Familial Alzheimer’s Mutations: Different Mechanisms, Same End Result
Quick Links
Though all autosomal-dominant AD mutations lead to Aβ pathology, they do so by disturbing γ-secretase processing of APP in different ways. In the April 12 Molecular Psychiatry, researchers led by Selina Wray of University College London compared the array of Aβ peptides secreted from neurons derived from carriers of different familial AD mutations. Mutations in APP affected where γ-secretase took its first bite, while mutations in presenilin 1 had three distinct outcomes. They hobbled the Pac-Man-like, processive activity of the protease, prevented its maturation, or reduced its expression. All mutations ultimately boosted the proportion of Aβ42, and most of the PS1 mutations also upped Aβ43, a peptide more recently found to be highly amyloidogenic. The study is the largest, most comprehensive analysis of Aβ peptides secreted from patient-derived neurons harboring different familial AD mutations to date.
- In iPSC-derived neurons, APP and PS1 mutations both upped Aβ42:Aβ40 ratios.
- The London APP mutation changed γ-secretase’s endopeptidase activity; PS1 mutations its carboxypeptidase activity or presenilin’s stability.
- In a carrier, the neurons’ Aβ38/42 ratio matched that seen in the person’s CSF.
“This is an exciting new study that demonstrates how human stem cell models can be leveraged to gain a more nuanced understanding of the impact of familial AD mutations on presenilin function and APP processing,” commented Celeste Karch of Washington University in St. Louis.
As the catalytic subunits of γ-secretase, the presenilins release Aβ peptides, including the infamous Aβ42, by snipping the transmembrane portion of the C-terminal fragment of APP produced by β-secretase. The proteolytic release of Aβ from β-CTF occurs stepwise. First, γ-secretase cuts, endopeptidase fashion, at either Leucine-48 or Threonine-49, leaving either Aβ48 or Aβ49 lodged in the membrane and releasing the APP intracellular domain. Then, carboxypeptidase fashion, γ-secretase chops the membrane-bound fragment three to four residues at a time. Depending on where the initial endopeptidase snip takes place, two predominant Aβ production lines emerge: Aβ49>Aβ46>Aβ43>Aβ40 and Aβ48>Aβ45>Aβ42 and even some Aβ38 (Takami et al., 2009; Fernandez et al., 2016).

Two Tracks. Endopeptidase (ε-cleavage) of APP at two sites leads to two production lines of Aβ peptides, each generated by processive carboxypeptidase trimming by γ-secretase. [Courtesy of Arbor et al., Molecular Psychiatry, 2019.]
In vitro biochemical studies found that APP mutations skew the endopeptidase cleavage site toward leucine 48, leading to more Aβ42, while PS1 mutations hobble the enzyme’s chopping capacity, leading to longer Aβ peptides (Chávez-Gutiérrez et al., 2012; Fernandez et al., 2014). Small studies using iPSC-derived neuronal lines from FAD mutation carriers have come to similar conclusions (Muratore et al., 2014; Sproul et al., 2014).
In the present paper, co-first authors Charles Arber and Jamie Toombs used a larger set of iPSC-derived neuronal lines. They obtained cells from two unrelated carriers of the APP-V717I mutation, five people who each carried a unique PS1 mutation, and five healthy controls for comparison. After the neurons reached maturity at around 100 days in culture, the researchers analyzed their secreted Aβ “peptidome” three times every two days, and once again at 200 days. Using immunoassays, they found large fluctuations in the absolute amount of Aβ peptides measured at different times, even within the same cell line. However, the relative amounts of one peptide to another held steady, so they focused on ratios throughout the study.
Across all familial AD mutations, the Aβ42:40 ratio was high, on average doubling that of non-AD controls. The researchers used the Aβ38:40 ratio to gauge the predominant γ-secretase endopeptidase site, finding that in the APP V717I, but not the PS1 mutant neurons, that ratio exceeded that of control cells. This suggested that the APP mutation shifts the endopeptidase cleavage toward the Leucine-48 site, which ultimately leads to Aβ42 and Aβ38. To assess how the mutations affected carboxypeptidase activity, the researchers measured Aβ42:38 ratios. They found higher ratios indicative of diminished trimming activity in all PS1 lines, while the APP lines had but modest increases. In all, the findings suggested that the APP mutations affect endopeptidase activity, triggering a chain of events that ultimately leads to Aβ42, while PS1 mutations hobble carboxypeptidase activity, leading to the release of multiple longer peptides including Aβ42.
What about Aβ43? Previous mouse studies indicated that the PS1 R278I mutation ups production of this longer peptide, and that it is even more amyloidogenic than Aβ42 (Jul 2011 news; Szaruga et al., 2017). Aβ43 has also been spotted in plaques of PS1 L435F mutation carriers, as well as in cell lines overexpressing various PS1 mutant proteins (Apr 2016 news). However, researchers had yet to detect Aβ43 in patient CSF, or secreted from patient-derived neurons. Using an immunoassay to measure the Aβ43:40 ratio, Arber et al. now report a marked increase in the culture medium of all PS1 mutant neurons except PS1-M146I, but not in the APP mutant neurons. Confirming mouse and in vitro studies, the Aβ43:40 ratio was elevated in PS1-R278I cells, which had the smallest increase over controls in the Aβ42:40 ratio.
At the AD/PD conference held last month in Lisbon, Portugal, Federica Perrone, who works with Christine van Broeckhoven at the VIB Antwerp Center for Molecular Neurology, presented her analysis of Aβ43 levels in CSF and virally transformed lymphocytes of APP and PS1/2 mutation carriers at her clinic. Perrone said that CSF Aβ43 was reduced with some presenilin and APP mutations, and that Aβ43 may help explain the mechanism of action of certain mutations that were deemed of unclear pathogenicity because they did not change Aβ42.
In toto, Arber et al.’s findings support the idea that regardless of which Aβ production line is initiated by the initial endopeptidase cleavage, mutations in PS1 slow carboxypeptidase activity, leading to more of the longer, amyloidogenic Aβ peptides.
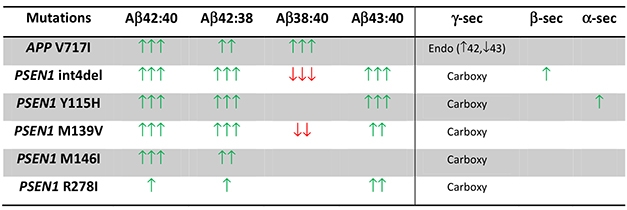
Revealing Ratios. The APP-V717I (London) mutation bumped up the Aβ38:40 ratio, indicating altered endopeptidase cleavage, while PS1 mutations upped Aβ42:38 and Aβ43:40 ratios, suggesting reduced carboxypeptidase activity. [Courtesy of Arber et al., Molecular Psychiatry, 2019.]
The researchers also used the iPSC-derived neurons to build three-dimensional organoids. Comprising masses of neurons and astrocytes, organoids are thought to better approximate AD pathological hallmarks than two-dimensional cultures (Raja et al., 2016). The organoids yielded similar ratios of the various Aβ peptides, supporting the idea that the mutations had consistent effects on the Aβ secretome.
Arber et al. also replicated their immunoassay findings with mass spectrometry, reporting similar ratios of the various peptides, except that Aβ43 was not detected via mass spectrometry. They did detect Aβ39, an alternative fragment generated from Aβ42, and found that its ratio to other Aβ peptides across the mutations was similar to that of Aβ38.
While all PS1 mutations impaired γ-secretase cleavage, they did so in different ways. Western blotting showed robust expression of full-length, immature PS1 in neurons harboring the PS1-R278I mutation, while PS1 expression was highly variable at different time points in the M139V and M146I mutant lines. This suggested that some PS1 mutations douse activity of the enzyme, and others prevent expression of its fully active form place.
“This study validates findings showing that different FAD mutations in PSEN1, PSEN2, and APP have differential effects on γ-secretase cleavage of APP,” commented Tracy Young-Pearse of Brigham and Women’s Hospital in Boston. “Importantly, this study uses human neurons derived from FAD mutation carriers to show effects on secreted Aβ.” Young-Pearse would like to know whether the mutations affect Aβ ratios differently depending on neuronal cell fate. Her work suggests that a given APP mutation can affect the Aβ repertoire differently in neuronal types that are selectively vulnerable than neurons that are resistant to Aβ accumulation (Muratore et al., 2017, altered Aβ peptides differently in rostral versus caudal-type neurons).
How did the spectrum of Aβ peptides released from iPSC-neurons relate to those secreted in the brain? To begin to address this, Arber et al. compared Aβ peptides in multiple biosamples from the same AD patient with an APP-V717I mutation. They saw similar ratios of Aβ38:40 in the CSF as in culture media and lysates from the iPSC-derived neurons, but were unable to detect Aβ38 in postmortem brain homogenate from this donor. The person’s Aβ42:40 ratio was lower in CSF than in the media from their cultured neurons or in the soluble fraction of brain homogenates, supporting the idea that amyloidogenic Aβ42 was sequestered in Aβ plaques. The Aβ42:40 ratio in neuronal lysates, which contain intracellular and membrane-bound Aβ peptides, was similar to that observed in CSF.

Brain to Culture. Researchers analyzed Aβ production in postmortem brain, three-dimensional organoids, and two-dimensional cortical neuron cultures from the same person carrying an APP V717I mutation. [Courtesy of Arber et al., Molecular Psychiatry, 2019.]
“Systematic comparison of different tissue/compartments from the same patient is a powerful approach to validate cellular models while exploring molecular mechanisms of pathogenesis in the patient’s brain,” noted Weiming Xia of Boston University, who also uses three-dimensional organoids to study FAD mutations. However, Xia noted that absolute measures of Aβ peptide concentrations would be more useful than ratios to draw quantitative comparisons between samples.
Co-senior author Henrik Zetterberg told Alzforum that he envisions using iPSC-derived neurons from FAD patients to screen for personalized treatments, such as γ-secretase modulators, that would most effectively restore the person’s Aβ repertoire to normal. Then they could monitor changes in the CSF in response to treatment. While this approach may not work once Aβ deposition has begun, thus skewing peptide ratios in the CSF, it could work for mutation carriers tested prior to Aβ deposition, he said.
To Daisuke Ito of Keio University in Tokyo, the study exemplifies how advances in stem cell technology have allowed investigation of patient-derived cells with sufficient reproducibility to facilitate drug discovery. “iPS cell drug discovery is taking off with clinical trials for other diseases,” he wrote. “Although bad news for clinical trials against AD continue, I really hope these findings and iPS cell lines in Arber’s study will lead to innovative drug discovery.”—Jessica Shugart
References
Mutations Citations
News Citations
- What’s Another Amino Acid? Aβ43 Drives Amyloid Pathology
- Pathogenic Presenilin Mutations Generate Aβ43
Mutation Position Table Citations
Paper Citations
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci. 2009 Oct 14;29(41):13042-52. PubMed.
- Fernandez MA, Biette KM, Dolios G, Seth D, Wang R, Wolfe MS. Transmembrane Substrate Determinants for γ-Secretase Processing of APP CTFβ. Biochemistry. 2016 Oct 11;55(40):5675-5688. Epub 2016 Sep 30 PubMed.
- Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014 Nov 7;289(45):31043-52. Epub 2014 Sep 19 PubMed.
- Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LN, Walsh DM, Selkoe DJ, Young-Pearse TL. The familial Alzheimer's disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet. 2014 Jul 1;23(13):3523-36. Epub 2014 Feb 12 PubMed.
- Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, Santa-Maria I, Zimmer M, Aubry S, Steele JW, Kahler DJ, Dranovsky A, Arancio O, Crary JF, Gandy S, Noggle SA. Characterization and molecular profiling of PSEN1 familial Alzheimer's disease iPSC-derived neural progenitors. PLoS One. 2014;9(1):e84547. Epub 2014 Jan 8 PubMed.
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
- Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, Tsai LH. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer's Disease Phenotypes. PLoS One. 2016;11(9):e0161969. Epub 2016 Sep 13 PubMed.
- Muratore CR, Zhou C, Liao M, Fernandez MA, Taylor WM, Lagomarsino VN, Pearse RV 2nd, Rice HC, Negri JM, He A, Srikanth P, Callahan DG, Shin T, Zhou M, Bennett DA, Noggle S, Love JC, Selkoe DJ, Young-Pearse TL. Cell-type Dependent Alzheimer's Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Reports. 2017 Dec 12;9(6):1868-1884. Epub 2017 Nov 16 PubMed.
Further Reading
Papers
- Moore S, Evans LD, Andersson T, Portelius E, Smith J, Dias TB, Saurat N, McGlade A, Kirwan P, Blennow K, Hardy J, Zetterberg H, Livesey FJ. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015 May 5;11(5):689-96. Epub 2015 Apr 23 PubMed.
- Hung CO, Livesey FJ. Altered γ-Secretase Processing of APP Disrupts Lysosome and Autophagosome Function in Monogenic Alzheimer's Disease. Cell Rep. 2018 Dec 26;25(13):3647-3660.e2. PubMed.
Primary Papers
- Arber C, Toombs J, Lovejoy C, Ryan NS, Paterson RW, Willumsen N, Gkanatsiou E, Portelius E, Blennow K, Heslegrave A, Schott JM, Hardy J, Lashley T, Fox NC, Zetterberg H, Wray S. Familial Alzheimer's disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol Psychiatry. 2020 Nov;25(11):2919-2931. Epub 2019 Apr 12 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.