CONFERENCE COVERAGE SERIES
Alzheimer's Association International Conference (AAIC) - 2022
San Diego, California and Online
31 July – 04 August 2022
CONFERENCE COVERAGE SERIES
San Diego, California and Online
31 July – 04 August 2022
While the Alzheimer’s field awaits pivotal results from three Phase 3 trials of anti-amyloid antibodies, data on this class of therapeutics continue to trickle in. At the Alzheimer’s Association International Conference held July 31-August 4 virtually and in San Diego, California, researchers expanded on previously reported negative topline results from the Alzheimer Prevention Initiative’s trial of crenezumab in Colombia. Crenezumab, made by Roche/Genentech, differs from other anti-amyloid antibodies in late-stage trials in that it targets Aβ monomers and oligomers rather than fibrillar forms. As had been the case with memory and cognitive co-primary endpoints, all secondary clinical and biomarker outcomes favored the drug, but the difference did not reach statistical significance. Dose-response curves, subgroup analyses, and plasma biomarkers are still being analyzed, with the goal to inform future studies in the Colombian autosomal-dominant AD kindred.
Meanwhile, the Phase 3 GRADUATE trials of Roche’s fibrillar-targeting antibody, gantenerumab, are expected to read out this fall. At AAIC, scientists presented a smidgen of new data from the open-label extension studies of the older, and negative, SCarlet RoAD and Marguerite RoAD trials, showing plasma biomarker effects, plus a potential clinical benefit when compared to the expected rate of cognitive decline in matched controls. Janice Smith leads the gantenerumab program. She noted that the drug has been in development for two decades, with more than 2,600 patient-years of testing. Both the gantenerumab and crenezumab programs represent an enormous investment of time and resources.

Consistent Trend? In the API Colombian trial, clinical and biomarker endpoints all favored crenezumab, though none reached statistical significance. [Courtesy of Roche.]
Crenezumab Data: Stepping-Stone to New Trials?
The API Colombian trial of crenezumab pioneered prevention studies for disease-modifying AD drugs, demonstrating it was possible to run such trials. It enrolled 169 people who carried the E280A Paisa mutation in presenilin 1; 85 of them received subcutaneous crenezumab, 84 placebo. The trial also included 83 noncarriers, all of whom received placebo. At baseline, almost half of carriers were amyloid-negative (Aug 2019 conference news). The study ran for eight years, with an astonishing 94 percent retention rate. At AAIC, Pierre Tariot of the Banner Alzheimer Institute in Phoenix quipped that it was a “huge small trial.”
Roche previously reported the trial was negative on both primaries, the Free and Cued Selective Reminding Test (FCSRT) and the API cognitive composite (Jun 2022 news). In San Diego, Tariot put numbers to this. On average, people on crenezumab declined 20 percent more slowly on the FCSRT than did carriers on placebo; on the composite, they declined 23 percent more slowly. On secondary clinical endpoints, the crenezumab group declined 9 percent more slowly on the CDR-SB, 8 percent on the CDR, and 44 percent more slowly on RBANS, a neuropsychological composite. That said, variability within the treatment groups was large, and none of the differences were statistically significant.
Progression to mild cognitive impairment or dementia also appeared to slow down a tad on drug, Tariot reported. Progression curves were identical for the first four years of the trial, and then separated, with the treatment group progressing 21 percent more slowly after eight years. As with the other endpoints, this difference was not statistically significant, and Tariot cautioned that the number of participants at later timepoints was quite small.
Biomarkers followed a similar pattern. FDG PET, which measures the brain’s glucose use, weakened 18 percent less on drug than placebo. MRI scans showed 8 percent less whole brain shrinkage on drug than placebo; regional MRI findings are still being analyzed. Tau PET was added to this trial late and done for only 83 mutation carriers. The PET signal rose 51 percent less in those on drug than placebo. Cerebrospinal fluid was drawn from about half of participants. CSF p-tau181, total tau, and NfL worsened 37, 29, and 18 percent less on drug than on placebo, respectively. For all these imaging and fluid biomarkers, the variability within each treatment group was large. None of the findings were statistically significant.
One exception was CSF Aβ42 and Aβ40 concentrations. Aβ42 stabilized on drug, but declined in controls; Aβ40 rose on drug and dropped in controls. Both changes were statistically significant. Eric Reiman at Banner noted that the uptick in these peptides may be due to slower clearance of antibody-bound Aβ from brain and CSF, indicating that crenezumab interacts with its target. The drop in Aβ40 in placebo controls was unexpected; Reiman speculated this could reflect its sequestration into vascular plaques.
Other CSF markers, including α-synuclein, neurogranin, and numerous inflammatory proteins, remain to be analyzed. Also ongoing is analysis of oligomeric Aβ, which will reveal how effective crenezumab was at engaging its target. The researchers did not present any data on how plasma and CSF concentrations of crenezumab differed, which would indicate how much drug reached the central nervous system.
API scientists showed only one subgroup analysis. They compared decline on the two primary endpoints in mutation carriers who were amyloid-positive versus amyloid-negative at baseline. This analysis hinted at a bigger benefit in the amyloid-negative group, but again, this was statistically insignificant. Yet to come are dose-exposure data and “spaghetti plots” showing individual trajectories. The large variation within small treatment groups could make personalized trajectories on all outcomes and biomarkers measured in a given person over time, from baseline onward, particularly revealing.
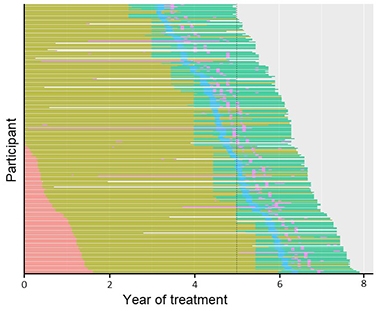
Dose Escalation. Dosing in the API Colombian study increased over time from 300 mg subcutaneous (pink) to 720 SC (olive) to 60 mg/kg intravenous (aqua); all participants missed doses due to the COVID shutdown (blue), with some missing other doses (purple). [Courtesy of Roche.]
Reiman noted several limitations. Partway through the eight-year trial, the field realized that antibodies needed to be given at much greater doses than originally thought. The researchers boosted the dosage twice and switched from subcutaneous injections to intravenous. At study start people received 300 mg crenezumab per month subcutaneously, and by the end, 60 mg/kg per month intravenously, which Roche researchers noted was an increase of more than sevenfold. Alas, participants were only on this highest dose for about two years. This means the drug may not have had enough time at its effective dose to generate a benefit. To complicate matters further, right after switching to the highest dose, COVID pandemic shutdowns caused participants to miss on average four doses, noted Rachelle Doody at Roche. Even so, over the whole trial, 88 percent of scheduled doses were delivered.
The trial had less statistical power than anticipated. Partly, this was because the cognitive composite was developed to detect decline in the larger observational cohort of the Colombian kindred. However, mutation carriers who enrolled in the trial were on average four years younger than the observational cohort, and twice as likely to be amyloid-negative. This meant the composite lacked sensitivity to detect early cognitive change in the participants.
Sensitivity analyses found that the FCSRT was a better measure. Tariot noted that the FCSRT had 63 percent power to detect a 30 percent slowing of decline in this trial, while the composite had only 6 percent. Future trials in this kindred should therefore use the FCSRT, and might need to enroll twice as many carriers, Tariot believes. While researchers decide on the appropriate drug and design for the next trial in this population, all mutation carriers in the current trial will receive crenezumab in an open-label extension, while noncarriers continue to receive placebo.
Gantenerumab Open-Label Data Hints at Cognitive Benefit
Like crenezumab, gantenerumab’s dosage was also boosted during the course of its trials. After two negative Phase 3s, SCarlet RoAD and Marguerite RoAD, Roche upped the amount they gave in the open-label extensions (OLE) fivefold, to 1,200 mg given subcutaneously. After three years at this higher dose, 80 percent of participants had fallen below the threshold for amyloid positivity (Dec 2017 conference news; Dec 2019 conference news).
At AAIC, Tobias Bittner at Roche reported plasma biomarker findings from these OLEs. Over three years, plasma Aβ40 and Aβ42 concentrations each nearly doubled, while the Aβ42/40 ratio nudged up by about 10 percent. Meanwhile, plasma p-tau217 and p-tau181 edged downward by about 10 percent over three years. The findings complement results from other studies, in which gantenerumab reduced total tau and p-tau181 in CSF and slowed the rise of the neurodegeneration marker NfL (Jun 2021 news).
Because OLEs have no placebo control, clinical effects of the treatment cannot be directly evaluated. To estimate them, statistician Paul Delmar at Roche used ADNI observational data as an external control. In his analysis, he included 147 OLE participants with mild cognitive impairment or mild AD dementia, defined as an MMSE of 18 or higher, CDR of 1 or lower, CDR-SB of 5.5 or lower, and ADAS-Cog13 of 38 or lower. He screened 1,530 ADNI participants to find 430 that matched the OLE cohort on multiple criteria, such as sex, age, APOE genotype, education, and cognitive scores. All participants were amyloid-positive and 50 or older. The ADNI participants who were most similar to RoAD participants were given more weight in the statistical analysis.
On the CDR-SB, the cohorts started to diverge at two years, with the RoAD OLE cohort declining 24 percent less than ADNI controls; by three years, this difference grew to 39 percent. Results were similar for the ADAS-Cog13 and the MMSE, with the OLE cohort declining 36 and 19 percent less than ADNI participants at three years, respectively. The findings were statistically significant. “These results provide context and support the potential clinical relevance of the biomarker findings,” Delmar said.
Delmar also reported that the cognitive benefit correlated with amyloid removal. In people with the most plaque clearance, CDR-SB changed little over three years, whereas in those with the least clearance, CDR-SB rose about 4 points. The correlation was weak, at r=0.19. However, in the 34 OLE participants with four-year data, correlation strengthened somewhat to r=0.44. “This speaks to the importance of considering the long-term effects of the drug,” Delmar said. Smith agreed, noting that the clinical benefit of gantenerumab appears to increase over time.
Other talks at AAIC attempted to model the long-term effects of anti-amyloid antibody treatment. Overall, they likewise concluded that progression may slow as treatment continues (see related conference story).—Madolyn Bowman Rogers
Alzheimer’s trials targeting β-secretase screeched to a halt when it turned out that the drugs slightly worsened cognition. Alarmed, pharmaceutical companies ditched their BACE programs, or put them on ice. Still, since the last such trial ended in 2019, a small cadre of scientists has been urging the dementia field not to forget these inhibitors. At the Alzheimer’s Association International Conference held July 31 to August 4 in San Diego, and at a BACE symposium on July 26 at the University of Connecticut School of Medicine, Farmington, these researchers honed their argument for reinvigorating these programs, claiming that a dose could be found that both is safe and would sufficiently slow or stop the trickle of Aβ that causes AD.
Some scientists outlined trial designs to test this hypothesis. Others suggested biomarker or cognitive tests that might track cognitive side effects. All agreed that these inhibitors are worth another look and that they could provide a simpler and cheaper alternative to long-term immunotherapy.
Though academics had voiced concerns about moving forward too quickly with BACE inhibition, the first clinical trials started more than a decade ago. Despite early setbacks with compounds that damaged the eye or the liver, a slew of candidates with good safety profiles soon emerged that looked promising in early trials. Then, one by one, BACE inhibitor programs were terminated in Phase 3. Merck’s verubecestat, Janssen/Shionogi Pharma’s atabecestat, Novartis/Amgen’s umibecestat, Biogen/Eisai’s elenbecestat, and Lilly's lanabecestat all fell by the wayside (Feb 2018 news; May 2018 news; Jul 2019 news; Sep 2019 news). The most obvious problem was that these drugs worsened performance on cognitive tests, though some people also lost weight or had brain atrophy as well.
What happened? Scientists at these two conferences believe that the cognitive loss directly resulted from BACE inhibition, not from some off-target effect. This is because in addition to APP, BACE cleaves dozens of other substrates, including seizure protein 6, aka Sez6-like (Sez6L), neuregulin, and NCAML1, any of which might be indispensable for synaptic activity. That these other substrates might be affected had always been a red flag for these programs, but because BACE1 knockout mice seemed mostly normal, drug sponsors had calculated that if they didn’t completely block the enzyme, pharmacological inhibition would be a net positive.
Some BACE afficionados believe it still can be. “I think that industry was too quick to abandon these inhibitors,” said Stefan Lichtenthaler, DZNE, Munich. “We know that clinical development of BACE inhibitors is on hold, but this is not automatically the end of the development. Let’s keep in mind that setbacks in clinical trials are completely normal,” he said. This is also the view of Riqiang Yan, University of Connecticut School of Medicine, who organized the mini symposium there, and Robert Vassar, Northwestern University, Chicago. At AAIC, Vassar co-chaired a focused topic session on BACE inhibition for the prevention of AD with Maria Carrillo of the Alzheimer’s Association. Vassar noted that statins were almost abandoned for cardiovascular disease because of side effects, yet went on to become widely used. “That’s the paradigm to keep in mind,” he said.
So, how to continue from here? Vassar recommended beginning with small, three-month, dose-finding trials in healthy controls, aiming to block 12, 25, or at most 50 percent of BACE activity. Most of the prior clinical trials inhibited the protease by around half, and in some cases by more than 70 percent. Scientists think this was too high and likely spelled doom for these trials.
Once a suitable dose is found, Vassar thinks the next step would be a Phase 2 trial in older, cognitively unimpaired people who are at risk for AD, testing about 100-200 people per dose. These volunteers might be homozygous for ApoE4, or, in a primary prevention trial, carry a familial AD mutation. If such trials can pinpoint a dose that show signs of efficacy without worsening cognition or causing other serious side effects, then a large Phase 3 secondary prevention trial akin to the AHEAD trials would be on order. This might take four to five years, or up to 10 years in a primary prevention trial in FAD carrier. “Despite the challenges, BACE inhibition offers exquisite sensitivity and specificity for Aβ lowering, and represents a powerful, practical, simple approach to AD prevention,” said Vassar. Others envision this playing out similarly. “Whatever BACE inhibitor we use, we have to determine if it is going to have that negative [cognitive] signal,” noted Bruce Albala, who is now at University of California, Irvine, at the AAIC. Albala had worked at Eisai during the elenbecestat trials.”
Reisa Sperling, Brigham and Women’s Hospital, Boston, agreed with this reasoning. She noted that in BACE trials, the greatest cognitive decline seemed to occur in people who were least impaired to begin with, suggesting this might be the best population in which to rule out detrimental effects on cognition. Though cognitive decline in BACE trials was apparent by three months, it might start earlier. “Three months was the earliest we looked, but we might actually see it at three to four weeks,” she suggested.
Alas, the same scientists agree that more work needs to be done on understanding BACE function before the pharmaceutical industry is likely to get on board with this type of plan. “What is the mechanism of the cognitive loss? That’s the key question for moving into low-dose clinical trials,” said Vassar.
There is some progress on that front. Working with Matthew Kennedy from Merck, Lichtenthaler has been analyzing samples from verubecestat trials to identify substrates that might have been improperly processed under BACE inhibition. Previously, he had reported that soluble ectodomains of Sez6 and gp130 plummeted in CSF shortly after people began taking the drug (Apr 2021 conference news). Sez6 is known for supporting synaptic spines, and expressed by neurons, gp130 forms a complex with interleukin 6 receptors that’s essential for IL-6 signaling. This cytokine can suppress appetite, and Lichtenthaler suggested that this might help explain the weight loss seen in clinical trials and in preclinical mouse studies of BACE inhibitors. He believes that the soluble ectodomains of Sez6 and gp130 in the CSF could serve as pharmacodynamic activity markers to track side effects in clinical trials.

Tracking BACE2. Levels of the soluble ectodomain of VEGFR3 fell in plasma of mice treated with verubecestat (left). Plasma sVEGFR3 also fell in four people who were treated with atabecestat (right). [Courtesy of Stefan Lichtenthaler.]
What about BACE2? Most inhibitors tested in trials cannot distinguish between the two BACE isoforms. Although BACE2 expression in the central nervous system is low, Lichtenthaler suspects that its inhibition might also affect cognition. Tracking BACE2 activity has been challenging because there are no easy redouts bar fur color in mice, which grays when BACE2 is blocked. This warning sign is not only slow and unquantitative, it also breaks a human trial's blind.
At the UConn symposium, which was attended by a few dozen people from a handful of leading BACE labs, Lichtenthaler reported a new substrate that could be a much better marker, even in furless humans. It turns out levels of the soluble ectodomain of vascular endothelial growth factor receptor plummet in the plasma of BACE2 knockouts compared to normal mouse plasma. Likewise, BACE inhibitors reduced VEGFR3 ectodomain shedding in cell culture and in vivo, verubecestat dose-dependently reduced sVEGFR3 in mouse plasma. In fact, its levels dropped by about 25 percent after only one day on a high dose of the drug; a week later, sVEGFR3 was down by 75 percent. The ectodomain dropped by about 60 percent in mice fed a low dose, even though their coats kept their hue. The data suggest that this ectodomain could be a sensitive and fast-responding marker of BACE2 inhibition.
What about in people? Preliminary data suggests there here, too, sVEGFR3 might be useful. Lichtenthaler's lab found that, in the plasma of four people who had been on atabecestat for a month, the protein level had dropped, in one person by up to 50 percent.
Do any of these substrate changes track with cognitive changes? That’s a question several labs are going after. In ongoing work, Lichtenthaler and Kennedy are correlating potential markers with cognitive outcomes from verubecestat trials. In Vassar’s lab, Elyse Watkins is hunting substrates responsible for cognitive deficits by genetically or pharmacologically knocking them down one by one and checking if that mimics the effect of BACE inhibition. “We hope to be able to correlate behavior with specific substrates to see if we can find a sweet spot that will significantly lower Aβ without affecting cognitive decline,” said Vassar.
Are these substrate cleavages responsible for the biological effect, or do they merely reflect a signaling change on the surface of the cells bearing the receptors? To address this question, Watkins plans to treat PDAPP mice with a BACE inhibitor, then inject adeno-associated virus expressing either the soluble shed fragments of various substrates or their plasma membrane C-terminal domains to see if any will rescue phenotypes that have been linked to BACE inhibition, such as loss of dendritic spines or mossy fiber disorganization (Dec 2013 conference news; Sep 2018 news). Watkins told Alzforum that ideally she would like to have a cognitive test for mice that better reflects the subtle changes seen in people on BACE inhibitors. Many scientists believe that the standard memory tests used for mice poorly reflect cognition in people.
For his part, Kennedy wants to skirt this problem by studying nonhuman primates. At Merck, Kennedy uses a colony of rhesus monkeys to monitor BACE substrate changes in the CSF, as well as cognition in a visuospatial paired-associate learning task. He wants to see if he can reproduce the cognitive worsening seen in people on BACE inhibitors, relate that to CSF markers of BACE substrate cleavage, and look for a dosing window where Aβ production can be reduced without worsening cognition. This work was interrupted by the COVID pandemic; Kennedy said it has now resumed, and the monkeys are relearning the cognitive task.
Even if a dose window can be found for these drugs, questions remain. Whom to treat, and when? At AAIC, Mathias Jucker, University of Tübingen, Germany, argued that BACE inhibition would have to start very early in the disease process, before plaques had become established, if it is to head off neurodegeneration. Scientists in his lab fed a BACE inhibitor to APPPS1 mice at various ages, then measured the effect on various markers of AD pathology, including Aβ, sTREM2, soluble tau species, and neurofilament light (NfL), a marker of neurodegeneration. They found that both chronic dosing and three-month stints on the inhibitor reduced Aβ, sTREM2, and tau, regardless of when the treatment was started. NfL, however, was another matter. If treatment began when plaques were already established, then NfL just kept rising, albeit at a slightly slower pace (see image below).
“This is reminiscent of what we have seen in clinical trials, that NfL just doesn’t really follow Aβ once the brain was full of Aβ,” said Jucker. He believes that once amyloid seeding activity reaches a plateau, which happens at about 50 percent maximal plaque load, then the brain reaches a tipping point and neurodegeneration proceeds independently of Aβ, in keeping with the proposed cellular phase of AD (De Strooper and Karran 2016). Eric McDade, Washington University, St. Louis, saw this as a problem for the field. “It is very concerning that in treating with just a BACE inhibitor after NfL has started to change, it is really difficult to have a dramatic effect in a positive direction,” McDade said.
In his talk, McDade made a case for using BACE inhibitors as an adjunct to immunotherapy. “We have a number of immunotherapies that will potentially be approved that significantly reduce amyloid pathology, but we have to know how we will use these therapies long-term,” he said. If those therapies are stopped, then plaque load would rebound by about 20 percent over 18 months. “Do we continue to dose at a low level, or do we combine with a therapy that will prevent amyloid production?” McDade asked.
Here is where the BACE inhibitors come in. By preventing Aβ production, they can keep amyloid from creeping back up. McDade sees primary prevention trials as an essential first step to establishing whether BACE inhibitors can be used safely. He thinks frequent cognitive and psychiatric testing, combined with monitoring by MRI, plasma markers of AD pathology, and other BACE substrate cleavages, now offer the necessary tools to make this happen.
Sperling agreed. She thinks some of these tests can be run now on existing samples. “We have really good tools now, but we should look hard at the plasma data we have from prevention trials to try to get hints at the pharmacodynamic levels of BACE inhibitor exposure and when we can we start to see biomarker changes,” she said. “We should even look to see if there is any evidence of a beneficial change on p-tau measures as well,” Sperling said.

BACE and DAMs. In microglia, BACE1 attenuates signaling through toll-like receptors. Blocking the protease activates PI3K and p38 MAPK pathways, facilitating the transition to the DAM state and enhancing phagocytosis of amyloid. [Courtesy of Singh et al., 2022, Science Advances.]
One surprise benefit of BACE inhibition came from Yan’s lab. At the UConn meeting, Neeraj Singh reported that microglia in 5xFAD mice increase expression of BACE1 as plaques accumulate, and that this restricts plaque clearance. When Singh conditionally knocked out the gene in microglia, the cells increased phagocytosis and degradation of amyloid. As a result, synaptic function improved, as did learning and memory. It turns that that BACE1 activity helps microglia transition from a homeostatic state to a more phagocytic disease-associated microglia. Similarly, Singh found that the transition from homeostatic to DAM cells correlated with reduced microglial expression of BACE1. Digging into the mechanism, Singh found evidence that BACE inhibition reduced signaling through interleukin 1R2 and toll-like receptors 2 and 4. Reduced cleavage of these receptors associated with activation of p38 MAPK and phosphatidyl inositol 3-kinase pathways and the expression of DAM genes. The findings were recently published in two papers in Science Advances (Singh et al., 2022; Singh et al., 2022).
Yan suggested that a microglial-specific BACE1 inhibitor would be better than one that systemically blocks the enzyme—a tough ask for medicinal chemists.—Tom Fagan
No Available Comments
A resolution revolution is happening to spatial transcriptomics. At this year’s Alzheimer’s Association International Conference, held July 31 to August 4 in San Diego, California, and online, Shane Liddelow of New York University presented stunning images of expression within mouse brain slices. Combining single-cell RNA sequencing and in situ hybridization to map gene expression in individual cells, his high-resolution method detected a small but specialized population of astrocytes that flock to the blood-brain barrier after inflammatory insult. He also zoomed in on expression changes around amyloid plaques, finding 400 plaque-induced genes, up from the almost 60 previously reported. The findings help researchers understand how dysfunction spreads from pathology to nearby cells to the entire brain.
“Location dictates biology—how cells talk to each other, how they coordinate functions—especially in an organ as sophisticated as the brain,” Andrew Yang, University of California, San Francisco, told Alzforum. “Both single-cell and spatial transcriptomics provide the solid biological foundation that the dementia field needs by characterizing the diversity of cell types, states, and locations in different diseases.”
![]()
Bayesian Breakdown. The BayesSpace algorithm improves spatial transcriptomics resolution by dividing pixels (hexagons) into sections (bottom), then comparing the transcriptomes of each section (red) to its neighbors (blue). [Courtesy of Zhao et al., Nature Biotechnology, 2021.]
The need for high-res spatial transcriptomics was born from an experiment exploring astrocyte subtypes in mice. Philip Hasel of Liddelow’s lab had curated a massive dataset of about 80,000 astrocyte transcriptomes from wild-type mice that had been injected with lipopolysaccharide to stimulate acute inflammation (Hasel et al., 2021). The astrocytes fell into 10 subtypes. One, accounting for just 2 to 3 percent of all reactive astrocytes, included cells that were unusually interferon-responsive, suggesting provocation by peripheral inflammation. Curious about where these interferon-responsive reactive astrocytes (IRRAs) resided in the mouse brain, the scientists first used in situ hybridization to locate expression of Igtp, an interferon response gene expressed by this astrocyte subtype. They found it in outermost cortical layer next to Cldn5-positive blood vessels.
In situ hybridization only captures a small window of the brain and limits detection to a single marker. Liddelow wondered if spatial transcriptomics would offer a better brain-wide view of the IRRAs. First, the scientists spatially mapped approximately 200 genes differentially expressed in the IRRAs. Hotspots of these differentially expressed genes (DEGs) appeared around the outer surface of the brain and in what seemed to be the brain ventricles, but the resolution was too poor to precisely discern these locations.
Next, the researchers reanalyzed the image with BayesSpace, a recently created algorithm that uses Bayesian statistics to enhance image resolution (Zhao et al., 2021). Rather than comparing gene expression between the pixels of a spatial-transcriptomics picture, the algorithm divides each pixel into portions and compares expression between each portion, effectively reducing pixel size (see image above). This improved the resolution dramatically, clearly showing that IRRAs reside around ventricles and in the outer cortical layer (see image below).
![]()
High Definition. To get a higher-resolution spatial transcriptomics image of IRRAs (yellow), researchers progressed from searching for just Igtp expression (top left) to all 200 IRRA DEGs (top right). Reanalyzing the images with BayesSpace vastly improved resolution of Igtp (bottom left) and DEG (bottom right) expression. [Courtesy of Shane Liddelow, New York University.]
Liddelow thinks the location of IRRAs at the blood vessels and ventricles suggests that they are responding to peripheral immune cells that secrete interferon. IRRAs also express genes involved in antigen processing and presentation, which may beckon other immune cells to the blood-brain barrier. Upregulation of similar peripheral sirens have been reported in other types of inflammatory astrocytes (Jun 2022 news). “We think the strategic location of IRRAs also hint that they are integral to the infiltration of peripheral immune cells,” Liddelow said. In some neurodegenerative diseases, T cells can pass through blood-vessel walls and hang out in the glia limitans, the thin layer of astrocytic feet that coat the cortex, or enter the cerebrospinal fluid by infiltrating the choroid plexus (Oct 2021 news; Jun 2021 news).
Yang agreed that these astrocytes may be sentinels of the brain, and was struck by their exquisite localization. Intriguingly, Hasel and colleagues also detected IRRAs in snRNA-Seq data from mouse models of amyloidosis and multiple sclerosis, suggesting that peripheral inflammation also drives astrocyte reactivity in those diseases.
Liddelow is tracking down similar cells in people. Jessica Sadick and Michael O’Dea in his lab ran RNA-Seq of single cells from 10 postmortem AD and six control prefrontal cortex samples, isolating 41,000 astrocytes and clustering them by their transcriptomes into eight subpopulations (Sadick et al., 2022). In a control who had vascular dementia, as determined by neuropathology and clinical evaluations, one cluster had a similar gene-expression profile to the IRRAs found in mice. The scientists are currently collecting more data and working on high-res human brain tissue spatial transcriptomics.
Plaque Transcriptomics
Could high-res images clarify the location of other gene-expression changes within the brain, such as those surrounding amyloid plaques? Previously, researchers led by Bart De Strooper, UK Dementia Research Institute, London, identified 57 plaque-induced genes by spatial transcriptomics. These PIGs were up- or downregulated in microglia and astrocytes surrounding amyloid plaques in old APP knock-in mice and in people who had had AD (Jul 2020 news; Aug 2019 news).
Hasel and Emilie Castranio of Mount Sinai, New York, took a similar approach, collecting single-cell and spatial transcriptomics data on brain tissue of 6-month-old PS/APP transgenic mice (Castranio et al., 2022). But they analyzed the data differently, using the spatial prowess bestowed by BayesSpace analysis. De Strooper clustered genes based on weighted gene co-expression network analysis (WGCNA), which groups genes that are up- or downregulated in concert. Hasel and Castranio grouped gene expression based on proximity. They compared gene expression between cells from wild-type and PS/APP mice to identify DEGs, then clustered pixels of the spatial transcriptomics image based on the DEGs expressed in each pixel.
Clustering of the DEGs revealed 27 subtypes of pixels. Each occupied a unique spatial niche in the brain. One subtype seamlessly overlapped with plaques labeled by the antibody 6E10 (see image below). Cells within these pixels shared 403 DEGs, which the authors named newPIGs.
![]()
High-Res PIGs. Combining snRNA-Seq and spatial transcriptomics of brain tissue from APP mice pinpoints DEGs surrounding plaques (red/yellow pixels, left). BayesSpace (middle) improves the resolution, showing PIGs (yellow) coinciding with plaques (red, right). [Courtesy of Shane Liddelow, New York University.]
What were these newPIGs, and which cells expressed them? In addition to the 57 original PIGs, many of the remainder are expressed in IRRAs and disease-associated microglia (DAMs). Wei-Ting Chen of KU Leuven, first author of the original PIGs paper, was pleased to see that the same genes she had identified in her study turned up here, too. Liddelow noted that the strong co-localization with PIG genes hint that DAM microglia and IRRA astrocytes interact around plaques (see image below).
![]()
Do PIGs Give a DAM? High-res spatial transcriptomics show co-localized expression of PIG (left), DAM (middle), and IRRA (right) genes around plaques. [Courtesy of Castranio et al., 2022, bioRxiv.]
One newPIG stood out to the scientists because of its stark specificity for plaques. CST7, which encodes the protease inhibitor cystatin F, was expressed in every pixel surrounding plaques and none away from plaques. This gene was also the most upregulated of the previously identified PIGs, Chen noted. Liddelow thinks it may be a novel plaque marker.
All told, high spatial resolution clarifies relationships between pathology and gene expression, affording a clearer understanding of disease biology. Another beauty of BayesSpace is that any researcher can use it right now—they just need to plug existing spatial transcriptomics data into the algorithm to get high-resolution images. “The most amazing thing is that we didn’t do anything different in how we generated the data; we just changed how we analyzed it,” Liddelow said. He encourages other researchers to use his lab’s spatial transcriptomics data, including the new set from the PS/APP mice.—Chelsea Weidman Burke
No Available Comments
Part 1 of 3
Second to an effective treatment for Alzheimer’s disease, a blood test for amyloid plaques and tau tangles lurking in the brain has been one of the field’s most urgent needs. And in recent years, it has started to be met. Whether by plasma Aβ42/Aβ40 or, more surprisingly, by a growing number of phospho-tau species, the presence of amyloid plaques in the brain can now be detected in the blood. That said, these discoveries come from research cohorts and, recently, clinical trials, with homogenous, largely white populations selected by way of various inclusion criteria. How will these biomarkers perform out in the real world, where people have diverse genetic, social, and cultural backgrounds, and live with different comorbidities? As the number of biomarkers, and assays to measure them, both continue to grow, implementation questions start to press in. How to choose among these tests? What type of patient should get them? Should they be restricted to memory clinics? Should any primary care doctor be able to order them? And how best to interpret results, and tell the patient?
The Alzheimer’s Association International Conference, held July 31-August 4 in San Diego and online, showcased these and other looming questions that confront the field at large. Blood-based biomarkers were a hot topic at the meeting. More than 100 presentations featured findings linking various markers and assays to amyloid plaques, tau tangles, and cognitive decline, while also grappling with practical matters of how to get them into clinical practice. Below, read on about how scientists at AAIC offered guidelines for recommended use of these tests, and charted a course from exploration to implementation. In Part 2, learn how head-to-head comparisons of the biomarkers in community-based cohorts reveal an abundance of robust assays. Part 3 of this story summarizes the logistical, technical, and ethical dilemmas of moving the tests into doctors' offices, including early probes into how common comorbidities might skew biomarker test results.
Recommendations for Appropriate Use
Oskar Hansson of Lund University, Sweden, set the stage by highlighting newly published recommendations for appropriate use of blood-based biomarkers (Hansson et al., 2022). The recommendations are a first stab at organizing a rapidly advancing field. Hansson said it may take several more years before the field can settle on more stringent criteria like those published for amyloid-PET and CSF biomarkers. Until then, Hansson estimates that these recommendations may be tweaked every nine to 12 months.
At this point in time, how do leading biomarker scientists suggest the tests be used? In the context of clinical trials, blood biomarkers could serve screening purposes, either to select participants for AD trials or to exclude participants from trials of other neurodegenerative diseases, such as frontotemporal dementia. For now, amyloid-PET or CSF should be used to confirm a person's amyloid status, though it will likely be possible soon to use plasma biomarkers as stand-alone proxies of brain amyloid. The authors also recommend using blood biomarkers to track how people responded to treatment in trials, and to inform decisions about moving from early to late-stage trials. That said, the authors believe blood tests should not be used as primary outcomes for pivotal trials.
What about in clinical practice? For now, the guidelines would limit blood biomarker use to people with cognitive symptoms who are seen in specialist memory clinics and, ideally, receive follow-up with amyloid-PET or CSF to confirm they really do have amyloid plaques in the brain.
In his memory clinic at Lund, Hansson envisions using the blood tests in the near future to reduce the number of lumbar punctures performed. Currently, about 80 percent of people who visit the Lund memory clinic receive a lumber puncture for CSF analysis, he said. This is the case in other large sites in Sweden, as well, such as at University of Gothenburg's Sahlgrenska Hospital. Using blood, CSF, and amyloid-PET data previously collected from patients visiting the clinic, Hansson and colleagues first sorted people into high, intermediate, or low probability of amyloid positivity based on an algorithm that combined plasma p-tau217 and ApoE4 status. By limiting CSF analysis to patients with an intermediate likelihood of amyloid positivity based on the blood-based algorithm, Hansson projected that clinicians could avoid two-thirds of the lumbar punctures they now perform while maintaining an accuracy of 91 percent in detecting amyloid. Further narrowing that window of uncertainty could spare even more patients the hassle of a lumbar puncture, with a minimal dip in amyloid detection accuracy, Hansson reported.

So Long, LP? Plasma p-tau217 plus ApoE4 status predicts how likely a person with MCI is to have brain amyloid. When lumbar puncture for CSF Aβ42/40 measurements are performed on people with intermediate probability (yellow), two-thirds of these pesky pokes could be avoided while still catching amyloid positivity with 91 percent accuracy. [Courtesy of Oskar Hansson, Lund University.]
“Of course, the real game-changer will be using blood biomarkers in primary care,” Hansson told the audience. The current restriction to specialized memory clinics stems largely from the need for further studies in more diverse settings.
Hansson called these first recommendations cautious by design, because much work remains to be done before blood biomarkers can be made widely available. Besides evaluation in diverse populations, blood tests need side-by-side comparisons of different assays and scrutiny of potential confounders that might influence results. They need prospective evaluation in trials, in specialist clinics, and in primary care, and the healthcare logistics of deploying biomarkers equitably still need to be solved.

Road to Implementation. The path of blood-based biomarkers to a medical clinic near you requires many steps, including prospective studies in different populations. [Courtesy of Teunissen et al., Lancet Neurology, 2022.]
Charlotte Teunissen of Amsterdam University Medical Center co-authored the appropriate-use recommendations. She stressed that much work remains before patients will be able to access them in the course of routine medical care. Teunissen outlined a recently published roadmap of five phases of biomarker development. Phase 1 involves exploratory, preclinical studies of biomarkers, while phase 2 includes the development and validation of clinical assays. In phase 3, researchers conduct retrospective and longitudinal studies. In phase 4, “real-world performance” of biomarkers is put to the test in prospective studies, and phase 5 involves ironing out all the practical and ethical details of their use in clinics and primary care (Teunissen et al., 2022). For the most part, phases 1 and 2 have been completed for Aβ, p-tau, and NfL. Work on phases 1 and 2 for GFAP, and on phase 3 for all blood biomarkers, is ongoing. Phases 4 and 5 still need to be addressed for all markers.
Teunissen and colleagues expect use of blood-based biomarkers in specialist memory clinics to be in full swing within the next three to five years, while implementing them as diagnostic aids within primary care could be expected within the next five to 10 years.
“It’s an impetuous field,” Teunissen said. “But we still have a lot of work to do.”
Many of the biomarker findings presented at AAIC focused on Phase 3 of Teunissen’s roadmap, as researchers pulled banked blood samples from different community cohorts to compare the top biomarker contenders directly to each other (see Part 2 of this series). Others looked forward to the two final phases, and delved into the technical, practical, and ethical issues that are sure to arise as the biomarkers become more widely available (Part 3 of the series).—Jessica Shugart
No Available Comments
No Available Further Reading
Part 2 of 3
Only five years ago, the advent of a blood test that would root out amyloid plaques lurking in a person's brain was just emerging on the horizon (Jul 2017 news). Now, multiple such tests—particularly the Aβ42/40 ratio and several phospho-tau species—have proven to be quite exquisite detectors of amyloid plaques, at least among well-characterized research cohorts. At the Alzheimer’s Association International Conference, held July 31-August 4 in San Diego, scientists presented new findings in more heterogenous community cohorts, and compared multiple assays head-to-head. Among p-tau assays, mass spectrometry-based tests came out on top, though a handful of immunoassays were not far behind. Researchers agreed that different combinations of biomarkers may suit different clinical stages of disease, with fewer needed to detect amyloid as symptoms worsen.
Charlotte Teunissen of Amsterdam University Medical Center laid out a five-phase roadmap of biomarker development (Part 1). Exploration and clinical assay development comprised the first two, while in phase 3, the biomarkers are put to the test in retrospective studies using banked blood samples from longitudinal studies, including population-based cohorts that tend to be more heterogenous than typical research cohorts. At least for the best-established biomarkers thus far, many of the data shown at AAIC focused on this third phase, as scientists tried out the most promising biomarkers in community cohorts.
Blood Biomarker Palette
Tim West from C2N Diagnostics in St. Louis presented biomarker findings from 221 participants in the ongoing Plasma Test for Amyloidosis Risk Screening study. PARIS is a prospective add-on to the community-based Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study. All participants in PARIS have cognitive symptoms; for 65 percent, their amyloid PET scan was positive. In prior research cohorts, C2N’s plasma Aβ42/40 ratio assay, a mass spectrometry test that is commercially available for order by dementia specialists, has predicted the likelihood of amyloid positivity with an area under the curve of above 0.9, West noted. AUC is a combined measure of specificity and sensitivity in which 1.0 is the highest score.
In the PARIS-IDEAS cohort, the blood test predicted amyloid-PET positivity with an AUC of 0.79, the researchers recently reported (Hu et al., 2022). Why the dip? IDEAS enrolls Medicare patients whose physicians have deemed them “difficult to diagnose,” making them candidates for amyloid PET. In contrast, most research cohorts include people who participate in longitudinal aging studies, enabling a clearer delineation of persons who are healthy from those who have MCI, or dementia due to AD. For this reason, the lower AUC of plasma Aβ42/40 in the PARIS-IDEAS came as no surprise, C2N’s Joel Braunstein told Alzforum. As in previous evaluations of this test, including a measure of age and ApoE prototype—the latter measured by mass spec—nudged up the AUC significantly, to 0.86 in this cohort. This combination of age, plasma Aβ42/40, and ApoE prototype—sold as PrecivityADTM—yields an amyloid probability score. C2N is currently marketing this combined test to dementia specialists. According to an August 23 press release, the company has joined forces with Eisai, Inc., to market blood biomarker tests in traditionally underserved communities.
A bigger improvement was to be had by adding plasma p-tau217. As many presentations at AAIC made clear, the plasma concentration of various phospho-tau species, including p-tau181, 217, and 231, are sensitive detectors of brain amyloid plaques, not of tau tangles as seen by PET. C2N has a mass spec test for p-tau217 and, at AAIC, West reported how it performed alone or in combination with Aβ42/40 in PARIS-IDEAS. On its own, plasma p-tau217 outperformed the Aβ ratio test, with an AUC of 0.92. Accuracy climbed to 0.95 when the ratio of phosphorylated to unphosphorylated tau at this residue was measured instead of the concentration p-tau217 alone. Combining this tau ratio with the Aβ42/40 ratio achieved an AUC of 0.96, although that was not significantly higher than the p-tau217/tau217 ratio alone.
“Even in a difficult-to-diagnose, real-world population of patients with MCI/dementia of unknown etiology, the combined test demonstrated outstanding performance,” Braunstein said.
Under what circumstances would clinicians need both biomarkers, if p-tau217 appears to do the job by itself? West said including plasma Aβ42/40 would make the diagnosis more accurate among people in the earlier stages of amyloid deposition. In PARIS-IDEAS, some participants whose plaque load was at the bottom of the range of amyloid-PET positivity were detected as being amyloid-positive via plasma Aβ42/40, but not by p-tau217, he reported.
“If, in fact, the goal of a disease-modifying treatment strategy is to intervene as early in the disease process as possible among cognitively impaired individuals with AD, then having Aβ42/40 to aid in that earlier detection should be clinically impactful,” Braunstein wrote. He added that the combined biomarkers may become even more helpful as the field evolves toward preventing disease in cognitively normal people.
Test evaluation in the Swedish BioFINDER cohort led to similar conclusions. Oskar Hansson of Lund University, Sweden, said that at different stages of AD, a different biomarker might best predict whether a person has brain amyloid. At the dementia stage, p-tau217 alone should be sufficient, whereas at the prodromal stage, p-tau217 might be paired with cognitive testing. At the preclinical stage, Aβ42/40 plus a measure of p-tau might prove most accurate at detecting amyloid plaques lurking in the brain. The question of which p-tau species might best detect early amyloid deposition at the preclinical stage is under intense investigation in several labs.
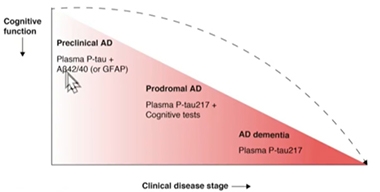
Biomarkers by Stage. As clinical disease worsens, fewer blood markers may be needed to detect brain amyloid pathology. [Courtesy of Oskar Hansson, Lund University]
In BioFINDER, a comparison of different combinations of plasma biomarkers among cognitively normal participants showed that WashU’s mass spec blood Aβ42/40 ratio together with Eli Lilly’s blood p-tau217 assay detected amyloid with an AUC of 0.91. Either alone did so at AUC 0.84.
Hansson also presented a side-by-side comparison of 10 plasma p-tau assays measuring different phospho-epitopes and using different measurement platforms among people at the MCI stage. Wash U’s mass spec assay was the most accurate, with an AUC of 0.95, while Lilly and Janssen’s assays, which use meso-scale discovery (MSD) and Simo immunoassays, respectively, also performed well, posting AUCs approaching 0.9. Regardless of the type of assay, p-tau217 outperformed p-tau181 or p-tau231 in detecting amyloid at the MCI stage.
To move from learnings in cohorts toward enabling prognosis in individual patients, the Lund researchers are weaving these biomarker findings into algorithms that include ApoE status and cognitive testing (Janelidze et al., 2021; Palmqvist et al., 2022).
The superiority of the mass spec-based p-tau217 assay jibes with what was previously reported for plasma Aβ42/40, where mass spec assays led the field in amyloid detection (Oct 2021 news). Other assays are nipping at mass spec's heels. “We now have assays that perform remarkably well. This will revolutionize diagnostics in clinics,” Hansson said.
In an encouraging sign of converging evidence, Marc Suárez-Calvet of Hospital Del Mar Medical Research Institute and Barcelonaβeta, both in Barcelona, Spain, came to a similar conclusion. In San Diego, Suarez-Calvet presented a head-to-head comparison of nine plasma p-tau biomarker assays in a longitudinal cohort that included patients with neurodegenerative diseases who visited his hospital and underwent lumbar puncture. This real-world cohort is heterogenous. Participants have received different clinical diagnoses, including MCI, AD dementia, vascular dementia, dementia with Lewy bodies, progressive supranuclear palsy, and primary progressive aphasia. Instead of amyloid-PET, Suárez-Calvet relied upon the CSF ratio of Aβ42/p-tau as the determinant of brain amyloid positivity.
Suárez-Calver’s lineup consisted of Janssen’s p-tau217; Lilly’s p-tau181, p-tau217, and total tau; Quanterix’s p-tau181; the University of Gothenburg’s in-house p-tau181 and p-tau231 assays; as well as p-tau181 and p-tau217 assays from ADx Neurosciences, a Belgian assay developer acquired this summer by Fujirebio (Bloomberg news). Suárez-Calvet's team evaluated the performance of each assay using plasma and CSF from 197 participants, including 127 who were amyloid-positive based on their CSF Aβ42/p-tau ratio.
In short, the scientists found that several plasma assays were highly accurate at distinguishing between AD and non-AD CSF status in this memory clinic cohort. With an AUC of 0.96, Janssen’s p-tau217 came in first in this comparison, while ADx’s p-tau181 and Lilly’s p-tau217 assays were close behind at 0.94. Quanterix’s p-tau181 assay discriminated amyloid from non-amyloid CSF profiles with an AUC of 0.8, while Gothenburg’s p-tau231 assay clocked in at 0.88.
Suárez-Calvet concluded that, at least among people at this Barcelona hospital, several plasma p-tau assays work well on their own at pinpointing who has amyloid in their brain. He also believes more work is needed to see how these results apply to different populations, and that different marker combinations might do better at different stages of disease. For example, p-tau231 might prove most sensitive among people with early stage amyloid deposition who are cognitively normal. Recently published studies and presentations at AAIC suggest that plasma p-tau231 rises before p-tau217 in response to amyloid accumulation.
The field seems to have no shortage of plasma biomarker assays that closely track with amyloid status. But a test's accuracy is far from the only factor to consider when rolling it out around the globe. For one, tests need to be scalable and accessible, Hansson noted. While mass spec tests appear to be the most sensitive of the lot, they are costly and require specialized facilities and highly trained technicians to run. According to Braunstein, C2N is currently able to process several thousand per week in its CAP-CLIA facility and is partnering with other mass spec laboratories to boost capacity.
Immunoassays could be more practicable for use on a mass scale but even these have limitations, said Edward Wilson of Stanford University. He noted that some tests are plate-based, meaning that the test is run on proprietary plates preloaded with the reagents. These are vulnerable to variation between plates, he said, and are not amenable to adding in other biomarkers. He also noted that some tests are incapable of running at high throughput, and require expertise of experimenters or specialized equipment. Typically in medical care, blood tests that are widely used in routine clinical care have been fully automated as part of their clinical standardization and certification process.
Wilson tried out Fujirebio’s Lumipulse platform and plasma p-tau181 assay in the Stanford ADRC and Stanford Aging and Memory Study (SAMS), longitudinal studies that collect cognitive, genomic, and biomarkers data on their participants. All components of this assay are commercially available, high-throughput, fully automated, and run on a mid-size desktop analyzer. Wilson found that the assay differentiated amyloid-positive from -negative participants, with increasing separation between groups as their clinical disease stages worsened. The test posted an AUC of 0.95 for distinguishing amyloid-negative, cognitively unimpaired and amyloid-positive symptomatic people, though Wilson acknowledged that this is the lowest-hanging fruit in terms of discrimination. Future studies will compare the Lumipulse assay head-to-head with others in the field, Wilson said.
Much of the blood-biomarker data at AAIC focused on Aβ and p-tau. Is that all scientists need? Or would other analytes add value for prognosis? Consider GFAP. Astrocytes crank up expression of this cytoskeletal protein in response to neuronal damage, as well as in the vicinity of amyloid plaques. Teunissen used data from the Netherlands Twin registry, a population-based cohort that includes pairs of monozygotic twins, to investigate the predictive value of plasma biomarkers, including GFAP, for amyloid-positivity in people approaching their 60s.
At baseline, when participants were still cognitively normal and averaged 68 years of age, they were tested for amyloid by CSF or PET. They had already given blood samples 10 years prior. In San Diego, Teunissen reported that among those who were amyloid positive at baseline, blood levels of p-tau181 and GFAP already had been elevated a decade before and rose sharply thereafter. What’s more, twins who were concordant for amyloid at baseline also tended to be concordant for p-tau181 and GFAP. Curiously, even twins who were mismatched for amyloid were concordant for GFAP. To Teunissen, this concordance suggests that shared environmental or genetic factors influence GFAP levels, suggesting this astrocytic protein could be some sort of a predisposition marker.
Among people with brain amyloid, plasma p-tau181 and GFAP tracked with subsequent cognitive decline on memory tests, with GFAP having the strongest link. These twin cohort findings jibed with recently published findings from cognitively normal participants in the Amsterdam Dementia Cohort, where plasma GFAP and, to a lesser extent, plasma NfL, correlated with future conversion to MCI (Verberk et al., 2021).
At AAIC, Teunissen reported that in the DIAN cohort, plasma GFAP begins to rise in mutation carriers about 10 years prior to their expected symptom onset. Unlike p-tau181, which levels off after symptoms emerge, GFAP continued to rise for years as symptoms worsened. GFAP also correlated with subsequent brain atrophy among mutation carriers in the DIAN cohort, Teunissen reported. The findings cast GFAP as both a predictive marker that symptoms lie ahead and cognition will decline, and as a marker of progression. “This indicates that there is really added value of GFAP, in the early and late stages of the disease,” Teunissen said.
Teunissen agreed with other investigators that despite the rapidly growing, and largely convergent, data demonstrating the power of blood tests for the diagnosis and prognosis of AD, much work remains before they will wend their way into practice, especially primary care. For more on the road to get there, see the next part of this series (Part 3).—Jessica Shugart
No Available Comments
No Available Further Reading
Part 3 of 3
An abundance of blood-based biomarker findings were on display at the Alzheimer’s Association International Conference, held July 31-August 4 in San Diego. Scientists presented findings directly comparing the top markers to each other in different cohorts, revealing a slew of markers that detect amyloid plaques in the brain and predict cognitive decline (see Part 2 of this series). With tests coming onto the scene, scientists are starting to grapple with the problems that remain to be solved before they can be used broadly in clinical practice, let alone in primary care settings without the help of memory specialists. Researchers at the meeting charted a path toward implementation, articulated the questions for this next phase, and proposed studies needed to answer them. One focus: initial data on how different comorbidities might tweak readings of some biomarkers.
Roadmap to Primary Care
Charlotte Teunissen of Amsterdam University Medical Center laid out a path of studies needed to develop Alzheimer's disease blood biomarkers, from initial preclinical exploration to routine use (see Part 1). It includes five phases, each requiring completion of studies in ever-more-heterogenous, diverse populations. The final phase concerns the ultimate, and most far-off, goal: to ready blood tests for routine use not only in specialist memory clinics, but also in primary care.
Douglas Galasko, University of California, San Diego, zeroed in on that phase. For starters, the role of primary care doctors in diagnosing and treating neurodegenerative disorders varies markedly by country. Some, such as England, France, and Spain, have established memory clinics all over the country; in others, including Australia, Belgium, Finland, Denmark, and Canada, primary care doctors manage Alzheimer's care with support from specialists. In the U.S., the balance between primary care and specialist care varies by state and health plan, Galasko said. This is to say nothing of the situation in most developing countries, where dementia specialists are few and far between.
Regardless of the structure of the healthcare system in which they work, physicians ordering a blood biomarker test will need to have a firm grasp of what the biomarker is measuring. They need training in what different results may and may not mean for an individual given their overall health, age, sex, and ApoE status, and in how to accurately communicate these nuances to the patient.
“This is not some simple, check-off-a-box blood test,” Galasko said. “One may need to discuss the test, its context, and possible outcomes with a patient before ordering it.” Once the results are in, an in-depth conversation about interpretation, prognosis, and potential treatment options will need to occur, Galasko said. As of now, such analysis and discussion are beyond the scope of most busy primary care doctors, and even some dementia specialists, Galasko said.
How can physicians help their patients decide if biomarker testing is right for them? Several groups are working on developing tools for physicians and patients to aid in decision-making. One is the Advisory Group on Risk Evidence Education for Dementia (AGREEDementia), an NIA-coordinated working group that previously developed a decision aid for people with MCI who were considering undergoing amyloid-PET. The group is now putting together a decision aid for blood biomarkers.
Biomarker scientists at AAIC broadly agreed that blood tests are best interpreted in the context of a cognitive assessment. The trouble with that? Cognitive tests often fall by the wayside in primary care, Galasko said. Many primary care doctors lack the time or expertise to test cognition consistently. Case in point: Although cognition is included in annual Medicare wellness exams in the U.S., doctors only need ask the patient in front of them about their cognition, rather than actually test it, to check this box.
“Cognition should be considered a vital sign,” Galasko told Alzforum. To take pressure off primary care doctors, the field could agree on digital tests, whose development has accelerated in recent years. A patient could take a brief cognitive test on a smartphone or tablet, either at home or in the waiting room prior to their annual wellness check, Galasko suggested. This would remove the burden from primary care doctors, standardize cognitive tests, and aid in decisions to order biomarker tests and interpret their results.
Work is already underway to figure out how best to combine quick cognitive tests with blood biomarkers in primary care. Oskar Hansson of Lund University described an ongoing prospective study that aims to derive easy-to-use algorithms based on combinations of blood biomarker assays and brief cognitive tests to improve the diagnosis and prognosis of AD in primary care (see image below). Thus far, it has enrolled 300 of what are to be 800 participants from 25 primary care centers across Sweden with subjective cognitive complaints, MCI, or mild dementia. Nurses at the primary care clinics collect blood and oversee cognitive tests that run on smartphones, iPads, and paper. To establish a reference standard of diagnostic accuracy, all participants in this study will also undergo the kind of extensive, specialized memory clinic work-up that is familiar to Alzforum readers—neuropsychological testing, CSF sampling, and PET. The winning mixture of blood biomarkers and primary care-level cognitive tests that best matches the memory clinic findings will then be considered for broader deployment in primary care.

Blood Tests in Primary Care. To move blood biomarkers from research cohorts into primary care, researchers are testing various combinations of blood biomarkers and brief cognitive tests in 25 primary care clinics in Sweden. [Courtesy of Oskar Hansson, Lund University, Sweden.]
Contending with Comorbidities
Even with a winning combination of cognitive tests and blood biomarkers in hand, physicians remain uncertain about the influence of genetics, race/ethnicity, and comorbidities on the test. Most blood-biomarker studies have been conducted in research cohorts that included predominantly white participants. A notable exception are the cohorts at Washington University, St. Louis. A recent study led by Suzanne Schindler there found that while plasma Aβ42/40 had comparable predictive value in African Americans and Caucasians in that cohort, plasma p-tau181 performed worse in African Americans (Apr 2022 news on Schindler et al., 2022).
The authors speculated that these differences likely reflect differences in underlying medical comorbidities and social determinants of health. For example, they noted that African Americans in the study had higher rates of hypertension and diabetes than their Caucasian counterparts. This jibes with recent studies reporting that heart and kidney disease influence biomarker levels (Syrjanen et al., 2022).
At AAIC, researchers dug more deeply into the potential influence of comorbidities on biomarker levels. Michelle Mielke, who is now at Wake Forest University in Winston-Salem, North Carolina, reported that among 1,329 participants in the Mayo Clinic Study of Aging, the effect of chronic kidney disease rivaled that of amyloid status on a person's level of plasma p-tau181 and p-tau217 (Mielke et al., 2022). Stroke and myocardial infarction were each associated with higher p-tau levels as well, whereas higher body-mass index (BMI) lowered the measured p-tau value. In her AAIC talk, Mielke described an example of one participant who had minimal AD pathology upon autopsy, despite having the highest plasma concentration of p-tau217 in the entire cohort. Mielke discovered that this participant also had sky-high serum creatinine levels, indicative of poor kidney function. “As a result of having chronic kidney disease, this person would have been a false positive,” Mielke said.
Mielke further reported that including people with myocardial infarction, stroke, or kidney disease in the cohort widened the normal range for these biomarkers, such that the cut point of abnormality for p-tau181 decreased from 1.75 to 1.5 pg/mL once these people were excluded. This effect was smaller for p-tau217, where it inched the cut point down from 0.26 to 0.25. Such findings suggest that, at the group level, such comorbidities have minimal influence on plasma p-tau217’s predictive power for amyloid. However, for an individual person with one of these comorbidities, researchers will need to figure out whether adjustments are needed to avoid misdiagnosing them.
Of the participants in the Mayo Clinic cohort, 95 percent are non-Hispanic white, so Mielke was unable to investigate the relationship between race, ethnicity, comorbidities, and biomarkers. She did note that African Americans have a higher prevalence of all the comorbidities that also increase biomarker levels, which could lead to disproportionate misdiagnoses in this group. Mielke highlighted a recent study in Mexican Americans, which reported that dyslipidemia, hypertension, and diabetes bumped up their AD plasma biomarker levels (O’Bryant et al., 2022).
Hansson also reported findings about how comorbidities might degrade the accuracy of plasma biomarkers. In the Swedish BioFINDER-1 and BioFINDER-2 cohort studies, the Lund team investigated whether potential confounds such as kidney function or BMI influenced the associations between individual plasma biomarkers and their CSF counterparts, or the ability of the plasma markers to predict a person's progression to dementia. Their finding? Creatinine levels correlated with higher plasma concentrations of NfL, GFAP and, to a lesser extent, p-tau181 and p-tau217.
As in the Mayo cohort, BMI had the opposite association in BioFINDER, probably reflecting the diluting effect of a larger blood volume. Importantly, however, when the researchers adjusted for creatinine or BMI in their models, they saw that these factors did not influence how well a given blood marker correlated with its counterpart in CSF, nor a blood marker's ability to predict subsequent dementia. Hansson concluded that while creatinine and BMI do hold sway over levels of certain plasma biomarkers, they are not clinically relevant confounds for most people.
In a comment to Alzforum, Mielke cautioned against using the term “confounder” to describe physiological factors that can affect the interpretation of the blood biomarkers. “In public health, a confounder is something associated with both the risk factor and outcome, and can be adjusted for,” Mielke wrote. “Chronic kidney disease is not associated with amyloid pathology, but [is associated] with neurodegeneration and vascular pathology. Therefore, we cannot simply ‘adjust’ for kidney disease or other factors shown to affect the blood biomarkers due to physiological reasons.”
Hansson said that how comorbidities influence the predictive value of blood biomarkers relates not only to the prevalence of these comorbidities in the population at hand, but also to how strongly the disease raises the biomarker concentration.
For example, in cognitively normal people, p-tau217 shoots up by 80-350 percent in those with brain amyloid, depending on the study. Among people with cognitive symptoms, p-tau217 is typically up a whopping three- to sevenfold in people with plaques. In the latter group, the influences of kidney dysfunction or high BMI are unlikely to cause a problem, Hansson said, adding, “But they might be a problem in population screening of cognitively normal individuals, especially in populations where kidney problems and very high BMI are common.” This was the situation with the Mayo Clinic cohort, in which most participants were cognitively unimpaired, and the incidence of comorbidities was higher than in the BioFINDER cohort.
Future prospective studies in ever more diverse cohorts will be needed to understand how comorbidities disproportionately affect biomarker effectiveness in different populations.—Jessica Shugart
No Available Comments
No Available Further Reading
As the mountain of whole-genome sequencing data grows, so does the likelihood of finding rare variants within it. Such mutations can have stronger associations with disease risk, or resilience, than do common variants, opening a window into the underlying biology.
This is true for both known and new risk genes. For example, the rare Christchurch and Jacksonville APOE3 variants, and the newly discovered APOE4 mutation R251G, seem to shield their carriers from cognitive decline (Jun 2022 news; Oct 2021 news; Nov 2019 news).
Trouble is, rare mutations may occur only in one person or a single family. “Among 20,000 people with whole-genome sequencing in the Alzheimer’s Disease Sequencing Project, 54 percent of the single nucleotide polymorphisms are seen only in one individual,” wrote Yann Le Guen, Stanford University, California (full comment below).
How, then, can scientists tell if such variants influence disease? When small sample size makes it hard to know if a given association with disease is meaningful, researchers perform functional or multi-omic analyses, Rita Guerreiro of the Van Andel Institute in Grand Rapids, Michigan, explained. At the Alzheimer’s Association International Conference held from July 31 to August 4 online and in San Diego, California, Guerreiro and Le Guen co-chaired a session on such efforts. Some geneticists looked for groups of rare AD risk variants within genes to identify those linked to disease. Others studied neurons cultured from cells of rare variant carriers, or analyzed anomalies within brain tissue from carriers.
Variant Clumps
One new way to show that rare variants are AD risk factors is to find genes containing many such mutations. “Clusters of rare variants identify novel genes associated with disease, or indicate which domain of a known disease-linked protein may be malfunctioning,” said Guerreiro.
Along those lines, Bowen Jin of Case Western Reserve University, Cleveland, Ohio, searched for groups of rare variants within proteins to pinpoint functional hotspots (Jin et al., 2022). She started with whole-exome sequencing data from about 5,500 AD cases and 5,000 controls in ADSP, identifying more than 1.6 million rare variants within almost 21,000 genes. Jin mapped the variants onto the structures of more than 6,000 proteins in the Protein Data Bank and nearly 17,500 putative protein structures from AlphaFold, an artificial intelligence system that predicts structure from an amino acid sequence.
Of the thousands of proteins scanned, only a handful of the corresponding genes had clusters of rare variants and rose above the threshold of statistical significance, indicating association with AD. Jin then searched for variant hotspots within these genes in the ADSP whole-genome sequencing dataset of 3,700 AD cases and 4,000 controls, an independent cohort from ADSP WES.
Three genes remained statistically significant. Two are known AD risk genes: the microglial membrane receptor TREM2 and the endosomal trafficking protein SORL1. The third, EXOC3L4, is a poorly understood protein predicted to be a component of the exocyst complex, which is involved in vesicle trafficking and exocytosis. In Jin’s analysis, 33 rare variants from cases and controls were found in TREM2, 56 in SORL1, and 68 in EXOC3L4.
Many TREM2 and SORL1 variants carried by people with AD clustered in each protein’s extracellular domain, both of which bind Aβ (see image below; Feb 2015 news). This suggests dysregulation of Aβ handling by both proteins in AD.

Nests of Variants. Rare variants from AD cases (red) pack into the extracellular Aβ-binding domains of TREM2 (top) and SORL1 (middle). The cluster in EXOC3L4 (bottom) dots its Sec6 domain, which may be involved in endocytosis. Rare variants from controls are in blue. [Courtesy of Bowen Jin, Case Western Reserve University.]
Fifteen EXOC3L4 variants in AD cases fell into the C-terminal Sec6 domain (see image). Rare variants within this domain had previously been linked to cortical glucose metabolism in AD (Miller et al., 2018). Jin thinks EXOC3L4 is a new AD risk gene that warrants further study.
“It is pleasing to see the AD genetics field rapidly expanding beyond traditional GWAS […] to the identification of biologically meaningful rare variants that functionally impact AD pathobiology,” wrote Rudolph Tanzi and Dmitry Prokopenko of Massachusetts General Hospital, Boston (full comment below).
SORL1 Cells
Brooke DeRosa at the University of Miami works on characterizing a rare SORL1 mutation in cell culture (DeRosa et al., 2022). DeRosa derived induced pluripotent stem cells (iPSCs) from two sisters with early onset AD. Each carried a frameshift variant of SORL1 that creates a truncated protein lacking 30 percent of its C-terminus (see Aug 2017 news). This is at the other end of the protein from the extracellular domain with the rare variant cluster reported by Jin.
DeRosa differentiated the iPSCs into neurons, then assessed synaptic trafficking by labeling the cells with the early endosome marker EEA1, a marker for APP, and the synapse marker Synapsin. At AAIC, she showed on a poster that, compared to control neurons, those carrying the SORL1 variant had 33 percent more early endosomes stuffed with APP and 35 percent fewer synapses. This suggests that the SORL1 variant contributes to dysregulated vesicle trafficking and synapse death. Taken together with the dementia diagnoses of its carriers, DeRosa believes that this rare mutation is pathogenic.
Trouble in the Tissue
Beyond cells, brain tissue from rare variant carriers can provide insight into how the mutation affects AD pathology. To this end, Elisabeth Hendrickx Van de Craen of University Hospital Antwerp, Belgium, studied hippocampal and cortical tissue from carriers of rare variants within ABCA7. Mutations in this lipid transporter increase the risk of developing AD (Oct 2020 news; Jul 2020 conference news).
Hendrickx Van de Craen searched for ABCA7 missense mutations and premature stop codons within whole-exome sequences of 491 early onset AD cases, 885 late-onset cases, and 976 controls. Of 102 ABCA7 mutation carriers, 14 had donated brain tissue samples.
Did these rare variant carriers have altered AD pathology? Labeling the tissue with the Aβ antibody 4G8, Hendrickx Van de Craen was struck by the hefty burden of cerebral amyloid angiopathy she saw, which exceeded the mild-to-moderate CAA typically seen in AD.
Curious about a potential genetic link between AD and CAA, Hendrickx Van de Craen searched for rare ABCA7 variants within whole-exome sequences from 90 Belgian people with CAA, then searched for functional mutations in ABCA7. Ten were carriers—more than double the prevalence in the AD cohort. Brain tissue from four carriers showed diffuse 4G8-positive amyloid plaques and AT8-positive neurofibrillary tangles.
Had the CAA ABCA7 carrier cases had cognitive problems late in life? Indeed, seven of the 10 were clinically diagnosed with AD. “ABCA7 rare variant carriers seem to have a spectrum of AD and CAA,” Hendrickx Van de Craen told the audience at AAIC. “Although these two diseases are distinct clinical entities, they may have a common genetic background.”—Chelsea Weidman Burke
Consensus is strengthening among Alzheimer’s researchers that antibodies that abolish plaque nudge down the rate of cognitive decline. Alas, the effect is so small—is it meaningful? At the Alzheimer’s Association International Conference, held July 31-August 4 virtually and in San Diego, California, several presenters quantified the clinical benefit of amyloid removal and attempted to forecast the effects of long-term treatment. They admitted that the cognitive benefit in trials has been tiny, but drug sponsors argued that the difference between treatment and placebo groups is likely to grow over time.
At Roche, Norman Mazer developed the “Quantitative ATN” (Q-ATN) model of AD to predict changes in cognition as a function of amyloid removal. He used data from numerous observational studies to derive mathematical relationships. The model’s predictions correlated well with actual clinical trial results. A simulated five-year course of the company’s anti-amyloid antibody gantenerumab forecast that the CDR-SB difference between the treatment and placebo groups would expand as the disease progression curve flattens. Evaluating a different metric, researchers at Eisai estimated that long-term treatment with their drug, lecanemab, could delay progression from mild cognitive impairment to dementia by more than three years. Upcoming trial results from both antibodies expected this fall will provide a reality check to these company models, and help refine their predictions.
Rachelle Doody at Roche believes the Q-ATN model could affect how such trial results are interpreted. “The clinical significance of the CDR-SB score at endpoint should be evaluated in the context of accrued benefits over time, especially once this model is prospectively validated with longitudinal data,” she wrote to Alzforum.

Stanch Progression? Roche’s mathematical Q-ATN model foresees a drop in plaque (upper left), followed closely by a drop in the tau PET signal (upper right). After a delay, the brain shrinkage slows down (lower left), stabilizing cognition (lower right). [Courtesy of Roche.]
Is the Cognitive Benefit Real?
Trials of early immunotherapies, such as bapineuzumab and solanezumab showed little effect on either plaques or cognition (Jan 2014 news; Dec 2016 conference news). Ditto for initial trials of later antibodies, such as the SCarlet RoAD and Marguerite RoAD studies of low-dose gantenerumab (Dec 2014 news; Nov 2015 conference news). In subsequent studies, however, high doses of four antibodies—aducanumab, gantenerumab, lecanemab, and donanemab—proved able to banish plaques, while lightly tapping the brakes on cognitive decline, by 20 to 40 percent (Aug 2018 conference news; Dec 2019 conference news).
At AAIC, Nicolas Villain of Sorbonne University in Paris, presented a meta-analysis of only the high-dose arms from the lecanemab and donanemab Phase 2 trials and the two aducanumab Phase 3 trials, EMERGE and ENGAGE. In the combined analysis, the drugs had a statistically significant cognitive benefit after 18 months, slowing decline on the CDR-SB by an average of 0.24 points, and on the ADAS-Cog by 1.25 points. Villain’s result contrasts with published meta-analyses of diverse amyloid-targeting therapies, many having little effect on plaque. Those analyses found no consistent cognitive benefit (Ackley et al., 2021; Richard et al., 2021). Villain said the size of the effects on both endpoints he looked at were far below what is considered the minimal clinically relevant difference, i.e., 1.63 points for the CDR-SB and 3.8 for the ADAS-Cog. In addition, about one in 200 participants in these trials developed serious symptomatic brain edema or microhemorrhages (ARIA). Thus, the risk/benefit ratio of short-term anti-amyloid immunotherapy is high, Villain concluded.
Other analyses also concluded that the cognitive benefit of amyloid removal is real. Yaning Wang of Createrna Science and Technology, Clarksburg, Maryland, a spin-off of the Chinese biotech firm Wuhan QR Pharma, previously worked at the Food and Drug Administration. He was part of the team that evaluated aducanumab’s licensing application. At AAIC, Yang defended the agency’s decision to grant accelerated approval despite a negative AdComs evaluation. After the FDA advisors had pointed out that there was no way to know whether the positive EMERGE or the negative ENGAGE aducanumab trial was the anomaly, FDA scientists analyzed available clinical trial results from all antibodies in this class. At the time, published data consisted of aducanumab, gantenerumab, lecanemab, crenezumab, bapineuzumab, and solanezumab trials. Wang said that the FDA found a consistent linear relationship between amyloid removal and cognitive benefit across these antibody trials, even for negative ones. ENGAGE was the only trial that did not fall on this line, leaving the FDA to conclude that it was the outlier.
Wang also talked about effect size. He noted that because cognition declines slowly at the prodromal stage of AD, the tiny, measured benefit on the CDR-SB in EMERGE represented a 25 percent slowing. To achieve the minimum clinically relevant difference on CDR-SB in 18 months, the drug would have had to completely halt progression, an unrealistic standard, Wang said.

Chewed Up Plaques? Amyloid deposits (brown) in a person treated with lecanemab (top) have a “moth-eaten” appearance compared to the denser plaques in an untreated AD patient (bottom). [Courtesy of Honig et al.]
Data at AAIC also helped lay to rest the niggling question of whether anti-amyloid antibodies truly clear plaque, or instead lower the amyloid PET signal in some other way, such as coating amyloid deposits and preventing tracer binding. This concern has not yet been definitively settled, since few people who have had high-dose anti-amyloid immunotherapy have passed away and had an autopsy. In San Diego, Lawrence Honig of Columbia University Irving Medical Center in New York City presented one such case. This man with mild cognitive impairment had received 10 mg/kg lecanemab monthly for 18 months as part of the Phase 2 trial, which reduced his amyloid PET signal from 45 to 26 centiloids. After a two-year gap, he joined the open-label extension and received 10 mg/kg biweekly for two more years, though Honig did not show PET scans from the OLE. The man died of an unrelated heart condition at the age of 85.
His autopsy showed that his brain contained almost no diffuse plaques, and only a few loose neuritic plaques that had a “moth-eaten” appearance. His brain had widespread neurofibrillary tangles of tau; likewise, these were not dense as they typically are, but had a thread-like consistency. The man’s brain was assessed as Thal Phase 2, indicating little amyloid, and Braak stage 6, indicating advanced tangle pathology. This combination is rare, making up only 2 percent of cases in the National Alzheimer’s Coordinating Center brain bank, Honig noted.
The results strengthen the case that anti-amyloid immunotherapy truly removes plaque. They dovetail with a previous report in the literature, of a woman who received 32 doses of aducanumab. At autopsy, she had little plaque and widespread tangles of low density (for paper and expert commentary, see Plowey et al., 2022).
Will It Grow?
Do benefits of amyloid removal build up over time, as might be expected for a disease-modifying therapy? To address this, Mazer and colleagues at Roche modeled the amyloid-cascade hypothesis. In its simplest form, it posits that plaques unleash tau aggregation, which in turn accelerates brain atrophy, degrading cognition. Mazer expressed these relationships mathematically, deriving equations that described the observed biomarker changes at each step. Natural history studies from several sources, including unpublished imaging data from the Harvard Aging Brain Study, tied plaques to excess tau production (Johnson et al., 2020). From data in multiple publications, the authors found a straightforward relationship between tangle accumulation in the medial temporal cortex and the rate of cortical thinning in that area. The link between thickness in the medial temporal cortex and the CDR-SB came from a study by Brad Dickerson at Massachusetts General Hospital, Boston (Dickerson et al., 2009).
Combining these equations allowed the authors to project how amyloid accumulation might affect the CDR-SB score. They tested their model on additional natural history datasets, and found that the simulated change in biomarkers and CDR-SB closely matched actual findings, suggesting the equations described real biological relationships (Delor et al., 2013; Kim et al., 2020).
But can this model predict the effects of amyloid removal? The researchers ran simulations of how amyloid clearance by aducanumab would affect tangle pathology and CDR-SB, and compared those to actual data from EMERGE and the tau PET substudy. The results agreed well, Mazer claimed. Extending the analysis to published trials of gantenerumab, lecanemab, donanemab, and bapineuzumab, the Q-ATN model accurately predicted CDR-SB change in five of the seven trials, with the discrepancies being the ENGAGE high-dose arm and the SCarlet RoAD trial. “The model is able to explain the lowering of tau PET,” Mazer told Alzforum. “As you remove amyloid, you dial down the inherent drive to generate tau tangles.”
Finally, the researchers used Q-ATN to forecast the results of five years of treatment with gantenerumab. The model predicts that plaques would recede over the first two years, sinking below the positivity threshold before tailing off over the next three years to reach zero. Tau PET would mirror this, but with a more gradual time course and a smaller overall reduction. The rate of cortical shrinkage would start to slow around two years, preserving the remaining thickness of the medial temporal lobe. CDR-SB decline would level off around the same time, reaching statistical difference from placebo of 0.87 points at 27 months, the stopping point for the Phase 3 GRADUATE trials of gantenerumab. The model projects that this difference would expand to 5.2 points after five years, as the placebo group continues to decline faster than the treatment group (see image above).
“Because the rate of cortical atrophy decreases, it bends the curve and leads to the growing separation between the untreated and treated CDR-SB curves,” Mazer said. Crucially, the Q-ATN model suggests that cognitive benefits will be delayed relative to amyloid removal. “There is an inherent lag in the system, because tau influences the rate of change of the cortical thickness. It takes a year or two to see a difference in thickness,” Mazer explained. The data have been submitted for publication.
Is It Meaningful?
How would such a subtle effect—slightly slower decline on cognitive tests—actually improve people’s lives? Amir Tahami Monfared and colleagues at Eisai addressed this using the AD Archimedes condition-event (ACE) simulator developed by the contract research organization Evidera (Kansal et al., 2018). Like the Q-ATN model, AD ACE simulates clinical progression based on biomarker changes; its equations were derived from ADNI observational data. Rather than focus on cognitive scores, AD ACE predicts when symptoms will worsen to the next stage of disease.
Eisai researchers selected 429 of the 1,735 ADNI participants in the AD ACE simulator database, choosing those who best matched the demographic characteristics of participants in the lecanemab Phase 2 trial. In this trial, treatment slowed progression on the CDR-SB by 26 percent. The matched ADNI cohort, with a mean age of 72 and MMSE of 26, provided a glimpse of how disease would progress in this population without treatment. Taking into account the typical remaining lifespan of AD patients and older adults, Eisai researchers projected that continuing lecanemab treatment until the end of a person’s life would result in 7 percent fewer people progressing to mild dementia, 13 percent fewer to moderate dementia, and 10 percent fewer to severe. Six percent fewer people would be institutionalized.
Looking at the data another way, Eisai researchers estimated that the time to each stage of dementia would be delayed by 2.5 to 3 years. People on lecanemab would remain in the community about one year longer and live one year longer on average, gaining 0.75 “quality-adjusted life years” (QALYs). The modeled benefits were greater in younger participants. In a subset with a mean age of 65, progression to mild and moderate AD was projected as being delayed by 3.3 and 3.4 years, respectively (Tahami Monfared et al., 2022).—Madolyn Bowman Rogers
FDA approval of the first anti-amyloid antibody to treat Alzheimer’s disease has left clinicians grappling with new questions. How are they to administer aducanumab safely? How will people cope with learning they have amyloid plaques in their brain, a prerequisite for treatment? What type of patient will benefit the most? And will the answers be the same or different for each anti-amyloid antibody? At the Alzheimer’s Association International Conference, held July 31 to August 4 virtually and in San Diego, California, speakers discussed ways to address these issues, sketching a roadmap for integrating anti-amyloid immunotherapy into clinical practice.
Specific topics included updates to aducanumab's appropriate-use recommendations (AURs), the consequences of disclosing amyloid status, and a new initiative, ALZ-NET, that is now set up to track how treatment works longer-term in the general population. Audiences seemed eager for the information, asking questions until time expired.
And no wonder. Eli Lilly’s anti-amyloid antibody donanemab this month was accepted for priority review under the Food and Drug Administration’s accelerated approval pathway, following Eisai’s lecanemab last month (Fierce Biotech story; Jul 2022 news). Roche’s Phase 3 studies of gantenerumab will read out this fall. This means that three more anti-amyloid antibodies could be approved next year, in an environment where most clinicians still have little experience with this new type of treatment.
Minimizing ARIA Risk
When the FDA granted accelerated approval to aducanumab last year, the terse label raised questions about how to administer the drug and track safety (Jun 2021 news). To fill this gap, six leading clinician-researchers developed AURs, specifying patient selection criteria and an MRI monitoring schedule (Aug 2021 news). Since then, however, concerns about the risk of the brain edema and bleeding known as ARIA have intensified (Oct 2021 news; Dec 2021 news).
These concerns have prompted a revision of the AURs. The original panel—Paul Aisen at the University of Southern California in San Diego, Liana Apostolova at Indiana University School of Medicine in Indianapolis, Alireza Atri at Banner Sun Health Research Institute in Sun City, Arizona, Jeffrey Cummings of the University of Nevada, Las Vegas, Stephen Salloway at Butler Hospital in Providence, Rhode Island, and Michael Weiner at the University of California, San Francisco—has become a working group. It added four members: Suzanne Hendrix of Pentara Corporation, Salt Lake City, Gil Rabinovici of UCSF, Marwan Sabbagh of the Barrow Neurological Institute in Phoenix, and Dennis Selkoe of Brigham and Women’s Hospital, Boston.
Introducing their new AURs in San Diego, Cummings said the main changes involve more stringent exclusion criteria and more frequent MRI scans. Both aim at reducing the risk of ARIA. Specifically, in addition to the previous exclusion of any person who is taking anticoagulants or has a history of bleeding disorders, the new AURs now advise against giving aducanumab to anyone who in the past has had seizures, a stroke, or transient ischemic attacks, as well as anyone who has an autoimmune or inflammatory condition such as lupus or psoriatic arthritis. All of these conditions can make a person more susceptible to ARIA.
Furthermore, the updated AURs recommend APOE genotyping. APOE is the main determinant of ARIA risk, with two-thirds of APOE4 homozygotes having developed ARIA during the aducanumab trials, compared to one-third of E4 heterozygotes and one-fifth of noncarriers. Knowing the genotype of the person before them could help the clinician devise the right management plan for that patient, Sabbagh said. This is the first time APOE genotyping has been recommended as a prerequisite for an AD therapy, and its clinical utility will need to be evaluated, Sabbagh added.
Other anti-amyloid antibodies have different risk profiles. In San Diego, Larisa Reyderman of Eisai reported a 23 percent risk of ARIA in E4 homozygotes treated with lecanemab. Heterozygotes had a 7 percent risk, not statistically different from the 5 percent risk in noncarriers.
For monitoring, the original AURs recommended MRI scans at baseline and prior to the fifth, seventh, and 12th monthly doses, as well as any time symptoms of ARIA appear. The new AURs add a scan before the ninth dose to minimize the risk of dosing through asymptomatic ARIA, and worsening it. The new guidelines also call for having a treatment plan in place to manage severe ARIA. This could include administering steroids or anti-epileptics. “The goal is to create a standard of care that will reduce the likelihood of serious outcomes,” Salloway said.
Audiences peppered the speakers with questions. Among other practical concerns, they wanted to know what factors predict ARIA, what strength scanner to use for MRI, and whether there are long-term effects from ARIA. Cummings noted that the first AURs have been downloaded 20,000 times, demonstrating high demand for this information. The working group also regularly discusses its findings with the FDA; the agency’s last update of aducanumab’s label, in April 2022, brought the label into closer concordance with the AUR. The updated AURs were published April 5 in The Journal of Prevention of Alzheimer’s Disease (Cummings et al., 2022).
Meanwhile, researchers led by Marco Lyons at Roche proposed an algorithm, dubbed Score of ARIA Risk (SoAR), for quantifying risk. The algorithm assesses a patient's number of E4 alleles as well as the presence of any microhemorrhages in the brain before treatment begins. In the gantenerumab Phase 2 studies, these factors seemed to have independent, additive effects, while other characteristics, such as age, sex, race, and education, did not change ARIA risk. The algorithm takes into account the hazard ratio of each contributing factor. People with at least two risk factors, such as E4 homozygotes, or E4 heterozygotes with microhemorrhages, ran the highest risk of ARIA. The SoAR algorithm requires further validation, but potentially could help doctors make decisions about treating with anti-amyloid immunotherapy, Lyons suggested.
Can You Handle the Truth?
Before starting anti-amyloid therapy, physicians need to know their patients have plaques in the brain. Amyloid positivity is a requirement of both the AURs and the updated FDA label. Alas, data on how people will handle knowing their amyloid status are sparse, though two studies that have looked at this reported no signs of increased anxiety or depression after disclosure (Grill et al., 2020; Largent et al., 2020).
In San Diego, several speakers presented data that strengthened the case for people taking this knowledge in stride. For example, Claire Erickson of the University of Wisconsin-Madison described a study of 101 cognitively healthy people with an average age of 72, many of whom had a family history of AD. They take part in longitudinal cognitive studies at UW and had undergone PiB PET scanning. After receiving training on the meaning of a positive and negative scan, they learned their results. Afterward, 88 percent said learning their status was useful; none expressed regrets.
Lindsay Clark at UW-Madison examined whether the knowledge changed these participants’ behavior. Before disclosure, all had received lifestyle counseling and set goals for making healthy changes, such as exercising more, eating better, improving sleep, and lowering stress. Six months later, 90 percent of participants had made some progress on these goals. The researchers found no difference in this progress between people who learned they had a positive and those who had a negative amyloid scan. The only difference in behavior between these two groups was that those with a positive scan reported slightly less social activity afterward, possibly indicating some withdrawal or fear of stigma.
“This can be empowering information,” Erickson said. However, she noted that this cohort was highly educated, informed, and engaged in research, hence was not representative of a general clinical population.
Other studies found study participants to be eager to learn biomarker results. Sara Feldman of the University of Michigan, Ann Arbor, described a cohort of 57 participant and partner pairs. Their average age was 74, they were racially diverse, and nearly half had amnestic MCI. In interviews, most said they wanted to learn their imaging, fluid, and genetic results, with 82 percent seeing no downside to doing so. Participants cited benefits such as the ability to participate in clinical trials, to make long-term care plans, and to adopt healthier habits. Of those who perceived negatives, most cited lack of treatment.
Likewise, Fred Ketchum of UW reported that in a telephone survey of 145 cognitively healthy African Americans in their mid-60s, 72 percent said they would be willing to tell their partner their amyloid scan results. Six percent expressed concerns about worrying their partner, another 6 percent feared social stigma (Ketchum et al., 2022).
“People want this information,” said Annalise Rahman-Filipiak of UMich. She said it will be important to develop educational materials that explain what biomarker results mean, and to assess whether people with MCI and dementia are able to understand and make decisions based on those results. Many researchers in this field participate in the AGREEDementia working group, which develops best practices for risk disclosure. It remains to be seen how well these findings translate to general clinical use, where people are less familiar with medical research.
Gathering Real-World Data
Because many questions remain unanswered about how anti-amyloid immunotherapy will work in practice, the Alzheimer’s Association has brought together a collective to track the benefits and harms of these drugs, as well as those of future disease-modifying AD therapies. Dubbed ALZ-NET, the registry is hosted by the American College of Radiology, with the American Society of Neuroradiology and Brown University’s School of Public Health providing additional support and data analytics, respectively, and the Critical Path Institute helping ensure the registry's data pass regulatory muster (Nov 2021 conference news). ALZ-NET has three co-primary investigators: Rabinovici, Michael Rafii of USC, and Maria Carrillo of the Alzheimer’s Association.
In San Diego, Rafii said that the study’s protocol has been finished and approved, and the IT infrastructure, including electronic case report forms, is in place. The first seven clinics have signed on and are ready to start enrolling. They are Butler Hospital in Providence, Rhode Island; the Neurology Center of New England in Foxboro, Massachusetts; Abington Neurological Associates, Pennsylvania; Coastal Neurology in Port Royal, South Carolina; First Choice Neurology in Aventura, Florida; Neurostudies Inc. in Port Charlotte, Florida; and Genesis Neuroscience Clinic in Knoxville, Tennessee. These sites represent a range of different types of practice, including psychiatric, neurology, and geriatric, Rafii said. Most of this first group are private practices, but hospital and academic clinics are joining, as well. More sites are needed, and interested clinicians can apply online to sign up. ALZ-NET plans to expand internationally in time.
How will it work? Patients will enter the study when they agree to receive anti-amyloid immunotherapy at a participating clinic. Physicians will collect a slew of baseline data, including demographics, medical history, APOE genotype, lifestyle factors, AD symptoms and diagnostics, and brain imaging and fluid biomarkers. In order to compare data between participants, the study requires either the MMSE or MoCA for a cognitive, and the FAQ for a functional assessment; other tests, including the AD8 and NPI-Q, are optional.
Sites will enter follow-up data on each participant every six months for the first two years, and annually thereafter. Safety will be a major focus, including MRI findings, adverse events, and hospitalizations. Participants will also be followed with amyloid and tau PET and FDG-PET. Clinics will partner with imaging sites, which can offer recommendations on detecting and managing ARIA. Patients will stay in the study until they choose to withdraw, or die. Even if they stop immunotherapy, study personnel will continue to track their long-term health.
ALZ-NET aims to answer questions such as how the effects of immunotherapy change over years, who responds best, and how well the treatment works in a diverse real-world population. In the future, as more disease-modifying AD treatments are approved, the data will help compare effectiveness of different drug classes, Rabinovici said. Data will be shared with the research community through USC’s Laboratory of Neuro Imaging (LONI) and the Global Alzheimer’s Association Interactive Network (GAAIN). The registry will work with drug sponsors to gather data they need, for example for Coverage with Evidence Development studies required by the Centers for Medicare and Medicaid Services.
“This is a paradigm shift. ALZ-NET will bring AD care into the modern era,” Rabinovici said.—Madolyn Bowman Rogers
Part 1 of 2
Intuitively, we all know that a life of movement, social connection, and a nutritious diet is good for us. But can lifestyle and behavioral interventions really stave off dementia in people whose cognition has already started to slide? While a definitive answer remains hard to come by for these types of studies, findings presented at the Alzheimer’s Association International Conference, held July 31-August 4 in San Diego, favor a “yes.”
A network of multimodal intervention studies is in full swing around the globe, with some reporting promising findings at the meeting. The Australian “Maintain Your Brain” study, which delivers digital personalized coaching in physical activity, diet, and brain health, boosted cognition in its participants over three years. In EXERT, a large Phase 3 trial that compared light versus moderate exercise in people with MCI, cognition held stable in both groups over a year. The study lacked a control group, but a matched group of participants from ADNI declined in that same amount of time, suggesting that both interventions may have done some good. A small South Korean study called SUPERBRAIN-AD also reported promising findings from its suite of lifestyle interventions among cognitively impaired people with amyloid plaques. Together, the findings suggest that participants respond well to both in-person and virtual formats of intervention, and that social contact and support, even when delivered remotely, plays a hand in the benefits.
Multimodal Intervention at Work Around the Globe
“Maintain Your Brain” and SUPERBRAIN-AD are part of the World Wide FINGERS network, a set of multimodal intervention trials. Each tailored to local culture and needs, the network started up around the globe in response to the success of the original FINGER trial, aka Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (Aug 2017 news; Rosenberg et al., 2020). The poster child of lifestyle modification studies, FINGER had boosted cognition and fended off chronic disease among participants in its intervention group (Jul 2014 conference news; Nov 2015 news; Marengoni et al., 2018).
At AAIC, scientists showed progress updates on six World Wide FINGERS network studies. Four are ongoing, hence had no outcome data to report. They are U.S. POINTER, MIND-China, the Latin American Fingers Initiative, and MET-FINGER—a version of FINGER with the drug metformin added in. Two of the studies had outcomes to report, and they were positive.
Maintain Your Brain (MYB) was one such study. It tested the cognitive effect of coached, multimodal interventions delivered entirely via a digital platform. Henry Brodaty of the Center for Healthy Brain Aging at the University of New South Wales in Sydney presented the primary outcome findings. Participants were recruited to MYB from the 45 and Up Study, Australia’s largest ongoing cohort study that includes some 250,000 participants, representing about 10 percent of the population of New South Wales (Bleicher et al., 2022). Initiated in 2005 to inform researchers and policy-makers about ways to improve health among an aging Australian population, the study tracks multiple factors related to healthy aging, including cardiovascular health, disabilities, social connections, housing and economic status, hospital visits, and medication use, to name a few. MYB recruited dementia-free, 55- to 77-year-olds from this cohort, who had at least two risk factors for dementia.
In the first year of the three-year trial, all participants received four 10-week modules to help them achieve healthy lifestyle changes such as physical activity, a Mediterranean diet, brain training exercises, and an online training program to reduce stress, anxiety, and depression. The experimental group received coaching for each of these modules. The coaching was personalized to each participant’s risk profile at baseline. The control group received static information for each module, based on publicly available Australian health guidelines.
For the next two years of the trial, participants received “booster sessions” for each module—once monthly for the coached group and once every quarter for the control group—to motivate them to keep up the healthy habits they’d learned. The researchers invited 96,418 participants to join the MYB study; 6,236 were ultimately enrolled and randomized equally to coaching versus control groups. Fewer than half—2,959 participants—completed the cognitive assessment at baseline and annually for all three years of the trial.
How did they do? The MYB’s primary outcome was change on a cognitive composite measured online with tests from COGSTATE and Cambridge Brain Science. On this composite, which included tests of visual memory, executive function, processing speed, and working memory, the coached group significantly outperformed the control group at years 1, 2, and 3.With its effect size of 0.1, the intervention was equivalent to delaying decline by one year, Brodaty said. For comparison, the effect size of the primary outcome for the FINGER trial was 0.04 over two years, he added.
“This was fresh data that none of us had seen before,” said Maria Carrillo of the Alzheimer’s Association following Brodaty’s talk.
Miia Kivipelto, Karolinska Institute, Stockholm, who had led the FINGER trial, was happy to see data suggesting that a digital intervention was effective. However, she noted that in FINGER, the social, in-person component of the intervention seemed crucial to the trial’s success.
“MYB trial results are impressive,” agreed Hiroko Dodge of Oregon Health and Science University in Portland. She told Alzforum that while the trial did not target socialization per se, participants in the experimental group likely received plenty of social interaction through coaching, adding, “I assume that this social interaction component played some role in the shown efficacy and also helped maintain high adherence.”
Dodge herself headed the I-CONECT study, which also presented outcome findings at AAIC. That trial found that even minimal social contact, delivered via phone or video chat, may bolster cognition and feelings of social connectedness among people with MCI. For details on those findings, read Part 2 of this series.
Laura Baker of Wake Forest University School of Medicine in Winston-Salem, North Carolina, presented the topline results of the EXERT study, an exercise trial with social benefits. Coordinated by the Alzheimer’s Disease Cooperative Study (ADCS), EXERT tested how exercise affects cognition among sedentary people with MCI. Participants were randomized into two intervention groups. One involved moderate-intensity aerobic exercise; the other, stretching, balance, and range-of-motion exercises. Both groups exercised at local YMCA health centers, where they met with coaches who had been trained to work with people with MCI. Participants were prescribed a 30- to 40-minute session four times per week—twice with their trainers and twice on their own. During the supervised sessions, the trainers asked participants specific questions to gauge how well they had complied with their solo assignments.
EXERT had 148 participants each in the stretching/balance/range of motion (SBR) and the aerobic exercise groups. Of the total 296, 257 started the program and completed at least one cognitive evaluation; 229—114 in the SBR and 115 in the aerobic group—completed the 12-month trial.
Baker reported no significant difference between the groups on primary outcome, which was change from baseline on the ADAS-Cog-Exec, a cognitive composite tailored for the trial that emphasizes executive function. By this measure, the trial missed its primary endpoint.
The researchers had anticipated this possibility, as neither intervention group was a control. To gauge how their participants’ cognitive trajectory might measure up to the natural course of decline, the researchers compared it to that of ADNI volunteers selected to match EXERT participants by age, sex, education, baseline cognitive status, and ApoE genotype. Baker reported that while these ADNI participants declined on the ADAS-Cog-Exec over 12 months, neither of the EXERT groups did. The difference between each EXERT intervention group and the matched ADNI group were statistically significant.
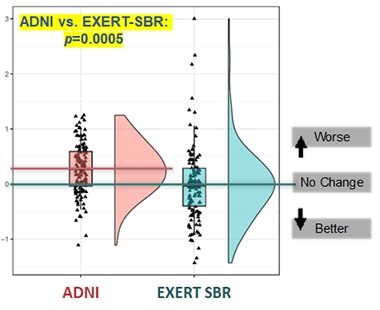
EXERTion Holds Off Decline? Cognition held stable for a year among participants in both EXERT intervention groups (stretching, balance, range of motion shown here); a closely matched group of ADNI participants slid in that time. [Courtesy of Laura Baker.]
Baker told Alzforum that in addition to remaining cognitively stable, EXERT participants maintained the size of their hippocampi throughout the study, whereas the hippocampus shrank over a year in the matched ADNI participants. Following the 12-month phase of supported exercise, participants were asked to continue their exercise routine on their own for six more months, and after that time underwent a final cognitive assessment. Those findings, along with blood biomarker measurements for Aβ, p-tau, and tau, will be presented at the Clinical Trials on Alzheimer’s Disease meeting in November, Baker said.
While the comparison to matched ADNI participants supports the idea that both interventions may have worked, it is not proof. Why didn’t EXERT include a typical control group? The answer comes down to right and wrong, and practicality. “It did not feel ethical to put people with MCI into a ‘do-nothing’ control group,” Baker said. She also feared that participants in such a control group might disproportionately drop out, realizing they were getting no benefit. Finally, Baker said that it was no easy task to recruit hundreds of people with MCI to participate in an exercise program four times per week for a year. “We had to do some selling,” Baker said. Guaranteeing that every participant would receive a bona fide intervention, including personal attention from a trainer, was a major draw for recruitment, Baker said.
Baker said that many of the participants developed meaningful relationships with their trainers, noting that during the COVID-19 pandemic, when gyms closed for a time and in-person sessions paused, many participants voiced concern about their trainers retaining their jobs. Many participants seemed hungry for social connection. When the whole study had to pause due to the pandemic, researchers called the participants every week to encourage them to continue their exercise at home. “Many said that this was the only phone call they got all week long,” Baker said.
If both groups did benefit, was it due to the exercise, to the social interactions with their trainer, or perhaps to the cognitive stimulation of leaving the house four times a week? Baker thinks it’s possible all of these factors contributed, although it’s difficult to tease them apart. In past studies in which people with MCI were given a trainer once per week and asked to exercise on their own three times per week, many only showed up for the sessions with the trainer. “I feel pretty confident that we would not see protection among people with MCI without support,” Baker said. “Unless there’s social engagement, they won’t do it.”
To Dodge, the finding that both intervention groups experienced the same cognitive stability suggests that factors besides physical exercise itself, such as socialization, might be playing an important role in the benefit. “Combining the recent I-CONECT results along with the findings of the EXERT trial suggests that enhancing socialization might be a good prevention strategy to delay cognitive decline,” Dodge said.
Besides EXERT, Baker also co-leads Protect through a Lifestyle Intervention to Reduce Risk (U.S. POINTER). This Phase 3 trial evaluates the combined power of physical and mental exercise, a healthful diet, and close management of cardiovascular health on cognition. The trial has enrolled about 1,600 participants out of a planned 2,000 so far, Baker told Alzforum, and results are expected in 2025.
In the meantime, Jeff Katula of Wake Forest updated AAIC attendees about how the COVID-19 pandemic, which emerged just months after the trial began, influenced design and adherence. The trial's interventions had been geared toward in-person meetings and exercise but, after a five-month hiatus at the start of the pandemic, the trial switched to an all-virtual format. It essentially yo-yoed back and forth between virtual and in-person formats as COVID-19 surges rose and fell. Participants weathered these changes remarkably well, adhering just as well to their regimens regardless of format, Katula reported. In fact, according to FitBit data, participants were slightly more active during virtual stints. In all, the findings suggest that even intensive, multimodal intervention studies can adapt to challenges like a global pandemic, and that participants are highly responsive to virtual communication, Katula concluded.
Finally, a small South Korean study posted promising results at AAIC. Eun Hye Lee of Ewha Womans University School of Medicine in Seoul presented findings from SUPERBRAIN-AD. This is an Alzheimer's-focused follow-up study to the previous SUPERBRAIN lifestyle intervention study (Park et al., 2020).
SUPERBRAIN-AD recruited 46 amyloid-PET-positive participants with MCI or mild dementia, and randomized them into three groups—two interventions and one control—for an eight-week trial. One intervention group received a suite of in-person lifestyle support, including help with managing metabolic and vascular risk factors, cognitive training and social activity, physical exercise, nutritional guidance, motivational enhancement, as well as a nutritional supplement drink. The second intervention group only received the nutritional supplement, a multivitamin drink with added ingredients purported to boost brain function, including omega-3 fatty acids, medium-chain triglycerides, phosphatidyl serine, and disodium 5' uridylate. The control group was put on a waitlist for future intervention, and received a booklet of lifestyle guidelines to prevent dementia.
The primary outcome was change in RBANS total score at eight weeks. Lee reported that participants in both the multimodal and supplement groups improved their scores over eight weeks, while the control group declined. The multimodal group improved substantially more than the supplement group. According to Lee, participants in the multimodal group also outperformed those in the other groups on secondary outcome measures, such as the Korean MMSE, and on exploratory measures including a boost in healthy bacterial species inhabiting the gut.—Jessica Shugart
No Available Comments
No Available Further Reading
Part 2 of 2
Alzheimer’s and related dementias are diseases of aging, yet the oldest among us tend to be excluded from most studies aiming to find treatments for them. Either directly, via exclusion criteria in drug trials, or indirectly, because they are homebound, frail, or lack family members to drive them to appointments, people older than 80 are both the most cut off from interventions to delay dementia, and arguably in greatest need of them. The I-CONECT project, led by Hiroko Dodge of Oregon Health and Science University in Portland, arose in response.
This internet-based conversational engagement trial gave people older than 75 easy-to-use tablets, through which they held regular video chats for a year. A control group received a brief weekly phone call to check in. At the Alzheimer’s Association International Conference, held July 31 to August 4 in San Diego, Dodge and other OHSU scientists reported that cognitive scores improved in people with mild cognitive impairment who chatted regularly via video, while feelings of social connectedness grew among all participants, even those who received only a weekly call.
The findings support the idea that social contact can stave off cognitive decline, said Dodge. She believes social engagement delays cognitive decline not by thwarting neuropathology, but by bolstering cognitive reserve. Functional MRI scans from a small subset of I-CONECT participants hint that a boost in synaptic connectivity may underlie this.
Other findings presented at AAIC supported the benefit of exercise, and of multimodal lifestyle interventions, in warding off cognitive deterioration (see Part 1 of this story). Even among these more intense interventions, it seemed that social engagement played a part. Might a simple conversation via video chat four times per week provide some benefit on its own? In a session dedicated to I-CONECT at AAIC, researchers presented findings on primary, secondary, and exploratory endpoints of this Phase 2 trial.
I-CONECT targeted socially isolated people with normal cognition, or with MCI, older than 75. The experimental group took 30-minute video calls for 12 months. Participants chatted with trained interviewers who used images and prompts to stimulate a free-flowing conversation. They occurred four times per week for the first six months, and twice weekly after that. This group also received once-weekly 10-minute phone calls to monitor their social activities, health, and mood. The control group only received these weekly check-in calls. A quick screen of cognition, as per the Montreal Cognitive Assessment (MoCA), served as the primary outcome. Secondary outcomes included measures of executive function, emotional well-being, and episodic memory, while changes in speech, medication adherence, brain connectivity, and cerebral blood flow were among exploratory outcomes.

Prescribed Chatting. I-CONECT participants talked with conversation staff several times per week for a year. The staff used images and prompts to break the ice, then let conversations flow naturally. [Courtesy of Hiroko Dodge, OHSU.]
Out of more than 1,100 telephone screenings, the researchers managed to recruit 186 people—86 who were cognitively normal and 100 with MCI—into the trial. Dodge said that the trial aimed to recruit African Americans, and that this group was particularly reticent to join. Ultimately, the trial enrolled 20 percent black participants, a higher proportion than most prior clinical trials.
What did the scientists find? First, the COVID pandemic interfered with the trial. Though its intervention was via video chat, the cognitive assessments were being conducted in person before the pandemic started, and over the phone after that. However, the telephone version of the MoCA—called “blind MoCA”—eliminates all items that require visual engagement, therefore its scores are not directly comparable to the in-person test, Dodge explained. Following recommendations from their data safety monitory board, the researchers limited assessment of the primary outcome to findings from 56 people who had finished their six-month assessment before the pandemic.
Among this much smaller group, which included 25 people with normal cognition and 31 with MCI, the researchers found a benefit of video chats among participants with MCI. While MoCA scores declined in the control group over six months, scores in the video chat group improved, resulting in a 1.75-point higher six-month score relative to controls. This difference is equivalent to the effect of 10 years of aging, Dodge said. At 0.73, the Cohen’s d value—a measure of effect size—was quite high. A boost in the memory domain drove this MoCA result, Dodge reported. Participants with normal cognition did not benefit.
Due to the MoCA mismatch, Dodge did not present 12-month MoCA scores. She did report outcomes on secondary measures of category fluency, immediate recall, and delayed recall among the 186 people who completed the trial. Among the 100 with MCI, Dodge reported a 2-point bump in immediate recall scores at 12 months in people who received the video chats relative to weekly calls. Among cognitively normal participants, Dodge reported a 2.56-point benefit of the video chat intervention at six months (but not at 12 months) in category fluency, a measure of executive function. Not all cognitive measures improved at both time points, nor were they corrected for multiple comparisons. Still, Dodge believes the findings suggest a benefit of social contact for older people, particularly those with MCI. OHSU’s Meysam Asgari reported a boost in the complexity of speech used by participants in the video chat group.
OHSU's Kexin Yu presented findings on psychosocial well-being, a secondary outcome measured using the NIH Toolbox Emotional Battery. Yu reported that neither video chatting nor phone calls influenced the participants’ negative affect, which includes feelings of anger, sadness, stress, and fear. However, measures of social satisfaction improved in both over the course of the 12-month trial. Yu interpreted the findings as indicating that even a weekly 10-minute phone call can boost feelings of connectedness.
Patrick Pruitt of OHSU presented functional connectivity findings collected from a small subset of I-CONECT participants. He hypothesized that social engagement influences network connectivity in the brain. If true, this could be picked up via fMRI. After excluding low-quality scans caused by participants wiggling in the scanner, the analysis netted 50 fMRI scans at baseline but just 15—six in the weekly call group and nine in the video chat group—at both baseline and six months. Pruitt detected an uptick in connectivity within the dorsal attention network (DAN) in the video chat group relative to the weekly phone call group. Connectivity in other networks, including the default mode, executive control, and salience networks, did not differ between groups. Furthermore, Pruitt reported that at baseline, the level of connectivity within the DAN, but not any other network, correlated with scores on participants' delayed and immediate story recall.
In discussion, David Morgan of Michigan State University in Grand Rapids said he was impressed that MoCA scores rose with social engagement. “As someone who’s been working to reduce the neuropathology of AD, my expectation is more of a stabilization of the disease symptoms, rather than an improvement,” Morgan said. That participants improved suggests that non-pathological effects, such as a boost in network connectivity, might underlie the benefit, he added.
Barry Greenberg of Johns Hopkins University in Baltimore, agreed. He added that behavioral and lifestyle interventions should be considered in conversations about combination therapy, which typically focus on different drugs. “A combination therapy could include cardiovascular health, an anti-pathology medication, together with these types of social interactions,” he said.
Morgan raised the idea of one day using robots to provide the social stimulus instead of live human beings, but Greenberg was skeptical. “We’re a social species,” he said. “The benefit of this intervention comes from the human interaction, with the full range of emotional expressions,” he said. For her part, Dodge agreed that artificial intelligence has come a long way in mimicking human interactions. It remains to be seen if the technology would be “human-like enough” to benefit socially isolated older people.
Whether delivered by a person or artificial intelligence, social interactions could prove to be a cost-effective way to delay the onset of dementia in populations most at risk for imminent decline, Dodge said. “Even delaying the onset of symptoms by a year would have an enormous impact,” she said. —Jessica Shugart
No Available Comments
No Available Further Reading
How early in the course of Alzheimer’s disease can a blood marker detect amyloid pathology, and which marker does it best? At this year’s Alzheimer’s Association International Conference, held from July 31 to August 4 in San Diego, California, and online, presenters converged on the same conclusion after comparing markers head to head: Plasma p-tau231 rises soonest after Aβ42/40 begins to fall in cerebrospinal fluid or after the locus coeruleus begins to atrophy. On the other hand, p-tau217 and p-tau181 tick up later, but they better track with amyloid plaques, neurofibrillary tangles, and cognitive decline. “With different p-tau forms changing at different time points during the AD process, there is the (theoretical) possibility of staging patients, but more studies are needed to confirm if this indeed is useful,” Henrik Zetterberg of the University of Gothenburg, Sweden, told Alzforum. People with multiple p-tau isoforms in their CSF also had more amyloid, tangles, and cognitive decline than those who tested positive for just one isoform.
Nicholas Ashton, also at UGot, previously reported that p-tau231 rises in the blood at the first signs of plaques on amyloid PET scans and that it predicts worsening on the Mini-Mental State Examination over one year (Feb 2021 news). At last year’s AAIC, he reported that plasma p-tau231 also rose when the Aβ42/40 ratio in the CSF fell, which happens years before plaques can be detected by PET and decades before cognitive symptoms (Aug 2021 conference news). But the marker had never been directly compared with other p-tau fragments in the blood, nor measured in many normal older adults. How would the markers compare, especially among cognitively normal people who might be on the cusp of early AD pathology?
Ashton and colleagues, led by Kaj Blennow of UGot and Marc Suárez-Calvet, Barcelonaβeta Brain Research Center, Spain, expanded their analyses to include the three most promising p-tau markers—181, 217, and 231—testing them in a large cohort of older, cognitively normal adults who are at risk for AD.
Ashton and Marta Milà-Alomà of Barcelonaβeta focused on 397 volunteers in the ALFA+ cohort, which comprises middle-aged volunteers who have normal cognition but a family history of dementia. One hundred thirty-five tested amyloid-positive by their CSF Aβ42/40. Of 339 volunteers who had an amyloid PET scan, 53 had signs of Aβ aggregation, having a Centiloid score above 12, while 26 of those were deemed to have established amyloid plaque pathology with a score above 30. A subset of 214 participants had taken the Preclinical Alzheimer Cognitive Composite test at baseline and at an average of three years later.
Because they had only cross-sectional data, the researchers used amyloid burden as a proxy for disease progression. As expected, p-tau231 was the first plasma p-tau marker to increase. Whether correlated to CSF Aβ42/40 or amyloid PET, it reached a concentration two standard deviations higher than that in amyloid-negative people before any other marker (see image below). P-tau217 rose next, followed by glial fibrillary acidic protein (GFAP), a marker of astrocytosis. Neither p-tau181 nor the neurodegeneration marker NfL crossed the 2-standard-deviation threshold. The researchers concluded that p-tau231 is the earliest p-tau marker of amyloidosis.

Early Riser. Using CSF Aβ42/40 (left) or Centiloids (right) as a proxy for disease progression (yellow), plasma p-tau231 (orange) ticks up earliest in AD. It is also the first to become abnormal, as defined by the marker level being two standard deviations above concentrations in people with normal CSF Aβ42/40, i.e. a ratio greater than 0.10. [Courtesy of Milà-Alomà et al., Nature Medicine, 2022.]
P-tau231 also had the tightest correlation with CSF amyloid positivity after the scientists adjusted for age, sex, and APOE4 status, with an area under the curve of 0.81. AUC is a statistical measure of sensitivity and specificity, with 1.0 being perfection.
“This work builds on the existing literature to understand the temporality of the blood-based AD-related biomarkers among middle-aged adults,” wrote Michelle Mielke, Wake Forest University, Winston-Salem, North Carolina (full comment below). Ashton and colleagues published their results in the August 11 Nature Medicine (Milà-Alomà et al., 2022).
What about cognitive decline? In her poster, Milà-Alomà showed that amyloid-positive people who had high p-tau231 or p-tau181, but not high p-tau217, in their plasma at baseline did worse on the PACC three years later. Among amyloid-negative participants there was no correlation with any baseline p-tau fragment and the PACC, although cognitive scores did trend down in people who had high baseline p-tau181. Indeed, considering both amyloid positives and negatives, Milà-Alomà found that the relationship between PACC change and plasma p-tau was driven by amyloid status only for p-tau231 (see image below). This marker, specifically, seems to associate with preclinical cognitive decline driven by early amyloid pathology, Milà-Alomà told Alzforum.
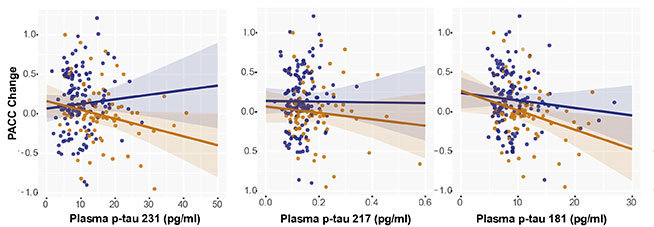
P-tau and PACC. In cognitively normal adults who tested positive for amyloid in CSF analysis (tan), high baseline plasma p-tau231 (left) and high p-tau181 (right) predicted decline on the PACC over three years. In amyloid-negative adults, there was no correlation (blue). Ditto for either group with p-tau217 (middle). [Courtesy of Marta Milà-Alomà, Barcelonaβeta Brain Research Center, Spain.]
Next, Milà-Alomà focused on 33 ALFA+ participants whose PACC score had slipped the most. In these, high baseline plasma p-tau181 and p-tau217—but not p-tau231—correlated with PACC change. Why not p-tau231? Though the small sample might be unreliable, Milà-Alomà hypothesized that the decliners had worsened to a disease stage where p-tau231 was less predictive than the other p-tau markers. This would be in keeping with p-tau231 rising then reaching a plateau before the other tau isoforms. She is now correlating how the plasma markers change over time with cognition.
Ashton did just that in two other cohorts, measuring the longitudinal change in plasma markers and MMSE scores. Oskar Hansson of Lund University, Sweden, presented the results at AAIC. The scientists, including Shorena Janelidze and Niklas Mattsson-Carlgren at Lund, measured the same six plasma markers—p-tau181, 217, and 231, and Aβ42/40, GFAP, and NfL—at baseline and an average of three times over six years among people who were cognitively normal or had signs of dementia at baseline. They analyzed samples from 388 cognitively normal participants and 187 people with mild cognitive impairment in the Swedish BioFINDER cohort and from 161 cognitively unimpaired participants from the Wisconsin Registry for Alzheimer’s Prevention. WRAP participants are at risk of developing AD because at least one parent had the disease.
First, the researchers divided participants into five groups based on baseline amyloid PET. Once again, the baseline data hinted that p-tau231 rises first in AD. This marker was high among people who clocked in with as few as 12 Centiloids, while p-tau217 or p-tau181 did not budge until Centiloid levels hit 36.
What about longitudinal change? Over the six years, p-tau217 rose the most of all the isoforms in amyloid-positive, cognitively normal people and in those with MCI who were amyloid-positive. The increase associated with accelerated cortical thinning and worse decline in MMSE scores (see image below). Again, why no correlation with p-tau231? Because it plateaued after early amyloid accumulation, while p-tau217 continuously rose with plaque load, Hansson noted.

P-tau217 Tracks Change. Compared to plasma p-tau231 (left) and p-tau181 (right), p-tau217 (middle) rose more in PET amyloid-positive (blue) versus negative (gray) cognitively unimpaired people (top row). In the amyloid-positive participants, cortical atrophy (middle row) and cognition (bottom row) worsened faster in people who had the highest plasma p-tau217 change (pink) than in those with the average (turquoise) or less change (purple). [Courtesy of Oskar Hansson, Lund University, Sweden.]
All told, both ALFA+ and BioFINDER/WRAP studies support the idea that plasma p-tau231 is the earliest isoform to detect amyloidosis in cognitively normal people, while p-tau217 and 181 are better predictors of longitudinal cognitive decline.
This held in yet more cohorts. At AAIC, Pamela Ferreira from the University of Pittsburgh reported that, compared to Aβ42/40, GFAP, and NfL, p-tau231 best indicated who among 138 cognitively normal volunteers in the Translational Biomarkers of Aging and Dementia (TRIAD) cohort in Montreal, Canada, were amyloid-positive by PET. The AUC was 0.85. Ferreira did not measure p-tau217 or p-tau181. Further, Hansson recently reported that among people with Down's syndrome, plasma p-tau217 correlates with plaques, tangles, and faltering cognition in people who do not yet have dementia (Jul 2022 news).
P-tau Predicts Atrophy, Degree of Pathology
Can plasma p-tau predict more about the brain than amyloid positivity? Milà-Alomà and Ashton correlated various tau isoforms in the plasma with the accumulation of plaques in different brain regions. They found that plasma p-tau231 and p-tau217 most strongly associated with the earliest PET signals, including those in the orbitofrontal cortices, cingulate gyri, insula, and precuneus.
Similarly, Heidi Jacobs, Massachusetts General Hospital, Boston, tied p-tau231 to early brain atrophy. She correlated plasma p-tau231, p-tau217, p-tau181, total tau, Aβ42/40, and NfL with high resolution 7T structural MRI scans and PACC scores from 99 cognitively normal people ages 30 to 85 in an MRI cohort study based at Maastricht University in the Netherlands (Van Egroo et al., 2021).
As in the ALFA+ cohort, Jacobs found that high plasma p-tau231 correlated with low PACC scores. From the MRI scans, she could see that the marker also tracked with atrophy in the locus coeruleus, picking up withering there in people as young as 55. This tiny area within the brain stem is one of the first sites to accumulate hyperphosphorylated tau in AD, and it shrinks 8.4 percent with each Braak stage (Braak et al., 2011; Theofilias et al., 2016). “These volumetric changes reflect the accumulation of tau pathology and loss of neuronal projections to the cortex, which may impact how the locus coeruleus communicates with cortical brain systems,” Jacobs said during her presentation.
Stijn Servaes of McGill University in Canada took a different approach to studying the various tau isoforms, asking what, if anything, the number of different p-tau isoforms in the CSF might reveal. First, using amyloid PET and CSF data from 60 amyloid-positive and 107 amyloid-negative TRIAD participants, he ran a machine-learning algorithm to establish cutoffs for amyloid positivity for different p-tau isoforms. Next, Servaes used those cutoffs to ask how the number of positive p-tau isoforms relates to plaques, tangles, and cognition.
Among 59 people with MCI or AD, and 73 age-matched controls, Servaes found that the more CSF p-tau species a person tested positive for, the higher their plaque and tangle load and the lower their MMSE score. People positive for two or more p-tau isoforms were amyloid-positive. Those positive for three different p-tau isoforms also tested positive for tangles. Finally, people with four p-tau species in their CSF had MMSE scores of 28 or less. The findings suggest that the different p-tau isoforms reflect cumulative damage in the brain.
Scientists hope to capitalize on these latest findings to develop better markers for AD diagnosis and staging. Researchers could choose which marker to measure based on which stage of preclinical AD they want to study, Suárez-Calvet said. For example, selecting people with high plasma p-tau231 would enrich for those who are just beginning to accumulate amyloid, while selecting for high p-tau217 would target those farther in the clinical trajectory at the threshold of robust plaque deposition. However, Mielke does not think it is that straightforward. “We are not at the point yet to determine what p-tau species should be used at each specific stage of the disease, either for diagnosis or prognosis of AD,” she cautioned.
This is, in part, because plasma assays are not yet reproducible or standardized (Oct 2021 news; Mielke et al., 2021). “[This is] a major problem in the field right now,” wrote Suzanne Schindler at Washington University in St. Louis. “It is difficult to conclude which analyte is best without considering assay performance. For example, it is possible that plasma Aβ42/40 changes before p-tau231, and the “earlier” changes in p-tau231 are artifactual because the p-tau231 assay is more sensitive or precise than the Aβ42/40 assay,” she added (comment below). For more on plasma marker development, see Part 4 of this series.—Chelsea Weidman Burke
Like so many others in her extended family with the misfortune of inheriting the pathogenic E280A variant in presenilin-1, Aliria Rosa Piedrahita de Villegas seemed destined to descend into Alzheimer's dementia before reaching her 50th birthday. Instead, she lived with her memory intact into her 70s, and passed away a month before her 78th birthday with only mild symptoms of dementia. Piedrahita de Villegas’ remarkable resilience likely stemmed from her inheritance of not one, but two copies of ApoE3 Christchurch—a rare, and apparently protective, mutation in this AD risk gene. But how did this apolipoprotein variant defend Piedrahita de Villegas against AD for nearly three decades? Some clues come from the postmortem examination of her brain, published in Acta Neuropathologica on July 15. Francisco Lopera of the Universidad de Antioquia in Medellín led the work, together with Kenneth Kosik of the University of California, Santa Barbara, and Yakeel Quiroz of Massachusetts General Hospital in Boston.
Speaking at the Alzheimer’s Association International Conference, held July 31-August 4 in San Diego, California, Lopera drew parallels between Piedrahita de Villegas from Angostura, Colombia, and Auguste Deter, a woman from Frankfurt, Germany, who died in 1906 at the age of 55 with what would later become known as Alzheimer’s disease, her brain riddled with plaques and neurofibrillary tangles. “These two women are most important in the study of Alzheimer’s disease,” Lopera said. “Auguste showed us how the brain is affected in AD, and Aliria showed us how the brain can be protected.”
The study identified an enormous burden of Aβ plaques, but an odd distribution of tau tangles. Piedrahita de Villegas’ frontal cortex was relatively free of tangles, while her occipital cortex was replete with them. Single-cell transcriptomics revealed that, intriguingly, regions of her brain that had managed to fend off tangles expressed more ApoE. Hinting at the power of the Christchurch variant, the researchers noticed that neurons considered exquisitely vulnerable to tau-induced neurodegeneration were doing fine in the frontal cortex and hippocampus. Tangle-free brain regions contained homeostatic astrocytes, while the occipital cortex was crawling with microglia of a distinctively inflammatory profile. Together, the findings suggest that the protective ApoE variant somehow severs an inflammatory cord linking Aβ plaques to tau tangles, buying at least this one homozygous carrier decades of life without dementia.
“This global resilience in the face of one of the most aggressive forms of AD underscores the apparently profound effect the APOE gene has on the time course of the AD pathophysiological process,” wrote Jacob Vogel of the University of Pennsylvania in Philadelphia, Rik Ossenkoppele of Amsterdam University Medical Center in the Netherlands, and Oskar Hansson of Lund University, Sweden, in a joint comment to Alzforum.
To neuropathologist Lea Grinberg of the University of California, San Francisco, the study speaks to the enduring importance of autopsy examination. “Methodological improvements in the last years have greatly enhanced the potential and impact of research using the human postmortem brain,” she wrote. “Hopefully studies like this will help to highlight the value of investing in autopsy programs in ADRD research.”

Affected, Protected. Auguste Deter (left) was the first woman to be diagnosed with what would later become known as Alzheimer’s disease. Aliria Rosa Piedrahita de Villegas was protected from the pathogenic AD mutation she carried by her unlikely inheritance of two copies of ApoE3-Christchurch mutation. [Courtesy of Francisco Lopera.]
At the age of 73 and again at 75, Piedrahita de Villegas traveled to Boston to undergo brain scans. Much to the shock of researchers at the time, her brain contained a higher amyloid burden than that of any other carrier of the PSEN1-E280A variant, also known as the Paisa mutation, within the extended Colombian kindred (Nov 2019 news). However, tau-PET scans revealed a minuscule tangle burden, confined mostly to the medial temporal lobe and occipital cortex. Similar to a different protective variant—ApoE2—ApoE3Ch poorly bound to low-density lipoprotein receptors, and hardly latched on to heparin sulfate proteoglycans (HSPGs). These extracellular matrix proteins are thought to facilitate tau propagation, offering a potential mechanism underlying the scant tau tangle burden in her brain.
Piedrahita de Villegas developed short-term memory loss at the age of 72, and was diagnosed with mild dementia at age 75. She died from malignant melanoma at 77. Within 200 minutes of her passing away at her home, her brain was removed and carefully prepared for neuropathological examination. This examination was done by scientists across the world. Co-first author Diego Sepulveda-Falla of University Medical Center Hamburg-Eppendorf, Hamburg, Germany, was one of them. A neuropathologist who has examined the brains of many Paisa mutation carriers, Sepulveda-Falla recalled what he felt when Piedrahita de Villegas’ extremely unlikely—and serendipitous—combination of mutations was first discovered several years ago. “I was surprised that she existed at all,” Sepulveda-Falla said. “Aliria and her family were very aware of how unique she was.”
What did the neuropathologists find in her brain? First, in keeping with her mild dementia, her brain was smaller than a typical 77-year-old’s, leading the Colombian scientists who removed the brain to classify it as having severe global atrophy. Atherosclerosis marred all major blood vessels. Co-first author Justin Sanchez of Massachusetts General Hospital in Boston and colleagues used immunohistochemistry to measure Aβ and tau pathology in 17 brain regions. In agreement with PET scans taken during life, Sanchez detected abundant Aβ plaques throughout the neocortex.
The distribution of tau tangles told a different story. Due to extensive p-tau accumulation in the isocortex, the researchers classified the pathology as Braak stage VI; alas, the overall distribution of tangles did not fit into the Braak staging model. The occipital cortex, which is typically affected in later Braak stages, had the greatest density of tangles, followed by the hippocampus and amygdala, which tend to deposit tangles much earlier on. Most strikingly, the frontal cortex, which typically becomes invaded by tangles in Braak stages V-VI, contained negligible tau pathology. Sepulveda-Falla noted that although tangles were found in the entorhinal cortex and hippocampus, their burden in these early Braak regions was mild compared to that observed in other Paisa mutation carriers.
This occipital-leaning distribution of tau pathology evokes a “posterior” subtype of AD, which is associated with a slower course of cognitive decline. It was recently reported by Vogel, Ossenkoppele, and Hansson (Apr 2021 news). “Future work will be needed to ascertain whether this similarity is a coincidence or if there is a consistent link between occipital phenotypes and slower clinical progression,” they wrote.

Aβ and Tau—A Severed Tie? Amyloid plaques (left panels) crowded Aliria Rosa Piedrahita de Villegas’ frontal cortex, and dotted her hippocampus and occipital cortex. P-tau aggregates were nearly absent from her frontal cortex, but abundant in her hippocampus and even more so in the occipital cortex. See map of p-tau regional intensity on right. [Courtesy of Sepulveda-Falla et al., Acta Neuropathologica, 2022.]
The scientists conducted a detailed accounting of the different morphological forms of each neuropathology throughout the brain. One result of this analysis? The occipital cortex was the lone neocortical region that contained Aβ plaques within blood vessels, aka cerebral amyloid angiopathy. CAA also cropped up in the amygdala and, where found, its extent correlated with the burden of tau pathology. This narrow range of CAA stands in contrast to the much broader distribution of CAA spotted in most other Paisa mutation carriers. The link between CAA and tau meshes with another recent postmortem study that tied CAA to tau and cognitive decline (Rabin et al., 2022).
Besides looking at Aβ plaques and tau tangles, the researchers also used immunohistochemistry to measure protein levels of ApoE and of glial markers. ApoE expression took on a plaque-like pattern, and was most intense in the hippocampus and occipital cortex. The microglial marker Iba1, along with the activation marker CD68, were both cranked up in the occipital relative to frontal cortex. Conversely, TMEM119, a marker of homeostatic microglia, gave the highest signal in the frontal cortex. The ratio of CD68 to TMEM119 was higher in the occipital relative to the frontal cortex, suggesting that microglia residing in occipital regions, where the tangles also were, tended to be more riled up than those in frontal regions.
ApoE3-Ch: Protector of Vulnerable Neurons, Blocker of Tangles?
To explore which molecular mechanisms might have been at play in different cell types, scientists in Kosik's lab conducted single-nuclei RNA sequencing from cells in the hippocampus, and frontal and occipital cortices. This analysis brought out different transcriptional clusters of excitatory and inhibitory neurons. Strikingly, a transcriptional cluster emerged of excitatory neurons marked by high expression of RORB, but only in the hippocampus and frontal cortex. RORB+ excitatory neurons earlier this year were reported to be exquisitely vulnerable to tau-induced neurodegeneration (Jan 2021 news).
Grinberg, a senior author on that study, commented that both studies highlight the importance of this neuronal class for AD pathogenesis. That these supposedly vulnerable neurons had been alive and well in the brain of a person with mild AD could reflect protection by the ApoE3-Ch variant, Sepulveda-Falla suggested. These RORB+ neurons abundantly expressed genes involved in neurodevelopment, while containing a dearth of transcripts encoding genes involved in synaptic function.
What about glia? Working with a single brain, the researchers had limited numbers of cells for the analysis, relying on 364 microglia, 598 astrocytes, and 1,250 oligodendrocytes to tell the tale of differential gene expression across three brain regions. They identified a single transcriptional cluster for each cell type, and then compared the expression levels of individual genes in each cell type by region. This identified subsets of genes that were differentially expressed in the frontal and occipital cortices. ApoE was prominent among them. In both astrocytes and microglia, ApoE expression was highest in the frontal cortex and lowest in the occipital cortex, with intermediate expression in the hippocampus. In other words, the more ApoE glia expressed in a given region, the less tau tangled there.
Curiously, another study, by Victor Montal of Hospital de la Santa Cru in Spain and Jorge Sepulcre-Bernad at Massachusetts General Hospital in Boston, recently uncovered the opposite association, reporting that regions of the brain with higher ApoE expression contained more tangles (Jul 2022 news). However, Montal and Sepulcre-Bernad believe that findings from the two studies complement each other. Both tied ApoE expression to tau accumulation. While ApoE3-Ch warded off tau tangles, common ApoE variants might beckon them to form. The MGH study did not distinguish between ApoE variants.
Sepulveda-Falla and colleagues also checked whether other genes might follow ApoE’s regional expression pattern in each glial cell type. For astrocytes, a cadre of homeostatic genes tracked closely with ApoE expression, such that astrocytes residing in the frontal cortex had a more homeostatic profile than astrocytes in the occipital cortex.
In microglia, genes involved in immune regulation tracked with ApoE expression. Focusing on microglia, the researchers found distinct profiles of immune genes expressed in the frontal and occipital cortices. These profiles did not match perfectly with those reported in mice. That said, microglia in Piedrahita de Villegas’ occipital cortex assumed a profile resembling a chronic inflammatory state reported in a mouse model of amyloidosis, while those in her frontal cortex expressed a suite of genes involved in acute immune responses (Sep 2017 news).
Sepulveda-Falla said it’s unclear what these different microglial profiles mean, and whether they were beneficial or harmful given the distinct neuropathological issues confronting microglia in each region. In the frontal cortex, microglia were dealing with extensive Aβ plaques, while in the occipital cortex, the cells faced both plaques and tangles.
Piedrahita de Villegas did ultimately develop characteristic symptoms of AD. That she did despite her atypical tangle distribution raises questions about the relationship between tau pathology and the clinical manifestations of AD. “Whatever was driving cognitive impairment in this case seems not to be defined by the distribution of tau pathology,” Sepulveda-Falla said. “Pathology is not the whole story.” He also wondered what role Piedrahita de Villegas’ malignant melanoma may have played in bringing on her Alzheimer's symptom. Though there were no tumors in her brain, systemic health crises are known to speed up the timeline of AD and other dementias.
Sepulveda-Falla and other neuropathologists are still carrying out more extensive comparisons of tau pathology in this unique brain to brains of AD cases who had other combinations of ApoE variants, including heterozygous carriers of ApoE3Ch. For more on the life and death of this remarkable woman, see Jennie Erin Smith's NYT story. —Jessica Shugart
The first reports from AMYPAD, a large European study of amyloid PET scans, agree with previous studies: The technology strongly affects Alzheimer’s diagnosis. At the Alzheimer’s Association International Conference, held July 31 to August 4 virtually and in San Diego, California, AMYPAD leaders said the scans changed doctors’ diagnosis of one in three people who came to memory clinics with cognitive complaints. However, during a six-month follow-up period, the scans made no difference in the patients’ quality of life, nor reduced their use of medical services, raising questions about the cost-effectiveness of this expensive imaging tool.
AMYPAD data also confirmed that a positive scan, or even subthreshold amyloid accumulation, predicts future cognitive decline. This decline occurred mostly in a person’s memory, with little impact on executive or visuospatial function. Other talks at AAIC noted that the scans’ predictive power can be sharpened by adding tau PET and MRI. In small studies, the majority of cognitively normal participants with positive amyloid and tau scans and hippocampal shrinkage on MRI developed mild cognitive impairment over the next two to three years, compared with about 10 percent of those who were positive for amyloid only. This strengthens the case for using biomarkers of amyloid, tau, and neurodegeneration in the clinic to determine prognosis, speakers said.
Better Diagnosis, But Not Better Health
Several previous studies have assessed how amyloid PET affects clinical care. In the U.S., IDEAS reported that scans changed treatment plans in two-thirds of its 11,409 participants (Aug 2017 conference news; Nov 2018 conference news; Apr 2019 news). Smaller European studies found a lesser benefit, with about one-quarter of diagnoses changed (Nov 2016 news; Jun 2018 news). Even so, the clinical usefulness of amyloid scanning has not been fully established. Additional studies, such as IDEAS 2 focusing on underrepresented minorities in the U.S., are ongoing.
In San Diego, Daniele Altomare of the University of Geneva offered a first look at AMYPAD data. The project consists of a diagnostic and patient management study (DPMS) and a prognostic and natural history study (PNHS). DPMS investigated the clinical use of amyloid scans (Frisoni et al., 2019). It recruited 840 people who sought an evaluation at one of eight participating memory clinics in France, Germany, the Netherlands, Spain, Sweden, Switzerland, and the U.K. between April 2018 and October 2020. Nearly all were white.
In this cohort, 244 people had subjective cognitive decline, 341 MCI, and 255 dementia. More cognitively impaired patients tended to be older, with average ages of 69, 72, and 75 years, respectively, for the three groups. The SCD group had somewhat more years of education on average than the other groups, while the dementia group was more depressed than the others. These findings are typical for memory clinic patients (Altomare et al., 2022).
Participants were randomized to one of three arms. In the first, they underwent PET scanning right away as part of their diagnostic workup, and had a follow-up scan after 12 to 18 months. In the second, they were scanned after eight months. In the third, physicians chose whether and when to use amyloid scanning; on average, this group underwent scanning six weeks after coming to the clinic. For the main outcome measure, researchers compared physicians’ initial diagnostic impression with the diagnosis they had settled on three months later.
In arm 1, diagnosis changed in 44 percent of cases. In arm 3, where PET scans were done slightly later, diagnosis changed in 29 percent of cases, not statistically different from arm 1. This contrasts with arm 2, which had no PET scans at this point; there, diagnosis changed in 11 percent of cases. Thus, about a third more of the cohort had an altered diagnosis when PET scans were part of their workup. Diagnostic changes were almost always consistent with the PET results, Altomare said. He noted that physicians altered their assessment more often after a negative than a positive scan, suggesting the biomarker is particularly useful for ruling out AD.
Physicians became more confident in their diagnosis once they had a scan in hand. In arm 1, physicians expressed high confidence in 40 percent of their diagnoses after three months. In arm 3, this was 37 percent; in arm 2, 11 percent.
Alas, a presumably more precise diagnosis did not translate to any effects on health outcomes over six months. Patients in each arm rated their own quality of life similarly, and beyond the cost of the PET scans themselves, the three arms were the same in their use of healthcare resources. This implies that PET scans are not cost-effective over the short term, Altomare acknowledged. In answer to an audience question, he suggested that prescreening with blood biomarkers could help manage costs. He speculated that longer-term follow-up might yet reveal more health differences. The study will continue to track participants for at least a year after their baseline visit.
What is the psychological impact of disclosing amyloid PET scan results to people? The scientists found that positive scans caused some distress, with participants reporting they had more intrusive thoughts and hyperarousal afterward, and avoided thinking about the results. These symptoms did not reach the threshold for clinical concern, and were lower than those reported after learning of other serious health conditions, such as cancer, Altomare said. The findings reinforce other studies suggesting that people can handle learning their amyloid status (see related conference story).
Predicting Progression
Separately, AMYPAD examined the prognostic prowess of amyloid PET (Lopes Alves et al., 2020). The PNHS part of AMYPAD enrolled more than 1,500 participants, most of whom were at the preclinical or early prodromal stage of AD. In San Diego, David Vállez García of Amsterdam UMC reported preliminary findings from the first 1,044 participants. These came from six research cohorts: EPAD, the ALFA+ study at Barcelonaβeta Brain Research Center, FACEHBI in Spain, EMIF Twin 60+ and EMIF 90+ in the Netherlands, and DELCODE in Germany.
Upon amyloid scanning, 650 of these participants proved to be amyloid-negative, with centiloid values around zero. Another 267 were in a “gray zone,” with an average of 10 centiloids of plaque. The remaining 127 were amyloid-positive, with varying amounts of amyloid plaque that averaged more than 50 centiloids. As might be expected, the amyloid-positive group was older, more cognitively impaired, and more likely to carry an APOE4 allele than the others.
Tracking participants’ cognition for up to six years, the scientists found that the amount of amyloid a person had at baseline predicted his or her global cognitive decline on the MMSE, as well as decline on tests of immediate recall, recognition, and visual memory. It also predicted functional decline on the iADL, as well as on one measure of attention, but had no impact on executive or visuospatial function over this time frame. On the MMSE, immediate recall memory, and attention tests, decline got steeper over time.
As expected, demographic factors such as age, sex, and education played into how fast a person declined. Education was protective, age made decline worse, and sex had complex, mixed effects. Having an APOE4 allele hastened slippage on the MMSE. Vállez García plans to further analyze how these risk factors interact with plaque burden to influence prognosis. All AMYPAD data will be made available to the research community via the ADDI FAIR platform.

Predictive Power. In four different cohorts—PREVENT-AD, HABS, AIBL, and Knight ADRC (left to right)—people who were A+T+N+ were most likely to progress to MCI (red), while the majority of people who were only A+ remained cognitively healthy (blue). [Courtesy of Cherie Strikwerda-Brown.]
The PNHS findings dovetail with other studies shown at AAIC. Cherie Strikwerda-Brown of McGill University, Montreal, presented findings from four longitudinal studies of people who were cognitively healthy at baseline. The full cohort included 251 people seen at the Knight ADRC in St. Louis, 153 in the Harvard Aging Brain Study, 128 from PREVENT-AD in Montreal, and 48 from the Australian Imaging, Biomarker, and Lifestyle (AIBL) study. At baseline, participants underwent amyloid and tau PET and volumetric MRI as the measures for the current ATN classification of preclinical AD. Because each of the four studies used somewhat differently methodology, Strikwerda-Brown, working with Sylvia Villeneuve at McGill, analyzed each separately.
Over up to three years of follow-up, people who had all three markers—amyloid, tau, neurodegeneration—were the most likely to develop mild cognitive impairment, while those with none were the least. In PREVENT-AD, 57 percent of A+T+N+ individuals developed MCI; in HABS, 71 percent; in AIBL, everyone did. In the Knight ADRC, which used a CDR score of 0.5 or more to measure disease progression, 43 percent worsened. By contrast, people who were positive for amyloid, but not tau or brain atrophy, tended to stay stable, with fewer than one in 10 going on to MCI. On sensitive cognitive measures such as the RBANS and the PACC5 cognitive composite, A+T+ participants diverged from the others, declining much faster. Strikwerda-Brown concluded from this that the ATN criteria have prognostic value.
Likewise, Karly Cody of the University of Wisconsin-Madison reported that amyloid and tau both affect cognitive decline. UW researchers led by Sterling Johnson previously developed an amyloid “clock” to predict the rate of cognitive decline based on when a person became amyloid-positive (Oct 2019 news). To find out if adding tau PET to the model made this clock more accurate, Cody and Johnson followed 386 cognitively healthy participants who underwent PiB PET and MK6240 tau PET at baseline. These scans showed a high variability in amyloid burden and in tangle burden in the entorhinal cortex.
Over eight years, people who had had plaques for more than a decade, putting them well along on the amyloid clock, and also had a lot of tangles in their entorhinal cortices, lost cognition much faster than did the other groups. EC tangles mark an early phase of tau pathology, Braak stage 1 or 2. A synergistic model of amyloid and tau interaction best fit the data, suggesting that both pathologies interact to harm cognition, Cody said.
Other talks at AAIC turned the spotlight on tau, parsing what factors affect the spread of tangles through the brain (see related conference story).—Madolyn Bowman Rogers
No Available Comments
The spread of tau tangles through the brain of a person who is coming down with Alzheimer’s disease correlates closely with his or her cognitive impairment, making this pathology a top therapeutic target these days. Even so, scientists still know little about what controls this process and how plaques promote it, a knowledge gap made painfully clear at the Alzheimer’s Association International Conference, held July 31 to August 4 virtually and in San Diego, California.
Taking a crack at the problem, several speakers shared the latest findings from tau PET studies, which are beginning to disentangle the various influences that come to bear on propagation. One talk implicated p-tau as a link between plaques and tangles; others focused on where tau aggregation starts, claiming its initial epicenters dictate the speed at which it travels through the brain. Genetic factors may underlie some of the heterogeneity in tangle accumulation as well. Overall, the emerging picture suggests manifold influences on tau pathology and the rate of disease progression.
“We are pretty sure the connectomic architecture of the brain dictates the spreading pattern of tau pathology in AD patients. Now we need to understand its modulators,” Nicolai Franzmeier of Ludwig-Maximilians University in Munich wrote to Alzforum. “We can use the tools we developed for predicting spreading patterns to zoom in and study what drives or attenuates tau spreading. This may give us more insight into how we could prevent it.”

Key Connections. One hypothesis holds that when tangles (red) form in hub regions (circles) of the brain, where many axonal highways meet, they are able to travel more rapidly to new areas, resulting in fast-advancing disease. [Courtesy of Nicolai Franzmeier.]
As people age, tangles accumulate in their entorhinal cortices and nearby regions, but in cognitively healthy people, tangles rarely move beyond that. Numerous PET studies have shown that amyloid plaques somehow unleash tangles, allowing them to rampage through the brain (Mar 2016 news; Aug 2016 news; Feb 2018 news). But exactly how do plaques spur tangle formation in distant brain regions? Some studies have suggested inflammation or synaptic connections as mediators (Sep 2021 news; Apr 2022 news). A better grasp of the mechanisms could help researchers design more targeted therapies.
Could the Culprit Be P-tau?
At AAIC, speakers offered many ideas. One early consequence of plaque formation is a rise in certain isoforms of phosphorylated tau, particularly p-tau217 and p-tau181, in cerebrospinal fluid and plasma (Aug 2019 conference news; Mar 2020 news; Jul 2020 conference news). Recently, researchers flagged p-tau231 as the earliest marker of amyloid aggregation (see related conference story).
Alexa Pichet Binette, working with Oskar Hansson of Lund University, Sweden, made a case that it is p-tau217 that brokers the relationship between plaques and tangles. In collaboration with Franzmeier, Pichet Binette investigated links between these two key pathologies in the BioFINDER-2 observational cohort. Her analysis included 204 amyloid-negative controls, 130 amyloid-positive people without dementia—half with MCI, half without—and 66 amyloid-positive with AD dementia.
Pichet Binette considered two possible ways in which a participant's amyloid load might influence his or her brain: via regional plaque accumulation or via p-tau217. Both related to the rise in their tau PET signal over time, but p-tau217 appeared to be the driving force. When both factors were entered into the model, the effect of amyloid dropped out statistically, leaving p-tau217 as the main predictor for the spread of tau tangles. It accounted for 54 percent of the association between plaques and tangles in non-demented participants.
This relationship between p-tau217 and tangles waned as disease advanced. In people with dementia, whose plaque load and soluble p-tau had plateaued, p-tau217 no longer had any effect on tangle accumulation. Metaphorically, p-tau217 may be the spark that lights the local fire of tangles, but once the fire is big enough, it spreads on its own through a self-fueling process, Franzmeier told Alzforum. The findings point to soluble p-tau217 as a potential therapeutic target in early, but not late, AD, Pichet Binette said. The article is available in preprint form (Binette et al., 2022).
Meanwhile, Davina Biel in Franzmeier’s group went a step earlier, investigating how plaques boost p-tau in the first place. She focused on the microglial activation marker sTREM2, because it has been linked to the rise in p-tau (Jan 2016 news; Dec 2016 news; Pascoal et al., 2021). To examine longitudinal changes, Biel stratified data from 402 ADNI participants into three groups: 131 amyloid-negative controls, 70 people who were amyloid-positive by CSF but negative by PET, i.e., “early accumulators,” and 201 who were amyloid-positive by both, i.e., “late accumulators.” In ADNI CSF data, only p-tau181 was measured, as newer markers were not yet available.
Plaques were associated with elevated sTREM2 and p-tau181, but the nature of this relationship evolved with disease stage, as a statistical tool called mediation analysis showed. In early accumulators, sTREM2 mediated the effect plaques had on p-tau181. This implies that plaques prompt microglia to become active in ways that somehow stimulate phosphorylation of tau at position 181, or allow p-tau181's concentration to rise. However, in late accumulators, rather than boosting levels of p-tau181 further, sTREM2 seemed to weaken p-tau181's effect on hippocampal atrophy.
Franzmeier presented this work in San Diego. He said the findings hint that microglial activation could be harmful at early stages of AD, but possibly protective later. This seems to contradict previous cross-sectional data, including some from Franzmeier, that report less cognitive decline in amyloid-positive people with high sTREM2, suggesting microglia activity helps early in disease (Aug 2019 news; Mar 2022 news). The issue is murky, however, with another longitudinal study linking sTREM2 to faster atrophy in preclinical AD (Apr 2020 conference news). Franzmeier noted that protective effects of sTREM2 have mostly been seen in people who already have symptoms of AD and thus are later in the disease trajectory. Moreover, microglia may have mixed effects, curbing plaque growth while boosting p-tau. More research will be needed to disentangle microglia’s effects at different stages, and reconcile these cross-sectional and longitudinal findings. “It’s highly complex, and we are just starting to understand how microglia play into AD biomarker and clinical trajectories,” Franzmeier told Alzforum.
Location, Location, Location
P-tau217 might somehow promote tangles, but that still does not explain how tangles spread in stereotyped patterns through a person's brain. Other talks in San Diego dissected the anatomy of tau propagation. Deborah Schoonhoven, working with Alida Gouw of Amsterdam UMC in the Netherlands, tested three models, one each for functional connections, structural connections, or diffusion through gray matter. Schoonhoven used data from 82 participants in the Amsterdam Dementia Cohort. All reported memory problems, but 25 were amyloid-negative, 16 were amyloid-positive and unimpaired on normed tests, 16 had amyloid-positive MCI, and 25 had AD dementia.
Schoonhoven constructed models of each potential pathway of tangle spread, then used those to predict how aggregated tau would spread in each participant’s brain based on where their tangles were at baseline. Functional connectivity maps for each participant were derived from magnetoencephalography, which records the magnetic fields produced by electrical activity in the brain. Schoonhoven compared the model predictions to actual tau PET scans from each person.
The functional connectivity model was most accurate, with a correlation of r=0.58 with the scan. The other two models both correlated at r=0.45, showing a weaker relationship. This suggests that both are involved, but functional relationships between brain regions are more important than structural ones in determining tau spread. Schoonhoven noted that functional connections always require a structural underpinning, but functional mapping provides another layer of information by showing which brain connections are most active and firing together most often. Thus, functional maps vary slightly from structural ones.
Curiously, some previous work had reached the opposite conclusion, finding a primary role for structural over functional connections (May 2020 news). Schoonhoven said methodological differences between the studies, such as the use of magnetoencephalography in hers, might explain the apparent contradiction. She acknowledged that her findings do not preclude that tau aggregates physically pass between axonal connections. Instead, the functional data simply suggest that neuronal activity plays a key role in promoting spread, she told Alzforum. Previous studies have recorded an increase in tau release upon neuronal stimulation (e.g., Feb 2014 news).
For his part, Franzmeier homed in on the role of brain hubs. He said that observational studies suggest that tangles spread at different rates in different people, and show some variability in their pattern of spread. Could the deciding factor be where tangles start? Franzmeier hypothesized that if tau first aggregated in a less-connected brain region, tangles would spread slowly, whereas if the aggregates first deposited in a highly connected hub, they would run like wildfire through the brain.
To identify hub regions, he mapped brain connectivity using resting-state fMRI data from 1,000 participants in the Human Connectome Project. Then he compared longitudinal tau PET scans from 242 ADNI and 57 BioFINDER participants against this map.
As predicted, when tangles in symptomatic AD patients were in hubs, the person’s subsequent tau PET signal increased more rapidly over time. Younger participants were more likely to have tangles in hub regions, while older participants tended to have them in limbic regions. Franzmeier believes this makes sense, because hub regions are crucial for complex cognitive processes, and therefore people with tangles in these areas are likely to show symptoms at a younger age.
In these cohorts, mediation analyses showed that the presence of tangles in hub regions was responsible for the faster accumulation of abnormal tau in younger than older symptomatic participants (Frontzkowski et al., 2022). Previous studies already noticed this age effect, where tau pathology is less pronounced in older people with AD (e.g., Apr 2018 conference news). Franzmeier noted that the big question remains: Why does tau aggregation start in distinct brain regions in different people?
Do Our Genes Have a Hand in This?
One driver behind the variability of tangle spread could be genetics. Anna Rubinski, working with Michael Ewers of Ludwig-Maximilians University, examined the role of known AD risk factors in tau pathology. Ewers’ group had previously tied a BIN1 risk variant to faster spread, and a protective klotho variant to slower (Franzmeier et al., 2021; Neitzel et al., 2021). To look at genetic factors more broadly, Rubinski computed a polygenic risk score (PGS) from 85 GWAS SNPs tied to AD, excluding APOE. In a cohort of 231 ADNI participants, having a higher AD PGS correlated with more rapid increase in one’s cortical tau PET signal at all Braak stages after 1. This genetic acceleration effect was stronger in people who had more plaque.
The same PGS was also associated with faster cognitive decline. This was only partially mediated by its effect on tangle propagation, suggesting that genetic factors may exert independent effects on cognition. A PGS comprising 85 AD risk SNPs likely reflects multiple mechanisms.
What about APOE4? This risk allele has been linked to tau pathology in transgenic mice (Sep 2017 news; Apr 2021 news). Beyond that, however, few studies have tied ApoE to tau thus far, whereas ample evidence exists for its effect on amyloid deposition. Elizabeth Mormino of Stanford University, Palo Alto, California, used data from 392 amyloid-positive, cognitively healthy participants in the A4 secondary prevention solanezumab trial to dissect the effect of this gene on tangles. In this cohort, 207 people had at least one E4 allele, and 31 an E2 allele.
The E4 carriers accumulated more tangles in multiple brain regions, particularly the medial temporal lobe, than did E3 homozygotes, while E2 carriers had less tau burden compared to E3s. Surprisingly, little of this effect was mediated by amyloid plaques. Instead, APOE4 appeared to directly affect tangle burden, in agreement with the mouse data. That said, E2's power to lower tangles was greater than E4's sway toward more tangles, as seen in 13 participants who carried both alleles. This highlights APOE2 as a key protective mechanism, Mormino said.
Exactly how APOE alleles affect tangles remains to be determined. Although amyloid is the strongest driver of aberrant tau accumulation, APOE may directly influence tau clearance, with E4 promoting and E2 diminishing tangles in amyloid-positive people, Mormino suggested.
In the bigger picture, Mormino believes it will be important to learn why tangle spread is so heterogenous from one person to another, and why even within an individual’s brain, some regions are spared. “It is striking that many amyloid-positive, clinically unimpaired individuals have low levels of tau. It is possible that these individuals have protective factors that prevent or slow downstream tau accumulation. Understanding resilient individuals and resilient brain regions could pave the way for preventative strategies,” Mormino wrote to Alzforum.
One such case, that of Aliria Rosa Piedrahita de Villegas, was discussed in San Diego. Her APOE3 Christchurch mutation protected her from otherwise certain autosomal-dominant AD for nearly 30 years; in part, it seems, by keeping tangles at bay (see related conference story).—Madolyn Bowman Rogers
No Available Comments
Almost 30 years after its discovery as the most prominent risk factor for AD, ApoE continues to serve more questions than answers to the field. As evidenced by the many ApoE-focused presentations at this year’s Alzheimer’s Association International Conference, held July 31 to August 4 in San Diego, California, the brain’s deceptively humble lugger of lipids is still holding mechanistic surprises.
One theme focused on the old question of how ApoE4—the AD risk isoform—wreaks havoc in the brain. Researchers reported that when churned out by microglia, ApoE4 shuts down neuroprotective responses in the brain’s immune cells, thwarting their clearance of Aβ or tau pathology. Early findings hint that the lipids ApoE4 carries are poorly processed by lipoprotein lipase within microglia, potentially scrambling their metabolism and function. In the meninges, the primary producers of ApoE were found to be myeloid cells. In this triple-layered membrane sheath surrounding the brain, ApoE4 had the surprising effect of enlarging lymphatic vessels responsible for immune regulation and brain drainage. E4 also skewed the trajectory of age-related changes in cells that make up the brain’s vast blood-brain barrier, offering clues about how risk alleles might accelerate its erosion.
Although astrocytes produce the majority of ApoE in the brain, microglia can be provoked to churn out the apolipoprotein, too. In fact, ApoE is a critical component of microglial gene-expression signatures documented in the face of different neuropathological insults, including amyloidosis (Jun 2017 news; Sep 2017 news). Recent studies have found that the ApoE4 isoform exacerbates tau pathology and neurodegeneration in transgenic mice, but only when microglia are around (Oct 2019 news).
These and other findings cast microglia as the cells doing the damage at the behest of ApoE4, though they stop short of proving that microglia themselves are also the source of the ApoE4. In fact, a recent study led by David Holtzman at Washington University in St. Louis concluded that removal of ApoE4 only from astrocytes dampened microglial responses to tau, and partially quelled tau-induced neurodegeneration (Apr 2021 news).
At AAIC, Neta Rosenzweig, a postdoc in Oleg Butovsky’s lab at Brigham and Women’s Hospital in Boston, presented findings that directly addressed the question of what role microglial ApoE4 plays in AD. She used a menagerie of transgenic mice to investigate this question in different pathological contexts.
First, Rosenzweig riled microglia by injecting fluorescently labeled apoptotic neurons directly into the brains of ApoE3- or ApoE4-knock-in mice, which only express the human isoforms of the protein. Rosenzweig sorted out microglia that had gobbled up the fluorescent, dying neurons, looking at what genes they expressed. This showed her that while phagocytic microglia in ApoE3-KI mice had switched on the neurodegenerative expression profile, called MGnD, previously described by researchers in Butovsky’s group, those in ApoE4-KI mice failed to flip this transcriptional switch (Krasemann et al., 2017).
Marked by ramped-up expression of ApoE, TREM2, and a handful of other inflammatory and lipid metabolism genes, the MGnD profile resembles the disease-associated microglia (DAM) described by Ido Amit and Michal Schwartz at the Weizmann Institute of Science in Rehovot, Israel, where both Butovsky and, later, Rosenzweig, completed their graduate work (Keren-Shaul et al., 2017). One lingering question in the field has been whether these microglial transformations are beneficial or detrimental. In support of the former, Rosenzweig found that the MGnD-reticent ApoE4-KI microglia were also sluggish in approaching the injection site. Furthermore, they appeared inept at handling the debris they had consumed, as Rosenzweig spotted a backlog of internalized contents in their lysosomes.
Strikingly, none of these problems surfaced when Rosenzweig performed the same experiments in ApoE4-KI mice in which ApoE4 had been conditionally deleted from microglia. This suggested that ApoE4 produced by the microglia themselves was responsible for squelching productive responses.
Rosenzweig drew similar conclusions when she deployed this experimental paradigm in the APP/PS1 model of amyloidosis or in P301S-tau mice. In both transgenic mouse lines, each crossed to different ApoE-KI backgrounds, and microglia in ApoE4-KI mice failed to switch on the MGnD profile in response to the transgenic pathology—whether it was Aβ or tau. This exacerbated the accumulation of either of these pathologies in ApoE4-KI relative to ApoE3-KI mice. Conditional deletion of ApoE4 from microglia corrected these deficits, suggesting that ApoE4 produced by microglia, not astrocytes, was the source of the subpar microglial responses. In the APP/PS1 mice, Rosenzweig also tied microglial ApoE4 to poor recruitment of astrocytes to plaques. In the tau model, nixing ApoE4 from microglia even prevented neuronal loss.
MGnD Repression Relieved. In APP/PS1 mice expressing human ApoE4 (left), microglia (white) around plaques (green) did not express the MGnD marker Clec7a (red). Removing ApoE4 from microglia only (right) restored microglial Clec7a and boosted recruitment to plaques. [Courtesy of Neta Rosenzweig, Brigham and Women’s Hospital, Boston.]
In all, the findings suggest that the MGnD transcriptional state is a beneficial one, and that ApoE4 production by microglia stymies the cells’ transition into it, rendering them less adept at handling neuropathology, Rosenzweig concluded.
To Butovsky, the finding that the MGnD state is a protective one in AD answers what has been a “Holy Grail” question in the field. However, that does not mean that this microglial response is always helpful. In fact, Butovsky and colleagues recently reported that in the context of glaucoma, a disease in which the ApoE4 genotype is protective, the microglial transition is a harmful one. The repression of MGnD by ApoE4 may explain why the isoform is protective in that disease (Margeta et al., 2022).
Jason Ulrich of Washington University called the findings consistent with previous work demonstrating a central role of microglia in ApoE4-related neurotoxicity. In the case of tauopathy, ApoE4 could influence the interplay between tau pathology, microglia, and neurodegeneration either by directly augmenting neurotoxic microglial responses, by somehow triggering the tau-related insult in neurons that leads to a neurotoxic microglial response, or a combination of these. “It will be important going forward to dig deeper into the different cell biological processes that microglia engage in their reactive state in the degenerating brain to determine which functions may be damaging and which may be protective,” Ulrich wrote to Alzforum.
Importantly, Rosenzweig spotted an echo of the dampened MGnD switch in ApoE4 carriers with AD. Using bulk RNA sequencing of microglia from postmortem brain samples, Rosenzweig found that compared to microglia in ApoE3/3 carriers, those in ApoE3/4 carriers expressed fewer genes from the MGnD playbook.
Of course, microglia in the human brain do not faithfully recapitulate MGnD, DAM, or any other mouse microglial profile (May 2019 news). The trend to name different glial gene-expression signatures—each detected in distinct experimental conditions, mouse models, or human disease states—recently prompted scientists across the field to call for a moratorium on christening such states with names, urging researchers to stick to detailed transcriptional and functional descriptions instead (Jul 2022 news).
One commonality between MGnD, DAM, and other previously reported disease-associated microglial states is the upregulation of genes involved in lipid handling, and these genes go far beyond ApoE. For example, they include lipid receptors such as TREM2, and the lipid hydrolyzer lipoprotein lipase (LPL). At AAIC, Kimberley Bruce of the University of Colorado in Aurora focused on LPL. This gene features prominently among the upregulated genes in microglia during development and in various disease states (Loving and Bruce, 2020). LPL is a secretory protein that, outside of the CNS, hydrolyzes triglycerides found in lipoproteins.
What it does in the CNS is less clear, as triglyceride-rich lipoproteins are typically rare there, Bruce said. However, she proposed that LPL could work in concert with lipoprotein receptors in the CNS to internalize all manner of lipid-laden debris. Myelin is one of them, as Bruce previously reported (Bruce et al., 2018). Genetic variants in LPL implicate this enzyme in AD, with loss-of-function variants upping risk, and gain-of-function variants reducing it (Bruce et al., 2020). Bruce previously reported that nixing LPL from microglia triggered the cells to transition into a pro-inflammatory state. Lipid droplets built up in them and cholesterol efflux plummeted, rendering the cells incompetent at processing Aβ, as well as lipids including myelin (Loving et al., 2021).
At AAIC, Bruce showed preliminary findings linking LPL to ApoE within microglia. First, she measured how efficiently LPL hydrolyzed lipids associated with different ApoE isoforms in vitro. When lipids were buddied up with ApoE4, LPL had a hard time processing them into fatty acids. A similar story played out in cultured microglial cells, where the cells poorly hydrolyzed lipids carried by ApoE4 relative to those carried by other ApoE isoforms. Bruce proposed that LPL is a critical modulator of microglial metabolism and inflammation. Together with ApoE, it plays a role in AD pathogenesis, she believes.
While Rosenzweig’s findings suggest a role for microglial ApoE in the brain parenchyma, other findings presented at AAIC hint that ApoE churned out by myeloid cells in the meninges may affect lymphatic drainage in that realm. Five years ago, scientists made the surprising discovery that the meninges—the triple layer of membranes that cradle the brain—house a lymphatic drainage system (Oct 2017 news). With age, the drainage becomes more stagnant (Jul 2018 news). The meninges also host a lively hub for immune cells, which enter and exit the brain via lymphatic vessels and, like satellites, communicate with immune cells residing in the brain parenchyma (May 2021 news).
At AAIC, Sandro Da Mesquita of the Mayo Clinic in Jacksonville, Florida, reported early findings about the role of ApoE this meningeal environment. Da Mesquita conducted some of his studies as a postdoc in Jonathan Kipnis’ group at Washington University in St. Louis.
Comparing the morphology of vessels crisscrossing the meninges in ApoE3-KI versus ApoE4-KI mice, Nikoleta Delivanoglou, a postdoc in Da Mesquita's lab, saw that, on average, lymphatic vessels in E4 mice were longer and wider than those in E3 mice, although Da Mesquita told Alzforum that some vessels in E4 mice retracted instead. ApoE knockouts had no expanded vessels, suggesting that the effect was due to a gain of function by ApoE4, Da Mesquita said. Notably, these E4-induced morphological changes were only significant in male mice. The researchers are currently investigating the mechanisms involved in this sexually dimorphic effect, and how the restructuring of meningeal vessels influences lymphatic drainage.
Using a spatial-transcriptomics technique called RNAScope to pinpoint which cells were expressing ApoE in the meninges, Da Mesquita pinned the vast majority of ApoE expression on resident macrophages there. Depleting these myeloid cells with a CSF-1R inhibitor drastically reduced expression of ApoE in the meninges.

In Meninges, Macrophages Express ApoE. Using RNAScope to image mRNA in the murine dura, macrophages (in green) can be seen expressing ApoE (red). Blood endothelial cells lining vessels express Pecam1 (blue). [Courtesy of Sandro Da Mesquita.]
Da Mesquita said that ongoing single-cell RNA-sequencing studies in the lab aim to decipher ApoE4-specific gene expression changes in these myeloid cells and other cell types in the meninges. Preliminary findings hint that macrophages in the meninges are more activated in ApoE4-KI mice, and that ApoE genotype more heavily sways the gene-expression profile of these macrophages than it does any other meningeal cell type.
Finally, according to studies led by Berislav Zlokovic, ApoE4 can promote the erosion of the blood-brain barrier. Previously, Zlokovic, University of Southern California, Los Angeles, had reported that with age, the blood-brain barrier becomes compromised, or leaky, in the hippocampi of ApoE4 carriers, and that this happens in a manner independent of Aβ or tau pathology (Jan 2019 news; May 2020 news). This degradation was fueled by activation of the CypA-MMP9 pathway in pericytes, which was detectable in the cerebrospinal fluid by way of the pericyte-injury marker sPDGFRb. (May 2012 news; Montagne et al., 2021).
At AAIC, Zlokovic presented fresh data on how ApoE4 alters gene expression in BBB-bolstering cells with age. Using single-nuclei RNA sequencing of brain endothelial cells and pericytes from the BBB, Zlokovic reported that different sets of genes in capillary endothelial cells and pericytes started to become dysregulated with age in ApoE4-KI mice relative to those in ApoE3-KI mice. The scientists saw differential expression of genes involved in cell junctions, cytoskeleton, and clathrin-mediated transport, as well as dramatic changes in the proteome and phospho-proteome—i.e., the landscape of phosphorylated residues among proteins. Together, these expression data implied that fundamental changes in BBB function were already afoot in relatively young, 2- to 3-month-old ApoE4-KI mice. These BBB changes preceded the onset of synaptic and memory deficits in 7-month-old ApoE4-KI mice relative to their ApoE3-KI counterparts, Zlokovic reported. He did not address which cells might have released the BBB-eroding ApoE4. However, he said that in ongoing experiments, researchers in his lab are testing how removal of ApoE from different cell types—including microglia and astrocytes—influences the effects on the barrier imparted by ApoE4.—Jessica Shugart
No Available Further Reading
When it comes to data, “bigger is better” is no empty phrase. At the Alzheimer’s Association International Conference held last month in San Diego, California, scientists presented their first analysis of two of the largest single-nucleus RNA sequencing efforts in Alzheimer’s research to date. They included 1.2 million cells from 84 people in the Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD) and 1.6 million cells from 478 participants in the Religious Orders Study and Memory and Aging Project (ROSMAP). These massive datasets taught scientists which neuronal subpopulations died early in AD, showed a subsequent rise in glia, and tied gliosis to memory decline in a subset of people.
“It’s exciting that different groups see similar abundance and gene-expression changes in single-nucleus data from AD donors, especially with distinct patient cohorts and sampling strategies,” Kyle Travaglini, Allen Institute for Brain Science, Seattle, wrote to Alzforum. “It gives us more confidence that our observations are robust and could lead to a better understanding of the disease process.” Evan Macosko of the Broad Institute of Harvard and MIT in Cambridge, Massachusetts, agreed. “The consistency makes me very optimistic about the future of single-cell analysis as a tool for studying disease mechanisms,” he wrote to Alzforum (full comments below).
Researchers at the Allen Institute, University of Washington, and Kaiser Permanente Washington Health Research Institute recently released the first fruits of their collaborative SEA-AD efforts. Published on the SEA-AD website, this database includes genotyping, single-cell transcriptomics, single-cell epigenomics, and spatial transcriptomics on cells from the middle temporal gyrus. The MTG is a portion of the temporal lobe that is important for learning and memory. “We wanted to start with an area that degenerates in mid-stage AD,” Eitan Kaplan of the Allen Institute told Alzforum.
The brain tissue came from people 65 years and older in two well-characterized cohorts, i.e., that of the UWash AD Research Center and the Adult Changes in Thought study led by UWash and Kaiser Permanente. The scientists sorted volunteers by their AD neuropathological change (ADNC) score, ending up with nine controls sans pathology and 12, 21, and 42 with low, intermediate, and high pathology, respectively. ADNC is a composite of Braak neurofibrillary tangle stage, Thal amyloid plaque phase, and CERAD plaque/neuropsychological testing score.
Travaglini presented the MTG snRNA-Seq data. The 1.2 million nuclei fell into 139 transcriptionally distinct clusters of 24 different brain cell types. Travaglini measured the proportion of each cell cluster in each participant, then assessed how cell-type proportions changed as AD worsened as per ADNC score. He used scCODA, a Bayesian algorithm that accounts for differences in all cell types simultaneously and “knows” that all cellular fractions must add up to one (Büttner et al., 2021). “If the proportion of a cell population increases, then all others must decrease to compensate,” Travaglini explained.
So which cells changed as AD pathology got worse? The proportion of layer 2/3 excitatory neurons, as well as that of two groups of inhibitory interneurons, oligodendrocytes, and oligodendrocyte precursor cells, all declined. The proportion of microglia and astrocytes grew. This suggests that the former cell types may be particularly vulnerable to plaques and tangles, while the latter may respond to the pathology.
To learn about how pathological and transcriptional changes might relate to one another, Victoria Rachleff, also at the Allen Institute, first performed a quantitative neuropathology analysis on cortical slices from all 84 SEA-AD participants. She labeled the tissue slices with antibodies against the neuronal marker NeuN, the microglial protein Iba1, the astrocytic glial fibrillary acidic protein, as well as pathology markers 6E10 for Aβ, AT8 for p-tau, and antibodies against α-synuclein or TDP-43. Using a machine-learning algorithm, Rachleff divided images of the slices into cortical layers, and then calculated how much of each marker was in each layer.
The researchers computed z-scores for each neuropathology variable, comparing the degree of pathology in each person to the average of all participants. Grouping similar z-scores together revealed how and when cell types increased or decreased in number as AD progressed. This process ordered participants from controls to those with extensive pathology according to their ADNC score. With increasing AD pathology came fewer neurons, smaller nuclei, more Aβ and tau accumulation, and more microglia surrounding amyloid plaques (see image below).

Picture of Progression. Grouping similar quantitative neuropathology data (rows) per participant (columns) paints a spectrum of changes, generally going from controls (dark blue, top left) to low (light blue), intermediate (dark orange), then high (light orange) AD pathology. [Courtesy of Kyle Travaglini, Allen Institute.]
A particular subset of participants, who had high ADNC scores, caught the researchers’ attention. Compared to the others, these people's brain samples had yielded consistently fewer nuclei for isolation and snRNA sequencing. They also had less DNA available for transcription as detected by ATAC sequencing, which measures genome-wide chromatin accessibility. Travaglini does not think these shortcomings were technical issues, because these measurements tracked with the participants' neuropathology data. For example, a person who had low-quality RNA and lots of transcriptional repression also had fewer layer 3 cortical neurons and had declined faster on the memory portion of the Cognitive Abilities Screening test (see image below).

Who Faded Fast? The worse a brain sample's quality of postmortem RNA, the fewer cortical neurons it contained (left). Participants with “bad” RNA also had faster memory decline (right, red) than controls (gray), or people with similar AD pathology but more intact RNA upon isolation (blue). [Courtesy of Kyle Travaglini, Allen Institute.]
Next, the scientists excluded these severely affected participants and once again deployed scCODA to recalculate how the cell proportions changed with pathology for the remaining groups. The same neuronal loss showed up, but the glial alterations no longer did. “This suggests that there is an underlying process driving neuron loss in all AD participants, and a secondary, inflammatory process driving the glial changes and, perhaps, worse cognitive decline in the severely affected donors,” Travaglini said at the conference.
Travaglini is now analyzing other brain regions affected at different stages of AD, including the medial entorhinal, dorsolateral prefrontal, and V1 visual cortices.
Gilad Green, Hebrew University of Jerusalem, Israel, presented another snRNA-Seq atlas at AAIC. His encompassed 1.6 million cells from prefrontal cortex tissue of 478 ROSMAP participants spanning the clinical spectrum from healthy to AD. Their average age was 89 years.
Green used an algorithm to sort the cells into 96 populations based on their gene expression, then correlated each with the presence of amyloid plaques, tau tangles, and cognitive decline. In his analysis, all three outcomes were associated with absence of a subset of somatostatin-expressing inhibitory neurons and presence of disease-associated microglia expressing markers of the so-called DAM state, e.g., APOE, TREM2, and GPNMB. In contrast, a population of astrocytes Green called disease-associated (DAA) were linked to tangles and cognition but not to plaques. This suggests that neuron loss and a microglial response of the DAM type occurs before reactive astroglia of this “DAA” type appear. In other words, the data suggest these inhibitory neurons might have died before astrocytes got worked up.
Increasingly, scientists are characterizing diseased glia by their gene expression, rather than using the acronyms of earlier studies (Jul 2022 news).
For a more complete sense of cellular changes during AD pathogenesis, Green measured the proportion of each cell population within each participant, summed them into a composite value, and then figured out how these whole-brain cell populations were related to one another, and to AD pathology. For this he used a pseudotime algorithm. To find out what that is, and what Green discovered, see Part 19 of this AAIC series.
Besides generating new, ever-larger RNA-Seq datasets, scientists are also finding success in compiling existing datasets into one big pool for more powerful analyses (see Part 18 of this series).—Chelsea Weidman Burke
No Available Further Reading
As some scientists create huge, new, single-nucleus RNA-sequencing datasets, others devise ways to better use existing ones. At the Alzheimer’s Association International Conference held last month in San Diego, California, Evan Macosko of the Broad Institute of Harvard and MIT in Cambridge, Massachusetts, presented his work combining small databanks into a large one. Akin to doing a meta-analysis, this approach boosts statistical power, allowing Macosko to test if transcriptome changes tied to AD progression are consistent across cohorts.
Macosko and colleagues first turned to a unique cohort by Ville Leinonen, Kuopio University Hospital, Finland. Leinonen and colleagues have collected prefrontal cortex biopsies taken from 700 adults with normal-pressure hydrocephalus, a condition where excess cerebrospinal fluid swells the brain ventricles. It is treated by placing a drainage shunt into the ventricles, and this procedure allows scientists to take a tissue sample during surgery. The tissue provides a snapshot into the living brain without the peri- or postmortem artifacts that can compromise RNA stability and skew transcriptional signatures.
The researchers chose samples from older participants, because some happened to have amyloid plaques and neurofibrillary tangles. By their neuropathology scores, 29 samples had neither (A-T-), 21 had plaques but no tangles (A+T-), and eight had both (A+T+). “The beauty of this dataset is that it allowed us to see cell vulnerabilities and transcriptional signals occurring at the earliest stages of amyloid accumulation in brain tissue,” Macosko wrote to Alzforum. SnRNA-Seq of these samples yielded 1.1 million cells that clustered into 92 populations.
This high-quality dataset subsequently guided cell population annotations of another 1.3 million cells from 29 published human and mouse snRNA-Seq datasets. Sixteen of those came from postmortem cortical tissue, six were from AD cohorts. This yielded a total of 2.4 million cells. To align them, the scientists used two analysis tools, LIGER, which the Macosko lab had developed, and Harmony. Both helped them cluster cells by type across these 30 datasets, by way of genetic markers (see image below; Welch et al., 2019; Korsunsky et al., 2019).
Reshuffle by Cell Type. Algorithms align cell clusters from different datasets (colors) to generate one cluster for each cell type (symbols). [Courtesy of Evan Macosko, Broad Institute.]
Which AD-linked cell changes jumped out of this combined dataset? At the earliest stage, i.e., in cases that had few amyloid plaques but no tau tangles, three neuronal populations tended to be already depleted from the cortex relative to pathology-free tissue: layer 1 inhibitory interneurons, layer 2 excitatory neurons, and layer 4 excitatory neurons. Moreover, interneurons from these amyloid-positive samples expressed fewer genes.
Notably, the excitatory neurons had been previously reported as depleted in entorhinal cortex tissue affected by AD, while the inhibitory ones had never been reported as MIA in AD, according to Macosko. Kyle Travaglini, Allen Institute for Brain Science, Seattle, said that he also saw these layer 1 interneurons die off, though interneurons in layers 2 and 3 degenerated more in his dataset. The Seattle Alzheimer’s Disease Brain Cell Atlas (SEA-AD) represents a new, large snRNA-Seq effort, containing 1.2 million cells from 84 people across the AD pathological spectrum (see Part 17 of this series).
In samples that had cortical tau tangles in addition to amyloid plaques, dubbed later-stage AD, microglial changes took center stage. Homeostatic CX3CR1-expressing microglia were depleted, while disease-associated microglia expressing the marker LPL were abundant. Not every individual dataset contained evidence of these DAMs because, on their own, they lacked the statistical power to detect such small cell subpopulations, Macosko said.
What about transcriptional changes across disease severity? In other words, do these disease-associated microglia and the vulnerable neurons express the same or different genes in early and later-stage samples? In this analysis, DAMs had the same differentially expressed genes (DEGs), many of which function in interferon signaling, throughout the progression of AD captured here, with expression higher in later-stage samples.
In contrast, layer 2 and 4 excitatory neurons changed. They overexpressed DEGs related to glycolysis, handling of reactive oxygen species, and synaptic vesicle processing—but only at the early stage. “This suggests that neurons are being taxed with hyperexcitability and are trying to manage and maintain homeostasis, which peters away at later stages of disease,” said Macosko. During later-stage AD when plaques and tangles are present, the same upper-layer excitatory neurons downregulate synaptic and neurotransmission genes, perhaps foretelling the impending loss of these neurons.
All told, Macosko thinks that the vanishing of specific inhibitory interneurons may cause overexcitability early in AD, eventually leading to the well-described neuron loss at later stages. Hyperactivation in early affected people has been described at other levels of investigation, for example by fMRI, but not at a single-cell transcriptional level (e.g., Dickerson and Sperling, 2008).
More analyses of these cumulative datasets are yet to come. “By incorporating more data as it comes out, we hope to make more and more insights into AD biology using this integration and analysis platform,” Macosko said.
Following SEA-AD’s lead, Macosko plans to make this dataset publicly available, together with tools that allow researchers to integrate their own snRNA-Seq data into the database.—Chelsea Weidman Burke
No Available Comments
No Available Further Reading
As the mountain of single-nucleus RNA-sequencing data grows taller, how can scientists extract meaning from it? One way is pseudotime analysis. In essence, this algorithm orders cells on a virtual timeline based on the similarity of their gene-expression patterns. “Cells that are alike are placed near each other along the spectrum of transcriptional changes,” explained Laura Heath of Sage Bionetworks in Seattle. Heath presented one of several pseudotime analyses currently being done in the Alzheimer's field at the Alzheimer's Association International Conference, held last month in San Diego, California.
The resulting diagrams look like trees. Scientists call branches healthy or diseased based on their expression of known markers. This, in turn, places each cell along the health to disease trajectory, exposing sequential gene expression patterns.
Pseudotime analysis allows scientists to turn cross-sectional data into “faux” longitudinal data to understand how cells change over time. This is important for Alzheimer’s, a disease that unfolds over the course of 30 years. Postmortem tissue offers but a snapshot of one time point, making it hard to discern when and how disease markers develop. Most brain transcriptomic data come from postmortem samples and are likewise difficult to interpret because it is hard to know if gene-expression changes are due to AD pathogenesis or organ damage associated with the end of life.
“Pseudotime trajectories offer a computational approach to model [transcriptomic changes], which can serve as a starting point for more detailed studies,” wrote Gregory Carter, Jackson Lab, Bar Harbor, Maine. Maria Wörheide agreed. She works at the Helmholtz Center Institute of Bioinformatics and Systems Biology in Munich. “Manifold learning algorithms, such as pseudotime, applied to cross-sectional data, have shown potential to provide novel insights into AD, although their robustness and scalability will require further investigation,” she wrote (full comments below).
At AAIC, four scientists showed how they use pseudotime analysis to wrangle RNA-Seq data. Two followed transcriptomic changes in astrocytes, describing a continuum of homeostatic to reactive cells in both healthy aging and AD. One plotted entire prefrontal cortex transcriptomes onto pseudotime trees, and one tied transcriptomic pseudotime to metabolomic changes in the AD brain.
Astrocyte Continuum
As a test of the methodology, Heath and colleagues first performed a pseudotime analysis on bulk RNA-Seq data from postmortem brain tissue of healthy and AD cases in the Religious Orders Study and Memory and Aging Project (ROSMAP) cohort (Mukherjee et al., 2020). “The modeled trajectory beautifully recapitulated neuropathology and clinical disease states, such that control samples were near the beginning and AD samples at the end,” she explained at AAIC.
Next, Heath used the algorithm on published single-nucleus RNA-Seq data from 80,500 prefrontal cortex cells of 48 ROSMAP participants, half controls and half AD, as well as 1.2 million cells from the middle temporal gyrus of 84 people ranging from healthy to AD in the new Seattle Alzheimer’s Disease Brain Cell Atlas (May 2019 news; see Part 17 of this series).
Aiming to draw pseudotime trajectories for each cell type, Heath first focused on astrocytes. She collected the transcriptomes of 3,400 astrocytes from the 48 ROSMAP participants and 47,000 astrocytes from nine controls and 47 AD cases in SEA-AD. Heath said she didn’t analyze all 84 SEA-AD participants to avoid bogging down the algorithm with too much data.
Heath identified about 3,000 differentially expressed genes in astrocytes from AD samples compared to controls, then plugged these DEGs into the pseudotime algorithm to generate proxy disease trajectories for both datasets. “Late” pseudotime correlated with a high degree of AD pathology via Braak and CERAD scores, though only in the ROSMAP dataset. Despite its much larger size, the SEA-AD data contained many more AD cases than controls, and Heath believes this might have obscured the initial branching off of a temporal pattern.
That said, astrocytic changes were consistent in both datasets. As pseudotime “went by,” the glia progressed through six distinct phenotypes (see image below). Heath called the first homeostatic and the sixth reactive because the former highly expressed genes essential to astrocytic function, such as APOE, clusterin, and glutamine synthetase-encoding GluL, while the latter barely expressed those genes. Also, astrocytes from three SEA-AD participants without AD pathology, men aged 29, 42, and 50, matched the first group, supporting the categorization.

Sprouting Subtypes. Pseudotime trajectories (left to right) of astrocyte transcriptomes (dots) from ROSMAP (top) and SEA-AD (bottom) datasets. Colors denote six states; the first is called homeostatic (red circle). [Courtesy of Laura Heath, Sage Bionetworks]
Notably, astrocytes from controls and AD cases seemed much the same. Though controls had slightly more homeostatic astrocytes and cases had a few more reactive ones, each participant had astrocytes in all six states at widely varying proportions (see image below). At first, this surprised Heath. “Given how essential astrocytes are to maintaining neuronal health, and how responsive they are to all kinds of signals occurring during aging, there must be a need for multiple types of reactive astrocytes in all or most aging brains regardless of overt neuropathology,” she reasoned.

Everyone Has Every Type. All six astrocyte subpopulations (colors) were present in varying proportions among controls (left) and AD cases with low (middle) and high (right) pathology. [Courtesy of Laura Heath, Sage Bionetworks]
Sudeshna Das, Massachusetts General Hospital, Charlestown, reinforced Heath’s findings. Her pseudotime analysis also rendered a continuous spectrum of change from homeostatic to reactive astrocytes. Her MGH colleagues, in collaboration with Abbvie Inc., sequenced single nuclei from the prefrontal, entorhinal, visual cortex, and inferior temporal gyrus of 32 participants from the Massachusetts Alzheimer’s Disease Research Center. Averaging 80 years old, they ranged from Braak stages 0 to VI; controls were defined as Braak 0, I, or II without amyloid plaques, intermediates as Braak II or III with plaques, and AD cases as Braak V or VI with plaques. The scientists did not present the pre-mortem clinical diagnosis in their study, only the neuropathology data.
About those “intermediate” astrocytes. Do they represent cells in a continuum between homeostatic and reactive? Das ran a pseudotime analysis organizing the cells from the former to the latter. Six clusters of genes with similar expression patterns appeared: two whose expression rose together from homeostatic to reactive, one whose expression fell, and three whose expression peaked somewhere in between. “This suggests that the different astrocyte subpopulations may not be specialized cells, but rather transcriptional states in a trajectory from homeostatic to reactive,” Das said. Again, Heath agreed, noting that she sees similar intermediate astrocyte clusters in her data.
Grouping People, Not Cells
Gilad Green, Hebrew University of Jerusalem, took a different approach. He ran pseudotime analyses on the combined transcriptomes of all brain cells from each participant. Heath noted that pseudotime modeling is flexible enough to work on noisy data like that.
First, Green created a sizable snRNA-Seq database of 1.6 million cells from prefrontal cortex tissue of 478 ROSMAP participants and defined 96 distinct cell populations (see Part 17). Then he calculated the proportion of each cell population for each person, combined them into one composite value, and used a pseudotime algorithm to plot each value based on how similar it is to others. This created a forked trajectory, much like Heath’s above, where each data point represents a person, rather than a cell. Green declined Alzforum's request to share a representative image.
In Green’s trajectory, a single mass of points, which he believes represent homeostatic cells, diverged into two, presumably disease-related, paths. As pseudotime “passed” in each path, the proportion of homeostatic glia decreased in each person, just as Heath and Das had found. One path became enriched with reactive, GFAP-positive astrocytes, the other with disease-associated microglia (DAM) and disease-associated astrocytes (DAA). Green named these cells in this way because of their strong upregulation in the presence of amyloid plaques and neurofibrillary tangles.
To relate these paths to AD, Green consulted neuropathological and clinical data. He matched each person's degree of amyloid or tau pathology, as determined by immunohistochemistry, and their rate of decline on the ROSMAP cognitive composite, with their placement on the pseudotime paths.
People situated on the DAM/DAA path had more plaques and tangles, and faster slippage, than those on the reactive astrocyte path. The farther along in pseudotime a person was, the worse their neuropathology and cognition had been. Moreover, the proportion of disease-associated microglia was highest at “early” pseudotime, while that of disease-associated astrocytes was highest at “later” pseudotime. This aligned with Green’s previous data that a “DAM” microglial response precedes “DAA” astrocytes (see Part 17). He concluded that this path modeled AD progression.
Then what is the reactive astrocyte branch? Green assumes it is not normal aging, as people on it comprise those with AD and a wide range of pathologies. He hypothesizes that it may be people with slow progression or mixed dementia.
To Pseudotime and Beyond
If pseudotime is not sci-fi enough, watch Wörheide take exploration of omics space a step further. Wörheide used Heath’s published pseudotime analysis of bulk RNA-Seq and related it to metabolic change. This identified how metabolites link up with disease progression.
Wörheide analyzed mass-spectrometry concentrations of 667 metabolites from prefrontal cortex tissue of 154 ROSMAP participants in Heath’s RNA-Seq pseudotime analysis. The metabolites ranged from lipids and carbohydrates to amino acids and nucleotides.
Because “later” pseudotime meant worse AD, Wörheide correlated pseudotime to metabolite level. The concentration of 89 molecules rose or fell in lockstep with pseudotime. The majority, 36, were amino acids and their metabolites, followed by 17 types of lipid and 10 nucleotides and their metabolites.
Wörheide then correlated each metabolite to data in the AD Atlas, a database her group created from genomic, transcriptomic, proteomic, metabolomic, and clinical data from ROSMAP (Wörheide et al., 2021). This atlas boasts such AD phenotyping on more than 20,000 protein-coding genes, 8,000 proteins, and nearly 1,000 metabolites.
Of the 89 statistically significant metabolites, the AD Atlas already contained 50. Thirty-four correlated with 619 genes mapped onto pathways such as amino acid metabolism and neurotransmitter transport. Wörheide then searched the atlas's transcriptomic and proteomic data for differential expression of those genes and their resulting proteins. She dredged up 193 DEGs and 39 differentially expressed proteins in AD cases versus controls. Differences in transcription were biggest in the temporal cortex.
Twenty-seven of these 34 AD-linked metabolites have already been correlated with plaque and tangle pathology, brain glucose uptake, or cognition (Batra et al., 2022). All in all, Wörheide believes that relating pseudotime to other omics data can shed new light on pathways that lead to AD dementia.
Heath agreed. “It is exciting when you see a convergence of signals toward a similar pathway among the different data types, because it strengthens the biological relevance of the pathway,” she said.—Chelsea Weidman Burke
No Available Further Reading
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.