Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
Quick Links
Faltering endosomal trafficking appears to be the common modus operandi by a range of familial Alzheimer’s mutations. In the August 14 Neuron, researchers led by Marc Tessier-Lavigne of Stanford University reported initial findings from a panel of isogenic stem cell-derived neurons carrying such mutations. The scientists found that, across the board, fAD mutations in APP or PS1 altered expression of genes involved in endosomal trafficking. They also led to a swelling of early endosomes. The researchers pinned the blame for the endosomal malfunction on β-C-terminal fragments (β-CTFs) of APP, not Aβ.
- Panel of isogenic iPSC-derived neurons with familial AD mutations: a new research resource.
- Endosomes were enlarged, and β-CTF—not Aβ—was the cause.
- FAD mutations changed expression of endosomal trafficking and other AD risk genes.
The findings expand on a wealth previous studies implicating β-CTFs in vesicular foul play in AD, Tessier-Lavigne told Alzforum. The new paper extends prior findings to multiple FAD mutations in iPSC-neurons on an isogenic background, allowing researchers to zero in on the effects of the mutations. The isogenic lines will be made freely available to the research community, Tessier-Lavigne said.
“This is an incredible resource,” noted Celeste Karch of Washington University in St. Louis. “These tools are already revealing critical insights into disease mechanism that have implications for the forms of APP that are being targeted therapeutically.” Karch herself oversees a growing number of fibroblast lines donated via skin biopsies from participants in the Dominantly Inherited Alzheimer’s Network (DIAN). DIAN currently has fibroblasts carrying 34 different APP and PS1 mutations. Karch plans to convert these into iPSC-derived neurons and use CRISPR to generate isogenic controls (Aug 2019 conference news). Well more than 200 mutations in APP, PS1, and PS2 have been tied to familial forms of the disease.
While Aβ peptides secreted into the extracellular space have hogged the limelight in AD research for years, much of APP processing goes on beneath the cell surface, within membranes of endosomes. There, BACE1, an enzyme that flourishes in endosomes’ low pH environs, cleaves APP, generating transmembrane β-CTFs. Then γ-secretase processes β-CTF into Aβ peptides of various lengths. Numerous studies have pegged β-CTFs as instigators of endosomal woes (Jul 2015 news; Xu et al., 2016; Takasugi et al., 2018). More broadly, clogged and dysfunctional vesicles in the endolysosomal system have been implicated in AD for decades (Cataldo et al., 2000; Feb 2005 news; Jun 2010 news).
In search of common consequences of familial AD mutations in human neurons, co-first authors Dylan Kwart and Andrew Gregg and colleagues first investigated how the mutations affect gene expression. They used CRISPR gene editing to introduce the two most widely studied AD mutations—APP-Swe (KM670/671NL) and PS1-M146V—into human iPSCs, then differentiated them into neurons. To accentuate effects of the mutations, they generated an iPSC line (called dAP) that is homozygous for both of the mutated genes. RNA sequencing of these respective lines revealed more than 4,000 genes that were differentially expressed relative to isogenic wild-type iPSC neurons in at least one of the mutant lines; more than 1,500 genes changed expression in more than two cell lines, and 406 changed in all of them.
Many differentially expressed genes played a role in endocytic vesicle pathways, and several are linked to AD risk by GWAS. Some of these AD risk genes were implicated in endosomal function, including ApoE, CLU, SORL1, and LRP2. Ribosomal profiling revealed many of the same changes at the protein level. In all, the gene expression analysis pointed to endosomal malfunction as a common consequence of at least these two familial AD mutations.
“That these pathways were consistently altered, regardless of vastly different fAD mutations, suggests that the endocytic network may be uniformly linked to both familial and sporadic AD pathogenesis and thus an early target for therapeutic intervention,” noted Jessica Young of the University of Washington in Seattle. Tessier-Lavigne believes the gene expression data could point to a common AD gene network.
Guojun Bu of the Mayo Clinic in Jacksonville, Florida, noted that many AD risk genes, such as ApoE, are expressed predominantly in glial cells. That familial AD mutations alter their expression in neurons could simply suggest that those genes are highly sensitive to perturbation, not necessarily that they are functioning as part of a common AD gene network, or that they are involved in endosomal malfunctions. Wim Annaert of KU Leuven in Belgium had a different interpretation. “The fact that they identified these protein networks in long-term differentiated human neurons underscores a crucial neuronal basis in early pathogenesis,” he wrote. “This is important to stress given that in the past few years a major focus and effort has shifted to microglia, and in particular to disease-associated microglia as major perpetrators.”
To gauge whether endosomal dysfunction is a shared consequence of multiple familial AD mutations, the researchers expanded their panel of fAD neuronal lines, using CRISPR to generate isogenic lines heterozygous for three APP and four PS1 mutations, as well as homozygous versions all of the APP and three of the PS1 mutations. Together with an APP knockout line, the dAP line, and an APP mutant line homozygous for a protective mutation, the researchers developed a total of 16 isogenic iPSC lines from eight mutations for this study (see table below).
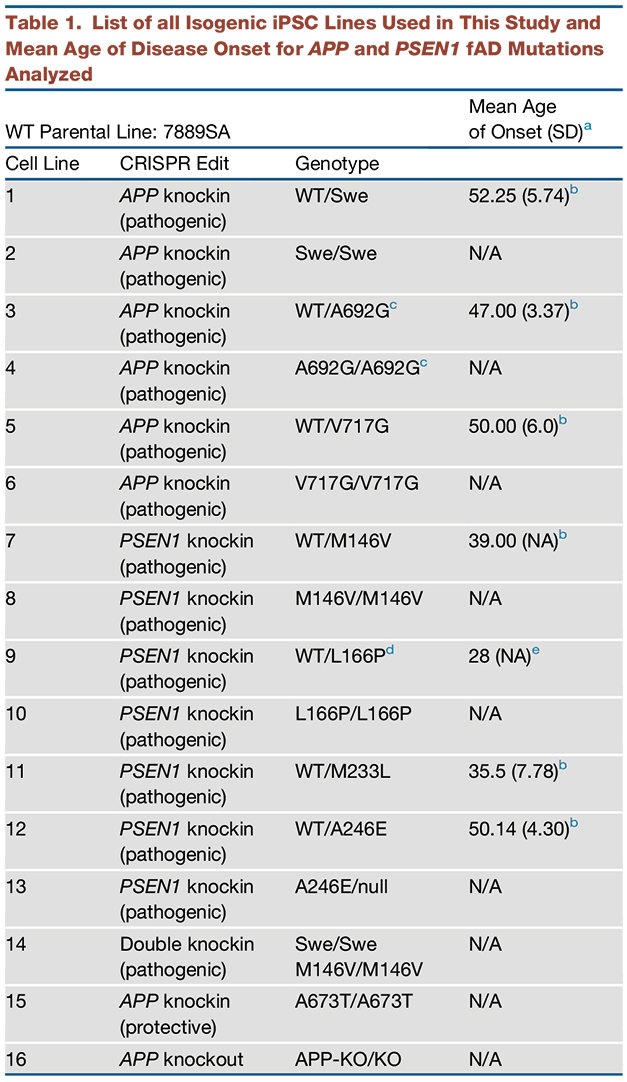
Isogenic Resource. A list of the isogenic lines developed for this study, which will be made available to researchers via the New York Stem Cell Foundation. APP-Swe, APP-A692G, APP-V717G, APP-A673T, PS1-M146V, PS1-L166P, PS1-M233L, PS1-A246E mutations are included. [Courtesy of Kwart et al., Neuron, 2019.]
The scientists report small increases in the size of Rab5+ endosomes in all heterozygous APP mutant lines, while only one heterozygous PS1 mutant—L166P—had significantly larger endosomes. However, every homozygous pathogenic mutant line had enlarged endosomes, suggesting that all of the mutations affected endosomal trafficking. dAP neurons, which carried two mutated copies of both APP and PS1, had the largest endosomes. A mutant line carrying two copies of APP-A673T, a protective mutation, did not have enlarged endosomes.

Ballooning Endosomes. Compared with wild-type iPSC-derived neurons, those carrying two copies of fAD mutations in APP or PS1 had larger endosomes. [Courtesy of Kwart et al., Neuron, 2019.]
To discern whether amyloidogenic processing of APP was involved in the endosomal phenotype, the researchers treated dAP neurons with a BACE inhibitor, which blocks cleavage of APP. The treatment stifled production of β-CTFs; endosomes were of normal size. An APP knockout iPSC-derived neuron line the researchers generated as a control had smaller endosomes than wild-type neurons did, suggesting that APP affects the size of endosomes. Kwart et al. also observed increased β-CTFs in all pathogenic mutant lines, and increases in α-CTFs in some. Notably, they found a strong correlation between endosome size and β-CTFs, not α-CTFs, across all their mutant lines.
Did the mutations influence the size of endosomes via β-CTF, or via the product of γ-secretase cleavage, Aβ? Before addressing this question directly, the researchers first assessed how each mutation affected the Aβ peptide repertoire. Confirming previous studies, they reported that all four PS1 mutations upped the ratio of Aβ42 to Aβ40, but reduced total Aβ production compared with wild-type neurons. All APP mutations enhanced total Aβ production, while APP-V717G also elevated the ratio of Aβ42 to Aβ40. dAP neurons—which carry two copies of both APP-Swe and PS1-M146V—had both increased total Aβ and Aβ42/40 ratio. All 14 cell lines used in this experiment did have one thing in common: a higher concentration of the longer Aβ peptides—Aβ42 and 43—in their culture supernatants than did their wild-type counterparts.
Were these long Aβ peptides to blame for the endosomal problems? Not according to experiments with a γ-secretase modulator (GSM), which skews γ-secretase toward producing shorter, non-amyloidogenic Aβ peptides without altering overall Aβ production. The researchers treated homozygous PSEN1-M146V, PSEN1-L166P, and dAP neurons with the modulator, because PS1 mutations sap the carboxypeptidase activity that the GSM targets. The PS1-L166P mutation did not change its repertoire of Aβ peptides in response to GSM treatment, a finding the researchers attributed to the severe effect the mutation has on γ-secretase activity. PS1-M146V and dAP neurons did respond by producing shorter Aβ peptides. However, endosomes remained enlarged. Notably, the GSM did not affect levels of β-CTFs, which were higher in all mutant lines than wild-type. The findings hinted that the endosomal phenotype may not be caused by longer Aβ peptides.
Next, the researchers treated wild-type iPSC-neurons with a broad γ-secretase inhibitor, which douses activity of the enzyme. This treatment blocked Aβ production, and led to an accumulation of both β- and α-CTFs. Endosomes grew enlarged. Adding a BACE inhibitor to the GSI treatment blocked all but α-secretase processing of APP. Endosomes stayed normal in size. When the researchers treated APP knockout iPSC-neurons with the GSI, endosomes stayed the same. Taken together, the findings tie β-CTFs, not Aβ peptides or α-CTFs, to the endosomal phenotype caused by familial AD mutations, the authors believe.
“This exciting study is an important contribution to the growing body of evidence that mutations in APP and PSEN1 lead to dysfunction of the endolysosomal network in human neurons, and that this is an early pathogenic event in monogenic Alzheimer’s disease,” commented Rick Livesey of University College London. The findings confirm Livesey’s recent work in a panel of patient-derived iPSC-neurons, including three APP and three PS1 mutations, in which he identified endolysosomal dysfunction and attributed it to β-CTF (Hung and Livesey, 2018). Tessier-Lavigne’s study builds on that work by examining a panel of mutations in isogenic lines, allowing researchers to more definitively link phenotype to mutation.
Livesey said the next question is how β-CTF interferes with endosomal trafficking. One possibility is by activating Rab5, as reported in a study led Chengbiao Wu of the University of California, San Diego (Xu et al., 2016). Wu reported that overexpression of β-CTF activates Rab5, leading to endosomal enlargement and disrupted retrograde axonal trafficking of nerve growth factor signals. Wu told Alzforum that Tessier-Lavigne’s findings agree with a large body of work showing that β-CTF disrupts endosomal trafficking. He believes β-CTF may be particularly prone to messing with endosomal trafficking because, unlike α-CTF, it retains its entire transmembrane domain.
In the brain, Aβ has been shown to damage synapses, stoke neuroinflammation, and interfere with vascular function. What about that? Tessier-Lavigne noted that his groups’ findings do not exclude a damaging role for Aβ, especially since the iPSC-derived neurons examined in this study—which are young cells, after all—do not undergo neurodegeneration. Gunnar Gouras of Lund University in Sweden wrote that while the new study convincingly ties β-CTF to enlarged endosomes in fAD neurons, it is difficult to rule out that Aβ disrupts vesicular trafficking. Previous studies, including Gouras’, implicated intracellular Aβ accumulation in endolysosomal trafficking disruption (Willén et al., 2017), whereas Tessier-Lavigne’s analysis was limited to Rab5+ early endosomes. Gouras speculated that both Aβ and β-CTF likely interfere with the endolysosomal system.
“This new work convincingly demonstrates that β-CTF suffices to drive major AD-related phenotypes, which may explain why therapies that target Aβ—but not β-CTF—have not been successful in the clinic to date,” wrote Rik Van Der Kant of VU University in Amsterdam (full comment below). BACE inhibitors, which target β-CTF, have thus far failed, as well (Jul 2019 news; Nov 2018 conference news). “One explanation for this failure might be that BACE inhibitors are effective in FAD neurons, in which β-CTFs accumulate downstream of APP and presenilin mutations as shown by Kwart et al., but not in sporadic AD neurons in which endosomal phenotypes are may be driven by other factors, such as ApoE genotype,” Van Der Kant added.
In a joint comment to Alzforum, Selina Wray and Charles Arber of University College London called familial AD an extremely heterogenous disease, at least from the perspective of how its manifold mutations affect the palette of Aβ peptides (Apr 2019 news). “It is noteworthy that, in addition to β-CTF, Aβ42 and Aβ43 are elevated in all lines tested, meaning that the contribution of these peptides to other pathological phenotypes cannot be discounted,” they added (full comment below).—Jessica Shugart
References
News Citations
- ASOs: Wave of the Future in Alzheimer’s Therapeutics?
- Partners in Crime: APP Fragment and Endosomal Protein Impair Endocytosis
- Varicose Axons: Traffic Jams Precede AD Pathology in Mice, Men
- Death of the Neatnik: Neurons Perish When Trash Clutters Their Space?
- Cognitive Decline Trips Up API Trials of BACE Inhibitor
- Bump in the Road or Disaster? BACE Inhibitors Worsen Cognition
- Familial Alzheimer’s Mutations: Different Mechanisms, Same End Result
Mutation Interactive Images Citations
Mutations Citations
- APP K670_M671delinsNL (Swedish)
- PSEN1 M146V
- APP A692G (Flemish)
- APP V717G
- APP A673T (Icelandic)
- PSEN1 L166P
- PSEN1 M233L (A>T)
- PSEN1 A246E
Paper Citations
- Xu W, Weissmiller AM, White JA 2nd, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, Gunawardena S, Ding J, Mobley WC, Wu C. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016 May 2;126(5):1815-33. Epub 2016 Apr 11 PubMed.
- Takasugi N, Araya R, Kamikubo Y, Kaneshiro N, Imaoka R, Jin H, Kashiyama T, Hashimoto Y, Kurosawa M, Uehara T, Nukina N, Sakurai T. TMEM30A is a candidate interacting partner for the β-carboxyl-terminal fragment of amyloid-β precursor protein in endosomes. PLoS One. 2018;13(8):e0200988. Epub 2018 Aug 7 PubMed.
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
- Hung CO, Livesey FJ. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer's Disease. Cell Rep. 2018 Dec 26;25(13):3647-3660.e2. PubMed.
- Willén K, Edgar JR, Hasegawa T, Tanaka N, Futter CE, Gouras GK. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener. 2017 Aug 23;12(1):61. PubMed.
Further Reading
Primary Papers
- Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, Tessier-Lavigne M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron. 2019 Oct 23;104(2):256-270.e5. Epub 2019 Aug 12 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
New York University School of Medicine/Nathan Kline Institute
This report on CRISPR-modified IPSC neurons expressing diverse APP and PSEN1 FAD mutations is a technical tour de force. It is a welcome confirmation of our previous findings on neurons in AD, FAD, and Down’s sydrome (DS) brains and human DS fibroblasts, establishing AD-specific early endosome dysfunction arising at the earliest disease stage and prior to amyloid deposition (Cataldo et al., 2000; Nixon 2006; Colacurcio et al., 2018).
The new data further substantiate rab5-endosome enlargement as a neuropathological hallmark of AD and confirm the specific role of APP- βCTF as the responsible APP metabolite driving endosome dysfunction (Jiang et al., 2010; Jiang et al., 2016). Enlargement of rab5-endosomes has been shown to reflect abnormally accelerated endocytosis driven mainly by pathological rab5 activation and its aberrant signaling via downstream effector pathways (Nixon 2017; Colacurcio et al., 2018; Chen et al., 2018).…More
The new IPSC neurons exhibit upregulated transcription of rab5 and endocytosis-related genes as expected based on human data (Ginsberg et al., 2010). Interestingly, GO analyses of DEGs from their transcriptomic data show significant congruence with ours in primary fibroblasts from controls versus individuals with DS (Jiang et al., 2019). This reflects considerable conservation of the disease responses to APP mutation or duplication in both neurons and fibroblasts, which extends to DS, another common cause of early onset AD.
Recent investigations of endosome dysfunction in AD strongly support its pathogenic significance. Beyond the well-recognized roles of early endosomes in sorting and routing internalized cargoes, new evidence is revealing diverse roles of early endosomes as complex signaling platforms and underscores the importance of rab5 as the critical initiator of endosome signaling cascades highly relevant to neuronal communication and survival, especially the survival of cholinergic neuronal populations (Chen et al., 2019; Nixon 2017).
Recently, the direct in vivo reproduction in mice of pathological rab5 upregulation and activation that was induced directly rather than by its usual trigger, APP-βCTF, recapitulated not only signature AD-like endosomal dysfunction but also a panoply of AD-related phenotypic features, including synaptic plasticity deficits, cholinergic neurodegeneration, tau hyperphosphorylation, and memory impairment (May 2019 conference news). This implicates the range of known rab5 functions as mediating a pathogenic cascade paralleling, but independent of, that induced following amyloid deposition.
Given the overwhelming evidence now implicating APP-βCTF neurotoxicity as a key player in AD pathogenesis, it is puzzling that the physiological and pathological roles of APP-CTFs continue to be largely ignored or even dismissed in interpretations of preclinical/clinical outcomes of putative AD therapeutics. Remarkably, BACE1 modulation outcomes in many preclinical studies are still interpreted solely in terms of effects on Aβ levels, frequently with CTF levels not even being measured.
In the failed clinical trials of BACE1 inhibitors, the objective seems so far to have been on maximally reducing Aβ at the expense of requiring strong BACE1 inhibition, even though preclinical studies would predict high risk of untoward effects on other BACE1 functions. Partial BACE1 reduction (e.g. BACE1 single-allele models) in preclinical models appears to be subtoxic, but was shown to be effective in ameliorating cholinergic neurodegeneration and other AD-related phenotypes linked to APP-βCTF toxicity in DS model mice (Jiang et al 2016).
In this regard, it is potentially significant that the “AD-protective” mutation of APP studied by Kwart et al. did not elevate βCTF levels (it actually reduced APP-C99) and was the only APP mutation studied that did not enlarge endosomes. This mutation is believed to act by reducing β-secretase cleavage of APP, even though its protective effect is invariably attributed to reduction of Aβ, not APP-βCTF. The protective effect of this mutation in a human population could well be considered proof-of-principle support for BACE1 clinical trials at subtoxic BACE1 inhibitory levels despite the acknowledged pharmacological challenges of titrating BACE1 activity reliably in a clinical setting.
References:
Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
Chen XQ, Sawa M, Mobley WC. Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic Biol Med. 2018 Jan;114:52-61. Epub 2017 Oct 12 PubMed.
Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer's Disease. Free Radic Biol Med. 2018 Jan;114:40-51. Epub 2017 Oct 6 PubMed.
Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, Che S. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010 Nov 15;68(10):885-93. Epub 2010 Jul 23 PubMed.
Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
Jiang Y, Rigoglioso A, Peterhoff CM, Pawlik M, Sato Y, Bleiwas C, Stavrides P, Smiley JF, Ginsberg SD, Mathews PM, Levy E, Nixon RA. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol Aging. 2016 Mar;39:90-8. Epub 2015 Dec 2 PubMed.
Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1630-5. Epub 2009 Dec 28 PubMed.
Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis. 2006;9(3 Suppl):277-89. PubMed.
Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J. 2017 Jul;31(7):2729-2743. PubMed.
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
This paper reproduces findings in human iPSC-derived neurons that others made decades ago in much simpler cell systems. These cell lines are therefore a nice and important resource for future research, but I would have expected a balanced discussion and acknowledgement of the earlier findings.
Institute of Neurology, University College London
UCL Queen Square Institute of Neurology
Induced pluripotent stem cell (iPSC)-derived cell types are widely used as physiologically-relevant models of human neurodegenerative disease, but challenges still remain around the inherent variability driven largely by differing genetic backgrounds. A particular strength of this paper is the large series of isogenic iPSC lines that have been generated, with heterozygous and homozygous mutations in both PSEN1 and APP, which have been carefully selected to span multiple regions along each gene and to investigate shared disease phenotypes. Although homozygous fAD mutations are not found in patients, here they provide a useful model to accelerate cellular phenotypes.
The main finding of the paper is that fAD mutations universally lead to increased levels of β-CTFs, which in turn leads to an abnormal endosomal phenotype that is independent of Aβ. This paper adds to increasing evidence that β-CTFs may play an important role in AD pathogenesis, particularly by mediating dysfunction of the endosome-lysosome system, as also suggested in patient-derived human neurons with PSEN1 and APP mutations (Hung et al., 2018; Israel et al., 2012. These findings significantly add to our understanding of the normal function of APP and its cleavage products. Relevant to this, C99 accumulation has been suggested to correlate with neurodegeneration in human brains (Puline et al., 2019) and, taken together, these data may prove important in designing the next generation of drug targets. It would be particularly interesting to extend this work to examine the impact of PSEN2 mutations on endosomal dysfunction, particularly given its unique subcellular location at the endosome as recently described (Sannerud et al., 2016; June 2016 news).…More
Although fAD mutations universally increase β-CTFs, it should be noted that monogenic AD is a clinically heterogeneous disease (Ryan et al., 2016). fAD mutations qualitatively impair APP processing resulting in a relative increase of longer Aβ peptides (Chávez-Gutiérrez et al., 2012), this is reflected in heterogenous Abeta profiles as demonstrated by multiple groups, including in our recent paper using a large series of patient-derived iPSC (Arber et al., 2019). As the authors state, the effects of these mutations on longer Aβ species such as Aβ45 and Aβ46 in human neurons is currently unknown, and as recent reports have highlighted the contribution of short Aβ species to attenuating toxicity (Moore et al., 2018), this study further highlights the need to study the full spectrum of APP-derived fragments. It is noteworthy that, in addition to β-CTFs, Aβ42 and Aβ43 are elevated in all lines tested, meaning that the contribution of these peptides to other pathological phenotypes cannot be discounted. It will be interesting to determine how changes in β-CTFs, together with diversity in Aβ profiles, other gamma secretase substrates, and downstream pathologies such as tau, can act in concert to result in clinically distinct presentations in fAD.
References:
Hung CO, Livesey FJ. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer's Disease. Cell Rep. 2018 Dec 26;25(13):3647-3660.e2. PubMed.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012 Jan 25;482(7384):216-20. PubMed.
Sannerud R, Esselens C, Ejsmont P, Mattera R, Rochin L, Tharkeshwar AK, De Baets G, De Wever V, Habets R, Baert V, Vermeire W, Michiels C, Groot AJ, Wouters R, Dillen K, Vints K, Baatsen P, Munck S, Derua R, Waelkens E, Basi GS, Mercken M, Vooijs M, Bollen M, Schymkowitz J, Rousseau F, Bonifacino JS, Van Niel G, De Strooper B, Annaert W. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell. 2016 Jun 30;166(1):193-208. Epub 2016 Jun 9 PubMed.
Ryan NS, Nicholas JM, Weston PS, Liang Y, Lashley T, Guerreiro R, Adamson G, Kenny J, Beck J, Chavez-Gutierrez L, de Strooper B, Revesz T, Holton J, Mead S, Rossor MN, Fox NC. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer's disease: a case series. Lancet Neurol. 2016 Dec;15(13):1326-1335. Epub 2016 Oct 21 PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Arber C, Toombs J, Lovejoy C, Ryan NS, Paterson RW, Willumsen N, Gkanatsiou E, Portelius E, Blennow K, Heslegrave A, Schott JM, Hardy J, Lashley T, Fox NC, Zetterberg H, Wray S. Familial Alzheimer's disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol Psychiatry. 2020 Nov;25(11):2919-2931. Epub 2019 Apr 12 PubMed.
Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, Ladd TB, Ran Y, Levites Y, Kukar TL, Kurian JJ, McKenna R, Koo EH, Borchelt DR, Janus C, Rincon-Limas D, Fernandez-Funez P, Golde TE. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018 Jan 2;215(1):283-301. Epub 2017 Dec 5 PubMed.
University of Washington
Recent failures of Aβ-centric clinical trials highlight the need for investigating early cellular phenotypes that cause dysfunction upstream of amyloid and tau pathology in Alzheimer’s disease. Endosome enlargement is an early cytopathology in AD, and multiple genes that confer increased risk for sporadic AD (sAD) are in loci attributed to endocytosis and vesicular trafficking. Understanding this pathway and how it becomes dysfunctional in central nervous system cells is imperative, as aberrant vesicular trafficking is a common theme in several neurodegenerative disorders.
In this elegant work, Kwart et al. use a carefully curated panel of gene-edited hiPSC-derived neurons to investigate cellular endosomal phenotypes. These cell lines harbor APP and PSEN1 mutations in an isogenic genetic background, thus reducing experimental variability. Intriguingly, they report the presence of enlarged Rab5+ endosomes that are dependent on cellular levels of APP β-CTFs, but independent of Aβ levels, which raises important considerations for testing drug candidates that alter different aspects of APP processing.…More
Following detailed molecular profiling, they show that hiPSC-derived AD mutant neurons show clear changes in gene expression networks regulating endocytosis and vesicular trafficking. That these pathways were consistently altered in all cell lines, regardless of vastly different fAD mutations, suggests that the endocytic network may be uniformly linked to both fAD and sAD pathogenesis and thus an early target for therapeutic intervention.
This study really highlights the strengths of using hiPSC-based approaches to interrogate dynamic cellular pathways that are relevant to increased AD risk and that are active early in disease manifestation. It will now be interesting to elucidate further the links between endosomal traffic jams and other pathologies indicative of cognitive decline such as tau pathology, which has also been linked to APP β-CTFs in stem cell derived neurons (Israel et al., 2012), and synaptic dysfunction.
Vrije Universiteit Amsterdam
This is a very impressive and exhaustive study by the Tessier-Lavigne lab showing that APP β-CTFs, but not Aβ, drives endosomal enlargement in human neurons from patients with familial AD (FAD). Using more than fifteen isogenic iPSC lines, the authors show that early endosomal enlargement driven by β-CTF accumulation is a common phenotype downstream of FAD mutations. What particularly stands out to me is the rigor and depth of the experimentation, as the authors test the effects of an array of different pharmacological interventions in various FAD and wildtype neurons as well as in isogenic APP knockout neurons.
The observation by first author Kwart and colleagues that β-CTF can drive AD-related phenotypes in an Aβ-independent manner is not a stand-alone finding. Others have previously reported similar effects of β-CTF on endosomal function in both mouse models and human iPSC-derived neurons (for example Hung & Livesey, 2018; Xu et al., 2016). Furthermore APP β-CTF has also previously been shown to drive the accumulation of phospho-tau (p-tau), in a manner independent of Aβ (Israel et al., 2012; Moore et al., 2015). This raises the question, to which extent are AD phenotypes previously attributed to Aβ actually driven by β-CTF? As a direct precursor to Aβ, β-CTF is (over)produced in equimolar concentrations as Aβ in the vast majority of existing AD animal models. This new work convincingly demonstrates that β-CTF suffices to drive major AD-related phenotypes, which may explain why therapies that target Aβ--but not β-CTF—have not been successful in the clinic to date.…More
If BACE inhibition reduces β-CTF accumulation, and BACE inhibition thereby prevents β-CTF dependent endosomal dysfunction (shown here) and p-tau accumulation, then why does BACE inhibition not provide clinical benefit in AD patients? In their paper, Kwart and colleagues argue that BACE inhibition might have failed because subjects were treated too late in disease (in prodromal stages). Unfortunately, even more recently, clinical studies with BACE inhibition in presymptomatic subjects at risk for AD have also been halted because treated patients showed faster cognitive decline than the placebo group. An alternative explanation for this failure might be that BACE inhibitors are effective in FAD neurons, in which β-CTF accumulate downstream of APP and presenilin mutations as shown by Kwart et al., but not in sporadic AD neurons in which endosomal phenotypes may be driven by other factors, such as ApoE genotype (e.g. Lin et al., 2018; Nuriel et al., 2017). In the future, it will be interesting to see whether β-CTF also accumulates in SAD genotype neurons and whether this also suffices to drive endosomal dysfunction.
Overall, this very elegant work by the Tessier-Lavigne lab, raises a number of interesting novel questions regarding the physiological role of APP in endosomal transport and the pathogenic role of βCTF in AD.
References:
Hung CO, Livesey FJ. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer's Disease. Cell Rep. 2018 Dec 26;25(13):3647-3660.e2. PubMed.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012 Jan 25;482(7384):216-20. PubMed.
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018 Jun 27;98(6):1141-1154.e7. Epub 2018 May 31 PubMed.
Moore S, Evans LD, Andersson T, Portelius E, Smith J, Dias TB, Saurat N, McGlade A, Kirwan P, Blennow K, Hardy J, Zetterberg H, Livesey FJ. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015 May 5;11(5):689-96. Epub 2015 Apr 23 PubMed.
Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, Ambat J, Levy E, Cookson MR, Mathews PM, Duff KE. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front Neurosci. 2017;11:702. Epub 2017 Dec 12 PubMed.
Xu W, Weissmiller AM, White JA 2nd, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, Gunawardena S, Ding J, Mobley WC, Wu C. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016 May 2;126(5):1815-33. Epub 2016 Apr 11 PubMed.
University College London
Pioneering work from several groups, most notably from Ralph Nixon and colleagues, identified dysfunction of the endolysosomal network in Alzheimer’s disease, and that this was due to abnormal processing of APP. This exciting study is an important contribution to the growing body of evidence that mutations in APP and PSEN1 lead to dysfunction of the endolysosomal network in human neurons, and that this is an early pathogenic event in monogenic Alzheimer’s disease.
The use of isogenic induced pluripotent stem cell (iPSC) lines and the characterization of iPSC-derived neurons carrying heterozygous and homozygous PSEN1 and APP mutations in multiple different ways, are real strengths of this work. Using patient-derived iPSCs, our group reported last year (Hung & Livesey, 2018) that heterozygous PSEN1 and APP mutations lead to lysosome and autophagy dysfunction that is rescued by BACE inhibition. That study also showed, by CRISPR knockout of APP in PSEN1 mutant iPSCs, that abnormalities in lysosome and autophagy function in PSEN1 mutant neurons are dependent on APP, and we implicated β-CTFs, rather than Aβ, as the key culprit. Here, Kwart and colleagues have used a combination of pharmacological treatments to dissect this further, generating compelling evidence that accumulation of β-CTFs is the primary problem leading to endosome enlargement.…More
A key emerging question is the mechanism by which accumulation of β-CTFs lead to malfunction of the endolysosome-autophagy network, which will probably also involve understanding the normal cellular function of turnover of β-CTFs by γ-secretase. Potentially related to this is the role of SORL1, which binds to APP, regulating its intracellular trafficking. Truncating mutations in SORL1 are a cause of monogenic dementia, and SORL1 variants that increase risk of AD are well-described in multiple GWAS. If loss of SORL1 function also leads to accumulation of β-CTFs and endolysosome dysfunction, it adds yet more weight to the author’s proposal that targeting APP or β-CTFs directly, as recently reported by the Roy group (Sun et al., 2018), is a potentially powerful therapeutic strategy.
References:
Hung CO, Livesey FJ. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer's Disease. Cell Rep. 2018 Dec 26;25(13):3647-3660.e2. PubMed.
Sun J, Carlson-Stevermer J, Das U, Shen M, Delenclos M, Snead A, Wang L, Loi J, Petersen A, Stockton M, Bhattacharyya A, Jones M, Sproul A, McLean P, Zhao X, Saha K, Roy S. A CRISPR/Cas9 based strategy to manipulate the Alzheimer's amyloid pathway. bioRχiv 310193; Apri 28, 2018
KULeuven & VIB
The authors did a tremendous job in creating a broad resource. Isogenic FAD/control iPSCs will likely set a new standard in the cellular modelling of AD-related phenotypes, and the lines reported here hopefully will be readily made available to a broader community.
Starting from an initial three cell lines, the authors identified dysregulated genes and proteins of which several are (i) clearly related to endocytic transport regulation and (ii) comprise several of previously identified risk genes.
It is becoming evident from both pathological studies, as well as in vivo and in vitro modelling, that dysregulation in the endocytic, and by extension endolysosomal, pathway may be intrinsically associated with early signs of disease. The fact that they identified these protein networks in long-term differentiated human neurons underscores a crucial neuronal basis in early pathogenesis.…More
This is important to stress given that in the past few years a major focus and effort has shifted to microglia, and in particular to disease-associated microglia as major perpetrators.
The authors highlight endosomal traffic dysregulation, notably enlarged endosomes, as a major effect of FAD mutations, but that does not exclude that other transport routes are affected, as well. There is quite some literature involving dysfunctional lysosomes in the disease pathogenesis. A recent similar paper of the Livesey group, using also differentiated human neurons, indeed showed dysfunctions in endosomes as well as lysosomes.
If the prior effect is on endosomal compartments, the observed lysosomal defects might be indirect. If so, it raises a question whether the observed lysosomal defects in PSEN-deficient cells are direct or indirect as well, another aspect that needs to be reinvestigated.
Both groups, Kwart et al. in a more elaborate way, now provide conclusive evidence that β-CTFs are the main culprits in the endosomal phenotype, not Aβ. The effects can be traced down essentially to β-cleaved CTFs, which is surprising given the small difference with α-CTFs and the fact that the latter are far more abundant.
This indicates that there is something peculiar about β-CTFs that renders them toxic to intracellular organelles. The nature of this remains to be investigated.
It also raises the question whether in the case of APP-CTF (as opposed to e.g. Notch processing) γ-secretase acts as a "proteasome of the membrane" (Kopan and Ilagan, 2004) to clean up CTF fragments. Although downstream signalling through AICD cannot be excluded, maybe these aspects should be re-nvestigated in these new models of human neurons.
References:
Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane?. Nat Rev Mol Cell Biol. 2004 Jun;5(6):499-504. PubMed.
Lund University
This is impressive work providing exciting new tools (a large library of isogenic APP and PS1 mutant iPSCs) to explore AD pathogenesis. They describe that both APP and PS1 mutant iPSCs have remarkably overlapping alterations in gene and protein expressions involved in endocytic trafficking. They then undertake a sophisticated attempt to pin down Rab5 endosome enlargement, which they see in all their APP and PS1 mutant iPSCs, to specifically APP β-CTFs and not to extended Aβ peptides. However, it is not easy to fully exclude a role of the latter. This and prior studies have relied on inhibiting γ-secretase to argue against Aβ but for APP β-CTFs in endosome enlargement. However, one should keep in mind that all PS1 mutant forms of AD still show massive accumulations of extended Aβ in their brains. Thus, certainly γ-secretase inhibitor (GSI) but also γ-secretase modulator (GSM) treatment might mimic AD causing β-CTF increases and endosome enlargement, but because of their Aβ lowering not adequately model PS1 mutant AD. It is, however, very interesting that early endosome enlargement correlated with β-CTF levels in their different iPSC neurons, which certainly strengthens the role for β-CTFs. It is also quite interesting that APP KO neurons have smaller Rab5 endosomes.…More
In their discussion the authors suggest that Aβ might still impact AD pathogenesis by alternative pathway(s) such as via inflammation, which is certainly possible and consistent with the growing importance of inflammation in AD. However, we did see that Aβ can lead to endosome enlargement (Willen et al., 2017). Moreover, extended Aβ peptides were seen to specifically accumulate and aggregate in endosomes of neurons prior to plaques, also in human Down's syndrome and AD brains (Takahashi et al., 2002; Takahashi et al., 2004; Cataldo et al., 2004). Thus, while the current paper is very compelling, it might not yet exclude a role for extended Aβ42/43 in Rab5 endosome enlargement.
References:
Willén K, Edgar JR, Hasegawa T, Tanaka N, Futter CE, Gouras GK. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener. 2017 Aug 23;12(1):61. PubMed.
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004 Apr 7;24(14):3592-9. PubMed.
Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004 Nov-Dec;25(10):1263-72. PubMed.
RIKEN Center for Brain Science
A simple question: Does CTF-β accumulate in preclinical and clinical AD patients? I believe that the findings must be confirmed in human brains.
Icahn School of Medicine
Accumulation of a presumptive β-CTF of APP is characteristic of the aging brain, without or with clinical Alzheimer's disease at the time of death (Nordstedt et al., 1991). While there is abundant evidence over the past 28 years that the hydrophobic Aβ and transmembrane domains of β-CTFs drive aggregation of β-CTFs, our experience would indicate that β-CTF accumulation is not a reliable biomarker for incident dementia.
Presumably, in the same way that there are resilience factors that sustain normal cognition in the face of full-blown Alzheimer's pathology (e.g., Driscoll and Troncoso, 2011), there might well be resilience factors that sustain normal cognition in the face of β-CTF accumulation. …More
References:
Driscoll I, Troncoso J. Asymptomatic Alzheimer's disease: a prodrome or a state of resilience?. Curr Alzheimer Res. 2011 Jun;8(4):330-5. PubMed.
Nordstedt C, Gandy SE, Alafuzoff I, Caporaso GL, Iverfeldt K, Grebb JA, Winblad B, Greengard P. Alzheimer beta/A4 amyloid precursor protein in human brain: aging-associated increases in holoprotein and in a proteolytic fragment. Proc Natl Acad Sci U S A. 1991 Oct 15;88(20):8910-4. PubMed.
RIKEN Center for Brain Science
As Sam Gandy pointed out, the APP fragment termed APP 113, or others, are presumptive CTF-βs (Nordstedt et al., 1991). It would be more informative to identify which protein fragments exactly correspond to CTF-β and CTF-α.
References:
Nordstedt C, Gandy SE, Alafuzoff I, Caporaso GL, Iverfeldt K, Grebb JA, Winblad B, Greengard P. Alzheimer beta/A4 amyloid precursor protein in human brain: aging-associated increases in holoprotein and in a proteolytic fragment. Proc Natl Acad Sci U S A. 1991 Oct 15;88(20):8910-4. PubMed.
Institut de Pharmacologie Moléculaire et Cellulaire
IPMC CNRS UMR72275
IPMC, CNRS, UMR7275
The paper by Kwart and colleagues is a nice piece of work. By CRISP/Cas9-generated knock-in in human IPSCs, they overall confirm previous data from several independent laboratories, i.e., the deleterious influence of the β-secretase-derived APP fragment C99 on endosomal-lysosomal system. Among previous studies concurring with this conclusion are our studies (not cited in the article) that demonstrate that C99 could accumulate early in neurons of various transgenic mice (Lauritzen et al., 2012) that it can trigger lysosomal dysfunction (Lauritzen et al., 2016). That these alterations occur independently of Aβ was demonstrated by comparing 3xTgAD (from Frank LaFerla’s lab that harbor mutated presenilin 1) and 2xTgAD (derived from 3xTgAD but harboring wild-type presenilins) transgenic mice. Thus, these mice accumulate early identical levels of C99 and display similar lysosomal dysfunction, although 2xTgAD mice did not harbor detectable levels of Aβ at this stage (Lauritzen et al., 2016). That C99 accounts for the deleterious phenotype was also strengthened by our observation that γ-secretase inhibitors worsen endo-lysosomal defects (Lauritzen et al., 2016). It should be said that at these early stages, an apathy-like phenotype (Bourgeois et al., 2018) reminiscent of that observed at early stages of AD pathology in patients has been reported.…More
Three direct and indirect lines of observations can be derived from the Dr. Tessier-Lavigne’s paper.
References:
Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, Fraser P, St-George-Hyslop P, Le Thuc O, Espin V, Chami L, Dunys J, Checler F. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci. 2012 Nov 14;32(46):16243-55a. PubMed.
Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, Checler F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016 Aug;132(2):257-76. Epub 2016 Apr 30 PubMed. Correction.
Bourgeois A, Lauritzen I, Lorivel T, Bauer C, Checler F, Pardossi-Piquard R. Intraneuronal accumulation of C99 contributes to synaptic alterations, apathy-like behavior, and spatial learning deficits in 3×TgAD and 2×TgAD mice. Neurobiol Aging. 2018 Nov;71:21-31. Epub 2018 Jul 12 PubMed.
The Rockefeller University
Multiple lines of evidence indicate that the processing of APP by β-secretase increases the risk for developing AD. For instance, several APP mutations increase BACE cleavage; PSEN1 mutations decrease C99 engagement, decreasing C99 cleavage by γ-secretase; a mutation in APP that decreases β-cleavage protects against cognitive decline; and increased BACE activity is found with aging. All these findings are commonly analyzed with regard to their effects on Aβ levels. However, this study, together with several earlier findings from the Nixon and Checler labs, point to deleterious effects mediated by C99, and not β-amyloid.
An interesting observation from the data in this study is that phospho-C99 seems to be equally abundant as its unphosphorylated form in “wild-type” neurons; however, PSEN1 mutations and some APP mutations increased the phosphorylated form. This raises the question of the potential pathogenic role of this phosphorylation. …More
Recent results from our lab show a remarkable correlation between C99 levels and cognitive decline in brain areas vulnerable to neurodegeneration. Interestingly, we found that C99 accumulates only in neurons presenting high levels of tau (Pulina et al. 2019), raising the possibility that C99 contributes to selective vulnerability in AD (Pulina et al., 2019).
Hospital de Sant Pau
The finding of this work that isogenic iPSC lines with autosomal-dominant Alzheimer’s disease (ADAD) mutations accumulate β-CTF replicates the previous findings observed in transgenic mouse models (Lauritzen et al., 2016; Saura et al., 2005) or after treatment with classical γ-secretase inhibitors (Mitani et al., 2012; Bittner et al., 2009). In these situations, APP CTFs accumulate at the presynaptic terminals, likely impairing synaptic plasticity and long-term memory.
Cellular models of AD are valuable if they can reproduce a cellular environment similar to the one observed in the diseased tissue. The findings reported by Kwart and Greeg et al. are important because they demonstrate that this model can recapitulate the findings observed in human brain in ADAD. In particular, our group published that ADAD cases show a prominent APP β-CTF accumulation in the brain when compared to age-matched controls and to sporadic AD cases (Pera et al., 2013). This abnormal feature was particularly associated with ADAD with APP and PSEN1 mutations, and was less evident in sporadic AD (Pera et al., 2013). …More
Although the precise cause of APP β-CTF accumulation in ADAD deserves further investigation, it is likely that this feature acts as an active component of the disease that may contribute to the synaptic and cytoskeletal derangement and neurodegeneration. Therefore, this APP metabolite could be of interest in trials in ADAD patients, and perhaps in sporadic AD. In any case, the confirmation by Kwart and Greeg et al. that β-CTF mediates alterations in early endosomes could definitely stimulate strategies to specifically target this fragment.
References:
Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, Checler F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016 Aug;132(2):257-76. Epub 2016 Apr 30 PubMed. Correction.
Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J. Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci. 2009 Aug 19;29(33):10405-9. PubMed.
Mitani Y, Yarimizu J, Saita K, Uchino H, Akashiba H, Shitaka Y, Ni K, Matsuoka N. Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J Neurosci. 2012 Feb 8;32(6):2037-50. PubMed.
Pera M, Alcolea D, Sánchez-Valle R, Guardia-Laguarta C, Colom-Cadena M, Badiola N, Suárez-Calvet M, Lladó A, Barrera-Ocampo AA, Sepulveda-Falla D, Blesa R, Molinuevo JL, Clarimón J, Ferrer I, Gelpi E, Lleó A. Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 2013 Feb;125(2):201-13. PubMed.
Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005 Jul 20;25(29):6755-64. PubMed.
Make a Comment
To make a comment you must login or register.