2022 Was Good to Basic Research, Too
Quick Links
Part 2 of 2
From the all-important treatment perspective, 2022 ended on a high note, with Phase 3 data from lecanemab setting up its FDA approval (see Part 1 of this review). There was plenty of action on the basic research front, too, from cryo-EM structures of amyloid fibrils, to the discovery of new protective ApoE variants, and a better understanding of the cellular shifts that underly this disorder. A lot happened, so we’ve summarized below in six sections: proteopathy; microglia; astrocytes; cell biology; genetics; and risk factors/lifestyle.
Proteopathy
Plaque Biology
Oligomers, protofibrils, diffusible fibrils—whatever the name, scientists have been searching for these elusive forms of Aβ for decades. November brought news that they can be coaxed from brain tissue by steeping it in buffer. The simplicity of the method could speed up research on these neurotoxic species, which bound lecanemab and twisted into the same core structures as do fibrils isolated from amyloid plaques.
The latter come in two types, cryo-EM showed, both comprising distinct S-shaped protofibrils of Aβ42.

Take Two. Type I (left) Aβ protofilaments predominate in sporadic AD, Type II (right) in familial AD and APP knock-in mice. [Courtesy of Yang et al., Science, 2022.]
Because the fibrils studied contained no Aβ40 or shorter species, it's unclear how they influence their structure. This question acquired new urgency last year because Aβ37 and Aβ38 fibrillize more slowly than do Aβ40 and Aβ42. When mixed, the shorter peptides “frustrate” fibrillogenesis of the longer ones. These are no longer just basic science findings. That shorter Aβ peptides retard AD pathology is reflected in cognition holding more steady in people whose CSF Aβ38 concentrations were high. The ratios of short to long Aβ peptides correlated better with age of disease onset than did the Aβ42/40 ratio.
How do these fibrils build plaques in the brain? And what causes the memory problems? In 2022, scientists focused on intraneuronal compartments and processes to answer these old questions. For example, several groups confirmed that Aβ accumulates in neuronal lysosomes when they become less acidic. Aβ-laden vesicles swell, infiltrating membranes of the Golgi apparatus and the endoplasmic reticulum. As the Aβ concentration rises, the peptides coalesce into perinuclear rosettes dubbed PANTHOS, aka poisonous anthos, from the Greek for flower. These seed fibrils and leak proteases into the cytoplasm. The theory is that these stressed neurons burst, leaving behind the Aβ rosettes, which coalesce into plaques.

Deadly Flower. The PANTHOS structure, comprising a nuclear center (black) surrounded by blebs of poorly acidified autolysosomes (yellow) forms in Tg2576 (left), 5xFAD (second), TgCRND8 (middle), PS/APP (fourth), and APP51 (right) mice. [Courtesy of Lee et al., Nature Neuroscience, 2022.]
Other work showed that plaques cause hundreds of adjacent axons to bud off spheroids, aka dystrophic neurites. They block action potentials, disrupting neurotransmission over long distances, even between the brain's hemispheres, hampering cognition. Advances in ex and in vivo high-resolution microscopy could help scientists track the spread of these plaques throughout the parenchyma.
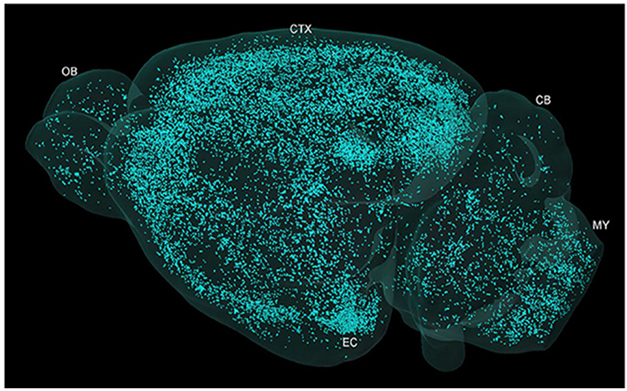
Galaxy of Plaques. Micro-optical sectioning tomography shows plaque distribution across the whole brain of a 5xFAD mouse. [Courtesy of Yin et al., Front Neurosci, 2022.]
Others reported that cerebral amyloid angiopathy—a disease of plaques within cerebral blood vessels that complicates safe administration of anti-amyloid therapeutic antibodies—can be triggered by a fragment of the cell adhesion protein lactadherin. Called Medin, it forms an amyloid in its own right, but also seems to promote fibrillization of Aβ40, the major component of CAA plaques. Mutations in the lactadherin gene MFGE8 truncate medin, and may protect against dementia.
April brought a surprise when, searching for TDP-43 fibrils, scientists instead plucked fibrils of TMEM106b from AD, FTD, ALS, synucleinopathy, and other tauopathy brain samples. TMEM106b protofibrils had a configuration dubbed “golf-course,” with 18 β-sheets meandering between adjacent N- and C-termini. How, or if, they factor into neurodegenerative disease pathogenesis is a question for 2023. This is not to let TDP-43 off the hook. It forms gel-like inclusions that stall proteasomes.

Fore! The intracellular domain of TMEM106b forms an 18 β-strand protofibril, with the N-terminus nestled deep within the center of the fold (left). The protofibrils stack upon each other (right). [Courtesy of Jiang et al., Nature, 2022.]
- Short Aβ Fibrils Easily Isolated from Alzheimer's Brain Fluid
- Cryo-EM Unveils Distinct Aβ42 Fibril Structures for Sporadic, Familial AD
- “Frustrated Oligomers” Slow Aggregation of Aβ42
- Does More Aβ38 Mean Less Cognitive Decline in Alzheimer’s?
- Ratio of Short to Long Aβ Peptides: Better Handle on Alzheimer's than Aβ42/40?
- Behold PANTHOS, a Toxic Wreath of Perinuclear Aβ That Kills Neurons
- Dystrophic Neurites Dampen Long-Range Neuronal Signaling
- Behold, Plaques Mapped in Whole Brain at High Resolution
- Meddling Medin: A Vascular Amyloid That Promotes CAA?
- Surprise! TMEM106b Fibrils Found in Neurodegenerative Diseases
- Death by Goo: TDP-43 Gels Paralyze Proteasomes in Neurons
The Tangle Angle
How amyloid plaques precipitate the spread of neurofibrillary tangles is a major conundrum in AD biology. Last year, analyses of PET and connectivity data from longitudinal cohorts indicated that plaques interact with tangles both remotely and up-close. The long-range disturbance occurs first, when cross-talk between neocortical neurons (weakened by amyloid) and entorhinal cortex neurons (weakened by tangles) force an exodus of tangles to other areas of the brain, including the inferior temporal gyrus. There, plaques and tangles meet face-to-face for the first time, and accelerate the spread of tangles to other areas. This follows the Braak pattern of tangle progression in most people, but in about one in 10 AD cases tau marches to the beat of a different drum. The findings bespeak the idea that there are different trajectories for AD that depend on how a person's brain is wired, i.e., his or her connectome. For example, tangles that form in axonal hubs spread faster through the brain (see also Apr 2021 news). Distinct conformations of tau fibrils may also cause rapidly progressing forms of AD.

Far and Near. Remote connections between regions with plaques (top left) and regions with tangles (bottom right) define a remote “amyloid influence metric.” Aβ and tau may also physically co-mingle. [Courtesy of Lee et al., Neuron, 2022.]
Other molecules besides Aβ can modulate the spread of tangles. In 2022, cholesterol, the synaptic protein Bassoon, and the kinase LRRK2 were implicated. The former slows tau's entry into neurons, blocking seeding of tangles, while pathogenic mutations in tau ramp up production of the lipid by astrocytes, perhaps as a defense. Bassoon seems to corral tau near synapses, perhaps explaining why tau hotspots appear near the cell membrane, and it expedites tau's release into the extracellular space where it might be taken up by other neurons. LRRK2 facilitates uptake of tau monomers; LRRK2 variants have been linked to primary tauopathies and tangle pathology in Parkinson’s.
- From Near and Far, Aβ Beckons Tau to Tangle in the Cortex
- What Drives Tangles to Spread? Answers Start Rolling In
- Does Fast-Progressing Alzheimer's Have a Whole Repertoire of Tau Conformers?
- Membrane Border Patrol: Cholesterol Stymies Tau Uptake, Aggregation
- Organoids Implicate Cholesterol Dysregulation in Tau Pathology
- Bassoon Heralds Spread of Toxic Tau
- Mysterious Tau Hot Spots Speckle Plasma Membrane
- At Tau2022: Unknown Functions Emerge for Tau, LRRK2
- Survey of Tau Partners Highlights Synaptic, Mitochondrial Roles
Brain Cleanse
A finer understanding of anatomy is helping scientists decipher how the brain filters large, toxic molecules. As it does with Aβ, the glymph system helps remove tau, slowing its aggregation and neurodegeneration, at least in mice. New to the glymph scene are perivascular macrophages. These keep the extracellular matrix around blood vessels supple, allowing small artery pulsations to squeeze interstitial fluid around the parenchyma. When these macrophages die due to age or disease, brain clearance slows. Pial membranes that surround blood vessels can also weaken with age and disease, allowing cerebrospinal fluid to seep in the opposite direction, into the parenchyma. On the other hand, amyloid can thicken these membranes, curtailing normal flow out of the CSF. To what extent these processes contribute to dementia remains to be seen, but in one specific disease, normal pressure hydrocephalus, the case seems solid. In this dramatic case of restricted CSF flow, dementia incidence exceeds that of the general population, tying inadequate brain clearance to dementia. In related research, the first-ever PET tracer for cholesterol debuted in 2022. It binds cytochrome P45046A1, the bottleneck in clearing cholesterol, hence might illuminate its exit from the brain.

Macrophages Doing Their Part. Brain of 3-month-old 5xFAD mouse (left) scattered with amyloid plaques (gold); absent perivascular macrophages (right), plaques are more abundant. [Courtesy of Drieu et al., Nature, 2022.]
More generally, revelations about the CSF and its protective membranes have taken the field by storm, showing how intimately the brain tissue communicates with the peripheral immune system. While immune cells surveilling the CSF become less inflammatory with age, in AD the opposite occurs, as microglia release chemokines that attract cytotoxic CD8-positive T cells to amyloid plaques. In Lewy body dementia, CD4-positive T cells infiltrate the brain in a similar manner and may exacerbate neuroinflmmation.
- Not Just Aβ—Glymphatic Flow Clears Tau, Too, Slowing Its Aggregation
- Perivascular Macrophages: New Target in Aging and Alzheimer’s Disease?
- Mesh, not Membrane: Pia Filters CSF, Gets Clogged by Aβ Fibrils
- In Hydrocephalus, Slow Drainage May Cause Dementia
- A New PET Tracer Could Track Cholesterol Clearance from the Brain
- In AD, CSF Immune Cells Hint at Mounting Mayhem in the Brain
- Intruder Alert: Inflammatory T Cells Lurk Near Lewy Bodies, Neurons
Microglia
Once considered bystanders, microglia took center stage in AD research once genetics implied that they can modulate amyloid load in the brain. Since then, scientists have been deciphering what roles these cells play in neuropathology. In autosomal-dominant AD, people with the highest concentration of soluble TREM2 in their CSF accumulate less amyloid, suggesting that cleavage of this microglial cell surface receptor helps clear plaques. Removing microglia from 5xFAD mice transformed the brain from one filled with dense-core plaques to one filled with diffuse and cerebrovascular plaques, suggesting that microglia condense amyloid. One idea is that activating this process could reduce pathology, and tests of TREM2 agonist antibodies for this purpose have begun.

TREM Down Those Plaques. Compared to controls (top), mice injected with Aβ18-TfR (bottom) had smaller plaques (red) ringed by more microglia (green). [Courtesy of Zhao et al., Science Translational Medicine, 2022.]
In mice, a tetravalent anti-TREM2 antibody clustered the receptor on the cell surface, upped its signaling 100-fold, and prompted microglia to surround and limit amyloid plaques. In keeping with this, Syk kinase, which is activated by TREM2 signaling, turns out to be essential for plaque clearance. Even so, targeting TREM2 may be tricky. One agonist antibody doubled the number of “disease-associated” microglia around plaques in mice; alas, plaques didn’t budge and more neurofibrillary tangles appeared. Curiously, knocking out TREM2 accelerated tangles in tauopathy mice, but only in those expressing the ε4 variant of human ApoE. Knocking out TREM2 also sped up neurodegeneration in a mouse model of frontotemporal dementia, despite reducing microglia hyperactivation. Confused? It appears TREM2 activation can have both protective and harmful effects, depending on the context.

With or Without. In 5xFAD mice (left), microglia (green) surround blood vessels (red lines) and diffuse amyloid deposits to mop up Aβ and package it into dense-core plaques (gray). Without microglia (right), the brain accumulates diffuse amyloid plaques (gray squiggles), cerebral amyloid angiopathy (gray branches), cerebral bleeds (red blotches), and calcium deposits (white). [Courtesy of Kiani Shabestari et al., Cell Reports, 2022.]
What genes microglia express in this state of affairs is surely important. The cells respond to AD pathology in multiple ways represented by antigen-presenting, cytokine-responsive, interferon-responsive, and DAM—aka disease-associated microglia—transcriptional states. DAMs ramp up expression of a slew of genes, including TREM2 and ApoE. If the latter is of the E4 variety, the microglia struggle to clear the debris they consume. Intriguingly, microglia failed to clear plaques when their cell-surface mechanoreceptor piezo1 was blocked. This sensor seems to sniff how stiff a fibril is, stimulating phagocytosis. Microglia also become more phagocytotic when forced to forgo glycolysis for burning fat, which generates more ATP.

Microglial Lineage. Data from chimeric mice delineate two pathways (brown, turquoise arrows) by which human homeostatic microglial (HM) react to amyloid. Cytokine response microglia (CRM) are different than human leukocyte antigen-presenting microglia (HLA), which transition through a DAM-like state. The former seem to be triggered by Aβ oligomers, the latter by plaques. [Courtesy of Mancuso et al., 2022.]
- Robust TREM2 Expression May Delay Alzheimer’s Disease
- Sans Microglia, Mice Develop CAA and Die Young
- Potent TREM2 Antibody Stirs Microglia to Prune Plaques in Mice
- SYK It To ’Em: Kinase Enables Microglia to Clear Plaques
- In Mice, TREM2 Antibody Mobilizes Microglia, Yet Worsens Tangles
- Sans TREM2, ApoE4 Drives Microgliosis and Atrophy in Tauopathy Model
- In Frontotemporal Dementia Model, Do Riled-Up Microglia Help?
- Human Microglia Mount Multipronged Response to AD Pathology
- Shooting Themselves in the Foot? Microglia Block “Good” State with ApoE4
- Piezo1 Steers Microglia Toward Rigid Aβ Plaques
- Forced Into Fat-Burning Mode, Microglia Gobble Up Plaques, Protect Neurons
Astrocytes
In 2022, the other “bystanders”—astrocytes—were accused of exacerbating early AD by ignoring hyperactive neural networks in the cingulate cortex years before amyloid plaques even start to deposit. Later in disease, astrocytes may blunt memory by churning out the inhibitory neurotransmitter GABA when, in response to Aβ, the glia fire up their urea cycle, which spits out the GABA precursor putrescine. In other words, deep dives into astrocyte biology have begun. Like microglia, astrocytes assume reactive states driven by specific gene-expression signatures. These states can oppose each other. Even in the healthy brain, astrocytes change shape, and some of the transcriptomes responsible include AD risk genes. High-resolution transcriptomics described astrocyte diversity, spotting subtypes such as interferon-responsive cells around blood vessels and ventricles of the brain, where they may liaise with peripheral immune cells. Neurofibrillary tangles can trigger production of interferon, but how this relates to these astrocytes has not been investigated.

High Def? To “see” transcriptomics (yellow), in reactive astrocytes at a decent spatial resolution, scientists went from searching for just expression of the interferon response gene Igtp (top left), to mapping expression of 200 genes differentially expressed by these glia (top right). Reanalyzing with a Bayesian statistics-based algorithm sharpened the images (bottom). [Courtesy of Shane Liddelow, New York University.]
With both the microglial and astrocyte fields growing rapidly, and new cell states being described with regularity, glia researchers around the globe have called for a moratorium on cute names, such as DAM, HAM, and ARM. They ask that, for the time being, scientists simply describe the states they observe, with an emphasis on function rather than only on the transcriptome.
- Hyperconnectivity in Cingulate Precedes Amyloid. Astrocytes to Blame?
- A Putrid Problem: Astrocytic Urea Cycle in Alzheimer’s?
- Astrocyte Reactivity: Opposing States Emerge
- Gene Networks That Control Astrocyte Diversity Linked to AD
- High-Res Spatial Transcriptomics Offers New Views of Mouse Brain
- Just Like Viruses, Tau Can Unleash Interferons
- Don’t Name It: Glial States Confound Easy Labels
Cell Biology
Single-cell and single-nucleus analysis have blossomed in recent years, allowing scientists to identify cell types and states that play good or bad roles in neurodegenerative disease. Early studies were based on small samples but, in 2022, data came in from snRNA-seq analyses of 1.6 million cells from the Religious Orders Study and Memory and Aging Project, and 1.2 million cells from Seattle Alzheimer’s Disease Brain Cell Atlas. They captured cellular changes that occur as AD worsens, including gliosis as a marker of rapid neurodegeneration and cognitive decline. Other research beefed up sample sizes using new tools to cluster cell types from combinations of prior data sets. This pegged cortical layer 1 inhibitory interneurons as early casualties of AD, followed by homeostatic microglia, which petered out as disease-associated microglia took over. Single-cell transcriptomics similarly pegged a subtype of dopaminergic neuron in the bottom part of the substantia nigra as being most vulnerable in Parkinson's disease. “Pseudotime” analysis, a way to order cells in time based on their relative similarity in gene expression, also tied these microglia to worsening neuropathology.
While such analyses identify changes in cell populations, others seek clues to pathogenesis amid changes in cell-to-cell communication. In AD, some cells changed gene expression in unison, and these coordinated responses correlated with plaques, tangles, and cognitive decline. Shifts in expression of receptor/ligand pairs indicated that microglial cross-talk with neurons changes the most, possibly to protect the latter. Curiously, microglia harboring AD risk variants in the TREM2 gene are less likely to strike up new “conversations” with other cells.

Synaptic Menagerie. Tau contacts all the synaptic vesicle proteins shown, except synaptobrevin, reinforcing the idea that it helps regulate exocytosis. [Courtesy of Tracy et al., Cell 2022.]
Lest the reader think 2022 was all about transcriptomics, the year also saw progress on proteomics, a methodology that is finally coming into its own. For example, one analysis of 8,600 proteins from 1,000 postmortem brains revealed big changes in extracellular matrix proteins in AD brain that had eluded prior transcriptomic studies. On a parallel track, studies of human induced pluripotent stem cells (iPSCs) identified an extensive tau interactome that highlighted roles for the microtubule binding protein in exocytosis and in mitochondrial metabolism. A stem-cell consortium introduced KOLF2.1J, a well-characterized iPSC cell line amenable to CRISPR editing and neutral for AD risk variants. If used widely, this reference cell line could standardize research across labs, which is poorly coordinated to date.
- RNA-Seq from 2.8 Million Cells Yields New Clues About Alzheimer's
- Meta-Analysis of RNA-Seq Data is Robust, Finds Key Changes in AD
- Single-Cell Sleuthing Nabs Neurons Prone to Perish in Parkinson’s
- Pseudotime Simulates Disease Progression from Case-Control RNA-Seq
- Eavesdropping on Cell-to-Cell Conversations in Aging, Alzheimer's
- Proteomics Highlight Alzheimer’s Changes in Matrisome, MAPK Signaling
- Survey of Tau Partners Highlights Synaptic, Mitochondrial Roles
- KOLF2.1J: The Mother of All iPSC Lines?
Genetics
In April, scientists published their haul from a massive GWAS meta-analysis; it identified 42 new common risk variants for late-onset AD, bringing the total to 75. The hits fell in loci linked to known AD-related biological pathways, including protein recycling, immunity, and amyloid precursor protein metabolism. In November, they piled data from the largest whole-exome analysis to date on top. It turned up new, rare variants in known AD risk loci, including SORL1 and TREM2, plus new risk genes involved in lipid metabolism. Likewise, a whole-genome analysis identified dozens of rare risk variants in SORL1 and TREM2, and in the new AD risk gene EXOC3L4, which encodes a protein involved in vesicle trafficking and exocytosis. New evidence implicated de novo somatic mutations in neurons, such coding changes and oxidized nucleotides that stress DNA repair mechanisms, allowing further mutations to accrue.
Beyond risk, scientists learned more about the functional effects of specific gene variants. For example, certain pPSEN1 mutations can stall endosome transport, causing axons to swell. Excitingly, scientists are finally making inroads into the longstanding question of how multiple variants in one person can modify each other's effect. For example, the autopsy of a woman who carried both the presenilin 1 Paisa mutation that causes early onset AD, and the protective Christchurch mutation in ApoE, showed that the latter restricted neurofibrillary tangles to the occipital cortex, where ApoE expression is low. This jibes with tau PET data suggesting that tangles spread more easily in areas where ApoE expression is high. Meanwhile, two new ApoE variants emerged that reduce risk by two- to threefold. One co-inherits with ApoE4 and neutralizes the extra risk brought on by this allele.

Aβ and Tau—A Severed Tie. In a woman who had both a PS1 and the ApoE Christchurch mutation, amyloid plaques (left column) dotted the frontal cortex, hippocampus, and occipital cortex. Tangles were mostly confined to her hippocampus and occipital cortex (right column). [Courtesy of Sepulveda-Falla et al., Acta Neuropathologica, 2022.]
Scientists reported a variant in the haptoglobin gene that tempers risk in ApoE2/E4 carriers but heightens it in ApoE4/4 carriers. The variant may have different affinities for ApoE isoforms.
Other variants reported to modify tau pathology include BDNF Val66Met, which turns out to accelerate tau phosphorylation, and the DRB1*04 subtype of human leukocyte antigen, which corrals aggregation-prone tau and reduces tangle load.
New tools emerged to help flesh out the effects of genetic variants. In mini pigs, knocking out SORL1, an AD gene that encodes the sorting receptor SorLA, boosted the amount of soluble Aβ and tau in the CSF of 2½-year old animals. Distended endosomes crowded neurons in their brains, suggesting that normal protein turnover was compromised. On that note, a neuron-specific retromer was discovered in the transentorhinal cortex that binds SorLA, supports neurotransmission, and tempers Aβ production. This retromer relies on a subunit called vacuolar protein sorting ortholog 26b, which was found to be deficient in some AD cases.
Gender effects grabbed some headlines in 2022, too. In women, a variant of the ILR1L1 gene protects against ApoE4, while the USP11 gene on the X chromosome may worsen tangles. ILR1L1 encodes an interleukin 33 receptor that activates microglia to clear plaques. The variant stifles a soluble form of ILR1L1 that acts as a decoy to disrupt IL-33 signaling and impede plaque clearance. USP11, on the other hand, encodes a peptidase that snips ubiquitin off tau, rendering it prone to aggregation. USP11 can evade X-inactivation in women, increasing their risk.

No USP11, Less P-tau. Cortical tissue from female USP11 knockout/P301S mice (bottom) had less p-tau202 (green) and less p-tau262 (red) than did P301S mice (top). [Courtesy of Yan et al., Cell, 2022.]
One aspect of genetics that has long stumped scientists is how to account for multiple genetic variations. In 2022 the MODEL-AD consortium debuted a plethora of polygenic mouse models. These include APP, MAPT, TREM2, ApoE, often in combination with other risk variants, such as in the ABCA7, MTHFR, and PLCG2 genes, and some target gene expression to microglia and astrocytes. For our part, Alzforum began curating an ApoE database, detailing the clinical phenotypes and molecular effects of 137 variants.
- Paper Alert: Massive GWAS Meta-Analysis Published
- Rare Variants in Lipid Transporter Genes Increase Risk for Alzheimer’s Disease
- Rare Variants Net New Alzheimer’s Gene, Links Between AD and CAA
- Somatic Mutations Accrue in Alzheimer's Neurons
- Presenilin Mutations Stall Endosomal Transport, Swell Axons
- In Brain with Christchurch Mutation, More ApoE3 Means Fewer Tangles
- Does Tangle Progression Follow ApoE Expression?
- BDNF Val66Met Hastens Tau Phosphorylation in Familial Alzheimer’s
- Major Histocompatibility Complex Curbs AD/PD Risk; Shows Tau to T Cells
- Two ApoE Mutations Decrease Risk for Alzheimer's Disease
- Up, and Down—Haptoglobin Moves APOE4 Risk in Mysterious Ways
- Receptor Decoy Raises Risk of Alzheimer’s—But Only in Women
- Ubiquitin Peptidase Linked to Increased Tau Pathology in Women
- In Mini-Pigs Missing SORL1, Endosomes Swell and Aβ Peptides Rise in CSF
- Neuron-Specific Retromer Identified—Does It Stave Off Alzheimer’s?
- Cornucopia: LOADs of New Mouse Models Available
- Introducing Alzforum Database of 137 APOE Gene Variants
Risk Factors/Lifestyle
In 2020, The Lancet had attributed 40 percent of global dementia cases to 12 modifiable risk factors. In 2022, analyses of a half-dozen nationwide surveys suggested a similar percentage in the U.S. Notably, obesity, hypertension, and physical inactivity weighed in as the strongest factors there, whereas hearing loss, low education, and smoking did in The Lancet report. Other studies backed these analyses. In the U.K. Biobank, a person's risk of dementia was 60 percent higher if they had two or more underlying health conditions, such as hypertension, heart disease, and diabetes, while it was 50 percent lower in those who walked 10,000 steps daily. Similarly, a U.S. study found that walking 8,000 steps per day protected people from obesity, hypertension, and Type 2 diabetes, aka dementia risk. A cataract surgery survey suggested that restoring vision could reduce dementia incidence by 30 percent. The take-home message—that lifestyle modifies risk for dementia—bore out in a PET study of identical twins in their 60s and 70s. Discordant patterns of neurofibrillary tangles accumulation correlated with lifestyle differences such that the twin with more tangles was less physically or socially active, or had more symptoms of depression.

Meet You at the Curve? Dementia risk was lowest at 10,000 steps per day (left) and 30 minutes at 112 steps per minute (right). [Courtesy of del Pozo Cruz et al., JAMA Neurology, 2022.]
These studies suggest that lifestyle intervention might reduce dementia incidence. Indeed, results from a growing set of multimodal and lifestyle intervention trials reported modest protection afforded by physical exercise, personalized coaching, and multimodal interventions that focus on vascular risk factors, nutrition, and cognitive, social, and physical activities. One new wrinkle from these trials is that people might do better when such interventions are tailored to a person's needs and preferences, rather than prescribed wholesale to a group. In general, these trials do not measure biomarker outcomes that indicate they are specific to Alzheimer's disease, but measure cognitive or functional outcomes of all-cause dementia more generally.
- In the U.S., 40 Percent of All-Cause Dementia Is Preventable
- As Health Problems Pile Up, Dementia Climbs on Top of the Heap
- Can 10,000 Steps a Day Keep Dementia at Bay?
- Another Study Links Daily Steps with Better Health
- Can Preserving Vision and Hearing Prevent Dementia?
- Identical Twins Share Tau Trajectory, but Lifestyle Can Make a Difference
- Lifestyle Interventions May Fend Off Decline; Social Contact Helps
- A Chat Every Other Day Keeps Dementia at Bay?
- Could Personalizing Multimodal Interventions Give Them Oomph?
—Tom Fagan
References
News Citations
- 2022, a Year in Review
- Short Aβ Fibrils Easily Isolated from Alzheimer's Brain Fluid
- Cryo-EM Unveils Distinct Aβ42 Fibril Structures for Sporadic, Familial AD
- “Frustrated Oligomers” Slow Aggregation of Aβ42
- Does More Aβ38 Mean Less Cognitive Decline in Alzheimer’s?
- Ratio of Short to Long Aβ Peptides: Better Handle on Alzheimer's than Aβ42/40?
- Behold PANTHOS, a Toxic Wreath of Perinuclear Aβ That Kills Neurons
- Dystrophic Neurites Dampen Long-Range Neuronal Signaling
- Behold, Plaques Mapped in Whole Brain at High Resolution
- Meddling Medin: A Vascular Amyloid That Promotes CAA?
- Surprise! TMEM106b Fibrils Found in Neurodegenerative Diseases
- Death by Goo: TDP-43 Gels Paralyze Proteasomes in Neurons
- Forget Typical Alzheimer's: AI Finds Four Types.
- From Near and Far, Aβ Beckons Tau to Tangle in the Cortex
- What Drives Tangles to Spread? Answers Start Rolling In
- Does Fast-Progressing Alzheimer's Have a Whole Repertoire of Tau Conformers?
- Membrane Border Patrol: Cholesterol Stymies Tau Uptake, Aggregation
- Organoids Implicate Cholesterol Dysregulation in Tau Pathology
- Bassoon Heralds Spread of Toxic Tau
- Mysterious Tau Hot Spots Speckle Plasma Membrane
- At Tau2022: Unknown Functions Emerge for Tau, LRRK2
- Survey of Tau Partners Highlights Synaptic, Mitochondrial Roles
- Not Just Aβ—Glymphatic Flow Clears Tau, Too, Slowing Its Aggregation
- Perivascular Macrophages: New Target in Aging and Alzheimer’s Disease?
- Mesh, not Membrane: Pia Filters CSF, Gets Clogged by Aβ Fibrils
- In Hydrocephalus, Slow Drainage May Cause Dementia
- A New PET Tracer Could Track Cholesterol Clearance from the Brain
- In AD, CSF Immune Cells Hint at Mounting Mayhem in the Brain
- Intruder Alert: Inflammatory T Cells Lurk Near Lewy Bodies, Neurons
- Robust TREM2 Expression May Delay Alzheimer’s Disease
- Sans Microglia, Mice Develop CAA and Die Young
- Potent TREM2 Antibody Stirs Microglia to Prune Plaques in Mice
- SYK It To ’Em: Kinase Enables Microglia to Clear Plaques
- In Mice, TREM2 Antibody Mobilizes Microglia, Yet Worsens Tangles
- Sans TREM2, ApoE4 Drives Microgliosis and Atrophy in Tauopathy Model
- In Frontotemporal Dementia Model, Do Riled-Up Microglia Help?
- Human Microglia Mount Multipronged Response to AD Pathology
- Shooting Themselves in the Foot? Microglia Block “Good” State with ApoE4
- Piezo1 Steers Microglia Toward Rigid Aβ Plaques
- Forced Into Fat-Burning Mode, Microglia Gobble Up Plaques, Protect Neurons
- Hyperconnectivity in Cingulate Precedes Amyloid. Astrocytes to Blame?
- A Putrid Problem: Astrocytic Urea Cycle in Alzheimer’s?
- Astrocyte Reactivity: Opposing States Emerge
- Gene Networks That Control Astrocyte Diversity Linked to AD
- High-Res Spatial Transcriptomics Offers New Views of Mouse Brain
- Just Like Viruses, Tau Can Unleash Interferons
- Don’t Name It: Glial States Confound Easy Labels
- RNA-Seq from 2.8 Million Cells Yields New Clues About Alzheimer's
- Meta-Analysis of RNA-Seq Data is Robust, Finds Key Changes in AD
- Single-Cell Sleuthing Nabs Neurons Prone to Perish in Parkinson’s
- Pseudotime Simulates Disease Progression from Case-Control RNA-Seq
- Eavesdropping on Cell-to-Cell Conversations in Aging, Alzheimer's
- Proteomics Highlight Alzheimer’s Changes in Matrisome, MAPK Signaling
- KOLF2.1J: The Mother of All iPSC Lines?
- Paper Alert: Massive GWAS Meta-Analysis Published
- Rare Variants in Lipid Transporter Genes Increase Risk for Alzheimer’s Disease
- Rare Variants Net New Alzheimer’s Gene, Links Between AD and CAA
- Somatic Mutations Accrue in Alzheimer's Neurons
- Presenilin Mutations Stall Endosomal Transport, Swell Axons
- In Brain With Christchurch Mutation, More ApoE3 Means Fewer Tangles
- Does Tangle Progression Follow ApoE Expression?
- BDNF Val66Met Hastens Tau Phosphorylation in Familial Alzheimer’s
- Major Histocompatibility Complex Curbs AD/PD Risk; Shows Tau to T Cells
- Two ApoE Mutations Decrease Risk for Alzheimer's Disease
- Up, and Down—Haptoglobin Moves APOE4 Risk in Mysterious Ways
- Receptor Decoy Raises Risk of Alzheimer’s—But Only in Women
- Ubiquitin Peptidase Linked to Increased Tau Pathology in Women
- In Mini-Pigs Missing SORL1, Endosomes Swell and Aβ Peptides Rise in CSF
- Neuron-Specific Retromer Identified—Does It Stave Off Alzheimer’s?
- Cornucopia: LOADs of New Mouse Models Available
- Introducing Alzforum Database of 137 APOE Gene Variants
- In the U.S., 40 Percent of All-Cause Dementia Is Preventable
- As Health Problems Pile Up, Dementia Climbs on Top of the Heap
- Can 10,000 Steps a Day Keep Dementia at Bay?
- Another Study Links Daily Steps with Better Health
- Can Preserving Vision and Hearing Prevent Dementia?
- Identical Twins Share Tau Trajectory, but Lifestyle Can Make a Difference
- Lifestyle Interventions May Fend Off Decline; Social Contact Helps
- A Chat Every Other Day Keeps Dementia at Bay?
- Could Personalizing Multimodal Interventions Give Them Oomph?
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.