Research presented at this year’s AAIC reflected an influx of new money into the field and, with it, a growing diversity of topics. There was new interest in government policy, NIA funding priorities, and underrepresented minorities, along with an even greater emphasis on lifestyle factors and technologies to lessen the impact of a dementia epidemic forecast by demographic change. No large clinical trials presented major data, while a session picking over the ruins of recent BACE inhibitor trials showcased some transparency and shared willingness among pharma companies to learn from failure. Genetics posted news, as did biomarkers, especially blood tests, and much effort being poured into preparing observational cohorts for prevention trials was evident. Imaging continues to light up the field, debuting a new amyloid staging system and offering head-to-head comparisons of tau ligands. Specific tracers for neuroinflammation and other proteinopathies are still out of reach.
New PET Staging Scheme for Amyloid?
Scientists are becoming more nuanced in how they use amyloid scans—not just to detect the presence of Alzheimer’s pathology, but also to pinpoint disease stage. At this year’s Alzheimer’s Association International Conference, held July 13–18 in Los Angeles, researchers led by Niklas Mattsson and Oskar Hansson at Lund University, Sweden, debuted a new staging scheme. Using longitudinal data from 741 participants in the Alzheimer’s Disease Neuroimaging Initiative, including cerebrospinal fluid Aβ42 as well as PET, the researchers defined four stages of amyloid accumulation. People at stage zero had a low risk of developing plaques, but those at higher stages were likely to move to the next-higher stage within a few years, suggesting the system reflects disease progression.
Their staging system differs notably from previous models derived from cross-sectional imaging and neuropathology data. It flags amyloid accumulation in the precuneus and posterior cingulate as the earliest signs of AD, long before the brain’s overall amyloid burden becomes positive on a PET scan. “If you wanted to run a very early AD trial, you could use this staging system to select participants, rather than a global amyloid cut point,” Mattsson told Alzforum.
Others agree. “This elegant approach provides critical information on how we can discern individuals in the amyloid-negative spectrum who are likely to progress to preclinical Alzheimer’s disease,” Heidi Jacobs at Massachusetts General Hospital, Boston, wrote to Alzforum (full comment below). Arthur Toga at the University of Southern California, Los Angeles, noted, “The approach could have significant utility for tracking disease progression in a clinical setting.” Mattsson and colleagues described the scheme in the July 17 JAMA Neurology.
To develop their scheme, the Lund group made use of their previous finding that CSF Aβ42 drops up to 10 years before an amyloid PET scan crosses the threshold for global positivity (Aug 2016 conference news; Palmqvist et al., 2016). These data suggested that CSF-positive people were accumulating amyloid in select brain regions. The researchers wondered if they could identify those regions of early buildup. In their initial study, CSF-positive yet whole-brain PET-negative ADNI participants indeed deposited amyloid only in specific regions, including the precuneus and posterior cingulate (Nov 2017 news).
Mattsson and colleagues extended those findings to develop a longitudinal staging system. First, they selected 641 ADNI participants who had CSF data and at least two florbetapir PET scans, and stratified them by amyloid positivity. Among this group, 288 were negative on both CSF and PET and were classified as non-accumulators; 69 were CSF-positive but PET-negative and were deemed early-stage accumulators; and 274 were positive on both—the late-stage accumulators. Ten discordant people were CSF-negative and PET-positive.
The researchers then examined longitudinal amyloid scans from the 69 early stage accumulators. They found six brain regions—the precuneus, posterior and isthmus cingulate, insula, and medial and lateral orbitofrontal cortices—where amyloid load was increasing compared to non-accumulators. Amyloid positivity in these regions, which form part of the brain’s default mode network (DMN), defined stage 1. Next, accumulation of amyloid in a large number of regions, including the parahippocampus, medial and inferior temporal lobes, inferior parietal lobe, and superior parietal, temporal, and frontal regions, marked stage 2. Most of these regions are known sites of pathology in early AD. Finally, in late-stage accumulators, amyloid piled up in precentral, postcentral, paracentral, lingual, and pericalcarine cortices. Amyloid in these regions defined stage 3 (see image above).
How did the ADNI cohort break down across these stages? For this, the researchers included another 100 ADNI volunteers who lacked CSF data but had at least two florbetapir scans, for a total of 741 participants. The full cohort comprised 304 cognitively healthy controls, 384 people with MCI, and 53 with AD dementia. Ninety-eight percent of participants fell cleanly into one of the four stages. More than half were at stage zero, three percent at stage 1, 11 percent at stage 2, and 30 percent at stage 3. The stages roughly corresponded to cognitive status, with 70 percent of controls at stage zero and 80 percent of AD patients at stage 3. Still, there were plenty of exceptions: 16 percent of controls were at stage 3, and 17 percent of AD patients were at stage zero.
The authors repeated the analysis with data from the Swedish BioFINDER cohort. This longitudinal biomarker study uses flutemetamol rather than florbetapir for amyloid scans. In a cross-sectional set of 306 healthy controls and 168 people with MCI, 98 percent of them fit unambiguously into one of the four stages. The percentages for each stage and cognitive group were similar to those in ADNI.
Notably, the staging patterns closely matched those seen in longitudinal PET scans from the Dominantly Inherited Alzheimer Network, where deposition occurred first in the precuneus, then in the posterior cingulate and medial orbitofrontal cortex (Feb 2018 news).
They also agree with other longitudinal PET data presented at AAIC. Michelle Farrell of Massachusetts General Hospital reported that among 265 cognitively healthy adults in the Harvard Aging Brain Study who had repeated PET scans, amyloid accumulated earliest in the precuneus, isthmus and anterior cingulate cortex, medial orbitofronal cortex, and middle and inferior temporal lobe. Likewise, Gemma Salvadó of Barcelonaβeta Brain Research Center in Barcelona, Spain, showed congruent data from the European Amyloid Imaging to Prevent Alzheimer’s Disease study. Her team developed a staging model based on PET data from 224 cognitively healthy participants in Barcelonaβeta’s Alzheimer and Families (ALFA) project, then applied it to 870 PET images from the ALFA, ADNI, ABIDE, and EMIF-AD cohorts. The first areas to accumulate plaques were the precuneus, anterior cingulate cortex, and orbitofrontal cortex.
On the other hand, the Lund group’s findings only partially overlap with cross-sectional staging schemes, including classic neuropathology data and a recent PET amyloid study from Michel Grothe and colleagues at the German Center for Neurodegenerative Diseases in Rostock (Braak and Braak, 1991; Oct 2017 news). Grothe detected the earliest amyloid deposition in neocortical regions such as the temporal lobe, parietal operculum, and anterior cingulate, but did not pick out precuneus and posterior cingulate as early sites.
“The strength of the Mattsson et al. approach is that they utilize longitudinal PET data, which can give a more dynamic picture than static states do,” Rachel Buckley at MGH wrote to Alzforum (full comment below).
Is this new staging scheme biologically meaningful? Several pieces of evidence argue that it is, Mattsson said. For one thing, the amyloid PET stages correlated with other biomarkers. People at stage 1 or higher had low CSF Aβ42 and high phospho-tau compared with controls. In those at stage 2 or higher, CSF total tau ramped up, while at stage 3 brain atrophy did. Cognitive decline began in stage 2.
Another indication that the staging system is valid is that it predicted progression. People at stage zero had a 15 percent risk of progressing to a higher stage over an average of four years, while those in stage 1 ran a 71 percent risk and stage 2, a 53 percent risk. This fast rate of progression may explain why relatively few people are found in stage 1 or 2 at any given time, the authors speculated.
A final piece of evidence is that the combined brain regions that define each stage had distinct patterns of gene expression. Using data from the Allen Human Brain Atlas, the researchers found classes of genes that were differentially expressed among the regions. They were linked to voltage-gated ion channels, neuropeptide and glutamate signaling, lipid transport, and axon guidance. The data hinted at biological factors that may underlie the selective vulnerability to amyloid accumulation of brain regions associated with different stages, Hansson said.
“The relationship to regional gene expression areas was interesting, as this may hint at the ‘why’ [of amyloid accumulation] as well as the ‘where’,” Toga wrote to Alzforum.—Madolyn Bowman Rogers
Physical Activity May Shield the Brain from the Onslaught of Aβ
Physical activity not only benefits the heart and the body, it may also shield the brain from the damaging effects of Aβ. That’s according to findings presented at the Alzheimer’s Association International Conference, held July 14–18 in Los Angeles. Active people in their 70s were less likely to suffer the cognitive and neurodegenerative consequences of Aβ deposition than their more sedentary peers, said Jennifer Rabin, Massachusetts General Hospital in Charlestown. Good vascular health also held Aβ-related damage in check, although it appeared to do so independently of physical activity, she said. The study, led by Jasmeer Chhatwal at MGH, was published July 16 in JAMA Neurology.
“This important study adds considerable weight to the growing amount of evidence indicating the importance of maintaining a physically active lifestyle for reducing the risk of cognitive decline and neurodegeneration,” commented Kirk Erickson, University of Pittsburgh.
This was borne out by several studies of large cohorts presented at this year’s AAIC. Researchers led by Katherine Bangen at the University of California, San Diego, reported that among 2,337 50-year-olds in the Framingham Offspring study, those who exercised less were more likely to become cognitively impaired seven years later. Tianya Hou, working with Dorina Cadar at University College London, reported that among 7,807 participants 50 and older in the English Longitudinal Study of Ageing, those who reported greater physical activity had slower cognitive decline and better maintained executive function over the ensuing eight years. In her talk, Cadar reported that in the same cohort, high physical activity reduced the risk of dementia over 12 years by 40 percent. Likewise, Priya Palta, Columbia University, New York, reported that in the Atherosclerosis Risk in Communities (ARIC) study, people who pursued moderate to high physical activity in midlife had 12 percent less cognitive decline over 14 years and about a 40 percent reduced incidence of dementia than their sedentary peers (Palta et al., 2019).
Wanqing Wu, Huashan Hospital, Shanghai, reported that among 1,370 50-plus-year-olds in the Shanghai Aging Study, those who exercised at a moderate to vigorous level were 45 percent less likely to get dementia over the next five years. Richard Mayeux’s lab, also at Columbia, reported that maintaining physical activity across the lifespan may be important for maximum benefit. First author Yian Gu’s analysis of 1,288 adults in the Washington/Hamilton Heights-Inwood Columbia Aging Project suggests that those who had exercised vigorously since their teenage years were 60 percent less likely to develop dementia. All is not lost for late starters. Participants who reported low physical activity between ages 12–25, but high activity after age 50, were still half as likely to get dementia as those who reported never being physically active.
How does exercise protect the brain? Earlier studies have implicated lower levels of brain amyloid, less hypometabolism and atrophy (Liang et al., 2010; Brown et al., 2012; Okonkwo et al., 2014). This, too, was borne out by presentations in Los Angeles. Katsuyuki Miura and colleagues from Shiga University, Japan, found that among 680 men in the Shiga Epidemiological Study of Subclinical Atherosclerosis, those 60 and older who walked a lot, as assessed by pedometer over seven days at baseline, had less brain atrophy five years later. The benefit appeared limited to the prefrontal cortex and left entorhinal cortex. Marta Milà-Alomà, Barcelonaβeta Brain Research Center, Spain, reported that in the ALFA+ cohort, who have family histories of AD and extensive biomarker data, physical activity came with reduced amyloid burden, as judged by CSF Aβ42. Among 257 cognitively normal volunteers, 26 percent of the physically active fell below the 1,098 pg/mL threshold versus 47 percent of the inactive group.
Even so, few studies have examined how physical activity modulates the relationships between Aβ deposition, brain volume, and cognitive decline.
Rabin and colleagues addressed this in the Harvard Aging Brain Study (HABS). They followed 182 cognitively normal participants who averaged 73 years of age at baseline. Each had worn a waistband pedometer for one week during waking hours, to measure the number of steps they were taking in a typical day. Also at baseline, the participants underwent Aβ-PET and MRI scans, took cognitive tests, and were assessed for vascular risk factors. Annual cognitive tests and MRI scans were then taken over an average of six and 4.5 years, respectively.
The participants averaged nearly 6,000 steps per day, a modest level of activity that did not necessarily include structured exercise, according to Chhatwal. The researchers found no relationship between physical activity and Aβ burden—as measured by the distribution volume ratio across a composite of frontal, lateral temporal and parietal, and retrosplenial regions—at baseline. As expected, more-active people had lower vascular risk.
The researchers also found that Aβ burden at baseline correlated with decline on Preclinical Alzheimer’s Cognitive Composite (PACC) in the following years. Baseline physical activity appeared to dramatically lessen that Aβ-related cognitive decline. In fact, among people with the highest Aβ burden—defined as a DVR above 1.9—those who had walked more than 8,300 steps per day did not decline on the PACC, while those who walked less than 2,900 dropped by about 1.25 standard deviations. People with the lowest Aβ burden, below 1.1 DVR, also did not decline regardless of their baseline physical activity.
The researchers observed similar trends for gray-matter brain volume. Though brain shrinkage occurred across the cohort, baseline Aβ burden correlated with a steeper drop in volume, and physical activity attenuated that effect. In particular, those who were more active had less Aβ-related cortical thinning in the entorhinal cortex, insula, lateral temporal, and medial parietal regions, all of which typically shrink over the course of AD. However, unlike some previous studies, they found no association between physical activity and hippocampal volume.
Chhatwal told Alzforum that although physical activity staved off brain atrophy in people with high Aβ deposition, that protection accounted for only about 20 percent of the cognitive benefits. He proposed that exercise improves the integrity of neural networks involved in memory, and improves circadian rhythms, both of which likely also contribute to the cognitive benefit.
Rabin also reported less cognitive decline and less brain atrophy in Aβ-positives who had low vascular risk. Chhatwal was surprised to find that vascular health and physical activity independently associated with these beneficial outcomes. This could suggest that physical activity protects neurons from Aβ-mediated damage by means other than improving vascular health, for example by boosting neurotrophic factors or strengthening neural circuitry. However, other researchers cautioned that it may be extremely difficult to disentangle the contributions of physical activity and vascular health.
Together, the findings support the idea that better physical fitness and better cardiovascular health can fend off Aβ-mediated degeneration, Chhatwal said.
This is borne out in the ARIC study. Also at AAIC, Palta reported that physical activity associated with less Aβ accumulation in the brain. She noted that prior studies on this association have been equivocal, perhaps because most examined few people over short intervals. In a subset of the ARIC cohort, Palta studied 326 participants who had florbetapir scans between 2011 and 2013, 25 years after the volunteers had entered ARIC. Each had described the duration and frequency of their four most common leisure activities during the year before their baseline assessment and also over the year before their brain scan. Palta and colleagues used the Compendium of Physical Activities to assign a metabolic equivalent (MET) to each activity (Ainsworth et al., 2000).
Mid-life METs per week correlated inversely with whole-brain florbetapir standard uptake value ratios (SUVRs). ARIC used an SUVR of 1.2 as a cutoff for amyloid positivity. Those below the cutoff reported a mean MET per week of 381 minutes, while those above only mustered an average of 270 minutes. Calculated as an odds ratio, those who were physically active in midlife and met 2018 physical activity guidelines (Piercy et al., 2018) were about 17 to 30 percent less likely to have brain amyloid. Though those ORs were not statistically significant, Palta said the trend was consistent. Late-life physical activity did not correlate with amyloid, likely because the two were measured within a year of each other.
Rabin’s findings add to the preponderance of evidence in the field that physical fitness and vascular health strengthen the brain’s resilience against all manner of insults, including Aβ, commented Eric Larson of Kaiser Permanente Washington Health Research Institute in Seattle. He added that improvements in overall health, including diet, exercise, and social connections, likely all help mitigate the risk of AD and related diseases.
Teresa Liu-Ambrose of the University of British Columbia in Vancouver agreed. “What is not known in this study is whether the level of physical activity observed at baseline was maintained by the individuals over time,” she added. “Understanding the relationship of physical activity levels over time in relation to changes in cognitive and brain outcomes is important. It may identify critical periods of prevention or intervention.”
Chhatwal acknowledged some study limitations, including the brief, one-week measurement of physical activity. The waistband pedometers available at the time had a short battery life, which limited the number of days they could be worn, and also did not measure the intensity of physical activity, he said. The researchers are now using modern wrist accelerometers, which give a more detailed accounting of activity and intensity over longer periods of time. He said future studies will include those data, as well as longitudinal measurements of both Aβ and tau deposition.—Jessica Shugart and Tom Fagan
Crenezumab Update: Baseline Data from Colombian Prevention Trial
A subset of the world’s largest known kindred of autosomal-dominant Alzheimer’s disease, near the Colombian city of Medellin, have enrolled in the Banner Alzheimer Prevention Initiative Autosomal Dominant Alzheimer’s Disease trial. It is treating presymptomatic mutation carriers with crenezumab or placebo (May 2012 news). It also represents the last ongoing trial for this anti-Aβ antibody (May 2019 conference news). At the AAIC conference held July 14–18 in Los Angeles, researchers presented the baseline demographic, cognitive, and biomarker data on 242 trial participants in this ongoing trial. At the start, carriers were on average five years younger than noncarriers but already performed worse on cognitive tests and also showed more variability.
Pierre Tariot of the Banner Alzheimer’s Institute kicked off a series of four data presentations from this trial. It evaluates whether crenezumab, which binds monomeric and non-fibrillar aggregates of Aβ, can stave off cognitive decline in asymptomatic E280A mutation carriers. People with genetic forms of AD make perfect subjects to study prevention, but there are so few of them in the world that they are hard to find. As the world’s largest characterized ADAD kindred at this time, the Colombian families offer a chance to find out whether early intervention can prevent disease, and do it in a trial large enough that it could lead directly to an approved therapy, Tariot said.
Desired Distribution. Amyloid burden at baseline in mutation carriers and noncarriers in the API Colombian ADAD trial. Half of the carriers (red) exceed the SUVR cutoff 1.1 for amyloid positivity.[Courtesy of Pierre Tariot.]
The trialists needed a design that did not compel participants to learn whether they inherited the dreaded mutation. To do that, the API team aimed to enrolled 200 mutation carriers, randomized equally to treatment or placebo, and 100 noncarriers, all of whom receive placebo (Tariot et al., 2018). DIAN trials do this, too. In total, the scientists in Colombia screened 365 people and randomized 252, including 169 mutation carriers and 83 noncarriers, somewhat less than the goal of 300.
Even though the researchers in Colombia had built a large prescreening registry containing more than 5,000 people, trial recruitment was difficult because many people who otherwise met inclusion criteria were either already mildly symptomatic or unable to take as much time off from work as the trial required, or delay pregnancy for the duration of the trial (Rios-Romenets et al., 2018). Even so, the trial maintained its statistical power because retention is high, at 92 percent, and because it was changed to end in a “common-close” design. The common close means that all participants will stop receiving treatment or placebo five years after the last person is randomized, which adds about 25 percent more observations to the primary analysis. The trial began in December 2013, the last person was randomized in February 2017, and the placebo-controlled treatment period will end in 2022.
API had contracted with crenezumab’s maker, Roche/Genentech, to share data and biological samples from the trial after it was completed. The agreement was subsequently tweaked to include baseline data, consistent with the principles set forth by the Collaboration for Alzheimer’s Prevention (July 2016 news). Tariot said it took years to work out exactly how to share the baseline data among the trial partners without unmasking the identities or genotypes of participants, or accidentally revealing who was receiving placebo or active treatment. When it came for public release, the trials’ small sample size presented a danger that simply knowing age and sex could be enough for individuals to identify which group they were in.
In the end, scientists pruned data on 10 participants older than 54, to allow age-range matching across groups and to avoid the risk of giving away anyone’s genetic status. Tariot showed data on the remaining 242, comprising 167 carriers and 75 noncarriers between the ages of 30 and 53. At baseline, carriers averaged 37 years old, younger than noncarriers, who averaged 42. The reason for this is that the trial enrolled cognitively normal carriers, and aging is strongly associated with MCI and dementia in carriers. Sex, education, and ApoE4 carriage were similar across groups. In general, all participants had fewer years of education than some other research cohorts, with an average of 8.5 years in school. In DIAN, that average is 14 years. No one had ARIA at baseline, per trial criteria.
Carriers and noncarriers had similar clinical dementia rating (CDR) global scores, but CDR Sum of Boxes and functional assessment staging were both worse in carriers. Tariot called the differences “small, but suggestive of something.” Carriers did worse than noncarriers on the MMSE and on many cognitive tests. By definition, they still fell in the normal range, because those who met criteria for MCI were excluded. Notably, carriers showed greater variation in scores, especially on measures of memory. Carriers and noncarriers did not differ on neuropsychiatric or depression measures.
Natalia Acosta-Baena, Universidad de Antioquia, scrutinized the baseline cognitive data in detail to try to understand age-related cognitive changes in this group. Looking at how age affected this cross-sectional analysis, she discovered that memory declined first, with mutation carriers demonstrating slippage by age 35. This confirms her earlier analyses for the onset of cognitive symptoms in this group (Acosta-Baena et al., 2011).
Other tests for language, attention, and visuospatial acumen, as well as RBANS total scores, showed no significant age-related differences between carriers or noncarriers at baseline. Further analysis is needed, with adjustment for education and ApoE status. Even so, Tariot said, “While generally confirming what has been seen before, I found it sobering to see the inescapable consequences of having this mutation.”
Yi Su, Banner Institute, presented data on brain imaging with florbetapir PET for amyloid, FDG PET to track hypometabolism, and structural MRI. As a group, the mutation carriers had more amyloid than noncarriers. At baseline, 46 percent of carriers exceeded the threshold for Aβ positivity on PET scans. This hit a benchmark the investigators had set of trying to enroll a carrier group who were not all amyloid-positive already, but spanned a range of SUVRs. None of the noncarriers were amyloid-positive.
Yakeel Quiroz, Massachusetts General Hospital, Boston, checked Aβ burden against memory performance in this group. She reported that cerebral amyloidosis correlated with lower episodic memory scores in the mutation carriers, even after adjusting for age. In other words, trial participants with more brain amyloid did worse on the CERAD word list and other memory measures.
Along with higher amyloid burden, the mutation carriers had on average lower FDG uptake than noncarriers, but similar hippocampal volume, Su reported. As a function of age, amyloid became abnormal first, then glucose uptake, then hippocampal volume. The time line largely matched previous work from this group, and DIAN data (Benzinger et al., 2013).
That said, changes in metabolism and hippocampal atrophy appeared later than expected. This could have been due to methodology, or this particular mutation. There may also have been selection bias, Su said. Because the current study excluded cognitively impaired participants, it may be enriched for people with protective factors, Su speculated.
Some of the baseline data is accessible via GAAIN. Researchers can apply to collaborate on analyses or seek access to the data at APIData@bannerhealth.com. For new results on plasma NfL in the larger Colombian ADAD cohort, see next story in this series. —Pat McCaffrey
At the Alzheimer’s Association International Conference, held July 14–18 in Los Angeles, a blood test indicating that neurons in the brain are degenerating generated considerable buzz. Eric Reiman, Banner Alzheimer’s Institute, Phoenix, presented data on plasma neurofilament light (NfL) that had been measured in 2,144 Colombian people aged 8–75. They all came from families plagued with a presenilin 1 mutation, whose carriers develop dementia in their 40s. The plasma NfL measure distinguished mutation carriers from noncarriers as early as age 22, which also happens to be 22 years before their expected age of symptom onset. Plasma NfL rose fastest just before symptoms began, and a rapid rise predicted declining cognition and brain shrinkage.
The data come on the heels of two smaller studies in autosomal-dominant AD (Jan 2019 news; Nov 2017 news), and the first longitudinal study showing that plasma NfL tracks disease progression in late-onset AD, as well (May 2019 news).
The Colombian cohort comprises 25 extended families who inherited the PSEN1 E280A, aka Paisa, mutation. The Colombian neurologist Francisco Lopera, Universidad de Antioquia, Medellin, together with Ken Kosik, University of California Santa Barbara, and others described the families 22 years ago (Lopera et al., 1997). Since then, scientists have learned that the brains of mutation carriers are subtly different starting in childhood, and their cognition starts to slide subtly in the late 30s, although there is some variability in age of onset. Mild cognitive impairment develops at 44 and dementia at 49 (Fuller et al., 2019).
In a study, a large collaboration of researchers measured plasma NfL in 2,144 mutation carriers and noncarriers drawn from the Alzheimer Prevention Initiative research registry in Medellin. This registry has become an extraordinary resource, having enrolled 5,846 people from Colombian families affected by the E280A mutation, including 1,192 carriers, 1,119 of whom are living, and six homozygotes (Kosik et al., 2015). The API researchers have blood and DNA samples for all, and clinical assessments for many. Some blood donors underwent brain scans, and donated CSF for biomarker analysis. For 504 participants, the scientists had multiple blood samples spanning an average of five years, and 399 of those had longitudinal clinical data as well.
Yakeel Quiroz, Massachusetts General Hospital, Boston, coordinated the study, selecting 1,070 carriers and 1,074 age- and sex-matched noncarriers whose plasma went to the lab of Kaj Blennow and Henrik Zetterberg at the University of Gothenberg, Sweden, and was analyzed using their ultra-sensitive single molecule array (Simoa) NfL assay. Both carriers and noncarriers averaged 30 years old, but included children as young as 8 and people in their 70s. The average plasma NfL in the mutation carriers was 18 pg/ml, versus 9 pg/ml for noncarriers. Despite large variation in both groups, the difference was statistically significant.
As expected, in the cross-sectional analysis NfL levels were higher in older people, but much more so in carriers. A linear regression analysis showed the curves diverged at age 22, or 22 years before the median age of MCI onset at 44. The investigators got the same result when they calculated the rate of change of NfL in the 504 mutation carriers and noncarriers for whom they had longitudinal data. NfL accumulation ramped up in carriers 22 years before the median age of MCI onset.
That is even earlier than in a previous study of 408 people in the DIAN cohort, which picked up an accelerated rise in NfL 16 years before symptom onset (Priesche et al., 2019). Reiman said the API team analyzed the Colombian data exactly as the DIAN researchers had done and, indeed, received help from Brian Gordon and Stephanie Schultz at Washington University, St. Louis. The differences may be due to the larger sample size and the fact that all the Colombian carriers have the same mutation, as opposed to the more than 40 different mutations across three genes represented in DIAN.
Plasma NfL foretold a person’s clinical course, too. In 38 cognitively impaired and 119 as-yet-unimpaired carriers who were followed clinically for about five years, the former had more than three times higher absolute NfL levels and 13 times the annual rate of change of the latter, who were themselves slightly higher than noncarriers. However, Reiman cautioned that the results need adjustment for age and sex. At 50 on average, the impaired carriers were older than the unimpaired carriers or noncarriers, who averaged 32 and 27 years old, respectively.
Likewise, in a cross-sectional analysis, higher baseline plasma NfL correlated with lower MMSE and word-list-recall scores among both impaired and unimpaired carriers. Longitudinally, both baseline plasma NfL and annual rate of change correlated with annual clinical decline rates. Again, neither is adjusted for age yet.
In response to audience questions, Reiman said the relationship between plasma NfL and amyloid status, ApoE4, and other factors in this group still needs to be examined. Previous work suggests that E280A carriers who have an Apoe4 allele suffer earlier clinical onset. “We now have the opportunity to use biomarkers such as NfL as endophenotypes to see if pathological changes are accelerated by ApoE4. These are exactly the kinds of studies one could leverage in this cohort,” he said.
Others in the audience asked how the rate of NfL accumulation changes over time. Reiman noted a dramatic increase in plasma NfL a few years before disease onset, and suggested that this may be the best time to detect changes in response to treatment. “We’ll have a better chance of seeing a reduction of high levels rather than slowing of a gradual increase. At age 22, we don’t have much power to see a change over time, so we’ll be doing studies in the late preclinical/early clinical stage.”
This NfL analysis represents the first biomarker study on this large collection of plasma samples. Next, API researchers will measure phospho-tau 181 in them.
These results are raising hope that NfL will provide a relatively cheap, quick, and easy readout to inform a person’s prognosis. Importantly, NfL could signal that a trial participant is responding to treatments aimed at slowing neurodegeneration. When axons degenerate, the NfL protein gets shed into the CSF, from which it makes its way into blood.
Separately at AAIC, Charlotte Teunissen, who leads a fluid biomarker lab at at Vrije University Medical Center, Amsterdam, presented ongoing work on a multicenter validation study on blood NfL assays. This kind of applied science is necessary to create reliable, certified assays that are suitable for routine clinical use.—Pat McCaffrey
Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, Gräber S, Kuder-Buletta E, LaFougere C, Laske C, Vöglein J, Levin J, Masters CL, Martins R, Schofield PR, Rossor MN, Graff-Radford NR, Salloway S, Ghetti B, Ringman JM, Noble JM, Chhatwal J, Goate AM, Benzinger TL, Morris JC, Bateman RJ, Wang G, Fagan AM, McDade EM, Gordon BA, Jucker M, Dominantly Inherited Alzheimer Network.
Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease.
Nat Med. 2019 Feb;25(2):277-283. Epub 2019 Jan 21
PubMed.
Further Reading
No Available Further Reading
Rare Luck: Two Copies of ApoE2 Shield Against Alzheimer’s
Mention ApoE and Alzheimer’s, and the conversation turns to the E4 allele, the strongest susceptibility gene for the disease. But ApoE has another side, in ApoE2. Though this isoform protects against AD, scientists have barely studied it. Now ApoE2 is attracting scrutiny as scientists are asking exactly how some people maintain their mental acuity into old age. At the Alzheimer’s Association International Conference, held July 14–18 in Los Angeles, a study of ApoE genotypes in 5,000 autopsy-confirmed cases of AD revealed that people with two copies of E2 see their risk of dementia plummet by a stunning 90 percent compared with those with the common E3/E3 genotype. Other work suggested that this could be because ApoE2 reduces amyloid and tau pathology, and boosts gray-matter volume in critical brain regions. E2’s benefits seem specific to Alzheimer’s, not generic to neurodegeneration.
ApoE is the major cholesterol-carrying protein in the brain. It has been studied since its discovery as an AD risk gene in the early 1990s, but is newly emerging as a hub for glial responses to amyloid and tau aggregate deposition (Aug 2018 news; Sept 2017 news). The gene exists as three polymorphic alleles—E2, E3, and E4—with a worldwide frequency of 8 percent, 78 percent, and 14 percent, respectively. Several mutated forms are also known, for example the so-called Heidelberg, Pittsburgh, and Christchurch mutations (Feussner et al., 1992; Kamboh et al., 1999; Wardell et al., 1987).
ApoE4 receives by far the most attention from AD researchers, because it boosts the risk of AD up to 15-fold depending on the study population, and occurs in 40 percent of people with AD. E2, the protective allele, has received scant attention, because it is the least common of the three and largely absent from AD samples. People with one copy of E2 have half the chance of developing AD compared with those with the more common E3/E3 genotype. But does the additional E2 have an effect beyond that? “We have not known whether E2 dose has a differential risk, i.e. whether E2/2 risk is significantly lower than E2/3,” Eric Reiman said in presenting the study.
To find out, Reiman and colleagues at Banner Alzheimer’s Institute, Phoenix, Gyungah Jun at Boston University, Joseph Arboleda of Massachusetts Eye and Ear, Yakeel Quiroz of Massachusetts General Hospital, and colleagues from the AD Genetics Consortium decided to look at data from a lot of brains. They analyzed the contribution of all three ApoE alleles to dementia risk and pathology in 5,007 brains from the ADGC. This sample included 4,018 autopsy-confirmed Alzheimer’s dementia cases, plus 989 pathologically and cognitively unaffected donors. Besides providing sufficient numbers to study E2/E2 homozygotes, this cohort avoids the confounding issue of misdiagnosis by eliminating dementias not due to AD, as well as people who had AD pathology at the time of death but no dementia.
As expected, ApoE2 homozygotes were rare—numbering just 24 out of more than 5,000 people, or 0.5 percent. Compared with other genotypes, they were far less likely to have AD. E2 homozygotes made up 0.1 percent of cases but 1.9 percent of healthy controls. In contrast, ApoE4/4 homozygotes accounted for 15.6 percent of cases and only 1 percent of controls. In other words, 19 of the 24 ApoE2/2s were cognitively healthy, but only 10 of 633 ApoE4/4s were.
E2 homozygotes had a 66 percent risk reduction compared even with E2/3 carriers, an 87 percent risk reduction compared to the most common genotype, E3/3, and a whopping 99.6 percent risk reduction compared to people who were E4/4. Basically, most people with E4/4 get Alzheimer’s dementia, while few with ApoE 2/2 do.
Having two E2 alleles correlated with less amyloid plaque and tau neurofibrillary tangle pathology. ApoE2’s protective effect on tau pathology was still apparent even when adjusted for amyloid plaque load. That mirrors recent animal data suggesting that ApoE4 acts to worsen tau pathology independent of amyloid (Shi et al., 2017).
The new study’s main finding echoes that of a previous study by Pieter-Jelle Visser, Maastricht University, the Netherlands, and colleagues. They had found 16 ApoE2/2 carriers in a sample of 7,583 people, of whom 10 were cognitively normal and amyloid-negative, whereas 301 of the 386 ApoE4/4 carriers in the sample had Alzheimer’s dementia. This sample, however, was not pathology-confirmed (Jansen et al., 2015).
How important is neuropathology confirmation? Reiman et al. compared the risk estimates derived from the autopsy-verified cohort to those calculated from a cohort of 23,857 living people who were clinically diagnosed as having probable AD dementia or being cognitively normal, and were of unknown amyloid status. In that analysis, the protective effect of 2/2, and the increased risk due to 4/4, both were underestimated. For example, the scientists found that the odds ratio associated with E4/E4 over E3/E3 in the clinical sample was 10.7, versus 31.22 in the pathologically confirmed cases. The dose effects for both E2 and E4 were also underestimated in the living sample. Thus, the autopsy analysis provides updated, and likely more accurate, risk estimates for all ApoE genotypes, Reiman and colleagues believe, at least for these non-Hispanic white research participants.
Life Extending. In a large epidemiological study combining six population-based cohorts of people with European ancestry, ApoE2 homozygotes were at lower risk of dying than other genotypes. ApoE4 homozygotes were at greatest risk. [Courtesy of Wolters et al., PLoS One 2019.]
ApoE’s impact on Alzheimer’s is known to differ among ethnic groups. This type of study is needed with more diverse subjects, more E2/2 homozygotes, and taking into account possible effects of the ApoE alleles on survival, which could skew the results. Nonetheless, Reiman said, “Our results suggest that ApoE2 homozygosity is associated with an exceptionally low risk of AD, that the impact of APOE and its variants on AD risk is significantly greater than previously appreciated, and that there is a compelling reason to discover treatments that promote this protective effect.”
That jibes with recent results from Sudha Seshadri, University of Texas Health, San Antonio, Frank Wolters, Erasmus Medical Center, Rotterdam, the Netherlands, and colleagues. They found a survival advantage for ApoE2 carriers in a study of 38,537 people from six population-based cohorts (Wolters et al., 2019). They identified 239 E2 homozygotes, who led the longest lives. The effect of E2 was only partly explained by its effects on blood lipids or vascular disease. E4 homozygotes had the highest risk of death, and this was largely accounted for by its association with dementia. Seshadri told Alzforum that the investigators will look at AD and cognitive change, and also at amyloid and tau PET measures in this sample.
Bit by Bit. Shading indicates areas with a significant stepwise, ApoE genotype-related increase in gray-matter volume, with E4/4 homozygotes having the lowest and E2/2s the highest volume. Graphic shows data for one brain region. [Image courtesy of Gemma Salvadó.]
How might ApoE2 bestow resilience? To find out, Gemma Salvadó,Barcelonaβeta Brain Research Center, Spain, presented a study where she and co-authors gathered together imaging data from different samples on as many older ApoE2 homozygotes as they could. Their goal was to compare the brain structure of E2/2s with that of other ApoE genotypes.
Previously, E2 had been linked to subtle changes in brain morphology in healthy people, including slower hippocampal atrophy in old age, and larger hippocampi in middle age (Chiang et al., 2010; Fennema-Notestine et al., 2011). In childhood, E2 carriers have been reported to have thicker entorhinal cortices than E3 homozygotes or E4 carriers (Shaw et al., 2007). But these studies all focused on people with one copy of E2.
To find out what that extra E2 would do, Salvadó collected and analyzed MRI data on cognitively unimpaired people in the ALFA study in Barcelona (Molinuevo et al. 2016), the Amsterdam University Medical Center cohort, OASIS open-access imaging studies, and ADNI. She found 28 E2/E2 homozygotes. She matched each of them with five other subjects from the same center on age, sex, and education level, and one of every other ApoE genotype. That gave a total of 168 subjects, with a mean age of 62.
ApoE2/2 Over 3/3. Red to yellow shading indicates brain regions where ApoE2 homozygotes had more gray matter than E3 homozygotes, including both hippocampi (inset). [Image courtesy of Gemma Salvadó.]
Compared with their E3/3 matches, the E2/2s had larger gray-matter volume in their hippocampi and other AD signature areas, including the medial temporal cortex, inferior temporal, temporal pole, precuneus, and superior parietal regions. E2 homozygotes also had more gray matter in areas related to cognitive resilience in aging, namely in the anterior cingulate and medial prefrontal areas (Arenaza-Urquijo et al., 2019; Harrison et al., 2018). When compared with E2/3 heterozygotes, the E2 homozygotes boasted few significant differences in gray matter, but they did have even larger hippocampi than the 2/3s.
Salvadó reported a stepwise, genotype-related increase in gray-matter volume, with E4/4 homozygotes having the lowest, E3/3s having intermediate, E2/3s higher, and E2/2s the highest volume (see image below).
ApoE2/2 Over 2/3. Compared with the next-protective genotype, ApoE2/3, E2 homozygotes have more gray matter only in a few small areas.
She believes the larger gray matter in strategic brain areas may help E2 homozygotes cope with AD pathology, if and when it appears. Because ApoE2 plumps up the entorhinal cortex already in childhood, resilience may spring from developmental processes, she said.
“These are very important findings,” Reiman commented. “They suggest that if atrophy is like eroding the tread in tires, E2 homozygotes may start with more tread,” he said. Salvadó is trying to expand the study to look at more homozygotes from additional cohorts, and other imaging modes.
Terry Goldberg, Columbia University, New York, has been studying potential mechanisms of ApoE2-mediated neuroprotection for years (Conejero-Goldberg et al., 2014). At AAIC, Goldberg presented results on the relationship of ApoE alleles with neuropathology in AD, and extended the analysis to other diseases.
He used data on 1,557 brains from the National Alzheimer’s Coordinating Center database that had both clinical and neuropathological assessments. Because he had so few E2 homozygotes, Goldberg grouped E2/E2 and E2/E3 genotypes together, for a sample of 130. In that combined group, one in four brains had AD neuropathic changes, compared with 40 percent of E3/3s, 65 percent of E3/4s, and 85 percent of E4/4s. Consistent with previous work, E2 carriers had the mildest amyloid pathology, fewest neuritic plaques, and mildest tau pathology. A statistical mediation analysis suggested ApoE2’s association with reduced tangles went partly through its effect on amyloid, and was partly independent of amyloid. That dovetails with Reiman’s results.
In this dataset, too, ApoE2 strongly protected, even if Goldberg did not parse homozygotes: E2 cut the risk of amyloid and tau pathology by half compared with E3, and by 90 percent compared to E4.
Which is more powerful, E2 or E4? Comparing E2/E4 and E3/E4 genotypes, Goldberg found comparable levels of pathology. If anything, the E2/E4 pairing was worse. This suggests that E4 overwhelms the protective effects of E2. “You could say E4 is toxic,” Goldberg concluded. That suggests therapies using viral constructs to introduce E2 into E4-expressing brain tissue may not be helpful, he said.
What about other neurodegenerative diseases?
The literature offers mixed results on E2 and E4 in frontotemporal dementia. Some studies suggest E2 promotes risk (Mar 2016 news) while others credit it with protection and blame risk on E4 (Mishra et al., 2017). At AAIC, Goldberg reported that E2 was associated with more severe TDP-43 pathology among 103 cases of frontotemporal dementia, and with more tau pathology in 28 cases of Pick’s disease and 51 cases of progressive supranuclear palsy. He cautioned that these numbers are so small, the results could be spurious. Goldberg doesn’t know if these patients had C9ORF72 or progranulin mutations, and TDP-43 accumulation may be age-associated. In cases of α-synuclein pathology, Goldberg found that ApoE4 promotes, and E2 reduces, the spread of Lewy body pathology outside of its origin in the midbrain, into limbic and neocortical areas
Together, these studies highlight a renewed appreciation of the enormous impact ApoE exerts on the pathogenesis of AD (for example, see Wu and Zhao, 2016). The profound protection afforded by E2 will likely rekindle interest among drug developers, who have tried before and failed, but may now see fit to revisit this target.—Pat McCaffrey
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging Initiative, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM.
ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy.
Nature. 2017 Sep 28;549(7673):523-527. Epub 2017 Sep 20
PubMed.
Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study Group, Aalten P, Aarsland D, Alcolea D, Alexander M, Almdahl IS, Arnold SE, Baldeiras I, Barthel H, van Berckel BN, Bibeau K, Blennow K, Brooks DJ, van Buchem MA, Camus V, Cavedo E, Chen K, Chetelat G, Cohen AD, Drzezga A, Engelborghs S, Fagan AM, Fladby T, Fleisher AS, van der Flier WM, Ford L, Förster S, Fortea J, Foskett N, Frederiksen KS, Freund-Levi Y, Frisoni GB, Froelich L, Gabryelewicz T, Gill KD, Gkatzima O, Gómez-Tortosa E, Gordon MF, Grimmer T, Hampel H, Hausner L, Hellwig S, Herukka SK, Hildebrandt H, Ishihara L, Ivanoiu A, Jagust WJ, Johannsen P, Kandimalla R, Kapaki E, Klimkowicz-Mrowiec A, Klunk WE, Köhler S, Koglin N, Kornhuber J, Kramberger MG, Van Laere K, Landau SM, Lee DY, de Leon M, Lisetti V, Lleó A, Madsen K, Maier W, Marcusson J, Mattsson N, de Mendonça A, Meulenbroek O, Meyer PT, Mintun MA, Mok V, Molinuevo JL, Møllergård HM, Morris JC, Mroczko B, Van der Mussele S, Na DL, Newberg A, Nordberg A, Nordlund A, Novak GP, Paraskevas GP, Parnetti L, Perera G, Peters O, Popp J, Prabhakar S, Rabinovici GD, Ramakers IH, Rami L, Resende de Oliveira C, Rinne JO, Rodrigue KM, Rodríguez-Rodríguez E, Roe CM, Rot U, Rowe CC, Rüther E, Sabri O, Sanchez-Juan P, Santana I, Sarazin M, Schröder J, Schütte C, Seo SW, Soetewey F, Soininen H, Spiru L, Struyfs H, Teunissen CE, Tsolaki M, Vandenberghe R, Verbeek MM, Villemagne VL, Vos SJ, van Waalwijk van Doorn LJ, Waldemar G, Wallin A, Wallin ÅK, Wiltfang J, Wolk DA, Zboch M, Zetterberg H.
Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis.
JAMA. 2015 May 19;313(19):1924-38.
PubMed.
Geneticists Seek Out Rare Contributors to Alzheimer’s
At one end of the Alzheimer’s disease genetic spectrum lie the catastrophic mutations in APP and presenilin that lead to autosomal-dominant, early onset AD. At the other end are dozens of common variants that each contribute a smidgen to a person’s risk of late-onset AD. Between the two ends lie rare variants—glitches in the DNA that appear less frequently than common variants, but pack a punch when they do appear. The Alzheimer’s Association International Meeting, held July 14-18, 2019 in Los Angeles, California, featured new rare variants. They came out of the whole-genome and whole-exome sequencing projects geneticists are currently focusing on, and from a deep dive into existing GWAS data. Other newly published work identifies two new risk genes near the ApoE locus. Both contribute to AD risk independent of ApoE, and they may help explain variations in risk seen with the ApoE4 allele in different ethnic groups (see below and Part 5 of this series).
Genetic Landscape: The spectrum of genetic influences on Alzheimer’s disease spans rare to common variants that add more or less risk. [Image courtesy of Richard Mayeux.]
Families with genetic forms of early onset AD have fueled a better understanding of the disease (Aug 2019 conference coverage). Scouting for additional genes that cause early onset dementia, Kenneth Kosik, University of California, Santa Barbara, looked to Colombia. Kosik has worked with Francisco Lopera, Universidad de Antioquia, Medellin, for close to 30 years studying the Colombian AD kindred, a group with early onset AD that comprises nearly 30 families with a presenilin1 E280A mutation, some of whom are now in a prevention trial (Aug 2019 conference coverage).
While recruiting for this trial, Lopera, who is famous in Colombia, made a public appeal for people with familial or early onset dementia to come forward, and more than 1,000 did. Kosik’s group sequenced their genomes, which yielded 11 different presenilin missense mutations in 13 families, including three (I162S, Q223K, I427V) that had never been seen before. Other mutations were new to Colombia, but had been reported elsewhere, such as the widespread H163R mutation.
The analysis included other types of dementia, as well. It yielded 14 missense mutations in eight genes in families with frontal temporal dementia and amyotrophic lateral sclerosis (FTD/ALS). Kosik found mutations in the tau gene MAPT, progranulin (GRN), TARDP, TBK1, CHMP2B, UBQLN2, SOD1, and TUBA4A. Some families span a range of clinical phenotypes. For example, the I383V mutation in TARDP causes mostly behavioral-variant FTD, but also PPA and ALS/FTD. In the newly sequenced Colombian families, Kosik also detected a slew of TREM2 variants, including some homozygous cases.
While FTD is the most common form of dementia in younger people, it remains rare, and researchers are constantly challenged to gather enough affected or at-risk people for research and trials. Much like the families with E280A AD, these FTD families provide an opportunity to study progression, and develop instruments for early detection. For example, among three families with a newly discovered P397S MAPT mutation, two of the sequenced cases were diagnosed with bvFTD, and one with atypical AD. In neuropsychiatric testing of one family, mutation carriers in their 50s and 60s scored up to two standard deviations below normal on language, memory, and functional tests, corrected for age and education. Noncarriers had no such deficits. The carriers had early changes on the geriatric depression scale, suggesting that relatively simple tests could be used to detect and follow their disease, Kosik said.
“All of these people, including the new presenilin families and those with FTD, offer us more opportunities for clinical trials. These families are aware of the clinical trial being run now, and would be keen on participating in new trials,” Kosik said.
Does Colombia have a concentration of presenilin mutations? To find out, Kosik considered founder effects. Spanish conquistadors and African slaves likely brought the presenilin 1 Paisa E280A and I416T mutations to the country, respectively (Lalli et al., 2014; Ramirez Aguilar et al., 2019). After 1492, the native population was decimated by diseases brought by the Spanish, and communities shrank into geographically isolated pockets. There, a rapid expansion occurred, as families numbering 10 to 15 children rebuilt the population. This likely allowed the proliferation of rare dominant mutations. Even today, mutations causing AD and other genetic diseases occur in regional clusters in Colombia.
“Why, at the time of massive population collapse due to disease, did a relatively few people survive?” Kosik asked. Did the presenilin mutations confer a fitness advantage? Kosik is examining genomes in search of a signature for selection during the time of rapid population expansion. At AAIC, he speculated that PS mutation carriers might be protected from infectious disease, given the postulated role of Aβ in the innate immune system.
Other whole-exome and whole-genome sequencing projects trawl for rare variants in late-onset AD. Rare variants interest scientists because, compared with the common SNPs that pop up in GWAS, they are more likely to change gene expression or protein function, revealing pathways that might yield therapeutic targets. So far, rare variants associated with AD include protein-truncating and missense variants in TREM2, SORL1, and ABCA7 (Aug 2017 news). The largest whole-exome sequencing effort published to date, from the Alzheimer’s Disease Sequencing Project (ADSP), added to that list the transcription factor ZNF655 and the long non-coding RNA AC099552.4 (Aug 2018 news).
More recently, Lindsay Farrer, Boston University, reported the discovery of a batch of new genes in ADSP participants selected for having a first-degree relative with AD (April 2019 conference news).
At AAIC, Richard Mayeux, Columbia University, New York, added PINX1. This gene encodes an enzyme that helps maintain telomeres. In previous work, Mayeux’s group had sought extremely rare variants, present in just one in 10,000 people, in whole-exome and whole-genome sequencing data from their multiethnic Washington Heights-Inwood Community Aging Project (WHICAP), the ADSP, and other cohorts. SORL1 came up as the only significant hit (Jun 2018 news).
To find more, the new study widened the lens. It included variants that were present in one in 20 people and likely affected expression or function, but used an analysis that counted multiple variants per gene toward the association. The combined cohorts totaled 15,030 cases and controls. This study recently reported multiple variants in PINX1 that, at the gene level, reached significance. No single variant reached genome-wide significance. TREM2 did, if the scientists considered only highly damaging variants that reduced the receptor’s expression at least 20-fold (Tosto et al., 2019).
The PINX1 news ties into a previous report that associated shortening of telomeres with aging, dementia and mortality (Honig et al., 2006). PINX1 has been found to be overexpressed in postmortem brain from people with late-onset AD (Myers et al 2007; Narayanan et al., 2014). However, it’s still unclear how the identified variants relate to either telomere length or PINX1 expression.
At AAIC, Mayeux described work defining the effects of rare variants in another gene, SORL1, using induced pluripotent stem cells and organoids. Current theory holds that reduced expression, or loss of function, of SORL1 in AD allows more secretion of Aβ (Campion et al., 2019). Mayeux’s collaborator, Andrew Sproul, also at Columbia, found that neurons derived from human IPSCs expressing the SORL1 mutant E270K process more APP, increase tau, and enlarge their endosomes, compared with isogenic control neurons with no mutation. Organoids derived from the cells reduce cell cycling and accumulate extensive Aβ. The group is currently converting the IPSCs into microglia to learn how the SORL1 mutation affects those cells, Mayeux said.
Also at AAIC, Adam Naj, University of Pennsylvania Perelman School of Medicine, talked about mining existing genotyping data to identify rare variants. Based on a newly released haplotype reference panel, scientists can now use genotypes at common SNPs to impute genotypes at linked, rarer SNPs.
An International Genomics of Alzheimer’s Project (IGAP) meta-analysis of genome-wide associations earlier this year identified 30 susceptibility loci, of which a majority were present in more than one in 50 people (Mar 2019 news). To identify rare variants, Naj imputed genotypes of SNPs present in less than 1 percent of the 64,859 subjects in this dataset. The top hits were in TREM2 and APOE, two genes that reached genome-wide significance, as did SORL1.
No other genes crossed this bar, but several SNPs in novel genes reached the p<10-5 significance level considered suggestive of an association. They include GCA, CTNND2, DYDC2, DYX1C1, B4GALT6, and PWP2. The implicated genes are involved in innate immunity, neuronal differentiation, or had previously been identified as candidate genes for LOAD, ALS, or brain development. Gene-based analysis implicated new signals in SORL1, plus SIRPD, a gene heavily expressed in macrophages; CYB561, which is associated with amyloid processing and inflammation; BLNK, which is coregulated with TREM2; and SLX4; which is involved in aging and telomere shortening. Replication is ongoing, Naj said.
Naj also looked for new common variants, present in more than 1 percent of the subjects, by imputation. He replicated almost all know IGAP loci, and identified one new genome-wide significant locus. The SIL1 gene encodes a protein involved in ER stress, and can reverse some types of tau hyperphosphorylation. They also found 43 other genes, albeit with lower significance, including almost all known IGAP loci. Most of the 18 new genes slotted into familiar pathways of cardiac-related metabolism, immunity, and neuron function.
In recent years, AD researchers have begun to grapple with how ethnicity affects AD genetics. Take ApoE. The risk imparted by its E4 risk allele is known to vary among ethnic groups. Its impact is greatest among Japanese people, intermediate in Caucasians, lower in Hispanics and lower still in African Americans (Farrer et al., 1997; Tang et al., 1998). According to work by Margaret Pericak-Vance and colleagues at the University of Miami Miller School of Medicine, the relative protection of African Americans seems to stem from outside the ApoE coding region, and involve the surrounding gene ancestry (Rajabli et al., 2018; Apr 2019 conference news).
Pericak-Vance and colleagues are trying to nail down just what those surrounding influences are. One hypothesis is that variants in the TOMM40 gene, which lies adjacent to APOE, could bestow the protection. Previous work suggested that an intronic polyT repeat in TOMM40 tended to be longer in non-Hispanic white people who had the ApoE 3/3 genotype and AD, compared to those without AD (Cruchaga et al., 2011). Alas, data presented at AAIC suggests repeat length is not what protects African Americans. Parker Bussies from the Pericak-Vance lab, measured the length of the polyT variant in cases and controls of 100 African Americans and 100 non-Hispanic whites. Half were ApoE3/E3, half E4/E4. The TOMM40 repeat had no length-dependent protective effect in E4/E4 African Americans.
To further proble local ancestry effects, Derek Dykxhoorn of the same group has made IPSC’s from people with African or European gene ancestry around the ApoE locus, with either E4/4 or E3/3 genotype, and will use the cells to look at molecular mechanisms underlying the ethnic-specific differences.
He may be helped by a recent publication from Nancy Ip, Hong Kong University of Science and Technology, China, and colleagues, which identifies AD causal variants in two genes near ApoE (Zhou et al., 2019). In that study, the authors fine mapped the APOE region using whole-genome sequencing and imputed array data from Chinese and non-Asian AD cohorts. They identify variants in two genes, PVRL2 and APOC1, that are associated with AD. The variants form extended haplotypes with ApoE, which appear more frequently in people with AD. The genes also act independently to raise risk in the absence of ApoE4. The extended risk haplotypes were associated with decreased cognitive performance, lower brain volume, particularly hippocampal, and with plasma and CSF biomarkers.
In the brain, the risk haplotypes were also associated with higher ApoE expression, which may play a role AD. Interestingly, the haplotype frequencies vary greatly among different ethnic groups. Almost no Africans carry the minor, risk haplotypes of PVRL2, ApoC1, or the extended minor haplotypes of ApoE, whereas 2 to 10 percent of Europeans do, perhaps explaining some of ApoE’s local ancestry effects.—Pat McCaffrey
Move Over Aβ, CSF P-Tau Tells Us There’s Plaque in the Brain
Just as labs around the globe are racing to start marketing plasma Aβ tests, along come new contenders to rattle the field. At this year’s AAIC, held July 14-18 in Los Angeles, scientists reported that certain amino acids on the protein tau are phosphorylated decades before AD symptom onset. Both Randall Bateman, Washington University, St. Louis, and Henrik Zetterberg, University of Gothenburg, told Alzforum that these tau species might reflect a response to Aβ pathology in the brain.
They, and other investigators, suspect that as amyloid accumulates in the brain, neurons begin to overproduce tau, which then becomes modified at specific sites, such as threonine 181 and serine 217. Those p-tau isoforms can readily be detected in the CSF—and, even more excitingly, in plasma—potentially making them early markers of AD pathology. Ironically, this even implies that a positive blood test for tau could indicate the presence of amyloid plaques in the brain. If these early findings hold up, they raise the future prospect of a single blood draw giving presymptomatic information on brain changes in Aβ and tau. It would be specific for AD as defined by Alois Alzheimer’s original definition of the disease.
Some of the evidence for this comes from Bateman’s own lab. At AAIC, Nicolas Barthélemy from WashU reported a detailed mass spectroscopy analysis of the various phosphorylated forms of tau in the brain and cerebrospinal fluid. Measuring p-tau in the CSF by mass spectrometry presents a challenge, because concentrations there are 1,000-fold lower than in brain lysates. Nonetheless, Barthélemy thinks the CSF may better reflect the state of tau phosphorylation in the brain because phosphatases that retain activity postmortem can change the profile of phospho-tau in tissue samples.
Phosphorylation Hot Spots. Degree of phosphorylation (circle diameters) varies among the normal brain, normal CSF, and AD CSF. Orange and red circles represent slight and high hyperphosphorylation, respectively, compared with normal brain. [Courtesy of Nicholas Barthélemy.]
Barthélemy’s data bore out his idea. He used nano liquid chromatography/high-resolution mass spectroscopy to improve detection of p-tau species in the CSF. Barthélemy immunoprecipitated total tau, digested it with trypsin, then tested for levels of each hypothesized phospho-tau amino acid. He measured the ratio of phosphorylated to unphosphorylated residues as a readout to adjust for fluctuations in total tau levels that might skew the data.
Barthélemy found 12 tau sites that are phosphorylated in normal human CSF. Some, for example p-T205 and p-S208, were unique to the spinal fluid, i.e. absent in brain lysates. Others, including threonines 111, 217, and 231, did show up in the brain, but were more highly phosphorylated in the CSF. Still other phospho-tau species, namely those at the N- and C-termini, only turned up in the brain. Barthélemy speculated that tau’s endpieces may never make it into the CSF because of how the protein is processed and cleared by proteolytic machinery.
What about AD? Here too, Barthélemy found specific changes. In CSF pooled from seven people who were amyloid-positive and had a clinical dementia rating of 0.5, tau was hyperphosphorylated at threonines 111, 181, 205, and 217, and at serine 208, relative to pooled CSF from five amyloid-negative, cognitively normal controls. Sites equally phosphorylated in AD and control CSF included S199 and T231. Serine 202 was slightly less phosphorylated in AD than in controls.
It was the timing of phosphorylation that drew the most attention in Los Angeles. Barthélemy assayed CSF samples from 639 people in the longitudinal DIAN cohort (for a DIAN update from AAIC, see Aug 2019 conference news). In this cross-sectional analysis, changes in phospho-tau emerged decades before symptoms. The earliest increase, at 21 years prior to onset, was for p-T217, followed at 19 years before onset for p-T181. Tantalizingly, Barthélemy reported that CSF p-T217 tau levels correlated with uptake of PiB in the precuneus, and that it predicted amyloid positivity with an accuracy of 97 percent. If this data holds up, it would place increases in p-tau at around the time of amyloid accumulation in the trajectory of AD pathology.
And hold up it did. Three days later at AAIC, Niklas Mattsson from Lund University, Sweden, painted a similar picture of CSF tau markers in the Swedish BioFinder cohort. Mattsson acknowledged earlier work from Barthélemy and Chihiro Sato, also in Bateman’s lab, who used stable isotope labeling kinetics to determine that tau production rates rise in early AD (Mar 2018 news). Based on this, Mattsson, Oskar Hansson also at Lund, and other colleagues in the BioFINDER group, hypothesized that changes in the metabolism and phosphorylation of soluble tau might mediate the relationship between Aβ fibrils and the subsequent development of tau tangles. Mattsson set out to test this.
First, Mattsson correlated p-tau levels with dementia severity. He found higher levels of CSF p181-tau, p217-tau, and total tau among 40 amyloid PET-positive, cognitively unimpaired people than among 18 amyloid PET-negative, cognitively normal volunteers. Phospho-tau levels crept higher still in 38 amyloid PET-positive people who also showed signs of mild cognitive impairment (MCI), and were highest in 35 people who had a diagnosis of AD dementia.
Levels of either phospho-tau species distinguished all four groups, suggesting the markers might prove useful for staging. That the p-tau markers were able to distinguish amyloid-positive from amyloid-negative individuals who all appeared cognitively normal suggests an advantage over tau PET, which becomes positive only once people become impaired. All told, 55 and 70 percent of the cognitively normal, amyloid-positive volunteers tested positive for p181- and p217-tau, respectively. These percentages rose to 90 and 100 among those who were cognitively impaired.
Having established that these tau species rise early in amyloid pathogenesis, Mattsson next asked how they change over time. He tested CSF samples taken two years apart from 32 amyloid-positive, cognitively unimpaired volunteers whose tau PET scans of the inferior temporal cortex would not turn positive for at least another 3.8 years. Even at this early stage, their CSF p181-tau rose by 8.7 pg/mL per year, and the p217 form rose even faster, at 13.8 pg/ml per year.
How does the timing of this CSF p-tau surge relate to early amyloid pathology? To address this, Mattsson correlated CSF p-tau levels with 18F-flutemetamol binding. Both p181- and p217-tau were already higher when a composite standard uptake value ratio of flutemetamol was still below 0.7. In this composite of brain regions where plaque deposition starts, an SUVR of 0.743 denoted amyloid positivity. In other words, these two p-tau CSF markers started to change before the amyloid PET scans were positive. In contrast, flortaucipir binding in the inferior temporal cortex only registered as positive on PET once the flutemetamol SUVR had reached 0.78. In toto, CSF p181- and p217-tau preceded amyloid PET, which preceded neurofibrillary tangle PET as sequential measures of AD pathology.
Other scientists at AAIC were impressed by Mattsson’s finding. Clifford Jack, Mayo Clinic, Rochester, Minnesota, said the CSF/PET relationship for p-tau was uncannily like that for Aβ, where dips in the CSF Aβ42 level precede positive amyloid PET scans. “It seems to be telling us that CSF may be a sensitive measure of what is going on in the brain,” he said.
Val Lowe, also from Mayo, asked why Mattsson focused on tau in the inferior temporal cortex. Lowe wondered if flortaucipir binding in a more sensitive region, such as the medial temporal, might tell a different story. Mattsson said he focused on the inferior temporal because ligand binding is most robust there, but noted that his group did the same analysis for the entorhinal cortex. “All the relationships with p-tau and tau PET remain very similar,” he said.
To learn how these new findings relate to p-tau species in the blood, see Part 8 of this series.—Tom Fagan
Move Over CSF, P-Tau in Blood Also Tells Us There’s Plaque in the Brain
From day one at this year’s AAIC, held July 14-18 in Los Angeles, conference halls and corridors buzzed with the sound of “p-tau.” Scientists from Randall Bateman’s lab at Washington University, St. Louis, reported that among volunteers in DIAN, certain species of phospho-tau inched up in the CSF as early as 21 years before symptom onset, many years before tangles can be detected in the brain by PET imaging. Niklas Mattsson from the BioFINDER group at Lund University, Sweden, showed that the same species, p181- and p217-tau, rose in the CSF even before plaques can be detected by PET (see Part 7 of this series). Researchers entertained the prospect of getting information on both early amyloid and early tau changes from one lumbar puncture.
What about blood? Given the recent success in developing plasma assays for Aβ that reflect ongoing brain amyloid, scientists wanted to know if this p-tau-amyloid link would hold up for a person’s blood as well. And there are tools to find out. Jeffrey Dage at Lilly Research Laboratories, Indianapolis, has developed an in-house antibody test for plasma p181-tau. Scientists at the University of Gothenburg are working on one, also.
Last year, Michelle Mielke and colleagues at the Mayo Clinic, Rochester, Minnesota, sent samples from their Mayo Clinic Study of Aging and from the Alzheimer’s Disease Research Center at Mayo to Dage for testing. Voila—increases in plasma p181-tau turned up, and tracked with AD severity (Mielke et al., 2018). In Los Angeles, Mielke was delighted to learn that other groups described complementary data.
In Synch. Levels of p181-tau in the plasma track with Braak staging as determined by tau PET. [Courtesy of Oskar Hansson.]
For example, Oskar Hansson and Shorena Janelidze from the BioFINDER group at Lund University, reported a tight correlation between plasma p181-tau and p181-tau in the CSF of amyloid-positive participants in this longitudinal cohort. Moreover, plasma p181-tau tightly correlated with flortaucipir uptake in the brain and predicted Braak staging, Hansson said (see image below).
To be sure, the concentration of tau in blood seems vanishingly small, but it is detectable. Average plasma levels of 1.25 pg/mL in tau PET negative controls compared with about 2.5, 3.8, and 6 pg/mL in those with tau PET scans indicative of Braak stages I/II, III/IV, and V/VI, respectively. This suggests this plasma marker could help with both diagnosis and staging.
In keeping with this idea, plasma p181-tau poorly correlated with flortaucipir in Aβ-negative people. Levels of p181-tau in plasma from people with a non-AD dementia didn’t budge above baseline, either, suggesting this marker might be quite specific for AD and help with differential diagnosis. Plasma p181-tau differentiated AD from other neurodegenerative disorders with an AUC of 0.95, a similar accuracy to tau PET and CSF p181-tau.
Elisabeth Thijssen, University of California, San Francisco, came to much the same conclusion. She compared plasma p181-tau and neurofilament light as potential markers for AD and frontotemporal dementias. Tau marker levels were higher in 39 AD patients than in any of 45 normal controls, 40 MCI patients, or 143 FTLD cases. Among the latter group, 36 people with corticobasal syndrome, 47 with progressive supranuclear palsy, 46 with behavioral variant FTD, and 14 patients with primary progressive aphasia all posted similar p181-tau levels as did controls. Plasma p181-tau distinguished these FTLD cases from AD with about 90 percent accuracy, Thijssen reported.
Why did the MCI group not have higher plasma p181-tau if this marker rises so early in AD pathogenesis? It turns out 21 of those 40 MCI cases were amyloid-negative. The amyloid-positive cases, on the other hand, did have three times more p181-tau in their plasma. Indeed, the marker identified amyloid PET-positives among the MCI group with 95 percent accuracy.
Like Hansson, Thijssen also found that plasma p181-tau correlated tightly with flortaucipir uptake in amyloid-positive people. Among 72 AD/MCI patients, plasma p181-tau also correlated tightly with atrophy of the temporo-parietal region of the brain, including the medial temporal lobe, the posterior cingulate cortex, and the precuneus.
Hansson believes that plasma p181-tau not only distinguishes AD from most non-AD dementias, but that it can also predict future development of AD. In a survival analysis, he found that for each standard deviation increase in this blood marker in a given group, their incidence of future AD rose 2.8-fold. And as a prognostic marker, p181-tau outshone other plasma markers in a multivariate analysis, including Aβ42/40, total tau, and NfL, none of which associated with risk of AD, he said. Hansson thinks plasma p181-tau, much like p181-tau in CSF, will be shown to nudge above baseline levels even before amyloid can be detected by PET imaging, and he foresees p181-tau being used as a very early marker of AD. He predicted a combination of plasma Aβ42/40 ratio and p181-tau might be optimal for detection of AD pathology during the early disease stages.
Paul Aisen, University of Southern California, San Diego, thought that p181-tau looked extremely interesting, but stopped short of calling it a marker of amyloidosis, as some suggested. “I think the better test for amyloid is the plasma Aβ42/40 ratio, particularly in presymptomatic individuals,” he said (for news on plasma Aβ42/40, see Parts 9 and 10 of this series).
Adam Boxer, University of California, San Francisco, agreed. “I think p181- tau reflects overproduction of tau in the AD state, but it is not a marker of plaques,” he told Alzforum. Boxer noted that p181-tau ticks up in certain FTLD patients who accumulate neurofibrillary tangles containing both three- and four-repeat tau—the same type of tangle found in AD. Some rare FTLD tau mutations, such as R406W, cause 3R/4R tangles. “I think p181 may be a marker for 3R/4R tangles, said Boxer.
This would agree with Mattsson’s data, which suggest that phosphorylation at threonines 181 and 217 associates with tangle formation. He used PET and CSF data to model how soluble forms of tau mediate the relationship between amyloid and tangles. To ensure he was capturing early changes, Mattsson analyzed data from people who were cognitively normal or only slightly impaired. He calculated that the direct effect of amyloid on tangles was small; rather, p217- and p181-tau mediated 68 and 80 percent of the effect, respectively.
Given this mediation, and their very early rise in AD, are p181- and p217-tau markers of tau processing rather than of tangles? “That’s the implication here. I would be surprised. I think we need to see more data on that,” said Aisen. Hansson also cautioned against overinterpretation. “We have a sensitivity issue with PET,” he said. “We can’t be sure that the p-tau increases do not come after someone has already got a handful of tangles in their brain that PET is not sensitive enough to detect.”
Noting that fluid tests tend to be more sensitive than PET, Kaj Blennow, University of Gothenburg, drew an analogy between the current state of p-tau research and the early days of Aβ42 CSF/amyloid PET concordance studies. Those studies found people who were CSF-positive but PET-negative, and these people became PET-positive over time. “That is accepted now,” said Blennow, “and the same could be the case with plasma tau.” In other words, if plasma and CSF p181-tau tests are more sensitive than tau PET, then people who are now testing positive on p181-tau fluid tests but negative on PET may well be positive on both in a few years.
This also means that the staging diagram as currently perceived may reflect differential sensitivity of the markers as much as true biological sequence of change, Blennow said.
Many researchers at AAIC suggested that plasma p181-tau might prove to be less fickle than the plasma Aβ42/40 ratio. The latter falls by only 10 to 15 percent in AD versus controls, and fluctuations caused by environmental factors such as sleep, exercise, and cardiovascular fitness might make that differential smaller still. In contrast, the change in p-tau is an order of magnitude above controls, and continues to climb with disease progression. “Yes, the differences in the Aβ ratio test may be small, but we are still able to achieve a robust distinction between normal and abnormal,” countered Aisen. “I think p181-tau is a measure of disease stage and of neurodegeneration, and is going to be extremely useful for tracking progression,” he said. Aisen believes it is time to start incorporating plasma markers into clinical studies. “Maybe p-tau is even better than NfL,” he suggested.
For her part, Thijssen might agree, at least on the differential diagnosis. She found that unlike p181-tau, plasma NfL levels were indistinguishable between normal controls and AD patients, while NfL levels in CBS, PSP, and bvFTLD patients were at least twofold higher than controls. For AAIC news on plasma Aβ, see part 9 of this series. —Tom Fagan
In the short space of two years, the erstwhile fantasy of a blood test for Alzheimer’s disease has become reality. Or so it seems. At the 2017 Alzheimer’s Association International Conference in London, Randall Bateman, Washington University, St. Louis, had wowed the audience by showing how an exquisitely sensitive mass spectroscopy assay of plasma detected a teeny drop in the Aβ42/40 ratio that correlated with positive PET scans for brain amyloid (Jul 2017 conference news). Scientists wanted to see this reproduced in other cohorts. At this year’s AAIC, held July 14-18 in Los Angeles, presentations on plasma Aβ measures abounded. Since last summer, as many as 11 different tests, both mass spec and immunoassays, have correlated plasma Aβ42/40 with CSF Aβ and amyloid in the brain. Is plasma Aβ ready for prime time? At AAIC, some researchers said yes, but others tempered their enthusiasm. Disagreement among mass spec and immunoassays emerged as a sticking point.
Paul Aisen, University of Southern California, San Diego, was in the first camp. “The plasma assays are great,” he told Alzforum. “When they came out, I thought, ‘Everything is different now but we need to confirm.’ Well, it’s been confirmed.” Aisen is ready to use plasma assays in clinical trials, though he sees room for growth. “These assays are not in their final state and every improvement will make a difference,” he said.
Bateman is also gung ho. “I’m convinced this will work,” he told Alzforum. “In fact, it is working. We can take samples from different centers and run them in our mass spec assay and find total agreement,” he said, noting the assay is so robust that data from different sources can be pooled for analysis. Bateman co-founded C2N Diagnostics, St. Louis, which develops plasma assays, including for Aβ.
Others were more cautious, citing a lack of correlation among immunoassays, and between them and mass spec tests. This could reflect methodological issues, or different forms or pools of Aβ in plasma, noted Henrik Zetterberg, University of Gothenburg, Sweden. If those pools react differently with different antibodies, that might imply something important about the underlying biology, some researchers suggested. Oskar Hansson, Lund University, Sweden, also takes this view. “We need large—i.e., many hundreds of cases—head-to-head studies comparing different plasma Aβ assays in the same population against the same standard of truth,” he told Alzforum.
Scattershot? Some examples from a round robin comparing 11 different mass spec and immunoassays. It found generally weak correlations for measurements of Aβ42. This peptide appears to be particularly fickle, as the same tests posted slightly better correlations for measuring plasma Aβ40. Dashed red lines represent perfect correlations. [Courtesy of Henrik Zetterberg and Kaj Blennow, University of Gothenburg.]
Assays Galore
Since 2017, Akinori Nakamura and Colin Masters and colleagues published a mass spec method to measure the Aβ40/42 ratio; it is being developed by Shimadzu Corp. The team at UGothenburg optimized their own IP mass-spec assay, and the Spanish company Araclon has been working on such a mass-spec test service for some years already. Various immunoassays exist, as well, some currently in-house, some commercially available (see table below). All are being tested on various cohorts.
At AAIC, Bateman reported that his group has tested plasma of a further 158 people, members of the fourth in a line of cohorts being followed at the Knight Alzheimer’s Disease Research Center at WashU. As was the case in a previous sample set of 164 people, the plasma Aβ42/40 ratio once again tightly correlated with both the CSF Aβ42/40 ratio and with brain amyloid assessed by PET. All told, the WashU plasma test predicted amyloidosis with a specificity and a sensitivity of 76 and 88 percent, respectively, Bateman said. In statistical receiver operator curve analysis, this amounted to an area under the curve of 0.88. The AUC improved to 0.94 when the researchers factored in age and ApoE4 status, making this plasma Aβ test as good as CSF or PET for diagnosing amyloidosis. Led by Suzanne Schindler in Bateman’s lab, this work came out August 1 in Neurology (Schindler et al., 2019).
This mass spec test had predictive power. Of eight people whose amyloid PET scans were negative at baseline but positive on follow-up, seven had baseline plasma tests that were already positive, falling under the 0.1218 cutoff value for the Aβ42/40 ratio. In other words, in this small sample, people who were amyloid-negative by PET but amyloid-positive by plasma mass spec were 15 times more likely to test positive on PET later than were people who were plasma-negative at baseline.
What about other cohorts? In LA, Bateman reported results of testing samples from the Australian Imaging and Biomarkers Lifestyle study, the Swedish BioFINDER study, and the Alzheimer Disease Neuroimaging Initiative. The plasma test predicted PET status with AUCs of 0.87, 0.83, and 0.85 for AIBL, BioFINDER, and ADNI, respectively. Once again, adding ApoE4 into the equation raised these AUCs to 0.93, 0.90, and 0.87. Aβ42/40 cutoffs for amyloid positivity worked out to be 0.1235, 0.1234, and 0.1269 for AIBL, BioFINDER, and ADNI, respectively, which is close to the 0.1218 ratio calculated for the WashU cohort. Combined, for a total of 468 subjects, these three replication cohorts returned a cutoff of 0.1234.
For their part, immunoassays are posting similar results. At AAIC, Hansson reviewed his groups’ analysis of Roche Diagnostic’s Elecsys plasma immunoassay. This data appeared June 24 in JAMA Neurology (Jun 2019 news), and Alzforum covered it in depth. Briefly, this assay predicted amyloid positivity among 842 people in BioFINDER with about 80 percent accuracy, slightly less than the WashU test. In a validation cohort of 237 volunteers in a prospective study in Germany, the Elecsys immunoassay performed better, yielding an AUC of 0.86. In the Swedish cohort, adding in ApoE increased the AUC to 0.87; in the German cohort, genotyping was unavailable. In LA, Hansson also reported that among 335 cognitively normal people, the Elecsys plasma assay predicted who would get dementia over the next six years.
Head-to-Head Comparison: A Little Hairy
How do different plasma assays compare? Two AAIC presentations broached this question. Hansson showed preliminary head-to-head data for four such tests: Roche’s Elecsys immunoassay; EUROIMMUN’s ELISA; Shimadzu’s mass spec assay; and Quanterix’s antibody-based single-molecule array (Simoa). All but the Quanterix assay similarly predicted amyloid PET status among 199 cognitively normal people. The Elecsys, EUROIMMUN, and Shimadzu AUCs were 0.81, 0.76, and 0.82, respectively; the Simoa assay posted a 0.59. Adding APOE genotype improved these values to 0.86, 0.84, 0.87, and 0.79.
However, when you take a given set of samples, are the different assays measuring the same absolute amount of Aβ in it? Here it gets tricky. Zetterberg presented a comparison of 11 different plasma Aβ42/40 assays conducted at 11 different sites (see table below). Co-led by Kaj Blennow, also at UGothenburg, this round robin study was a project of the Global Biomarkers Standardization Consortium. The idea was to select plasma samples across a wide range of Aβ concentrations and send 0.25 milliliter aliquots to each participating lab, which tested these identical aliquots on their respective platforms. Unlike the work summarized in this story so far, this study did not evaluate any given test’s diagnostic accuracy against amyloid PET. Rather, it determined how the different tests correlated with each other when all were presented with the same amount of a given analyte—Aβ40 and Aβ42—in identical samples.
Who Participated. A round robin compared 11 different assays for plasma Aβ being conducted at 11 different sites. [Image courtesy of Henrik Zetterberg and Kaj Blennow, University of Gothenburg.]
So how did the tests match up? In short, not well, at least for Aβ42. Presenting slide after slide of pairwise correlations, Zetterberg showed results that scattered widely rather than crowding neatly around a line. For example, Elecsys values for Aβ42 matched poorly with MS data from UGothenburg, Shimadzu, and WashU, with the latter achieving the highest correlation of only 0.22 (see image below). Correlations of 0.27 and 0.46 between Elecsys and Simoa assays used by UPenn and UAmsterdam, respectively, were slightly better, but still weak. The best correlations, of around 0.6, emerged among the UGothenburg, Araclon, and Shimadzu mass spec assays. Calculating the Aβ42/40 ratio did not help, Zetterberg told Alzforum.
Tighter correlations, in the 0.6 to 0.7 range, emerged for the measurement of Aβ40, which is less sticky and occurs at 10 times higher concentration in plasma than does Aβ42. Still, Aβ42 is the key driver of amyloid pathology and the indicator of amyloidosis.
Head to Head. In this overview of correlations from the plasma Aβ round-robin study, green denotes tight correlations between any given two tests; red, weak correlations. [Courtesy of Henrik Zetterberg and Kaj Blennow, University of Gothenburg.]
Overall in this round robin, the mass-spec methods showed less variance among each other than the immunoassays did, Zetterberg said. This prompted debate about whether immunoassays or mass spec will be better. With the former, different antibodies might detect different pools of Aβ. The latter are harder to scale up, and dependent on at least one antibody for immunoprecipitation prior to mass spectrometry. Larger head-to-head comparisons with different clinical groups are next. “Before such studies have been done, I would not dare to say that all MS methods are superior to all immunoassays,” Hansson said.
Jonathan Schott, University College London, also found disagreement between MS and immunoassays. Working with Zetterberg and Blennow, Schott’s team compared the Quanterix Simoa to mass spec analyses run at UGothenburg. The plasma came from the 1946 British Birth Cohort, which is unique in that its participants, drawn from across the U.K., were all born within the same week in March of that year. They have been followed clinically since birth, and in the past few years, fluid and PET biomarkers were added.
For each volunteer, blood was collected at the same time of day, under the same fasting conditions, and treated and frozen the same way, Schott said. The scientists then measured Aβ40 and Aβ42 in plasma from 414 cognitively normal volunteers in the cohort.
For Aβ40, Simoa and mass spec data agreed fairly well, just like in the round robin. For the complete sample set, Quanterix returned a median of 288 pg/mL and mass spec 284 pg/mL, and comparing sample by sample, the two assays correlated with a coefficient of 0.44.
For Aβ42, the story was different. Mass spec measured higher values, giving a median of 28.4 pg/mL versus 19.5 pg/mL for Quanterix, and sample-by-sample correlation had a meager coefficient of 0.22. Regardless of whether a participant had a positive or negative amyloid PET scan, the MS measured more Aβ42 in the blood than the Quanterix test.
These differences are important. Schott found that the Aβ42/40 mass spec assay predicted PET positivity better than the Quanterix Simoa assay, with an AUC (uncorrected for age or ApoE status) of 0.82 over 0.61.
Why do the methods not agree? Zetterberg thinks it is not only the assays themselves, because these same assays did show tight correlations when used to measure Aβ42 in the CSF. Scientists at AAIC said the problem is likely the plasma. For one, plasma presents difficult matrix effects, meaning that the complex mixture of different solutes in the solution can cause spurious results. Diluting the plasma to get a more CSF-like matrix might help, but then the Aβ concentrations will be extremely low, requiring even more sensitive assays, Zetterberg said. It could also be that some assays are particularly sensitive to details of sample handling, which vary across centers. The immunoassays may be measuring different pools of Aβ; and at 10 to 40 picograms per milliliter, the Aβ42 concentration in plasma is always hovering dangerously close to the limit of detection.
Bloody Complicated. Plasma Aβ comes from platelets, the brain, and peripheral organs. Systemic conditions influence plasma Aβ concentration. [Courtesy of Yan-Jiang Wang, Nature 2017.]
Does it matter how well assays correlate so long as they diagnose amyloidosis? Scientists seemed divided on this point. Blennow sees trouble down the road. “When different labs use different assays on identical aliquots that contain a given amount of Aβ42, and they measure quite different amounts—that is a problem,” he said (see Blennow Q&A). Colin Masters, University of Melbourne, Australia, echoed the sentiment, as did Ralph Martins from Edith Cowan University, Perth, Australia. They said that because the difference in plasma Aβ42 between controls and amyloid-positive people is so small to begin with, any biological phenomena that influence the blood Aβ concentration could tip the measurement. These phenomena include high blood pressure, diabetes, and hypoxia, and lifestyle factors, such as exercise.
Martins investigated the effect of physical activity on plasma Aβ42, and came up with a surprising result. Although evidence is strong that physical activity can protect the brain from dementia, among all healthy controls in AIBL, those who recorded high physical activity over the preceding seven days had less Aβ42 in their plasma than those who had reported low or medium activity. Less plasma Aβ42 could be taken to mean these people had more amyloid in the brain than people who exercised less. “The trend is exactly the opposite of what you might expect,” said Martins.
The trend held in healthy controls who tested negative for brain amyloid by PET, but in people who were amyloid-positive, physical activity seemed to have no effect on plasma Aβ42. Martins called the results perplexing, and surmised that exercise might promote Aβ degradation. “The take-home message is that lifestyle factors, particularly exercise, can influence plasma Aβ levels and need to be controlled for,” said Martins.
Those concerns prompted a study at WashU, where 1,000 people with common diseases such as hypertension, cancer, and diabetes are being enrolled for monitoring, periodic blood draws, and an amyloid PET scan in an effort to learn how various health conditions affect their blood Aβ42/40 ratios and its ability to predict their brain amyloidosis.
Meanwhile, companies are pushing to commercialize plasma assays. C2N and Shimadzu are already selling mass-spec tests to a few groups, and the other companies are not far behind. The stakes are high. Plasma assays will usher in a new era of diagnostics and prognostics in the AD field. Scientists at AAIC were giddy over the prospects of screening more effectively and cheaply for clinical trials. They envision running shorter trials with shifts in plasma Aβ as readouts, or of going back to thousands of blood samples stored from previous trials to tease out biological effects that may have been missed. Even basic questions such as Alzheimer’s incidence in developing countries will be easier to answer with a reliable blood test.
Bateman and Hansson both stressed the savings offered by plasma tests. Bateman calculated that in a trial such as A4, which used amyloid PET as an inclusion criteria, a plasma test could cut screening time from three years to six months and slash costs 10-fold. Hansson estimated that a plasma Aβ test could halve the number of PET scans needed to enroll 1,000 people, saving about $4 million. For his part, Schott based his estimate on a price tag of $3,000 for a PET scan and an assumed $400 for a blood test, calculating a saving of about $3.5 million for a trial of 500 participants.
That price assumption may prove optimistic. At AAIC, companies were marketing their plasma tests to customers such as pharma and large academic groups who are planning treatment trials and need to bring down screening costs. The companies are feeling out what they will be able to charge, but when asked about price, they were circumspect. When pressed, Shimadzu representatives allowed that they were currently selling their service in Japan at a list price of about $1,000 per test and an actual negotiated rate of between $500 and $900. Shimadzu intends to start selling for research use in the U.S. this fall. Its representatives told Alzforum that the pricing structure was not set yet, but would be somewhat lower than in Japan.
Like Shimadzu and Araclon, C2N sells its test as a service, where plasma gets shipped to its central lab and analyzed on the company’s mass-spec machines. C2N representatives told Alzforum that the current price for pharma and research use comes to roughly $700 per sample. Like Shimadzu’s, it is negotiated. C2N’s price varies depending on how many tests are being purchased, and whether the customer shares back data that C2N could use in its application for FDA approval. C2N has FDA breakthrough validation for its mass spec plasma Aβ assay, and is preparing an approval submission.
“This is our coming-out party,” Joel Braunstein of C2N told Alzforum during AAIC. “We are getting ready to stand behind our product. We have 30 meetings here to feel out what price pharma and academic groups find acceptable. Everyone is asking us what it will cost! We are envisioning below $1,000 per analyte.” For a Q&A on developing robust blood tests in Alzheimer's, see part 10 of this series. —Tom Fagan and Gabrielle Strobel
Why Bother With Round Robins on Blood Tests? Q&A with Kaj Blennow
At the AAIC conference held July 14–18 in Los Angeles, Kaj Blennow of the University of Gothenburg had his hands full trying to get researchers in the field engaged in the nitpicky drudgery of validation, standardization, and commutability studies when all they wanted to do was grab one of those fancy new blood tests and run with it. Blennow, a world leader in clinical chemistry in Alzheimer’s research, has been guiding the work of the Global Biomarkers Standardization Committee (GBSC) for the past 10 years, starting with a quality-control program for CSF Aβ and tau assays. Convening academic leaders in AD biomarker development and scientists from companies that make tests for biological fluids such as CSF and blood, this group has been meeting in person and over the phone regularly, under the auspices of the Alzheimer’s Association.
Kaj Blennow
Their work is not the stuff of Nature, Science, and Cell journals. It’s not glamorous. It’s complicated in a tedious sort of way. But it’s essential if the field is to move beyond tests that only work in the inventor’s lab, or are sold “research use only,” toward having a choice of automated, affordable, and clinically certified tests that are routinely used for millions of people in doctors’ offices, hospitals, and central labs—tests that perform identically, year after year, be it in London, Sydney, or anywhere else. The GBSC has had some success standardizing tests of CSF Aβ42 and tau, but given that the field is generating promising new AD tests, its work is far from done. In 2018, the group took up quality control of CSF NfL tests. In 2019, it started—with a first, much-debated round robin—to try to bring some order to a suddenly burgeoning plasma test scene. The round robin showed generally weak correlation among the tests. Even so, a handful of contenders are rushing to market (see Part 9 of this series). Is that good enough?
Q: What is your long-term goal for Alzheimer’s blood tests?
A: I foresee us having robust blood tests for clinical Alzheimer’s diagnosis and care. Ideally, multiple tests that are in agreement, as we have for cholesterol, blood sugar, cystatin C, hemoglobin etc. We need this for blood Aβ, and blood tau, too. I am trying to engage the field in the validation processes that are necessary to create this landscape. This means we have to look beyond just a single purpose for a favorite test, i.e., predicting brain amyloid deposition as a prescreen for clinical trials. We need tests that also work for screening, or even diagnostics, in clinical care.
Q: Is this realistic?
A: Of course. For CSF Aβ42, we are almost there. Fully automated, approved tests on the Fujirebio and Elecsys machines are being used in Europe in routine clinical care, spreading out from tertiary care centers, and may be approved in the U.S. next year.
Q: At AAIC, neurologists from different countries stressed the need for blood tests in primary care, where many dementia diagnoses are made—often the wrong diagnoses. Do you agree?
A: Absolutely. And it’s better if the world has more than one assay that measures the same thing and gives the same answer. It inspires confidence, and competition keeps prices down. As a research and clinical community, we want several assays that are equally good at measuring a given analyte.
Q: Is this happening?
A: Yes. The plasma round robin is a good example of a precompetitive collaboration between companies. For the CSF assays, companies have collaborated for a decade within the Alzheimer’s Association GBSC group, and are right now in the final stage of harmonizing assay readouts. So this is a favorable situation. But we need to continue, since there is much work waiting ahead.
Q: Why is the round robin result on the current plasma Aβ42/40 ratio tests important?
A: There is great excitement about the plasma Aβ42/40 ratio tests, and some have posted promising results. The round robin is a first step toward having standardized blood Aβ tests in clinical practice. For that to happen, we will need a certified reference material (“gold standard” plasma pools with known and exact Aβ levels) for the different vendors to calibrate their tests. To get a reference material, you also need a reference measurement procedure (one or more “gold standard” methods to measure Aβ levels) to establish a traceability chain. It’s a process, and requires multiple round-robin studies, including so-called commutability studies (in which candidate reference materials are tested out) along the way. The International Federation of Clinical Chemistry in Medicine (IFCC), the EU’s Joint Research Center, and the Institute for Reference Materials and Measurements (IRMM) guide us in this work, but it requires a sustained effort on the part of many academic and vendor labs.
Q: This is not something most people in our field focus on …
A: True, but it still needs to be done. It’s best learned from the above-mentioned fields in diagnostic clinical chemistry that went through these same steps on their way from selling a few research-grade tests to having tests approved for clinical practice. The EU’s Joint Committee for Traceability in Laboratory Medicine and the National Institute of Standards and Technology show what clinical development entailed for cholesterol and other tests we use in medical care all the time. We want JCTLM/NIST-approved materials for blood Aβ tests, and we will need to go through these steps, too. As part of this process, you need to show that different tests measure the same analyte, be it Aβ, tau, or NfL.
Q: If the round robin had shown that they do—it didn’t—then what would have happened?
A: Then we could have started the clinical development process I indicated, i.e., we could have initiated a standardization project and made a certified reference material. But the data show we are not fully ready for that. We should instead work on getting the methods better harmonized first.
Q: Not everyone here at AAIC thinks it’s a problem that the round robin showed poor correlations across the different test methods.
A: For a narrow application, or within one lab’s studies, that might be true. But for clinical use, it is not. Say you have a number of methods that have been shown to predict something, be it brain amyloidosis or liver disease or myocardial infarction. If these different methods that are supposed to measure the same solute do not correlate well, that usually means there are issues with some of them. It tells me to expect problems later on.
Q: Like what?
A: Test-retest performance, for one. Let’s do longitudinal studies in the same individuals. If you have one person with an Aβ42/40 ratio of 0.21 and examine the same person three or six months later, will they still read 0.21? We need data on that. This is especially important given that the reduction in the ratio in positive versus negative people is not more than around 15 percent, which is really minor.
Q: And this worries us because …
A: When you are working within a range as narrow as that, it is extremely important to have a very high-performing assay so that you can place a firm cutoff. Even if AUC values are high, the cutoff has to go right through spread of values in the control (amyloid-negative) group, and when your signal is only a 10 to 15 percent difference, the tests must be extremely robust.
Thin Blue Line? The difference in the plasma Aβ42/40 ratio between amyloid-positive and -negative people is less than 15 percent. [Courtesy of Palmqvist et al., JAMA Neurology 2019.]
For example, when you look at the blue line in Oskar Hansson’s data using the Roche test, it tells you there is very little separation to draw a cutoff. People close to that line may get false results. And it’s not just the Elecsys test. The Aβ42/40 ratio signal is small in the other tests, as well.
In CSF, by comparison, Aβ42 is 50 percent down from control, and the Aβ42/40 ratio values don’t even overlap much between control and amyloid-positive groups. This means placing the cutoff is easier and few people will get the wrong result. In CSF, the cutoff can accommodate a bit of variance between tests; in blood, it cannot.
Q: What problems could this cause?
A: You’ll have some people who are positive at first test but negative upon repeat even though they did not change, just because so many people are close to the cutoff. For use in clinical trial prescreening, that is one thing, but imagine this situation if screening in clinical care: “You have brain amyloid.” Three months later: “Sorry, we were wrong. You don’t.”
Q: What to do?
A: At a minimum, we could do test-retest studies with a predefined cutoff, to evaluate how big a percentage of controls individuals and patients will be negative on the first test and positive on the repeat test taken, e.g., a month later (and vice versa). This type of study needs to be done prospectively, with test results reported continuously, to mirror the real-life situation. Maybe this could be a study governed by the Alzheimer’s Association, and I also hope the vendors would agree to participate.
Q: I have heard criticism that the round robin should compare tests directly on the ratio, not on Aβ42 and 40 separately. What do you say to that?
A: It is definitely important to compare the ratio, but since it is built on two components, I find it essential that Aβ42 and Aβ40 correlate across assays.
Q: What do you want to see done next, as a result of the round robin?
A: One option would be to take a step back, work on the methods to improve their performance, and redo this kind of round robin with second-generation tests. Another is to work on larger cohorts. Oskar Hansson is doing that, and the FNIH [Foundation for the National Institutes of Health] are planning a round-robin study with the plasma tests on ADNI samples. This round robin we did is just a first indication. We now need a round robin on samples from 300 to 400 people who are amyloid-positive or negative. We’ll see if the tests correlate among each other, and also if they concur with amyloid positivity. For CSF Aβ42/40, concordance with amyloid PET has been shown in about 20 studies; the concordance rate is 90 percent, and this data became part of the appropriate use criteria for clinical CSF tests.
Q: What will the larger cohorts do that the smaller ones did not?
A: For example, they’ll help us walk the line between sensitivity and specificity. With high sensitivity, the risk is that you tell people that they have brain amyloid who don’t, especially when the range leaves no room for error. With high specificity, the risk is that you miss a lot of people who do have amyloid. When you look at plasma studies, e.g. the Roche data published by Palmqvist et al., 2019, the ratio changes so little between controls and cases that the optimal cutoff leads to a sensitivity and specificity in the 70 percent range.
Sensitivity and specificity. Findings from one evaluation of the Roche Ab immunoassays. [Courtesy of Palmqvist et al., JAMA Neurology 2019].
Q: Does the Aβ42/40 ratio correlate between plasma and CSF?
A: Rather poorly in many studies. And we don’t know what this lack of correlation tells us. What is the cause? Peripheral production of Aβ? Pre-analytics could have been a possible explanation for Aβ42, but then Aβ40 levels do not correlate between plasma and CSF. We need to learn more on the physiology of plasma Aβ.
Q: A company getting ready to sell their plasma test will not like to hear that correlation between their and their competitor’s test is important.
A: That’s possible, but when different labs use different assays on identical aliquots that contain a given amount of a well-studied analyte, and they measure two different amounts, that indicates a problem which need to be addressed.
Q: Any formal effort to get control of this issue?
A: Yes, we invite labs and companies to join us in the Alzheimer’s Association quality-control program of their blood-biomarker tests. It has worked well for the CSF tests, where it is still ongoing so labs can track their performance not just against others’ but over time, too. We will start with blood tests next year.
Q: Do you think p-tau will outperform the Aβ42/40 ratio?
A: It looks as if there will be a plasma p181-tau or p217-tau test that will really work. They could be better than the Aβ ratio because the difference between groups is much larger, around a 200 percent increase, not a 10 to 15 percent decrease. That makes everything easier.
Q: Could a plasma p181-tau test replace the Aβ42/40 ratio tests?
A: In the best of worlds, it will be complementary. My dream is that we soon can use blood NfL to screen for neurodegeneration, p181-tau to screen for tau pathology, and Aβ42/40 ratio for amyloidosis. And other tests are in development. But a lot of work lies ahead to get them from research-grade to clinically certifiable.
Q: Where is blood NfL in this certification process?
A: We started last year, with nine labs in an IFCC working group. It includes five companies selling NfL test on different platforms, with different antibodies. The first commutability study toward a reference material is getting underway.
Q: What was the most hopeful news to you at AAIC this year?
A: The data on plasma p181-tau, that is really very promising!
Synaptic Proteins in CSF: New Markers of Cognitive Decline?
Plasma tests for Aβ and phospho-tau may have stolen the show at this year’s Alzheimer’s Association International Conference, held July 14-18 in Los Angeles, but proteomic and synaptic markers were not that far behind. A neural pentraxin, NPTX2, emerged as a contender for a marker of synaptic function and cognition. Other researchers debuted an ELISA for the synaptic vesicle protein SV2A, and showed it can be measured in the cerebrospinal fluid. Unbiased proteomic analysis of the CSF turned up these and other markers, which could parse subtypes of Alzheimer’s disease or reflect a person’s susceptibility based on his or her polygenic risk score (see Part 12 of this series). “It was incredibly exciting to see the field moving on from Aβ to other markers that could be functional readouts,” noted Beth Stutzmann, Rosalind Franklin University, Chicago. “I think NPTX2, SV2A, and other synaptic markers could prove very valuable, since what we are really interested in is cognitive function,” she said.
Pentraxins are a family of proteins that bind AMPA glutamate receptors on the cell surface. By controlling the receptors’ maturation and recycling, pentraxins regulate synaptic plasticity. Expressed primarily in parvalbumin-positive inhibitory neurons, they help control network excitability (see image below). Previously, in collaboration with Doug Galasko’s group at University of California, San Diego, researchers at PaulWorley’s lab at Johns Hopkins Medical School, Baltimore, reported that NPTX2 levels fall in the brains and cerebrospinal fluid of AD patients (Xiao et al., 2017). Because NPTX2 levels appeared normal in cognitively unimpaired people who had amyloid plaques and neurofibrillary tangles in their brains at autopsy, the researchers reasoned that the protein might track with cognition.
Pentraxins and Plasticity. Neuronal pentraxin 1 (NP1) and 2 (also called Narp) and neuronal pentraxin receptor (NPR) form complexes on the cell surface that regulate AMPA receptor recycling on the postsynapse. [Courtesy of Cho et al., 2017 and Neuron.]
DenisSmirnov in Galasko’s lab set out to test this. Again collaborating with the Worley lab and with ADx Neurosciences, Ghent, Belgium, Smirnov correlated a variety of CSF markers with change in cognitive performance among 46 people with AD and 57 with mild cognitive impairment. All were patients at the UCSD Alzheimer’s Disease Research Center. They had been tested repeatedly on the Clinical Dementia Rating scale sum of boxes (CDR-sb), the Mattis Dementia Rating Scale (DRS), and for immediate and delayed recall in the California Verbal Learning Test (CVLT). Smirnov tracked cognitive scores against baseline CSF levels of total tau, NPTX2, and the synaptic markers neurogranin and SNAP25.
Over three years, people with MCI or AD who had high CSF total tau declined faster on the DRS and CDR-sb than those whose tau was low. Similarly, high SNAP25 correlated with faster decline in delayed and immediate recall, while those in the high-neurogranin group declined faster only on delayed recall. Strikingly, people who had low levels of CSF NPTX2 declined faster on all four measures (see image below).
Predicting Cognitive Decline. People with low levels of NPTX2 in their CSF declined faster than those with higher levels. [Courtesy of Doug Galasko, UCSD.]
Smirnov repeated the analysis using ADNI data. He readily conceded that this was not a true replication because some fluid marker assays were different, and ADNI uses the ADAS-Cog rather than the DRS and the Rey AVLT instead of the CVLT. Still, a similar picture emerged. Among 80 people with AD dementia and 108 with MCI, low NPTX2 meant faster decline on all four cognitive measures. High neurogranin correlated with faster decline on the CVLT delayed-recall test; SNAP25 did not. ADNI also measures CSF levels of neurofilament light (NfL), a marker of neurodegeneration. Curiously, high CSF NfL did correlate with faster decline on the ADAS-Cog and CDR-sb, but less so than did low NPTX2, and NfL correlated with neither recall test.
Smirnov thinks that CSF NPTX2 could improve prediction of cognitive decline and disease progression based on other markers, such as Aβ and tau. In fact, survival analysis of the ADNI data indicated that people with MCI who have low CSF NPTX2 are three times more likely to develop dementia over five years.
In separate presentations, David Salmon, also from UCSD, and Eugeen Vanmechelen from ADx detailed how combinations of CSF markers improve diagnosis and prognosis. Analyzing data from 89 normal controls, 54 people with MCI, and 44 with mild AD, all from the UCSD ADRC, Salmon found that CSF NPTX2 alone predicted AD with an area under the curve (AUC) of 0.71, which was better than predictions based on SNAP25 or neurogranin. When Salmon combined markers, he got better results. The NPTX2/tau ratios identified AD patients with 94 percent accuracy (see image below); in fact, it distinguished AD from controls better than the Aβ42/tau ratio did. The NPTX2/tau ratio best predicted CVLT and DRS scores.
Vanmechelen analyzed CSF samples from 17 healthy controls, 36 patients with MCI due to AD, and 50 with AD, all from the Biobank of Institute Born-Bunge, Antwerp, Belgium. In this set, the NPTX2/tau ratio distinguished AD from controls with an AUC of 0.84 compared with 0.91 for Aβ42/tau. The NPTX2/tau ratio correlated with cognitive decline as judged by the mini-mental state exam.
CSF Predictors. Among synaptic markers, the NPTX2/tau ratio best predicted a diagnosis of dementia, with an AUC of 0.94. NPTX2 also popped up in unbiased proteomics of synaptosomes and CSF (see Part 12 of this series). [Courtesy of David Salmon, UCSD.]
Then there is synaptic vesicle glycoprotein 2A (SV2), another synaptic marker that has attracted interest of late. Most synaptic vesicles contain two to five copies of this transmembrane protein, which binds the PET ligand UCB-J. In brain-imaging studies, patients with epilepsy take up less of the ligand in affected brain areas, and AD patients bind less UCB-J in the hippocampus than do cognitively normal controls (Jul 2016 news; Aug 2018 conference news). Does SV2A end up in the CSF, and would levels there correlate with cognitive decline or AD? Nicholas Ashton, University of Gothenburg, Sweden, developed an immunoassay to find out.
Ashton tested a variety of antibodies against SV2A. One, which binds to an epitope on the N-terminal, gave a CSF signal. He tested the antibody in cohorts from Lund University, University of Gothenburg, and Paris Diderot Hospital. The Lund CSF samples came from 20 AD patients and 20 controls, UGothenburg had 42 AD patients and 50 controls, and Paris, 81 AD patients, 35 controls, 51 people with a different neurodegenerative disease, 30 people with MCI due to AD, and 49 with non-AD MCI.
Across all three cohorts, people with either clinically diagnosed AD or AD determined by biomarker criteria (Dubois 2014) had less SV2A in their CSF than controls did. So did FTD patients. However, people with Lewy body and vascular dementia seemed to have normal CSF SV2A levels.
Next Ashton correlated CSF SV2A against A/T/N staging scheme markers (Aug 2016 conference news; Jack et al., 2016). In the Lund and Paris cohorts, people whose CSF p-tau levels were above 80 pg/mL had less CSF SV2A regardless of Aβ positivity as determined by a cutoff of less than 550 pg/ml Aβ42. The data suggested that SV2A falls when p-tau rises. In agreement with this, when Ashton treated ATN markers as continuous variables, p-tau and total tau correlated with SV2A in both the AD patients and controls in both cohorts. Correlation with Aβ42 was weak. While CSF SV2A did correlate with neurogranin, SNAP25, GAP43, and synaptotagmin across the whole population, only the neurogranin correlation held up in AD patients. There was no correlation with NfL.
Scientists at AAIC were encouraged by the data, though with some niggling questions. How does this transmembrane protein get into the CSF? Why is it truncated? Ashton had shown Western blots indicating that SV2A in CSF appears slightly smaller than SV2A from solubilized brain extracts. He said he was trying to figure out the difference between the two using mass spec. Why did SV2A drop in FTD? Ashton does not know, but noted that those patients had no increase in CSF tau. “It’s possible SV2A reflects synaptic density in these patients,” he said.
Yet others wanted to know if Ashton had compared SV2A with NPTX2. He has not, but noted that NPTX2 levels correlate with other synaptic markers whereas SV2A does not, suggesting the latter might add predictive value. He did find that SV2A correlated with MMSE scores in AD patients. In short, Ashton believes that that SV2A might be a good CSF marker of synaptic impairment.—Tom Fagan
Proteomics Uncovers Potential Markers, Subtypes of Alzheimer’s
In the last three decades, scientists have made strides in using biomarkers to diagnose Alzheimer’s disease and track its progression. Along with cerebrospinal fluid Aβ, tau, and phospho-tau, a handful of new markers, such as neurofilament light, neurogranin, and YKL40, seem poised to add diagnostic value. Newcomers such as NPTX2 and SV2A may also prove helpful (see Part 11 of this series). But what of the thousands of other proteins in the CSF and in the brain? This year’s AAIC, held July 14-18 in Los Angeles, showcased the power of proteomics, a set of methods that after years of generating diffuse, head-scratching results, is now coming into its own.
Researchers showed how proteomics methods can identify functionally related groups of proteins in the CSF that change with disease state, and might reveal insight into the underlying etiology or identify new markers of pathology. Others used the approach to identify clusters of proteins that appear dysregulated in specific groups of patients, suggesting AD subtypes might exist. Still others used proteomics to tease out synaptic changes in AD.
Becky Carlyle, who works at the lab of Steven Arnold at Massachusetts General Hospital, Charleston, took the latter approach. Carlyle investigates the synaptic underpinnings of resilience in AD. In other words, she teases out why some people stay cognitively intact despite having extensive plaque and tangle pathology.
Carlyle compared proteomes from four different groups of patients within the ROSMAP cohort at Rush University. First, she purified synaptosomes from brain tissue to avoid confounds caused by changes in cell volume, which can skew whole-tissue proteomics. Then she analyzed the synaptosome fractions using tandem mass-tag spectroscopy, a more sensitive and accurate version of mass spectrometry. Among samples from 100 people, she found 9,558 different proteins in the synaptosome fractions. Nearly half of those were present in all of the samples, so she focused on those, correlating them with pathology and cognitive status.
Carlyle found no differences in the composition of known pre- and postsynaptic markers in synaptosomes from amyloid-positive people with dementia and from those with dementia who were resilient. For example, SNAP25 and synapsin 1 levels in synaptosomes from AD patients were similar to those in synaptosomes from normal controls and from people Carlyle defined as cognitively frail—meaning they had no obvious brain pathology but were cognitively impaired. However, using pathology and cognition as continuous variables, Carlyle teased out some correlations.
Seventeen synaptic proteins associated with pathology and 24 with global cognitive score. One, neurosecretory protein VGF, tracked with both pathology and cognition. Proteins strongly linked with pathology only included VEGF receptor 1 and SPARC-related modular calcium-binding protein 1. SMOC1 is a member of the SPARC family of cell-surface proteins, of which SPARCL1 was recently reported to bestow synaptogenic properties on blood from young animals (Jun 2019 news). Of the proteins that associated only with cognition, NPTX2 correlated most strongly. Chromogranin A, a synaptic protein that previously had been linked to AD, did as well (Mar 2019 news; Mattsson et al., 2013).
Because ELISA or mass-spec assays are established for VGF, VEGFR1, NPTX2, chromogranin A, and SMOC1, these should be tested further as biological markers, said Carlyle. In fact, NPTX2, SMOC1, and VGF were among the hits in a proteomic analysis led by Nick Seyfried at Emory University in Atlanta. “It was very encouraging to see the similarity across our datasets,” he told Alzforum. “This bodes well for the rigor and reproducibility of these proteomic measurements.”
Seyfried took an unbiased, integrative, network-based approach to search for proteomic changes in the CSF and brains of dementia patients that overlap and might help to stage AD progression. He had previously identified more than 40 modules of related proteins in the brain using weighted co-expression network analysis of deep proteomic datasets, i.e., those containing more than 9,000 proteins (Jul 2017 conference news). For CSF, he compared protein levels among 20 AD patients and 20 controls using multiplex tandem mass-tag spectroscopy, and related those differences to the brain co-expression modules.
Seyfried identified about 43,000 peptides from 3,927 proteins across all 40 samples. Of these proteins, 228 were more abundant in AD CSF than controls, while 148 went the opposite way. The increased included TREM2, tau, and neuregulin, while NPTX2 and VGF were among the decreased. These differentially expressed proteins overlapped with 10 network modules previously identified in the brain. These modules covered five basic functional categories: synapses; humoral immunity; myelin; glia and injury response; metabolism. “We were particularly interested in CSF proteins that mapped to the glia modules in the brain because these contained tau, microglial proteins, and other proteins related to neuroinflammation, including SMOC1,” said Seyfried.
To validate specific proteins within these modules, Seyfried and colleagues relied on “parallel reaction monitoring.” This is a more quantitative type of mass spec where internal standards are spiked into the sample before analysis. From the glial, myelination, and synaptic modules, SMOC1, osteopontin, and UCHL1, respectively, were more abundant in AD versus control CSF. To see how SMOC1 changed during the course of disease, Seyfried analyzed 96 individual CSF samples from the Emory Goizueta Alzheimer’s Disease Research Center. Of these, 32 with asymptomatic AD had more SMOC1 in CSF, on average, than did 32 controls, and 32 AD patients had even more than the asymptomatic cases. “We need more groups looking into new targets, like SMOC1, that are being replicated independently. They could have high value as biomarkers or therapeutic value,” said Seyfried.
Other groups use proteomic analysis to distinguish subtypes of late-onset AD. Because the disease is so heterogeneous in presentation and progression, scientists believe that different triggers may cause or modulate its pathology. Betty Tijms and colleagues at Amsterdam University Medical Center reported three subtypes of AD based on CSF proteomics.
Alzheimer's Types? People (dots) fall into one of three subtypes (blue, green, and orange) that best fit correlations among proteins and individuals in the ADNI and EMIF data sets. Higher axes values indicate better matches to those subtypes. [Courtesy of Betty Tijms, VU Amsterdam.]
The researchers analyzed proteomic data from CSF of people in ADNI and, in collaboration with Johan Gobom at the University of Gothenburg, Sweden, from CSF in the EMIF-AD Multimodal Biomarker Discovery (MBD) cohort. From mass spectroscopy and ELISA of 127 controls and 425 AD patients, Tijms identified 705 proteins that were either up- or downregulated in AD, and simultaneously clustered them by protein and by patient to identify proteomic subtypes.
Most people in ADNI and EMIF-AD MBD fell into one of three subtypes (see image at right). In subtype 1, synaptic proteins were enriched in the CSF, suggesting a hyperplasticity, said Tijms. Subtype 2 was marked by elevated levels of immune proteins in CSF. In contrast, subtype 3 CSF had less synaptic proteins but increased levels of proteins involved in blood-brain barrier integrity. Subtype 3 also had the least CSF tau and p-tau, while subtype 1 had the highest.
What does this mean? Scientists at AAIC wondered if these groups represent true etiological types, or people who deteriorate at different rates. Either way, they agreed that stratifying subjects in this manner might tease out differences in responses to treatment. Tijms hopes that subtyping might allow for a more personalized approach to medicine.
There were few demographic differences among these three subtypes. Still, ADNI participants with subtype 2 tended to be older and more were male than those with subtype 1 or 3. Could genetic background explain the difference? APOE4 was equally represented across the groups, but Tijms did not correlate subtypes with other genetic markers. Lianne Reus, who works with both Tijms and Pieter Jelle Visser at Amsterdam VUMC, took up the challenge.
Reus correlated CSF proteomics with polygenic risk scores. PGS have become powerful gauges of AD risk, but they are difficult to interpret mechanistically. Reus believes coupling PGS with proteomic analysis could yield information about the underlying biological processes associated with genetic risk.
Reus determined polygenic scores for each person based on summary statistics from the genome-wide association study conducted by the International Genomics of Alzheimer’s Project (Jul 2013 conference news; Lambert et al., 2013). By using different significance thresholds for polymorphisms, she came up with 14 different polygenic scores for each of 250 subjects in ADNI, including 61 people with AD, 116 with MCI, and 73 controls. She related those scores to levels of peptide fragments in the CSF, looking for correlations with clusters of proteins.
From a query set of 412 peptides, Reus found that 199 correlated with PGS. They fell into three clusters. The first, comprising 68 proteins, was polygenic, associating with mostly high-risk single-nucleotide polymorphisms. This cluster contained proteins involved in Aβ pathology and complement, and correlated tightly with ApoE genotype. After correcting for ApoE, it still associated with polygenic risk scores. This means that more genes beyond ApoE contribute to Aβ pathology, said Reus. Twenty-one proteins made up cluster 2, which was intermediate on the polygenic scale. This cluster suggested no specific pathways, but it included markers associated with neuronal injury, including neurogranin, tau, phospho-tau, YKL40, and FABP. It also correlated with ApoE. Cluster 3 was the most polygenic. It did not correlate with ApoE genotype. It comprised cell-adhesion molecules and the AD-associated markers NfL, secretogranins 1, 2, and 3, chromogranin A, and VGF. Functionally, this pointed to cytokine-cytokine interactions.
“The data suggest that multiple genetic mechanisms lead to changes in AD CSF,” said Reus. She said it will be interesting to test how these CSF clusters can be modulated. “If we better understood the genetics behind these changes, we might be able to use them to more reliably monitor changes in clinical trials,” she said.
Choices, Choices … What to Do?
With all these potential markers at various stages of exploration, how can scientists determine which ones could become bona-fide markers? At AAIC, a collaboration of 13 biomarker leaders at five academic centers plus researchers at Lilly, Roche Diagnostics, and Genentech described their “NeuroTool Kit,” project. Led by Richard Batrla at Roche Diagnostics and Kaj Blennow, University of Gothenburg, Sweden, it aims to evaluate candidate markers in a coordinated way, so that the field can decide if they will work reliably in the clinic or should be abandoned.
The idea is to jointly choose candidate biomarkers based on current research needs, and then use standardized approaches to obtain comparable data for each of them across several well-defined cohorts. Such a dataset makes it easier than lots of individual single-center studies to determine a marker’s ability to differentially diagnose or track disease, or to monitor response to an investigational treatment.
In the end, a NeuroTool Kit could encompass all fluid markers validated for neurodegenerative diseases, much like scientists are building genotyping platforms that contain all known neurodegenerative risk variants for use in research and, eventually, in clinical diagnostics (e.g., Blauwendraat et al., 2017).
Which Ones Work? Twelve different markers, plus four ratios, were measured side-by side, in the same way, in the same normal (blue), MCI (yellow), and AD (orange) dementia samples of the WRAP cohort. Each sample is further subdivided as p-tau/Aβ42 negative (left) or positive (right). [Courtesy of Kaj Blennow.]
At AAIC, the scientists presented 16 side-by-side tests done on samples from the University of Wisconsin, Madison, ADRC research cohort. The 16 comprise 12 markers—including Aβ, p-tau, total tau, neurogranin, NfL, GFAP, sTREM2, YKL-40—plus four combinations. The same markers will be tested in additional cohorts, including the ALFA Plus cohort in Barcelona, Spain, the Parkinson’s Progression Marker Initiative, and Roche/Genentech’s ABBY and BLAZE clinical trial cohorts. Other markers up for consideration include NPTX2 (see Part 11 of this series), SNAP-25, TDP-43, BDNF, osteopontin, and various phosphorylated epitopes of tau.—Tom Fagan and Gabrielle Strobel
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Morón FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossù P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer's Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer's Disease, Alzheimer's Disease Genetic Consortium, Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O'Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH Jr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P, Wang J, Uitterlinden AG, Rivadeneira F, Koudstgaal PJ, Longstreth WT Jr, Becker JT, Kuller LH, Lumley T, Rice K, Garcia M, Aspelund T, Marksteiner JJ, Dal-Bianco P, Töglhofer AM, Freudenberger P, Ransmayr G, Benke T, Toeglhofer AM, Bressler J, Breteler MM, Fornage M, Hernández I, Rosende Roca M, Ana Mauleón M, Alegrat M, Ramírez-Lorca R, González-Perez A, Chapman J, Stretton A, Morgan A, Kehoe PG, Medway C, Lord J, Turton J, Hooper NM, Vardy E, Warren JD, Schott JM, Uphill J, Ryan N, Rossor M, Ben-Shlomo Y, Makrina D, Gkatzima O, Lupton M, Koutroumani M, Avramidou D, Germanou A, Jessen F, Riedel-Heller S, Dichgans M, Heun R, Kölsch H, Schürmann B, Herold C, Lacour A, Drichel D, Hoffman P, Kornhuber J, Gu W, Feulner T, van den Bussche H, Lawlor B, Lynch A, Mann D, Smith AD, Warden D, Wilcock G, Heuser I, Wiltgang J, Frölich L, Hüll M, Mayo K, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Singleton AB, Guerreiro R, Jöckel KH, Klopp N, Wichmann HE, Dickson DW, Graff-Radford NR, Ma L, Bisceglio G, Fisher E, Warner N, Pickering-Brown S.
Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease.
Nat Genet. 2013 Dec;45(12):1452-8. Epub 2013 Oct 27
PubMed.
Blauwendraat C, Faghri F, Pihlstrom L, Geiger JT, Elbaz A, Lesage S, Corvol JC, May P, Nicolas A, Abramzon Y, Murphy NA, Gibbs JR, Ryten M, Ferrari R, Bras J, Guerreiro R, Williams J, Sims R, Lubbe S, Hernandez DG, Mok KY, Robak L, Campbell RH, Rogaeva E, Traynor BJ, Chia R, Chung SJ, International Parkinson's Disease Genomics Consortium (IPDGC), COURAGE-PD Consortium, Hardy JA, Brice A, Wood NW, Houlden H, Shulman JM, Morris HR, Gasser T, Krüger R, Heutink P, Sharma M, Simón-Sánchez J, Nalls MA, Singleton AB, Scholz SW.
NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases.
Neurobiol Aging. 2017 Sep;57:247.e9-247.e13. Epub 2017 May 17
PubMed.
Further Reading
No Available Further Reading
Technology Brings Dementia Detection to the Home
The past decade has seen increasing use of technology in dementia research. Computer games that provide cognitive training have become part of prevention studies, and trialists are working to move cognitive testing out of the clinic and into everyday life, replacing pen and paper with tablets and smartphones. A plethora of new tools are being tried out. Alas, adherence is a problem and researchers grapple with how to get people to check in and complete tests day after day. At the Technology and Dementia Preconference, held in advance of the Alzheimer’s Association International Conference on July 14-18 in Los Angeles, scientists discussed roadblocks and efforts to boost engagement. They are transforming cognitive tests into fun games, and dangling bonuses to encourage participation. As an alternative, passive monitoring equips seniors and their homes with sensors to track activity around the clock, and to flag changes that signal dementia. Part 14 of this series describes the current status of some of these so-called digital biomarkers.
Failure to Launch. More than one-third of FINGER participants completed none of the trial’s screen-based cognitive training sessions. Lack of experience with computers was the main reason. [Courtesy of Turunen et al., 2019, PLoS One.]
Cognitive training by way of computer games is increasingly accepted as part of a dementia-prevention lifestyle (May 2019 news). At AAIC, Alina Solomon, University of Eastern Finland, reported on the experience to date from the use of computerized cognitive training in the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability. FINGER enrolled 1,260 healthy people aged 60–77, who were at risk for dementia, and offered them a multifaceted, two-year intervention of exercise classes, diet plans, computer work, and social activity, plus management of metabolic and vascular risk factors. The control group got standard health advice. This intervention boosted cognitive scores (Nov 2015 conference coverage; Ngandu et al., 2015) and is now expanding to other countries as Worldwide FINGERS (Aug 2017 conference news).
This summer, the researchers published data on adherence to FINGER’s at-home cognitive training regimen aimed at boosting mental speed, memory, and executive function (Turunen et al., 2019). The results were discouraging. Of the 631 participants—a self-selected, presumably motivated group of elders—37 percent did no training at all. Only 20 percent of participants completed at least half of the possible 144 sessions; 12 percent completed all. A dose-response analysis suggested that the benefit of cognitive training increased with the number of sessions, then leveled off after 40–50 sessions, Solomon said. A minority of participants hit this mark.
The primary factor associated with starting cognitive training, and with completing more sessions, was familiarity with a computer. People who did not use a computer before were unlikely to start the training as part of FINGER.
Fortunately, the problem of poor computer literacy appears to be solving itself. Compared with when recruitment for FINGER began in 2009, the researchers now see more familiarity and use of technology in elders joining the study. “Age 60-plus 10 years ago is not the same as 60-plus now,” Solomon said. Her impression is borne out by statistics. In Finland today, 60 percent of people between 64 and 75 own a smartphone, whereas in 2011 only 10 percent did. More than a quarter of people over 75 have a smartphone. Solomon said her group is currently surveying tech use in people older than 85.
For computerized cognitive training, a person’s stage of disease matters, Solomon said. FINGER targeted at-risk elders in the general population, but scientists are now piloting a similar intervention in prodromal Alzheimer’s. In this program, 120 people are receiving regular care, the FINGER intervention, or FINGER plus a medical food. Solomon said these participants are easily stressed, so the researchers simplified training tasks, made the sessions fewer and shorter, and added social support. Participants needed more time to get acquainted with the program and to understand the difference between cognitive training and cognitive testing. Many were worried they’d forget to do the training at home. Now, the participants take the training as a group, each with his or her own laptop, before exercise sessions at the gym. So far, adherence is higher than in FINGER, Solomon said.
The move toward computerized cognitive tests to diagnose and track Alzheimer’s started on terminals in clinics, then moved to tablets, and is now bursting out of the clinic setting entirely (see 2012 news series). Jason Hassenstab, Washington University, St. Louis, pioneered the use of smartphones to measure cognition. His Ambulatory Research in Cognition (ARC) app transforms testing from quarterly, hour-long clinic sessions into frequent, brief bouts that participants perform on their personal cell phones in the course of their daily lives (Aug 2018 conference news; Dec 2017 conference news). ARC is being used in prevention trials run by the Dominantly Inherited Alzheimer’s Network (DIAN) (Aug 2019 conference news). For those trials, the app enables the researchers to collect data from participants who hail from all over the world and who are younger, working and raising children, and hence unwilling or unable to travel to a clinic frequently, Hassenstab said.
At AAIC, Hassenstab shared some lessons learned from this work. He told the audience that developing a smartphone measure of cognition that could be used in a clinical trial had been arduous and costly. “Smartphone apps are a commitment, and a lot of work,” he said. Regulatory issues and documentation requirements are extensive, complex, and indeed became overwhelming. Hassenstab hired a consultant, and recommended that his fellow app developers consider early audits by such experts. To ensure privacy, ethics committees and IRBs require strict encryption and data-protection practices. Collecting data via phone cameras, and recording eye tracking or swiping behavior, are highly scrutinized. That became even more true after 2018, when the European Union implemented its general data-protection regulation (GDPR). All this makes development expensive. Hassenstab said that developing ARC for use in a trial cost $1 million.
Once deployed, the biggest challenge Hassenstab ran into was compliance. “How do we get people to do this, and to keep doing it over time?” he asked. The problem is not unique to Alzheimer’s. Adherence to electronic reporting is terrible across the board. Trials for Parkinson’s disease, asthma, and arthritis all suffer from attrition rates of 80 to 90 percent after several months, Hassenstab said.
Motivational Moves. To encourage people to stick with ARC testing, WashU researchers help participants track their progress (left), and pay them (right). [Courtesy of Jason Hassenstab.]
In DIAN, 95 percent of participants will take the tests on the app once, 50 percent do it twice, and down it goes from there. What to do? Hassenstab held tutorials to help participants learn the app. After that, feedback tends to keep people engaged. To that end, ARC tracks and shows participants their progress in the overall scheme of the study. Gamification uses elements taken from computer games—think confetti and badges—to boost engagement.
DIAN scientists are also testing money as an incentive, offering 50 cents per test, plus bonuses for streaks and overall completion rates. They nudge users with reminders that invoke altruism (“We are one step closer to solving AD”) or competition (“Seventy-five percent of people have completed this—how about you?”). Hassenstab’s team is starting to collect data on how well the different techniques work.
Hassenstab offered advice for others aiming to develop a smartphone app for cognitive testing. Trying to replicate a paper test on the smartphone is difficult in practice. Simple tests work best. These rely on uncomplicated visual stimuli, animation, and easy response selection. For older people, swiping and scrolling can be challenging; games should not require a stylus. Vocal responses are possible, as is using the camera for eye tracking, but these technologies are nascent and costlier to develop.
Once a test is developed, professional beta testers can help validate psychometrics, accessibility and engagement. Hassenstab collected feedback from 1,000 users in two weeks this way, which allowed him to optimize the ARC app and deploy it on iPhones and android phones.
From the start, Hassenstab advocated a bring-your-own-device model, where people use the app on their own phones. This, too, is easier said than done. To date, 19,000 different smartphone models circulate worldwide. They differ in tap sensitivity and other parameters. If an ARC test depends on response time, the variability between phones in their respective lag between tap and recording can render results uninterpretable. After much effort building phone-tapping robots to measure lag times and standardize test results across devices, Hassenstab is now de-emphasizing response time in his data collection, instead emphasizing correct responses. The alternative—having the study provide a dedicated device—cuts development cost, eases validation, and boosts privacy. Alas, some people tend to dislike carrying two phones, and symptomatic participants in particular struggle to learn a new device and remember to keep two phones charged.
Could phone apps be used to screen for preclinical AD in the general population? That is the goal of the GameChanger project, a collaboration between the lab of Chris Hinds at the University of Oxford and partners at the Alzheimer’s Society, U.K. It tests whether the app Mezurio can detect subjective cognitive impairment and presymptomatic AD. Intended to be freely available, Mezurio contains three game-like tasks designed to measure cognitive processes that are among the first to change in AD: a paired-association learning, an executive function, and a speech-production task. The tests require five minutes per day for 30 days.
In LA, Claire Lancaster in Hinds’ group presented an update on Mezurio. Since the app’s launch in 2018, more than 16,500 people across the U.K. have used it and contributed data to the study. Users range from 18 to 92 years old, 77 percent are women, and 42 percent have a family history of dementia. They are motivated; 95 percent reported a willingness to take part in future digital health research related to dementia. To promote adherence, the researchers built reminders and encouragement, daily facts about AD research, and periodic emails into the app. And users responded: Among the first 10,000 participants, more than 3,000 completed all 30 days of testing, and most rated the experience as enjoyable. The results suggest that it is feasible to use Mezurio in the general population, Lancaster said.
Swipe Left. In Mezurio’s GalleryGame association task, participants learn to pair swiping in a particular direction with pictures of familiar objects, and are asked to remember the pairing for up to 13 days. [Courtesy of Claire Lancaster.]
What were the initial results on detecting subjective cognitive decline? Besides its gaming features, Mezurio prompts users to self-report on their mood, sleep, and other parameters. It also collects information about “cognitive slips” on six behaviors known to discriminate preclinical AD from normal age-related cognitive change: forgetting names of familiar people or objects, word-finding difficulties, spatial disorientation, losing one’s train of thought, temporal disorientation, or trouble following a conversation or simple instructions. Every day for eight days, the app queries users to say how often they had such slips in the past six months and the past 24 hours. These queries are more specific than existing measures of subjective cognitive decline, which ask whether people or their caregivers perceive a change in memory in the last years or months that affect daily function and worry them.
At AAIC, Lancaster reported that, among the first 10,000 users, the frequency of these six types of slips in the previous six months correlated with scores on a traditional subjective cognitive complaints index. In these users, age and family history significantly predicted subjective cognition changes and frequency of slips in the past six months. Now, the scientists are looking at whether frequency of slips in the previous six months or the past 24 hours are better at predicting cognitive change when people are followed up at 12 or 24 months.
Additional cognitive test data on these 10,000 people will be presented in the coming months, Lancaster said. Besides following up with longitudinal data, she is forming collaborations with existing U.K.-based AD cohorts to compare performance on the app with medical records, genetics, and standardized neuropsychiatric testing scores.
The scientists are validating the Mezurio tests in pilot groups of older people at risk for AD by comparing them with traditional cognitive measures. The Gallery Game deployed on the Mezurio app is similar to lab-based delayed-recall tests, but assesses much longer recall intervals than possible in the lab, from one to 13 days. In one task, participants memorize the association between a photo of a real-life object and a swipe direction. A recognition task asks if the user has seen the object before. Among a test population of 36 cognitively normal people, Lancaster found significant forgetting occurred starting after three days for the association test, and five days for the recognition test, validating the use of extended testing periods. Outcomes correlated with established memory tests, according to a manuscript of the study posted on bioRχiv.
Picture This. A smartphone app from the Boston Remote Assessment of Neurocognitive Health (BRANCH) study at Brigham and Women’s Hospital uses real-life examples for at-home cognitive testing. [Courtesy of Kate Papp.]
For her part, Kate Papp of Brigham and Women’s Hospital, Boston, described how her group is exploring at-home testing in the Harvard Aging Brain Study. Previously, they had developed the computerized cognitive composite (C3) to assess subtle cognitive decline in the A4 prevention trial of the anti-Aβ antibody solanezumab. Done on an iPad using modified Cogstate software, C3 includes the Cogstate brief battery, plus two other tests of recognition and recall. One asks participants to memorize face-name associations; the other measures their ability to recall differences between similar but distinct objects, such as pairs of socks in various colors. The test comes in six different versions, which vary the test items. This allows the investigators to control for practice effects, where people test better if they see the same face-name pairs or objects over and over.
In A4, C3 serves as a secondary outcome, but participants take the test every three months in the lab. Papp wants to transition C3 to the home so she can collect data more frequently. In a pilot study, Papp gave iPads to 22 amyloid-positive and 58 amyloid-negative HABS participants to complete C3 twice a month at home. Both amyloid-positive and -negative participants performed the same at baseline, and if they repeated the same version of the test each month, everybody improved. However, amyloid-positive people improved more slowly than did amyloid-negative people. This difference emerged after four months. If the participants did a different version of the C3 each month, the learning effects disappeared, and both amyloid-positive and -negative people performed the same.
Like Hassenstab and Lancaster, Papp, too, wants to move cognitive testing to smartphones to allow even more frequent assessments on personal devices in this preclinical population. She is co-developing a new app designed like the C3 to detect subtle changes in AD-specific cognitive processes, and distinguish them from normal aging. Papp’s app draws on everyday experiences: recognizing street signs, judging prices of vegetables and, of course, face-name pairs. This app is currently being piloted in volunteers.—Pat McCaffrey
Smart Homes Open Doors to Insights into Aging, Dementia
Replicating existing paper-and-pencil tests on tablets or smartphones is one thing (Part 13 of this series), but what about taking technology itself to the next level? What are the prospects for continuously collecting information on people’s daily behavior, and using that wealth of data to discover entirely new biomarkers for dementia? Jeffrey Kaye, Oregon Health and Science University, Portland, and his group have pursued this approach for nearly a decade (Dec 2012 news), and at the 2019 AAIC conference in Los Angeles, Kaye gave a progress update. In his center’s Collaborative Aging Research Using Technology (CART) Initiative, Kaye’s group has been using remote-monitoring technology to collect data on people in their homes. The hope is that it will detect cognitive decline and track the effects of interventions. In homes and cars, nonintrusive remote-sensing devices track a participant’s movement, computer use, medication adherence, driving habits, and more, with the data continuously being fed into a central database.
So far, Kaye said the project is monitoring 274 people in four communities, including low-income participants in Portland, Oregon, veterans in rural Oregon, African Americans in Chicago, and Hispanic participants in Miami. Kaye noted that all of these can be challenging groups to reach and recruit into traditional clinic-based studies.
À LA CART: The smart home project pairs multiple passive monitoring technologies with conventional assessments to discover digital biomarkers for dementia. [Courtesy of Jeffrey Kaye.]
One such study addressed whether changes in sleep could flag cognitive decline. Most sleep and dementia studies take place in sleep labs, over a night or two, with people Kaye called “advantaged adults.” His group tracked total sleep time per day for up to five months in 126 CART participants living on their own in Oregon, 84 of whom were cognitively healthy and 42 of whom had MCI. They were asked to wear an activity-tracking watch round the clock. They did not seem to mind the watch overly much, as compliance with this request came in at 86.5 percent. People with MCI wore the watch less, but still on more than 80 percent of study days, Kaye said.
All groups posted about seven hours of sleep per night. Unadjusted sleep duration was the same between the groups, but when the investigators controlled for age, sex, and education, shorter sleep was significantly associated with MCI status.
Importantly, monitoring sleep for only two weeks did not predict MCI, nor did self-reported sleep time. “By monitoring for longer times, we can start to see differences,” Kaye said. “Normally, monitoring might be two weeks. Our volunteers did months with high compliance.” Kaye said CART is now analyzing sleep in its other participating groups. In the coming months, CART will move to more passive methods of sleep assessment, such as embedded infrared sensors or bed mats, Kaye said.
Other warnings of early cognitive impairment might come from tracking how people take their medications. Nora Mattek of OHSU collected data from 64 adults who used electronic pillboxes that reported by way of livestreaming each time a compartment was opened. Compared with cognitively intact older adults, people who were developing MCI started to take their pills later in the day, and varied their pill-taking time more. This in-person variability grew during the time up to and after the diagnosis of MCI. The authors suggest medication-taking behavior could be an early and predictive digital biomarker of emerging MCI.
Yet another harbinger of mild cognitive impairment can be when people start to move more slowly around the house, Kaye’s group had previously found (Dodge et al., 2012). New data from Antoine Piau of the University of Toulouse, France, now suggests that slowed walking also predicts impending falls. Working in Kaye’s lab, Piau analyzed data from 125 adults who were monitored continuously at home with a series of ceiling-mounted activity monitors. He discovered that people’s walking speed slowed significantly in the three months prior to a fall. Day-to-day variability in speed was also lower in the month and week prior to a fall (Piau et al., 2019). Such sensor-based monitoring could open up new opportunities for targeted and timely interventions to prevent falls, Piau said.
By this point in his longstanding research program, Kaye can tie in-home monitoring to neuropathology data. In LA, he showed a comparison of autopsy results and lifetime data from 41 participants who had lived with the activity-monitoring platform in their homes for more than a decade. Their average age at death was 92 years. Half of them passed away while still living in their sensor-equipped homes; for the others, the average amount of time between when they left their monitored home and when they passed away was one year. At autopsy, amyloid plaque load was scored as none, sparse, or moderate. An increasing plaque score was accompanied by stepwise decreases in markers of cognition (computer use), mobility (walking speed), and increased sleep time. Socialization waned at moderate levels of plaque burden, as measured by time spent out of the house. The same relationships held for Braak scores of tau pathology.
This is the first data showing a correlation between digital biomarkers and neuropathology in older people, Kaye said. Going forward, he said CART will continue to collect data from its diverse populations, to further define connections between the monitored behaviors and brain changes.—Pat McCaffrey
Can Induced Neurons Identify Early Signs of Neurodegeneration?
Researchers have linked dozens of genetic loci to late-onset Alzheimer’s disease and found dozens more that cause familial forms of AD and other neurodegenerative diseases. Teasing out how each contributes to pathology has proven a little more difficult. The effects of one single nucleotide are easily drowned out by the contributions of its 3 billion neighbors in the human genome. Enter induced pluripotent stem cells and CRISPR gene editing. Scientists are now using these technologies to make isogenic cells lines whose genomes often differ by just that one single-nucleotide polymorphism. At this year’s Alzheimer’s Association International Conference, held July 14–18 in Los Angeles, researchers showed how they used isogenic cell lines to uncover deficits in GABAergic signaling and lysosomal function caused by mutations in tau, and to identify new endosomal pathways taken by the cell-sorting protein SORLA, a key player in recycling of the amyloid precursor protein.
Trouble with Tau
In LA, Celeste Karch, Washington University, St. Louis, described how she used isogenic cell lines to study tau mutations. Karch’s overarching goal is to explore how genetic variants that cause familial AD and frontotemporal dementia affect neuronal cell biology. Karch focused on the R406W mutation that causes autosomal dominant FTD. This variant of the disease carries the pathological hallmarks of a typical tauopathy, namely tangles in neurons and glia, but clinically manifests with progressive memory loss much like Alzheimer’s disease. Karch wants to identify molecular events downstream of the mutation that might be responsible.
Collaborating with Carlos Cruchaga and Oscar Harari, both also at WashU, Karch’s group started with skin fibroblasts donated by a R406W tau carrier who receives care at the Knight Alzheimer’s Disease Research Center. The researchers de-differentiated her fibroblasts into induced pluripotent stem cells, then used CRISPR to correct the tau gene back to wild-type in some of those cells. Next they converted the wild-type and R406W iPSCs into cortical neurons and used RNA-Seq to look for transcriptional differences (Jiang et al., 2018).
Common Changes. Comparing patient and control tissue against mutant and wild-type isogenic cell lines, researchers found common changes in expression of 61 genes. [Courtesy of Jiang et al., Translational Psychiatry 2018.]
The scientists found that expression of 328 genes differed between wild-type and R406W-tau-induced neurons. Which of those explain neurodegeneration in an older adult with FTD? Since iPSCs have been genetically “deprogrammed” back to an early embryonic state, some expression changes in neurons derived from iPSCs might reflect developmental rather than neurodegenerative states.
To address this problem, Karch’s team combined differential expression analysis of the iPSC-derived neurons with similar analyses of human tissue from the insular and parietal cortices of healthy controls and from people with the R406W mutation. This process identified 61 genes that are differentially expressed in both iPSCs and disease brain (see image above). Because the cell lines are isogenic, these expression differences are likely driven directly by the tau mutation.
What do the 61 do? Functional annotation analysis predicted that half of this cadre interacts with tau. Pathway analysis highlighted many genes related to calcium-dependent synaptic function and GABAergic signaling. The GABA-associated genes SNAP-25 and SYT1 emerged as hubs of an interaction network (see image below).
“Our data indicated that even though our [iPSC-derived] cells may still be ‘young,’ essentially patterning like developing fetal cells, tau mutations are sufficient to induce changes that are detectable in an older brain,” said Karch.
Dysfunctional Network? The R406W mutation in tau alters expression of a network of genes linked to calcium signaling and GABAergic transmission. [Courtesy of Jiang et al., Translational Psychiatry 2018.]
Knowing this, the researchers wondered what other changes the R406W variant might evoke. Sidhartha Mahali, a postdoc in Karch’s lab, noticed a curious phenotype when he grew iPSC-derived neurons in two-dimensional cell cultures. LAMP1-positive vesicles, aka lysosomes, spread much farther from the soma of R406W-tau neurons than they did from wild-type soma. This distribution might change how lysosomes function, said Karch. Other groups had shown that the farther lysosomes are from the nucleus, the weaker their capacity for degrading protein (Yap et al., 2018). Indeed, Mahali found that lysosomes in R406W-tau neurons were less acidic. They ramped up expression of proteolytic enzymes, yet had no appetite for degrading protein. The uptick in proteases seems to be an attempt to compensate for poor proteolysis, said Karch.
And how does tau fit in? In LA, Mahali reported that in R406W-tau neurons, both total tau and p231-tau accumulated in LAMP1-positive lysosomes. He was also able to recapitulate the lysosomal distribution phenotype and the tau co-localization when he generated neurons from iPSCs containing either the P301L or IVS10+16 tau mutations. All told, the data suggest that tau mutations somehow lead to a redistribution of lysosomes in the cell that may cause them to malfunction.
The talk sparked a lively discussion. Some wanted to know whether degradation of tau itself was affected by the lysosomal shift, and if there were any functional changes to the neurons. Karch has no data yet. She plans to use SILK isotope labelling to quantify tau production and clearance rates, and also plans to test if the mutations and the lysosome dysregulation affect neuronal excitability.
Others asked whether tau accumulates in lysosomes because the organelles clear too little of it, or if lysosomes malfunction because of the tau mutation. “That’s a great question,” said Karch. “We know tau gets degraded through several mechanisms in the cell, including through autophagy. However, whether mutant tau triggers degradative dysfunction remains uncertain.”
Joanna Jankowsky, Baylor College of Medicine, Houston, speculated that mutations in the microtubule binding protein disrupt transport of lysosomes. Karch said she was unsure about that. “It could be a transport issue or maybe a component of some signaling pathway is missing or malfunctioning. We are trying to parse that out,” she said.
Beth Stutzmann, Rosalind Franklin University of Medicine and Science, North Chicago, Illinois, suspects signaling. “We see calcium dysregulation upstream,” she told Alzforum. “When we restore Ca2+ levels we clear p-tau accumulation and return phagosome levels and lysosome function to normal.” Stutzmann, with first author Sarah Mustaly and others, reported that calcium dysregulation tones down lysosomal acidity, which in turn leads to accumulation of protein aggregates, including tau. They studied induced neurons derived from AD patient fibroblasts and in a mouse model of tau tangle and amyloidosis.
For the latter, Mustaly examined the hippocampi and cortices of 3-month-old 3xTG mice which express human APP and tau transgenes. She found reduced expression of the vacuolar ATPases that pump protons into the lysosome to keep their pH acidic. ATPase subunit loss came with more numerous mature autophagosomes, a sign lysosomes are not processing them properly. Levels of p262-tau also rose. Injecting the mice with the ryanodine receptor (RyR) modulator Ryanodex once daily for 30 days corrected not only levels of Ca2+, but also of V-ATPase, autophagosomes, and p-tau. RyRs sit in the endoplasmic reticulum membrane and pump Ca2+ into the cytosol. “We think that as the V-ATPases pumps protons into the lysosome, Ca2+ must be pumped out to maintain electrical balance,” said Stutzmann. “If the levels of Ca2+ in the cytosol increase, then the lysosome has to pump Ca2+ against a stronger gradient, and if they can’t then they become more alkaline,” she said. In keeping with this, Mustaly showed that a RyR agonist raised the lysosomes pH; this was countered by Ryanodex.
Might something similar be going on in the human brain? Mustaly generated induced neurons from fibroblasts donated by AD patients and healthy controls, and treated them with the V-ATPase inhibitor bafilomycin. This led to tau accumulation in the AD-derived, but not the control-derived neurons. The data suggest that neurons in AD might be more susceptible to subtle changes in lysosomal pH balance.
What about Aβ?
In Stutzmann’s induced neurons, blocking the V-ATPase left Aβ unaffected. That’s not to say this peptide is insulated from changes in vesicular trafficking. Scientists know that amyloidogenic processing of amyloid precursor protein (APP) takes place on late endosomes. This process is enhanced by genetic variants, for example loss-of-function mutations in SORL1 (Aug 2019 news; Jun 2018 news). SORL1 encodes the protein SORLA, part of a cellular-protein-sorting conglomerate called the retromer. Retromers traffic APP away from late endosomes and to the Golgi, protecting it from cleavage by γ-secretase. Alas, in LA, Jessica Young, University of Washington, Seattle, reported that things are far more complex than that, revealing new pathways by which SORLA can shunt APP.
Like Karch, Young also uses human iPSCs and CRISPR editing. Her lab has knocked out or overexpressed SORL1, and tested how induced neurons would respond. At AAIC, Allison Knupp from Young’s lab reported that SORL1 KO cells had more APP in early endosomes and less in the trans Golgi, as would be expected. But there was also less APP in Rab9+ endosomes that recycle to the Golgi independently of the retromer. In keeping with this, when colleague Swati Mishra overexpressed SORL1, she found more APP in Rab9+ endosomes cycling to the Golgi, and also more APP in Rab11+ endosomes that recycle to the cell surface. “The data suggest there are more ways to less amyloidosis than through the canonical retromer,” Young told Alzforum. Indeed, she reported that SORL1 knockout and overexpression increased and decreased, respectively, production of Aβ1-40 and Aβ1-42 in induced neurons.
Karch and Young acknowledge that reprogramming cells to make iPSCs erases the epigenetic modifications cells have accrued during aging and thereby rejuvenate the stem cells relative to their fibroblast “ancestors.” For this reason, Young also experiments with direct conversion. This entails transmogrifying fibroblasts directly into neurons, bypassing the pluripotent stem cell step (Jun 2011 news; Jan 2013 news). Researchers led by Fred Gage at the Salk Institute, La Jolla, California, had previously reported that this process may preserve the transcriptomic age of the donor cells (Oct 2015 news).
Young, in collaboration with Dirk Keene and Suman Jayadev, both at UWashington, is testing this approach. They are starting out with fibroblast-like cells from the leptomeninges of autopsy-confirmed AD patients. They will run both iPSC and direct-conversion protocols side by side on the same tissue sample, then characterize the derived neurons to determine which best models Alzheimer’s pathology. Eventually, Young wants to use patient-derived neurons to test how a person’s genetic background influences endosomal trafficking, and defects thereof.
“It’s a huge collaborative effort,” Young said. “We plan to determine single-nucleotide polymorphism burden. We’ll analyze all the SNPS in endosome-related genes, group them by high and low risk, then see how they correlate with defects in trafficking, ultimately relating the phenotypes back to the genetics.” Young thinks this could identify very early changes in AD pathology that are not detected at autopsy. “You may have 100 people with a diagnosis of AD, but how that came about and progressed at the cellular level may be very different. If you subdivide based on genetics and cell biology, then you might be able to target treatments accordingly,” she said. “This is where derived neurons are really useful.”—Tom Fagan


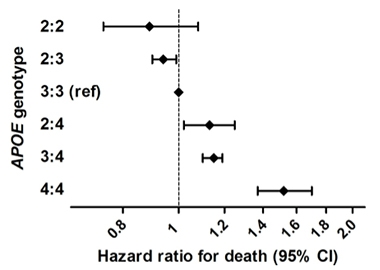
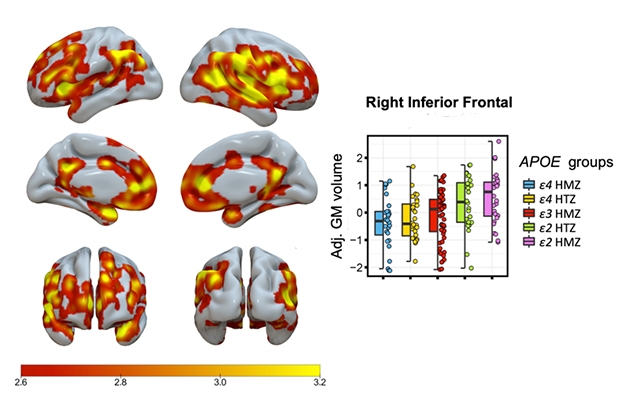



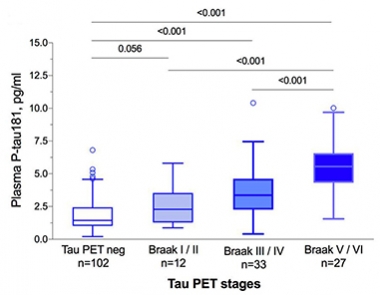
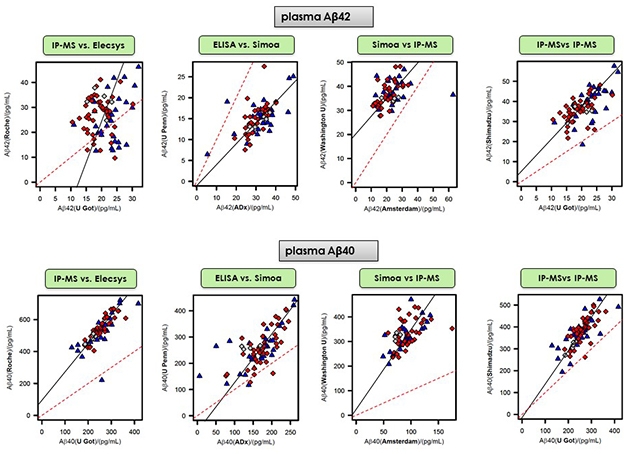


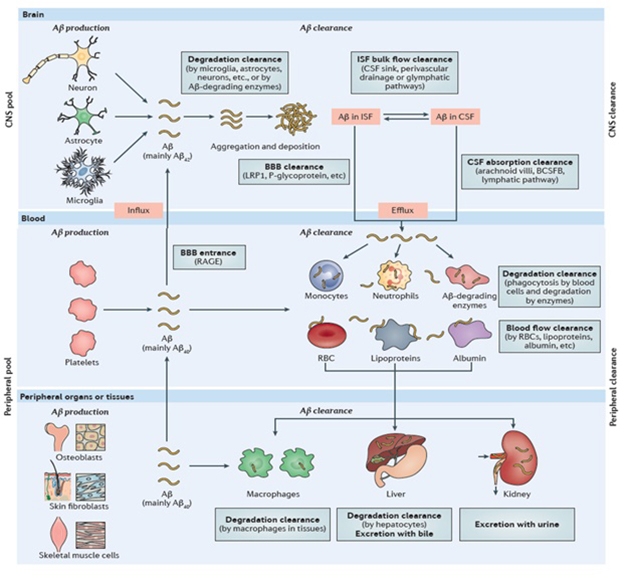





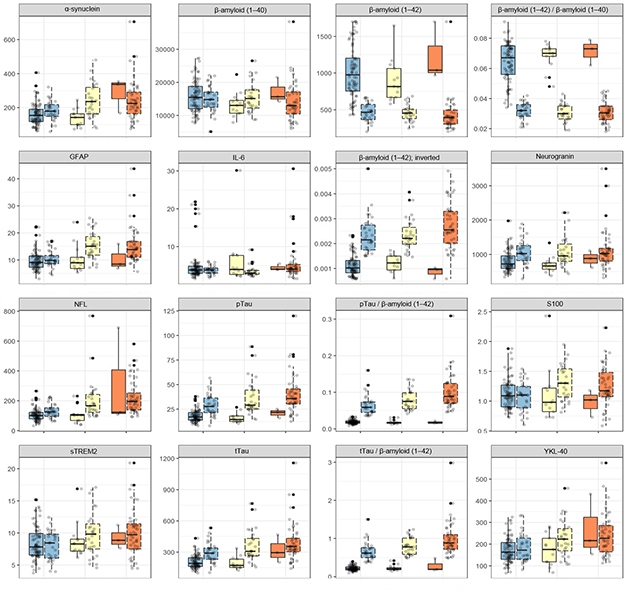




Comments
No Available Comments
Make a Comment
To make a comment you must login or register.