs come from mammals such as camels or llamas, whose antibodies contain a single variable heavy domain (VHH), unlike the double heavy and light chains of conventional antibodies. Also called a nanobody or llamabody, an isolated VHH domain weighs in at 13 kDa, compared to the 150 kDa of its big brother IgG. sdAbs have high affinity and specificity, Sigurdsson said. They are stable, enter the brain well and—importantly—can find epitopes that larger antibodies cannot reach.
To make tau-specific sdAbs, Sigurdsson’s group immunized llamas with the longest isoform of recombinant human tau, then boosted with paired helical filaments of tau from postmortem human brain. After screening a library of the resulting antibodies, the scientists selected a candidate, 2B8, that bound tangles in human brain. They injected fluorescently labeled 2B8 into tauopathy mice as a probe, finding that the signal correlated strongly with tau protein as seen by Western blot, at r=0.94. The probe ended up in endosomes and lysosomes, showing that it was internalized by neurons and positioned to influence tau degradation (Congdon et al., 2022).
But does this sdAb clean up aggregated tau? When expressed in tauopathy flies that make mutant R406W human tau, 2B8 greatly extended lifespan. A similar tau sdAb, 1D9, restored their lifespan to wild-type levels (image above).
Pursuing the same strategy to make sdAbs against aggregated α-synuclein, Sigurdsson's group found a candidate that halved the amount of α-synuclein in the brain of a synucleinopathy mouse. When the scientists conjugated this sdAb to thalidomide to bind E3 ubiquitin ligase and trigger degradation of the complex, the treatment nearly eliminated aggregated α-synuclein. Sigurdsson believes sdAbs have potential as both therapeutics and PET ligands, and has a paper coming out on the latter later this month.

Trim the Tau. Brain lysates from tauopathy mice (upper left) seed tau aggregates (green, arrowheads) in cultured cells, and in the absence of the E3 ubiquitin ligase TRIM21 (bottom left). Treating mice with an anti-tau antibody (upper right) suppresses seeds—but not if TRIM21 is missing (bottom right). [Courtesy of Mukadam et al., Science/AAAS.]
Intriguingly, a recent paper found that engaging the E3 ubiquitin ligase TRIM21 is essential to enabling antibodies to clear tau seeds. In the March 31 Science, researchers led by William McEwan at the U.K. Dementia Research Institute, Cambridge, reported that a tau-clearing antibody became ineffective in tauopathy mice if they lacked TRIM21. Cytosolic TRIM21 binds to full-size antibodies via their Fc domain, stimulating proteasomal degradation of the tau-antibody complex (Mukadam et al., 2023).
McEwan’s finding bolsters the idea that antibodies operate inside neurons, rather than extracellularly, to eliminate aggregated tau. It also implies that antibody fragments lacking the Fc domain might be less potent at abolishing tau, unless researchers activate the E3 ubiquitin ligase as Sigurdsson did.
Research on anti-tau sdAbs is also happening in the lab of Luc Buée of the University of Lille, France. At AD/PD, Buée likened these small fragments to Legos that allow scientists to build whatever tools they wish. His group searched a humanized library of VHH fragments to find sdAbs that recognize the hexapeptide sequence in tau’s microtubule-binding region (MTBR) that controls formation of paired helical filaments (von Bergen et al., 2000).
One candidate the scientists found was able to enter cells and bind tau, but did not stop aggregates. They then tweaked this sdAb to make a new version dubbed VHH Z70. It bound with micromolar affinity to the hexapeptide sequence and prevented tau aggregates in yeast. In THY-tau30 tauopathy mice, it halved tau deposits, as judged by AT8 staining. Buée believes that VHH Z70 blocks the growth of fibrils by preventing recruitment of tau into PHFs, but does not break apart existing strands (Danis et al., 2022).

Fibril Affinity. The peptide P5R (gray) recognizes heparan sulfate proteoglycans, allowing it to bind any type of amyloid fibril (green). [Courtesy of Attralus.]
One Treatment to Clear Them All?
In many neurodegenerative diseases, multiple types of aggregated protein build up in the brain, and scientists are seeking ways to remove these mixed deposits. In Gothenburg, Suganya Selvarajah of Attralus Inc., San Francisco, detailed her company’s strategy for homing in on such pathologies.
Instead of an antibody, Attralus generated a peptide sequence that selectively binds to heparan sulfate proteoglycans found on amyloid fibrils. Dubbed P5R, this peptide has a random shape in solution, but when it comes in contact with the dense, negatively charged surface of fibrils, it coils into a helix, attaching itself to the fibril via multiple side chains. Because this binding does not depend on the fibril's amino acid sequence, P5R can recognize multiple types of amyloid, including Aβ, tau, and α-synuclein, Selvarajah told the audience.
To build a therapeutic, aka peptibody, the researchers linked the peptide to Fc antibody fragments. The idea is to opsonize fibrils and tempt microglia to eat them. The hybrid molecule, AT-04, consists of two linked Fc fragments, each attached to a P5R peptide. It weighs 59 kDa. AT-04 binds Aβ with 0.2 nM affinity, similar to aducanumab’s binding strength, and orders of magnitude higher than that of VHH Z70. Unlike aducanumab, however, AT-04 also recognizes tau fibrils with 0.2 nM affinity, and α-synuclein fibrils at 0.8 nM. Because these latter two are intraneuronal, peptibodies would have to enter cells to access them.

Modular Molecule. Peptibodies combine a peptide (blue) with an antibody Fc fragment (tan), plus sometimes a variable antibody fragment (green) to recognize the transferrin receptor in brain blood vessels and enhance uptake. [Courtesy of Attralus.]
To test AT-04 in vivo, the researchers infused 50 mg/kg into a vein of 5XFAD mice three times over the course of 10 days. The peptibody bound plaques in mouse brain, although Selvarajah did not say if it promoted clearance. AT-04 also recognized plaques in postmortem AD brain tissue.
The researchers made another peptibody, AT-07, for which they fused Fc domains to VNAR domains, variable heavy chain fragments made in sharks. Because the VNAR domain chosen for AT-07 recognizes the transferrin receptor expressed by blood vessels in the brain, this construct enters the brain better than does AT-04.
Scientists for some years have exploited the ability of transferrin receptors to shuttle large molecules past the blood-brain barrier (Hultqvist et al., 2017; Mar 2021 conference news; Jan 2023 news). About 1.4 percent of AT-04 passes the blood-brain barrier, slightly above the 0.5 to 1 percent uptake of conventional antibodies, while AT-07 uptake is 10-fold higher, with 15 percent of it reaching the parenchyma. At 80 kDa, AT-07 is larger than AT-04. It also binds Aβ fibrils with 0.2 nM affinity, and recognizes plaques in AD brain.
An audience member asked how the peptibodies will choose between amyloid, tau, and α-synuclein if all three types of aggregate are present in brain. “That’s the million-dollar question. We don’t know yet,” Selvarajah said, referring to ongoing in-vivo studies. Attralus peptibodies for peripheral amyloidoses are in Phase 1 (AT-02; AT-03).

Stop Tau Seeds. In a biosensor cell line (top), tau (green) aggregates into punctate deposits (bottom) when exposed to seeds. Polyclonal antibodies generated by Vaxxinity compound A inhibited this, while compound F fractions performed similar to the purified monoclonal semorinemab. [Courtesy of Justin Boyd, Vaxxinity.]
Rallying the Body’s Own Defense
For long-term treatment, coaxing the body to produce its own antibodies would be far cheaper than therapeutic antibodies. Several groups are working on tau vaccines, including Axon Neuroscience, AC Immune, and an academic collaboration that develops the recombinant protein vaccine AV1980R/A (Jun 2021 news; Dec 2022 conference news).
In Gothenburg, Justin Boyd of Vaxxinity, Merritt Island, Florida, described his company's strategy. To make a tau vaccine, or “vaxxine” as they call it, company scientists link a short synthetic peptide that mimics a portion of tau to another that activates CD4+ T-helper cells. The latter helps stimulate an immune reaction and boost antibody production. The company has one against α-synuclein in Phase 2 (UB-312; Apr 2022 conference news).
For tau, 100+ peptides were screened to find those that induced the best immune response. Two candidates emerged: vaxxine A, which mimics part of tau’s N-terminus, and F, from the MTBR. Both produced antibodies that bound full-length tau monomers as well as tau fibrils from Alzheimer’s brain sections.
Differences between the two became apparent in cellular assays. Antibodies generated by vaxxine F better inhibited tau aggregation in the tau biosensor cell line developed by Marc Diamond of University of Texas, Southwestern Medical Center, Dallas, than did those generated by A (Oct 2014 news; Apr 2023 conference news). Compared head-to-head, polyclonal fractions of F antibodies slashed aggregation by 80 percent; A, 20 percent. For comparison, Genentech’s anti-tau monoclonal antibody semorinemab suppressed aggregation by 60 percent in this assay. On the other hand, A outdid F at blocking tau seed uptake by neuroblastoma cells. It diminished uptake of tau fibrils by three-quarters, compared to about 60 percent for F. In this assay, semorinemab was similar to A, at 80 percent.
How do the vaxxines perform in vivo? Boyd reported that vaccination with A slashed tau accumulation in the cortex of P301L mice almost to wild-type levels. Lysates from the treated P301L brain contained half as many tau seeds as untreated.
Rather than pick one of the two to take forward, company scientists decided to combine them. In cell cultures, A and F together better blocked tau uptake than did A alone, and this version, dubbed VXX-301, is now being evaluated in P301L mice. The company also intends to test a multivalent vaxxine against both tau and Aβ.
Likewise, Chad Swanson of Prothena introduced PRX123, a vaccine that produces antibodies against both Aβ and tau. In a talk devoted mostly to Prothena's anti-Aβ antibody PRX012 (Part 12 of this series), Swanson mentioned that Prothena will submit an Investigational New Drug application for PRX123 this year, but offered no details about the molecule. Given how easily vaccines can target multiple epitopes, there will likely be many more such efforts.—Madolyn Bowman Rogers
No Available Comments
With the emergence of COVID-19 three years in the past, the lingering neurological effects after initial illness remain nebulous. At the recent International Conference on Alzheimer’s and Parkinson’s Diseases in Gothenburg, Sweden, scientists presented data from ongoing longitudinal trials studying the neurological effects of long COVID. Others tied the APOE4 genotype to higher odds of brain bleeds post-COVID. And there is data on how a protein from SARS-CoV-2, the virus that causes COVID, can gather within, and maybe inflame, the brain.
AD/PD featured a dedicated session on this topic, plus presentations throughout. The upshot? While certain neurologic symptoms are being reported subjectively, not all are measurable objectively. Affective issues present before infection appear to play a role, as well, making the problem difficult to define.
“The session emphasized the complexity of research into concrete neurological and pathophysiological sequelae of SARS-CoV-2 infection, as distinguishing de novo damage and predisposition is only possible in studies with well-characterized pre-morbid status,” wrote Michael Schöll, University of Gothenburg, who co-chaired the session.
Co-chair Michael Schlossmacher, Ottawa Hospital Research Institute, Canada, thinks that exploring COVID-related effects on the nervous system has implications for other neuropsychological disorders that result from interactions between viruses and genetic susceptibility. “The COVID-19 pandemic has unearthed the tip of an iceberg. What would Constantin von Economo say had he lived 100 years later?” Schlossmacher wrote. In 1917, this Greek-Austrian neurologist described encephalitis lethargica in the wake of that period's influenza pandemic (Kaya et al., 2016).
Best known for its respiratory symptoms, COVID can cause neurological changes, such as transient neurovascular damage and a spike in cerebrospinal fluid markers of neuronal injury, like neurofilament light (NfL) and glial fibrillary acidic protein (GFAP; Jan 2021 news).
At AD/PD, Jennifer Cooper in Cheryl Wellington’s lab at the University of British Columbia, Canada, reported seeing such a spike in NfL and GFAP in half the blood samples taken from 237 adults, ages 49 to 72, within a week after they were admitted to the ICU with COVID. High NfL and GFAP predicted who had neurological complications, such as ischemia or hemorrhage on CT scans, or who was likely to die, with areas under the curve of 0.77 and 0.82, respectively. AUCs measure sensitivity and specificity, with 1 being perfect. Cooper is following up these people at six and 18 months after hospitalization, collecting blood samples, MRI and PET scans, and testing their cognition.
Previously, some labs had reported that plasma NfL, GFAP, Aβ42, and total tau normalize six months after mild to severe COVID. Others still saw abnormal markers at six months in people who had had severe COVID, particularly with neurologic symptoms (Sep 2021 conference news). A recent systematic review of blood biomarker studies found elevated NfL and GFAP three to six-plus months post-infection in people with lingering neurological symptoms (see image below; Lai et al., 2023).
Also called “post-COVID condition” or “post-COVID syndrome,” long COVID broadly refers to continuation of symptoms, or appearance of new symptoms, three months after infection. Complaints include fatigue, shortness of breath, headaches, and difficulty with thinking or memory, aka “brain fog.”

Busy Biomarkers. People with long COVID have more NfL (red); inflammatory markers IL-6, TNF-α, CCL2; and SARS-CoV-2 antibody (Aβ) than people who fully recovered or never got sick (left). Biomarker changes depended on a person's symptoms, but high CRP was common to all (right). [Courtesy of Lai et al., 2023, Front Med (Lausanne).]
Is Long COVID Tied to Dementia?
The rise in neuronal injury markers begs the question of whether COVID increases one's odds of dementia later in life. Contracting herpes or influenza has been shown to increase the risk of developing dementia and other neurodegenerative diseases over 15 years (Feb 2023 news).
Fifteen-year data on COVID is a long way off, but some one-year data is in. Mild or severe bouts of COVID can, in some people, worsen existing neurological problems and speed cognitive decline in the year after infection (Apr 2021 conference news; Mar 2022 news). A study led by Ziyad Al-Aly at the Veterans Affairs St. Louis Health Care System last November reported much the same (Xu et al., 2022). Among 154,000 veterans who had COVID and 11.5 million who did not, the former were 1.4 times likelier than controls to have a neurologic condition—memory problems, a stroke, neuropathy, mental health disorders, migraines, or seizures—in the year following the illness. This was true for all COVID cases, from mild to severe.
Is there a link between “brain fog” during acute illness and lingering neurological problems? On May 5, Neil Wenger and colleagues at the University of California, Los Angeles, reported that among 766 adults, average age 60, who'd had COVID, 36 percent self-reported forgetfulness or trouble concentrating on the Perceived Deficits Questionnaire 30 days after hospitalization or outpatient clinic visit for COVID (Liu et al., 2023). Two months later, half of those people still reported neurological symptoms; one-quarter of people whose acute COVID had been without cognitive symptoms newly reported them. Prior depression, anxiety, or cognitive complaints were associated with these self-reports, leading the authors to propose an affective component to long COVID. This comports with an observation Schlossmacher noted, i.e., that a large fraction of patients with long COVID symptoms linked to nervous system dysfunction also had psychiatric symptoms, such as anxiety, depression, and adjustment difficulties.
To measure cognitive change in long COVID, Ann-Katrin Schild and colleagues at the University of Cologne, Germany, assessed global cognition and five domains in 42 adults, average age 45, with long COVID. All self-reported subjective cognitive impairment. Screening their cognition with the MMSE or MoCA picked up only one or 10, respectively, as impaired. This was true at three and nine months post-COVID. A composite of five cognitive domains—learning and memory, attention, executive function, visuoperception, and language—detected mostly mild impairment in 60 percent of participants at three months and 40 percent at nine. Both times, people had the most trouble with memory and executive function. When asked at AD/PD about the discrepancy between the greater subjective than objective cognitive assessment, Schild had no explanation.
The most comprehensive ongoing study of brain structure and function shown at AD/PD is that of Schöll and colleagues. They are measuring a broad range of dementia-related outcomes in 40 adults hospitalized with COVID and 20 controls who had never had it, all in their late 40s to mid-50s. Of the COVID patients, half were enrolled one month after admission to the hospital, half were enrolled between three and 20 months after hospitalization when they had been discharged but returned to an outpatient clinic for lingering neurological symptoms.
Participants underwent blood draws, lumbar punctures, structural and functional MRI scans, FDG PET scans, and extensive neuropsychological testing. The latter included the MoCA to measure global cognition, the Repeatable Battery for the Assessment of Neuropsychological Status for memory, and Trail Making tests to measure speed/attention and executive function.
At AD/PD, Schöll presented 12-month data on all but the fluid markers. At baseline, MoCA scores were the same between the three groups. After 12 months, the long COVID participants did marginally worse than controls, scoring an average of 25 versus 27, respectively. This indicated slightly impaired global cognition in those with PCC, as a score of 26 and above is normal.
In individual domains, people with long COVID had worse memory, executive function, and speed/attention than controls at baseline. People who had recently gotten COVID scored slightly worse only on the latter two. Speed/attention scores improved somewhat by 12 months; the other deficits did not change significantly (see image below).

Slooowly Getting Better? Over a year, speed/attention (right) improved significantly, memory (left) and executive function (middle) trended upward in people who had gotten COVID a month before enrolling in the study (top) or who had long COVID (bottom) before enrollment. The latter started with worse scores than the recently ill. [Courtesy of Michael Schöll, University of Gothenburg.]
MRI scans showed no differences in gray-matter volume in any group. People who more recently had COVID had larger white-matter hyperintensities than the earlier-discharged cases or controls at baseline; the number remained steady over 12 months. Intriguingly, radiologists noted that these hyperintensities were mostly widened perivascular spaces, which these people may have had before they contracted COVID. Schöll speculated that people with such hyperintensities might be predisposed to neurological symptoms.
On FDG PET, those with earlier COVID, but not the more recent cases or controls, at baseline had spots of hypometabolism in their precuneus, and clusters of hypermetabolism in their parietal cortices. In contrast, spots of glucose hyper- and hypometabolism popped up in the acutely ill, but not in the long COVID participants or controls, on their 12-month scans (see images below). Schöll noted that other researchers see similar patterns, but no one yet knows what to make of them. "Is it real? Is this compensating for an inflammatory response?" he wondered.
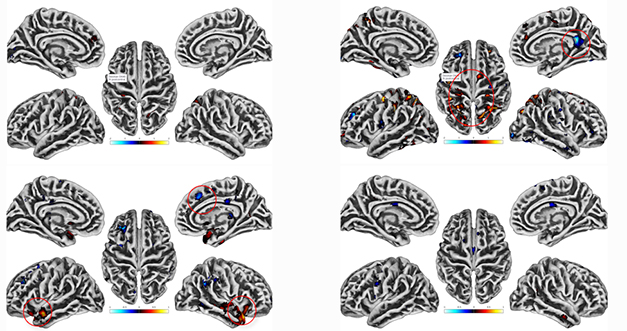
Perplexing PET. Compared to controls at baseline (top), people with long COVID (right) had spots of hypo- and hyperactive glucose metabolism (red circles), while people who'd had COVID recently did not (left). The opposite was seen at a 12-month follow-up (bottom). [Courtesy of Michael Schöll, University of Gothenburg.]
Overall, both Schild and Schöll found slightly worse memory and executive function in some people with long COVID, though the latter thus far is unable to find structural or functional explanations for the differences.
APOE and COVID
Pondering dementia risk invariably evokes the APOE4 allele, being, as it is, the largest genetic hit for sporadic AD. Some studies have shown that APOE4 carriers are likelier to show symptoms, become gravely ill, and die from COVID than APOE3 carriers, but others saw no influence of APOE genotype on COVID severity or outcomes (Jan 2021 news).
At AD/PD, data from Sophie Stukas, also in Wellington's lab at U British Columbia, fell into the latter group. Stukas genotyped blood samples from the same cohort of 237 people hospitalized with COVID that Cooper studies. Of those, 57 carried at least one APOE4; 180 did not. According to Stukas, an allele frequency of one-quarter is on par with E4 in the general Canadian population, meaning that APOE4 carriers were no more likely than noncarriers to get severe COVID.
Stukas found no differences in COVID symptoms, treatments, complications, length of hospital stay, or mortality based on APOE genotype. That said, E4 carriers were twice as likely to show neurological complications, such as ischemia or hemorrhage, on CT scans. Stukas did not say whether they had stroke symptoms. "This population may be more vulnerable to cerebral injury," she concluded.
This could be due to inflammation in the brain, at least in mice, said Ling Li, University of Minnesota, at AD/PD. Compared to human APOE3 knock-in mice infected with SARS-CoV-2, E4 knock-ins overexpressed RNA encoding the cytokines IL-6 and CCL2 and other proteins associated with the innate immune response to viruses. Li detected no SARS-CoV-2 viral RNA in the infected mouse brains, indicating that this inflammation occurred without direct viral brain infection.
Similarly, the SARS-CoV-2 spike protein, sans virus, was enough to trigger inflammation in cultured human brain cells treated with the viral glycoprotein. At AD/PD, Huyen Ngo in Hansang Cho’s lab at Sungkyunkwan University, Suwon, South Korea, reported that the spike protein binds to toll-like receptors 2 and 4 on the outer membranes of cultured human microglia and astrocytes. Binding evoked glial activation and anti-viral interferon signaling within the cells.
Li and Ngo's data mirror findings in people, namely the absence of detectable SARS-CoV-2 virus in postmortem COVID brain tissue despite a neuroinflammatory viral response (Jun 2021 news).
Tales from COVID Tissue In infected mice, the spike protein—again going solo—was spotted crossing the blood-brain barrier (Rhea et al., 2020). Could this be true in people? If so, could this explain some of the neuroinflammatory response to COVID?
Yes, according to researchers led by Ali Ertürk, Helmholtz Center Munich. Ertürk's group recently reported accumulations of the SARS-CoV-2 spike protein in cortical tissue, meninges, and skull bone marrow from 27 adults who had died from COVID (Rong et al., 2023). Mass spectrometry-based proteomics of their skull marrow showed downregulation of complement proteins and upregulation of pro-inflammatory cytokines compared to 10 control tissue samples. SARS-CoV-2 RNA was detected in only half the meningeal and skull samples that had spike protein. This led the scientists to suspect specific uptake, or a longer half-life, of this viral protein than the virus itself in the human brain.
Surprisingly, the scientists also found the spike protein in skull samples from 10 of 34 people who had died from something other than COVID in 2021 or 2022. This means they likely had had COVID in the past, and the spike protein lingered in the brain for the remaining months of their lives. Ertürk thinks that this loiterer might contribute to the neurological symptoms of long COVID.
Along similar lines, Ina Vorberg of the German Center for Neurodegenerative Diseases, Bonn, reported that the SARS-CoV-2 spike protein, as well as proteins from endogenous retroviruses, can help shuttle tau seeds between brain cells, possibly implicating the viral protein in tauopathies (Oct 2021 news; Apr 2023 news).
That said, spike must form trimers on cell membranes to enable tau spreading. “I do not find it likely that spike taken up by brain cells from the circulation would assemble into functional trimers,” Vorberg wrote to Alzforum. “However, a reservoir of spike protein in meninges and skull marrow could affect inflammatory responses in the CNS, which could affect protein aggregation and spreading by other means.”
Did people who perished after COVID have more neuropathology? At AD/PD, Schlossmacher shared early data from ongoing work. In piriform cortex and olfactory bulb tissue from 47 people who had died from COVID complications two to 12 weeks after infection, his group spotted tau tangles in 23 people and α-synuclein aggregates in 12; among 19 non-COVID autopsy controls, 11 had tangles, and none had α-synuclein aggregates. Schlossmacher said it's too soon to know if this was due to infection or age. The prevalence of olfactory bulb tau and α-synuclein aggregates in these Ottawa COVID cases was similar to that of a larger series of normal older adults from the Banner Sun Health Body Donation Program (Tremblay et al., 2022).
All told, the connection between COVID, neuropathology, and dementia risk remains murky despite efforts around the world to characterize the question. Longitudinal studies on neurological sequelae are continuing.—Chelsea Weidman Burke
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.