Mutant Presenilin Knock-in Mice Mimic Knockouts, Stir Old Debate
Quick Links
Mutations in presenilin 1 (PS1) cause the majority of familial Alzheimer’s disease cases, but researchers still don’t quite agree on how the mutations do their dirty work. Most scientists accept that they partially inactivate PS1, which alters processing of amyloid precursor protein (APP) and leads to a build-up of toxic peptides such as Aβ42. Jie Shen of Brigham and Women’s Hospital and Raymond Kelleher of Massachusetts General Hospital, both in Boston, maintain that at least some mutations render PS1 nonfunctional and unable to attend to its many substrates, wreaking havoc on the brain independently of Aβ. The researchers claim that their newest study, published March 5 in Neuron, supports their hypothesis. Not all commentators agree.
PS1 is the catalytic subunit of γ-secretase, a multi-subunit, transmembrane protease. While β-secretase snips off the extracelluar domain of APP, PS1 attacks what's left from within the membrane in two major steps (see Takagi-Niidome et al., 2015). The first cut, known as ε-cleavage, removes the intracellular domain of APP and frees the transmembrane stretch for further trimming. PS1 then kicks into a processive mode known as γ-cleavage, in which it munches off roughly three amino acids at a time and produces multiple Aβ fragments, ranging in size from 46 to 38 amino acids (see Gu et al., 2001). These fragments are then shed from the cell surface.
Biochemical studies have shown that PS1 mutations causing autosomal-dominant familial AD (FAD) reduce this processing activity, resulting in an overabundance of incompletely cleaved products, including Aβ42 (see Chavez-Gutierrez et al., 2012, and Fernandez et al., 2014). In support of this idea, PS1 mutation carriers reportedly have higher levels of Aβ42, and a greater ratio of Aβ42 to Aβ40 in their cerebrospinal fluid than do non-carriers (see Jun 2013 news). While researchers agree that, biochemically, PS1 mutations result in a partial loss of enzymatic function, some researchers have referred to the resulting phenotype of elevated Aβ42 as a toxic gain of function. This semantic argument was active a decade ago but has since faded.
Now, Shen, Kelleher, and colleagues report that new knock-in mice expressing mutant human PS1 have essentially the same phenotype as older PS1 knockout animals. First author Dan Xia and colleagues chose two known AD mutations—L435F and C410Y. In vitro, both mutations inactivated PS1 (see Heilig et al., 2013). Like PS1 knockouts, mice expressing two copies of either mutated gene had profound neurodevelopmental defects and died soon after birth. The γ-secretase Aβ40, Aβ42, and Notch intracellular domain were undetectable in embryonic brain extracts. This suggested to the authors that the homozygous mutations abolished γ-secretase activity.
Animals with one normal and one mutant copy of the gene lived normal lifespans. Though they had less Aβ in the brain than wild-type mice, the heterozygotes had deficits in long- and short-term synaptic plasticity and performed poorly in cognitive tests of spatial memory. The researchers propose that loss of PS1 stifled neuronal function independently of Aβ in these mice.
To zero in on why Aβ levels were reduced in the heterozygotes, the researchers used the APP fragment C100 as a substrate to measure γ-secretase processing. They report that production of Aβ40 and Aβ42 dropped by half in brain extracts from the heterozygous mice compared to extracts from normal mice, and conclude that the mutated PS1 was unable to produce Aβ. Steady-state levels of both Aβ species were reduced in the animals’ brains; however, the ratio of Aβ42 to Aβ40 increased by about 15 percent.
How would the mutant PS1 affect amyloid deposition? To find out, the researchers crossed the L435F presenilin knock-in mice with J20 mice, which overexpress mutated human APP and start to develop plaques at around 5 months of age. Amounts of Aβ40 in insoluble brain extracts from 9-month-old crosses were slightly lower and the Aβ42/40 ratio slightly higher than in the J20 controls. The plaque burden in the cortex more than quadrupled in J20/PS1 mutant mice, as compared with J20 mice. The researchers attributed the elevated Aβ42/40 ratio and subsequent rise in plaque deposition to an alteration in the clearance, rather than the production, of Aβ42, though clearance rates were not measured.
Did expressing a mutant copy of presenilin affect neurons and cognition? The researchers measured this by comparing cognitive performance of L435F heterozygous mice to mice with two normal copies of PS1. They did these experiments on a presenilin 2 (PS2) knockout background, because PS2 has been reported to compensate for loss of PS1. Mice expressing L435F PS1 underperformed in the learning phase of the Morris water maze test and made more errors in finding hidden food in a radial-arm maze, suggesting that their hippocampal spatial memory was impaired. Hippocampal neurons in the heterozygous mice had reduced short- and long-term potentiation.
Shen and Kelleher proposed a model in which PS1 mutations prevent the enzyme from cleaving crucial substrates, triggering synaptic and cognitive impairment, and eventually neurodegeneration and dementia. The study did not name the crucial substrates or characterize the effect of PS1 mutations on their processing. In a parallel pathway, PS1 dysfunction also reduced Aβ production and elevated the ratio of Aβ42 to Aβ40, which eventually promoted amyloid accumulation.
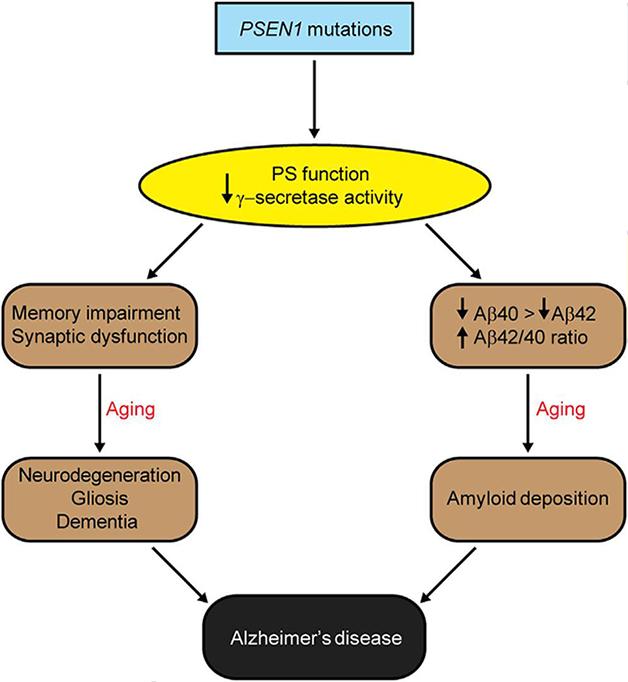
Loss of presenilin function could promote neurodegeneration and amyloid accumulation via two distinct pathways, according to Jie Shen and Raymond Kelleher. [Image courtesy of Xia et al., Neuron, 2015.]
Some commentators took issue with the conclusion that PS1 mutations completely inactivate the enzyme. Bart De Strooper of KU Leuven in Belgium and John Hardy of University College London wrote that the field has long accepted the idea that FAD-associated PS1 mutations diminish PS1 function (see Lewis et al., 2000, and De Strooper, 2007).
De Strooper commented that while known PS1 mutations vary in their ability to abolish ε-cleavage, they all have in common a malfunction in the processive γ-cleavage that produces Aβ peptides. He interpreted Kelleher and Shen’s findings as evidence of residual PS1 activity in the mutated copy, though the researchers detected no Aβ40 or Aβ42 in the homozygous knock-in mice. Michael Wolfe at Brigham and Women’s Hospital in Boston told Alzforum that the researchers might have detected longer Aβ peptides, such as Aβ43, if they had looked. Takaomi Saido’s group at RIKEN Brain Institute in Wako, Japan, reported that a PS1 knock-in mouse with the R278I mutation produced an abundance of amyloidogenic Aβ43, but not Aβ40 or Aβ42 (see Jul 2011 news). De Strooper and Lucia Chavez-Gutierrez, also at KU Leuven, wrote that in their hands, the R278I mutation actually does make Aβ, noting that the contrasting results may reflect different sensitivities of γ-secretase assays (see full comment below). Kelleher said they are currently investigating how the PS1 mutations affect levels of the longer peptides.
If loss of PS1 function causes AD, as Xia and colleague suggest, then why, several commentators asked, don’t people with acne inversa, a skin condition caused by mutations that wipe out γ-secretase expression, develop AD? The disorder is caused by heterozygous mutations that lead to haploinsufficiency of PS1 or other γ-secretase subunits (see Pink et al., 2013). Kelleher responded that, unlike the missense mutations that lead to AD, the acne inversa mutations ablate the γ-secretase complex. AD only happens, he proposed, when the single point mutations in PS1 that abolish protease activity leave the γ-secretase complex to fulfill other unknown roles that would alter the course of disease. PS1 has been touted as a calcium leak channel in the endoplasmic reticulum (see Jun 2010 news).
Kelleher and Shen pointed out that they chose two mutations that were particularly adept at derailing PS1 function. Of the 200 or so presenilin mutations linked to AD, it is possible that some promote partial loss of function while others wipe it out, they said. They conclude that the importance of their findings is that a complete loss of PS1 protease function in one copy of PS1 triggers deficits in cognitive and synaptic function in the heterozygous mice. They propose that the neuronal and cognitive deficits elicited by this loss of function were more proximal to the AD phenotype than the parallel rise in Aβ42/40 ratio. However, the relative contribution of Aβ accumulation to the AD phenotype could vary in humans and between different mutations, they say.—Jessica Shugart
References
Alzpedia Citations
News Citations
- In Familial AD, Aβ Production Up, Clearance Down
- What’s Another Amino Acid? Aβ43 Drives Amyloid Pathology
- Perplexing Presenilins: New Evidence for Calcium Leak Channels
Mutations Citations
Research Models Citations
Paper Citations
- Takagi-Niidome S, Sasaki T, Osawa S, Sato T, Morishima K, Cai T, Iwatsubo T, Tomita T. Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of γ-secretase. J Neurosci. 2015 Feb 11;35(6):2646-56. PubMed.
- Gu Y, Misonou H, Sato T, Dohmae N, Takio K, Ihara Y. Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem. 2001 Sep 21;276(38):35235-8. PubMed.
- Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ. J Biol Chem. 2014 Nov 7;289(45):31043-52. Epub 2014 Sep 19 PubMed.
- Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ. Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Aβ production. J Neurosci. 2013 Jul 10;33(28):11606-17. PubMed.
- Lewis PA, Perez-Tur J, Golde TE, Hardy J. The presenilin 1 C92S mutation increases abeta 42 production. Biochem Biophys Res Commun. 2000 Oct 14;277(1):261-3. PubMed.
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007 Feb;8(2):141-6. PubMed.
- Pink AE, Simpson MA, Desai N, Trembath RC, Barker JN. γ-Secretase mutations in hidradenitis suppurativa: new insights into disease pathogenesis. J Invest Dermatol. 2013 Mar;133(3):601-7. Epub 2012 Oct 25 PubMed.
Other Citations
Further Reading
No Available Further Reading
Primary Papers
- Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ 3rd. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. Neuron. 2015 Mar 4;85(5):967-81. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UK Dementia Research Institute@UCL and VIB@KuLeuven
In 2007, I wrote: “From a genetic point of view, these mutations seem to result in a gain of toxic function; however, biochemically, they result in a partial loss of function in the γ-secretase complex, which affects several downstream signaling pathways. Consequently, the current genetic terminology is misleading. In fact, the available data indicate that several clinical presenilin mutations also lead to a decrease in amyloid precursor protein derived amyloid β-peptide generation, further implying that presenilin mutations are indeed loss-of-function mutations” (De Strooper, 2007). This statement was based on the review of work of at least five different groups, including mine, and performed in cells, C. elegans, and mice, and all published in the 10 years before.…More
The remaining question was, what aspect of the loss of function of the presenilin mutations causes Alzheimer's disease? Shen and Kelleher seem to argue that abnormal Aβ generation is an epiphenomenon. However, our previous work (Chávez-Gutiérrez et al., 2012) clearly points out that the consistent feature of presenilin mutations is an effect on the carboxypeptidase-like activity of γ-secretase, and thus on Aβ generation. The effects of the pathogenic mutations on other aspects of presenilin function are variable. So while in my view these other aspects of presenilin dysfunction might contribute in variable ways to the disease phenotype, they are not essential to cause AD.
Indeed, if the hypothesis of Shen and Kelleher were right, one would expect to find real loss-of-function mutations of γ-secretase in FAD. In contrast, however, every FAD mutation identified is a missense mutation limited to the catalytic subunit presenilin. Real loss-of-function mutations of γ-secretase that, indeed, have also been found in humans cause the skin disease hidradenitis suppurativa (Wang et al., 2010; Chen, 2015), not familial AD.
References:
De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007 Feb;8(2):141-6. PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, Chen M, Sun M, Shen Y, Zhang X. Gamma-secretase gene mutations in familial acne inversa. Science. 2010 Nov 19;330(6007):1065. PubMed.
Chen S, Mattei P, You J, Sobreira NL, Hinds GA. γ-Secretase Mutation in an African American Family With Hidradenitis Suppurativa. JAMA Dermatol. 2015 Jun;151(6):668-70. PubMed.
View all comments by Bart De StrooperInstitute of Neurology, UCL
It is vexing to be set up as the straw man suggesting that PSEN1 mutations act as a gain of function. In fact, not only did our quoted review (Hardy and Selkoe, 2002) not say this anywhere, but I have always thought that PSEN1 mutations were a subtle loss of function. Indeed, in 2000 we assessed the effects of the PSEN1 C92S mutation, which is the homologue of the sel12 loss-of-function mutation, and showed that it behaved like an Alzheimer-causing mutation. This genetic experiment suggested (if one accepts the premise of homology) that PSEN1 mutations are loss of function (Lewis et al., 2000). Our abstract from that paper states:…More
"To investigate the effects of the loss of function sel12 mutation on Aβ production in mammalian cells, we analyzed Aβ production in transfected H4 neuroglioma cells expressing the PS1 homologue of the sel12 C60S mutant, PS1 C92S. This analysis revealed that PS1 C92S increased Aβ42 levels in a similar fashion to other pathogenic Alzheimer's disease (AD) PS1 mutations. Significantly, the PS1 C92S mutation has recently been identified as the pathogenic mutation in an Italian family with FAD. Thus, placing a mutation that results in loss of function in C. elegans into a context whereby its effect on mammalian cells can be evaluated suggests that all FAD-linked PS1 mutants result in increased Aβ42 production through a partial loss of function mechanism."
These genetic experiments have, however, been rendered immaterial by the more recent papers of Chávez-Gutiérrez et al., who have shown exactly by what mechanism presenilin mutations alter APP metabolism through partial loss of function (Chávez-Gutiérrez et al., 2012). The work by Xia and colleagues is thorough in execution, but not in referencing the literature.
References:
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353-6. PubMed.
Lewis PA, Perez-Tur J, Golde TE, Hardy J. The presenilin 1 C92S mutation increases abeta 42 production. Biochem Biophys Res Commun. 2000 Oct 14;277(1):261-3. PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
View all comments by John HardyK.U.Leuven and V.I.B.
How mutations in PSEN1 cause AD is definitely an unresolved fundamental question in the AD field that Shen, Kelleher, and colleagues bring into discussion.
The authors propose that pathogenic PSEN mutations cause AD via a loss of essential PSEN/γ-secretase function and “loss of PS function conferred by PSEN mutations promotes amyloid pathology by inhibiting γ-secretase activity (meaning wild-type enzyme), decreasing Aβ production, and increasing the Aβ42/40 ratio due to a greater relative reduction in Aβ40 accumulation.” As summarized by Karen H. Ashe, a major implication of “The Presenilin Hypothesis” is that changes in Aβ peptides are not the key pathogenic trigger for AD.…More
Shen and Kelleher base their model on two FAD-linked PSEN1 mutations (L435F and C410Y), which, according to their data, are completely inactive (Heilig et al., 2013; Xia et al., 2015). These mutants are able to form the γ-secretase complex but interfere with the activation of PSEN1. Interestingly, Shen and colleagues described a third pathogenic mutant (R278I-PSEN1) that causes a very similar effect on the γ-secretase. In their 2013 paper they report that “NTF and CTF were not detectable for R278I and L435F, and were reduced to <10% of WT levels for C410Y.” In addition, “For the L435F and R278I mutations, AICD production was at or below the limit of detection, comparable with the effect of the catalytically inactive D257A mutation, indicating complete inactivation of APP proteolysis.” And, “The levels of Aβ40 and Aβ42 produced by PS1 bearing the L435F and R278I mutations were at or below the limit of detection, indicating virtually complete loss of proteolytic activity.” While we do not have kinetic data on the L435F and C410Y PSEN1 mutants, we have analyzed the R278I PSEN1 mutation. In our hands, the R278I mutant is active. It reduces substantially the activation of PSEN1 (measured as the generation of N- and C-terminal fragments) and consequently activity is very low; however, it is detectable and quantifiable. Remarkably, we found an increment in Aβ42/Aβ40 for the R278I mutant, relative to the wild-type enzyme. Most likely, our and Shen/Kelleher's results conflict because of differences in the sensitivity of the respective activity assays for γ-secretase.
I would also like to draw attention to the fact that inactivation of PSEN/γ-secretase is not always seen in FAD-linked PSEN mutations. We performed a rigorous kinetic analysis and showed that some pathological PSEN mutations may inactivate PSEN/γ-secretase (endopeptidase/ε cleavage) to different extents, but others are as efficient as the wild-type PSEN/γ-secretase in the endopeptidase/ε cleavage (Chavez-Gutierrez et al, 2012). Significantly, what seems to be consistent across pathological PSEN mutants—and independent of their effect on the ε cleavage—is impaired efficiency of the carboxypeptidase-like activity (γ-cleavages), which translates into production of longer N-terminal fragments from γ-secretase substrates, meaning longer Aβ peptides from APP and likely longer N-terminal peptides from other substrates (Chavez-Gutierrez et al., 2012).
I would like to finish by stressing that more than 200 mutations in PSEN1 are pathogenic and importantly, their location in the primary structure of PSEN1, the nature of the substitution, and their endopeptidase (ε) activity levels vary substantially. This report studies a particular subset of pathogenic mutations and a pertinent question is if the observations by Shen, Kelleher, and colleagues can be extrapolated to a general mechanism in FAD.
References:
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ. Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Aβ production. J Neurosci. 2013 Jul 10;33(28):11606-17. PubMed.
Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ 3rd. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. Neuron. 2015 Mar 4;85(5):967-81. PubMed.
View all comments by Lucia Chavez-GutierrezThe University of Tokyo
The effect of familial Alzheimer’s disease (FAD)-linked mutations in presenilin genes has been enigmatic. However, deposition of Aβ42 in the brains of FAD patients is more severe than that in sporadic AD patients (Mann et al., 1996; Mann et al., 1997). Here Xia et al. reported that homozygous L435F and C410Y knock-in mutations in PS1 resulted in complete loss of γ-secretase activity. Heterozygous mice, which would mimic the situation in FAD patients, showed increased Aβ42:Aβ40 ratio and accelerated amyloid deposition, although total Aβ production was significantly reduced. In fact, almost the same results already have been reported by Saito et al. with different mutations (Saito et al., 2011). Thus, proportion of aggregation-prone Aβ species (i.e., longer Aβ), not quantity, is critical to amyloid deposition, and FAD-linked mutations in presenilin caused a loss-of-function effect on the intrinsic proteolytic activity of presenilins, as suggested in enzymatic analysis (De Strooper, 2007; Wolfe, 2007; Shimojo et al., 2008; and Chávez-Gutiérrez et al., 2012).…More
On top of this, Xia et al. now report that FAD-linked mutations caused memory impairment and neurodegeneration in a Psen2 null background. In combination with the previous reports by the same group, these results suggested that FAD-linked mutation caused defects on the processing of several γ-secretase substrates, presumably including Notch. However, it remains unclear that this is the case in patients, since they still carry normal PS2 genes, and a single wild-type PS1 allele. Longitudinal analysis of young presymptomatic FAD-linked carriers such as those carried out in DIAN or API should provide important information on the effect of PS1 mutations on neuronal function and degeneration (Bateman et al., 2012; Reiman et al., 2012). In addition, loss-of-function mutations, including nonsense or frameshift mutations, in γ-secretase genes have been identified in patients with hidradenitis suppurativa (acne inversa) (Wang et al., 2010), although no further information on the patients has emerged as yet. Nevertheless, these data also strengthen recent hypotheses that modulation, but not inhibition, of γ-secretase activity by small compounds (i.e., γ-secretase modulators) is one therapeutic approach for AD (Chávez-Gutiérrez et al., 2012; Takeo et al., 2014; Dang et al., 2015).
References:
Mann DM, Iwatsubo T, Cairns NJ, Lantos PL, Nochlin D, Sumi SM, Bird TD, Poorkaj P, Hardy J, Hutton M, Prihar G, Crook R, Rossor MN, Haltia M. Amyloid beta protein (Abeta) deposition in chromosome 14-linked Alzheimer's disease: predominance of Abeta42(43). Ann Neurol. 1996 Aug;40(2):149-56. PubMed.
Mann DM, Iwatsubo T, Nochlin D, Sumi SM, Levy-Lahad E, Bird TD. Amyloid (Abeta) deposition in chromosome 1-linked Alzheimer's disease: the Volga German families. Ann Neurol. 1997 Jan;41(1):52-7. PubMed.
Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J, Nishimura M, Iwata N, Van Broeckhoven C, Ihara Y, Saido TC. Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci. 2011 Aug;14(8):1023-32. PubMed.
De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007 Feb;8(2):141-6. PubMed.
Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007 Feb;8(2):136-40. PubMed.
Shimojo M, Sahara N, Mizoroki T, Funamoto S, Morishima-Kawashima M, Kudo T, Takeda M, Ihara Y, Ichinose H, Takashima A. Enzymatic characteristics of I213T mutant presenilin-1/gamma-secretase in cell models and knock-in mouse brains: familial Alzheimer disease-linked mutation impairs gamma-site cleavage of amyloid precursor protein C-terminal fragment beta. J Biol Chem. 2008 Jun 13;283(24):16488-96. Epub 2008 Apr 21 PubMed.
Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JB, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012 Dec;11(12):1048-56. PubMed.
Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, Chen M, Sun M, Shen Y, Zhang X. Gamma-secretase gene mutations in familial acne inversa. Science. 2010 Nov 19;330(6007):1065. PubMed.
Takeo K, Tanimura S, Shinoda T, Osawa S, Zahariev IK, Takegami N, Ishizuka-Katsura Y, Shinya N, Takagi-Niidome S, Tominaga A, Ohsawa N, Kimura-Someya T, Shirouzu M, Yokoshima S, Yokoyama S, Fukuyama T, Tomita T, Iwatsubo T. Allosteric regulation of γ-secretase activity by a phenylimidazole-type γ-secretase modulator. Proc Natl Acad Sci U S A. 2014 Jul 22;111(29):10544-9. Epub 2014 Jul 9 PubMed.
Dang S, Wu S, Wang J, Li H, Huang M, He W, Li YM, Wong CC, Shi Y. Cleavage of amyloid precursor protein by an archaeal presenilin homologue PSH. Proc Natl Acad Sci U S A. 2015 Mar 17;112(11):3344-9. Epub 2015 Mar 2 PubMed.
View all comments by Taisuke TomitaUT Southwestern Medical Center at Dallas
A number of presenilin- and APP-based models of familial Alzheimer's disease (FAD) have been previously generated, but most of these rely on transgenic overexpression. An APP-knock-in (KI) mouse was generated recently at RIKEN (Saito et al., 2014). Only a single KI model of a mutated PS1 (M146V) has been available so far (Guo et al., 1999). Now, Xia et al. report the characterization of two novel PS-KI mouse models—C410Y and L435F. Surprisingly, the phenotypes of these mice are much more severe than the phenotype of the PS1-M146V KI mouse.…More
In our previous studies we suggested that the M146V mutation primarily acts by disrupting the endoplasmic reticulum (ER) Ca2+ leak pathway (Tu et al., 2006; Zhang et al., 2010) and that the C410Y mutation acts by affecting γ-secretase activity (Nelson et al., 2010). We also argued that mutations such as M146V correlate with dense core plaques and mutations such as C410Y correlate with cotton-wool plaques activity (Nelson et al., 2010). We have not analyzed the L435F mutation in our Ca2+ signaling studies. Comparing PS1-M146V KI and PS1-C410Y KI mouse phenotypes is very informative, as it opens the possibility to compare the contribution of ER Ca2+ dysregulation and amyloid dysregulation pathways in AD. In recent studies, we demonstrated loss of mushroom synaptic spines in PS1-M146V KI neurons as a result of dysregulation of the neuronal store-operated Ca2+ entry (nSOC) pathway (Sun et al., 2014). It will be very interesting to determine if spine loss and nSOC pathway dysregulation also occurs in PS1-C410Y KI mice.
References:
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014 May;17(5):661-3. Epub 2014 Apr 13 PubMed.
Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999 Jan;5(1):101-6. PubMed.
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006 Sep 8;126(5):981-93. PubMed.
Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I. Role of presenilins in neuronal calcium homeostasis. J Neurosci. 2010 Jun 23;30(25):8566-80. PubMed.
Nelson O, Supnet C, Liu H, Bezprozvanny I. Familial Alzheimer's disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J Alzheimers Dis. 2010;21(3):781-93. PubMed.
Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL 3rd, Bezprozvanny I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron. 2014 Apr 2;82(1):79-93. PubMed.
View all comments by Ilya BezprozvannyBrigham and Women's Hospital, Harvard Medical School
The debate seems to miss the point—our genetic studies suggest that loss of presenilin’s essential functions is a more potent cause of neurodegeneration than accumulation of Aβ species. This conclusion is based on the findings that 1) loss of presenilin function leads to neurodegeneration; 2) FAD mutations in the presenilin genes are loss of function mutations; 3) numerous APP transgenic mice overproducing massive amounts of Aβ failed to develop neurodegeneration. The mouse results are crystal clear, and human clinical trials aimed at reducing Aβ have so far been uniformly disappointing. Based on our mouse data I cautioned 12 years ago against the use of γ-secretase inhibitors as an AD therapy, and unfortunately this concern was realized when GSIs worsened the cognition of human patients in phase III clinical trials. Further human data are forthcoming from prevention trials currently being conducted with presenilin mutation carriers in Colombia and elsewhere. Time will soon tell whether removing Aβ will be sufficient to prevent neurodegeneration and dementia in FAD.…More
I’m glad that the loss of function idea has steadily gained popularity. I still remember the days when this idea was considered "radical" and a "lighting rod." If truly everyone had thought PSEN mutations cause FAD via a loss of function mechanism, it would have been inconceivable for big pharma to spend billions on developing γ-secretase inhibitors.
Massachusetts General Hospital
We appreciate the comments about our paper and the resulting discussion of disease mechanisms.
1. Hardy and Selkoe (2002) ascribed FAD pathogenicity to an enhancement (i.e., a gain) of presenilin function, stating that “... AD-causing mutations in PS1 and PS2 enhance the processing of APP to form amyloidogenic Aβ ...” Indeed, intense interest in the therapeutic potential of γ-secretase inhibitors—prior to their recent clinical trial failures—was justified by the view that all FAD mutations increase the production of Aβ42 (Annaert and De Strooper, 2002; Hardy and Selkoe, 2002). If investigators in the field truly believed that loss of presenilin function was responsible for disease pathogenesis, it would have made little sense for them to advocate therapeutic approaches, i.e. γ-secretase inhibitors, that would exacerbate this loss of function. Conversely, we showed that loss of presenilin function reproduced AD-like phenotypes, and raised concern about the potential clinical hazards of γ-secretase inhibition, more than a decade ago (Saura et al., 2004).…More
2. The assertion that simple loss-of-function mutations are compatible with the presenilin hypothesis is inaccurate. Rather, we have proposed and provided experimental evidence that pathogenic PSEN1 mutations act through a dominant-negative rather than a simple loss-of-function mechanism (Heilig et al., 2013; Kelleher and Shen, 2010; Shen and Kelleher, 2007). Dominant-negative action provides the most parsimonious explanation reconciling the dominant inheritance and missense nature of PSEN mutations in FAD with their allelic heterogeneity and evidence that they cause a loss of protein function. Dominant-negative action may also explain how a single mutant PSEN1 allele can lead to a global impairment of presenilin and γ-secretase activity. Notably, recent work from the Shen lab shows that partial loss of presenilin function is sufficient to trigger neurodegeneration in the cerebral cortex (Watanabe et al., 2014). Dominant-negative effects of PSEN1 mutations on wild-type PS1 and/or PS2 activity may be brain-specific, just as Aβ deposition is brain-specific, and PSEN1 mRNA is subject to brain-specific splicing (Watanabe et al., 2012).
3. The relevance of mutations identified in familial acne inversa (FAI) to disease mechanisms in FAD is questionable. It remains uncertain that FAI and FAD are truly allelic disorders as initially suggested by Wang et al. (2010). Whereas there have been numerous independent replications of NCSTN (nicastrin) and PSENEN (Pen-2) mutations in FAI, the reported association of PSEN1 with FAI has not been replicated and remains limited to a single reported family. Mutations in NCSTN and PSENEN have not been associated with FAD. The single reported family with a PSEN1 mutation in FAI includes only a single affected individual over age 50, and the paper states that “the diagnosis of AD or dementia was excluded by a dermatologist [!?] based on the lack of recognizable symptoms or signs,” which falls well short of a rigorous clinical assessment.
4. The Lewis et al. (2000) publication cited by John Hardy has no bearing on the question of whether pathogenic PSEN1 mutations cause a gain or a loss of function. The paper presents only measurements of the Aβ42/Aβ40 ratio for wild-type or mutant PS1 expressed in a glioma cell line. The actual levels of Aβ40 and Aβ42 are not presented, nor is any other measure of γ-secretase activity. Based on the data in the paper, it is impossible to discern whether the observed changes in the Aβ42/Aβ40 ratio are due to a gain or a loss of function. The authors’ inferences regarding PS1 function are drawn from Iva Greenwald’s previous demonstration that the corresponding sel12 mutation, as well as a series of FAD mutations, reduces biological activity in C. elegans (Levitan et al., 1996; Levitan and Greenwald, 1995). In another previously published study (which was not cited by Lewis et al.), Jie Shen and Bruce Yankner provided the first evidence that FAD mutations impair PS1 function and γ-secretase activity in mammalian cells (Song et al., 1999). Therefore, we cited the papers by Levitan et al. (1996) and Song et al. (1999), but there was no reason to cite the paper by Lewis et al. (2000).
5. We are not suggesting that PSEN1 mutations cause FAD through an Aβ-independent mechanism (see our mechanistic model reproduced above). On the contrary, our findings don’t specifically address this issue but merely raise it as a possibility. Our results suggest that loss of PS function may be more proximate to disease phenotypes, particularly neurodegeneration. We have previously proposed, for example, that longer forms of Aβ could act as endogenous inhibitors of γ-secretase (Shen and Kelleher, 2007). It remains to be determined whether APP or Aβ is required for the impairments in synaptic and cognitive function and neuronal survival caused by the pathogenic PSEN1 mutations in our study.
References:
Annaert W, De Strooper B. A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol. 2002;18:25-51. PubMed.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353-6. PubMed.
Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004 Apr 8;42(1):23-36. PubMed.
Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ. Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Aβ production. J Neurosci. 2013 Jul 10;33(28):11606-17. PubMed.
Kelleher RJ, Shen J. Genetics. Gamma-secretase and human disease. Science. 2010 Nov 19;330(6007):1055-6. PubMed.
Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007 Jan 9;104(2):403-9. PubMed.
Watanabe H, Iqbal M, Zheng J, Wines-Samuelson M, Shen J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J Neurosci. 2014 Nov 26;34(48):15912-22. PubMed.
Watanabe H, Xia D, Kanekiyo T, Kelleher RJ, Shen J. Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. J Neurosci. 2012 Apr 11;32(15):5085-96. PubMed.
Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, Chen M, Sun M, Shen Y, Zhang X. Gamma-secretase gene mutations in familial acne inversa. Science. 2010 Nov 19;330(6007):1065. PubMed.
Lewis PA, Perez-Tur J, Golde TE, Hardy J. The presenilin 1 C92S mutation increases abeta 42 production. Biochem Biophys Res Commun. 2000 Oct 14;277(1):261-3. PubMed.
Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1996 Dec 10;93(25):14940-4. PubMed.
Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995 Sep 28;377(6547):351-4. PubMed.
Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci U S A. 1999 Jun 8;96(12):6959-63. PubMed.
The University of Adelaide
Xia and co-authors should be commended for examining the function of familial AD (FAD) mutations in PSEN1 in a very physiologically relevant model system. Their observation of loss of γ-secretase activity for FAD mutations engineered into in mouse Psen1 is provocative and raises many questions regarding the true mechanism by which mutations in human PSEN1 cause FAD.
A simple loss of γ-secretase activity model cannot explain the available data. As others have pointed out, the result of reduced γ-secretase activity appears to be the disease acne inversa and it is remarkable that FAD mutations occur only in PSEN1 and PSEN2 rather than in other γ-secretase complex members. Xia et al.’s results indicate that an FAD mutation of PSEN1 can cause complete loss of γ-secretase activity of the mutated protein product. Unless there is interaction between mutant and normal PSEN molecules within γ-secretase complexes, it is unclear how carboxypeptidase-like activity but not epsilon-cleavage activity could be affected in cells heterozygous for these mutations. As I understand the field, current thinking favors independent action of individual γ-secretase complexes although, apparently, there is possible structural evidence of tetrameric organisation (Li et al., 2013) as well as molecular genetic evidence for interaction between PSEN1 and PSEN2 (e.g., Nornes et al., 2008).…More
It is fascinating to note that one function of PSEN1 that is apparently independent of other members of the γ-secretase complex is the disputed role of the PSEN1 holoprotein in delivery of the v-ATPase V0a1 subunit to lysosomes (Lee et al., 2010). Lee et al. showed that lysosomal acidification did not appear to be affected by chemical inhibition of γ-secretase activity or by loss of the γ-secretase complex component NICASTRIN. If FAD mutant PSEN1 holoprotein molecules could dominantly interfere with PSEN1’s role in lysosome acidification, it would explain why all the many FAD mutations of PSEN1 maintain the open reading frame while nonsense or frameshift mutations do not appear to cause the disease (but can cause, e.g., acne inversa by loss of γ-secretase activity).
More evidence that loss of γ-secretase activity does not explain FAD comes from another study by Shen and Kelleher (Watanabe et al., 2012) examining a mouse model of the human PSEN1 mutation G183V that is suspected of causing Pick disease but not FAD. There, the mutation caused brain-specific aberrant splicing leading to nonsense mediated decay of the mutant transcripts. The reduced non-mutant mouse Psen1 transcript levels lead to reduced γ-secretase activity. (However, note that we have argued that the Pick disease-specific phenotype of the G183V mutation may be due to translation of the residual levels of the aberrantly spliced PSEN1 transcripts in the brain giving truncated peptides with dominant and γ-secretase substrate-specific activity [Newman et al., 2014].)
I agree with Shen’s comment that loss of PSEN1’s essential functions may be a “more potent cause of neurodegeneration than accumulation of Aβ species.” Xia et al.’s heterozygous loss-of-function mouse model shows how both Aβ40 and Aβ42 production is decreased and it is intuitively difficult to believe that such a minor change in the ratio of these two species could be pathological—especially when one considers that Aβ levels fluctuate diurnally and in response to oxygen levels.
References:
Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013 Jan 3;493(7430):56-61. PubMed.
Nornes S, Newman M, Verdile G, Wells S, Stoick-Cooper CL, Tucker B, Frederich-Sleptsova I, Martins R, Lardelli M. Interference with splicing of Presenilin transcripts has potent dominant negative effects on Presenilin activity. Hum Mol Genet. 2008 Feb 1;17(3):402-12. PubMed.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146-58. PubMed.
Watanabe H, Xia D, Kanekiyo T, Kelleher RJ, Shen J. Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. J Neurosci. 2012 Apr 11;32(15):5085-96. PubMed.
Newman M, Wilson L, Verdile G, Lim A, Khan I, Moussavi Nik SH, Pursglove S, Chapman G, Martins RN, Lardelli M. Differential, dominant activation and inhibition of Notch signalling and APP cleavage by truncations of PSEN1 in human disease. Hum Mol Genet. 2013 Oct 6; PubMed.
Institute of Neurology, UCL
In my scientific career, I have been wrong about many things and I take no shame in being wrong. However, I have not been wrong about whether pathogenic presenilin mutations are gain- or loss-of-function (and as De Strooper comments, these genetic definitions are moot now we know precisely, for APP at least, what change in function occurs). In the 1990s, with colleagues, we tested as best we could, in two experiments, whether the mutations were gain- or loss-of-function and in both we came to the conclusion they had an element of loss of function. One is the Lewis et al. paper cited already, where we showed that a mutation known to be a loss-of-function mutation behaved as an Alzheimer-causing mutation in its APP processing properties. The other was Refolo et al. 1999, in which we used antisense to knock down PSEN1. I quote the entire abstract because it captures my view at that time:…More
"Autosomal dominant mutations in the presenilin 1 (PS1) gene are associated with familial, early-onset Alzheimer's disease. Although the pathogenic mechanism of these mutations is unclear, their common feature is that they lead to an increased concentration of amyloid beta-peptide (Abeta) 42 in the plasma of early-onset patients, in the conditioned media of transfected cells, and in the brains of transgenic mice that overexpress mutant PS1. To address the mechanism(s) by which the pathogenic PS1 mutations increase Abeta42, we constructed human cell lines expressing a doxycyclin (dox)-inducible antisense PS1 RNA and measured its effects on the levels of PS1, amyloid precursor protein (APP), and Abeta. In time course experiments, we observed a statistically significant (p = 0.0038) more than twofold elevation in secreted Abeta42 as early as 12 days after addition of dox. This correlated with an 80% decrease in the 46-kDa PS1 holoprotein and a 30% decrease in the 26-kDa N-terminal fragment (NTF). Furthermore, there was a significant fivefold (p = 0.002) increase in Abeta42 after 14-day dox treatment; this correlated with a >90% decrease in PS1 holoprotein and 60% decrease in NTF. At no time point did we observe significant changes in Abeta40, APP holoprotein, presenilin 2, or tubulin. Ten days after the removal of dox, we observed a return to constitutive levels for Abeta42, PS1 holoprotein, and NTF. These results suggest that in human cell lines, the reduction of normal PS1 activity results in the increased production of Abeta42. Furthermore, our results are consistent with a loss of function or dominant negative mechanism for the pathogenic PS1 mutations."
Clearly, I did not believe at that time that presenilin mutations were a gain-of-function, and if anyone else did think that, then Shen and Kelleher should cite their papers that say so and not incorrectly assign those views to me. This is a slippery debate, however, which hinges on how the word “function” is defined. I do think that production of more Aβ42/43 is the key to their pathogenicity.
References:
Refolo LM, Eckman C, Prada CM, Yager D, Sambamurti K, Mehta N, Hardy J, Younkin SG. Antisense-induced reduction of presenilin 1 expression selectively increases the production of amyloid beta42 in transfected cells. J Neurochem. 1999 Dec;73(6):2383-8. PubMed.
Medical University of South Carolina
I would like to point out that loss and gain of function are really definitions dependent on the phenotype being assessed. We have always held that presenilin mutations represented an impairment of γ-secretase activity, and recognized a parallel between several γ-secretase inhibitors that actually increase Aβ42 at low doses and antisense RNA that also increases Aβ42, as indicated by John Hardy above. However, the system used in the tests also matters a lot. More recently, we found that low doses of a γ-secretase inhibitor, paradoxically, increased Aβ1-40 and Aβ1-42 simultaneously and equally in SH-SY5Y cells, but only reduced Aβ in Chinese hamster ovary cells. We also showed that the paradoxical stimulation of Aβ40 was prevented by inhibition of ECE, an Aβ-degrading activity (Barnwell et al., 2014). Moreover, overexpression of all γ-secretase subunits in HEK293 cells increased the yield of total Aβ, but simultaneously reduced the Aβ42/40 ratio showing that stimulation of γ-secretase does the opposite of FAD mutant presenilins (Marlow et al., 2003).…More
References:
Marlow L, Canet RM, Haugabook SJ, Hardy JA, Lahiri DK, Sambamurti K. APH1, PEN2, and Nicastrin increase Abeta levels and gamma-secretase activity. Biochem Biophys Res Commun. 2003 Jun 6;305(3):502-9. PubMed.
Peking Union Medical College
Dr. Baoxi Wang at Peking Union Medical College, the first author of our Science paper (Wang et al., 2010), has performed evaluations, with neurologists, on all available affected individuals in the six studied families and additional families with acne inversa. No confirmed diagnosis of AD could be made in the individuals affected with acne inversa.
References:
Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, Chen M, Sun M, Shen Y, Zhang X. Gamma-secretase gene mutations in familial acne inversa. Science. 2010 Nov 19;330(6007):1065. PubMed.
Make a Comment
To make a comment you must login or register.