SYK It To ’Em: Kinase Enables Microglia to Clear Plaques
Quick Links
Activating the cell surface receptor TREM2 rallies microglia to clear amyloid plaques. Can activating a kinase downstream of TREM2 do the same? Two groups of scientists say yes. In the October 26 Cell, researchers led by Marco Colonna, Washington University School of Medicine, St. Louis, reported that mouse microglia cannot phagocytose Aβ or condense plaques if their gene for SYK kinase is knocked out. Amyloid accumulation and memory loss accelerated. Meanwhile, in the October 17 Cell, John Lukens, University of Virginia, Charlottesville, and colleagues reported much the same. SYK knockout mice had fewer disease-associated microglia (DAMs), more plaques, and worse memory. In mouse models of multiple sclerosis (MS), knocking out microglial SYK worsened myelin loss and hind-limb paralysis. Activating the kinase in either amyloidosis or MS mice enhanced microglial responses to toxic debris.
- Activated by TREM2, SYK kinase regulates microglial responses in mice.
- Without SYK, they fail to compact plaques, exacerbating amyloidosis and memory loss.
- In a multiple sclerosis model, demyelination and paralysis worsen.
- Anti-CLEC7A activates SYK directly, rescuing TREM2 risk variants.
“Syk is an indispensable regulatory molecule for maintaining these [DAM] subsets in the degenerative milieu,” wrote Josh Morganti of the University of Kentucky in Lexington. Ana Griciuc and Rudolph Tanzi, Massachusetts General Hospital, Boston, agreed. “Both papers suggest that, in addition to TREM2 signaling, the SYK pathway in microglia may represent a novel therapeutic target for AD,” they wrote (comments below).
Activation of TREM2 signaling evokes the DAM signature, enabling these cells to surround and engulf amyloid plaques. Researchers typically gauge such microglial activation by measuring phosphorylation of SYK, which lies downstream of TREM2 and other cell surface signaling receptors, such as CLEC7A and CD33. What if SYK was knocked out?
To study this in the context of amyloidosis, both groups turned to 5xFAD mice, which develop amyloid plaques and gliosis by 2 months old. The scientists crossed them with conditional knockouts whose Syk expression can be turned off in their immune cells, including microglia. One-month-old crosses were fed tamoxifen for two to four weeks to silence Syk. Then, in the Lukens’ lab, first author Hannah Ennerfelt studied the effects in 5-month-old mice, while first author Shoutang Wang of the Colonna Lab evaluated 9-month-olds.
Both groups found similar changes. Compared to 5xFAD controls, tamoxifen-fed animals had one-third fewer microglia in the cortex and hippocampus at 5 months old and three-quarters fewer at 9 months. Around plaques, the dearth was more pronounced. Ennerfelt found that 5-month-old knockouts had 60 percent fewer microglia surrounding amyloid plaques, while in 9-month-olds Wang found almost none (see image below). Furthermore, any Syk-negative microglia that had sidled up to plaques expressed less of the DAM markers CLEC7A, TREM2, and CD11c than did microglia in 5xFAD controls (Jun 2017 news). SYK knockouts also had more Tmem119-positive homeostatic microglia. To the scientists, all this data suggested that microglia need SYK to mount a DAM response to amyloid plaques.
Without microglial SYK, Aβ clearance and neuropathology worsened. The microglia engulfed three-quarters less Aβ than did control microglia in both 5- and 9-month-old mice. Knockout mice had twice as much soluble Aβ40 and Aβ42, half as much insoluble Aβ42, and more diffuse plaques in the cortex, a sign that the microglia poorly compacted amyloid. Near plaques, knockouts had more phospho-tau, dystrophic neurites, and dying neurons than did control animals. Knockouts at both ages also took longer to find a hidden platform in a water maze and spent more time exploring the open arm of an elevated maze than age-matched 5xFAD mice, suggesting worse memory and increased risk-taking behavior. Both research groups concluded that microglia need SYK for optimal protection against AD pathology.
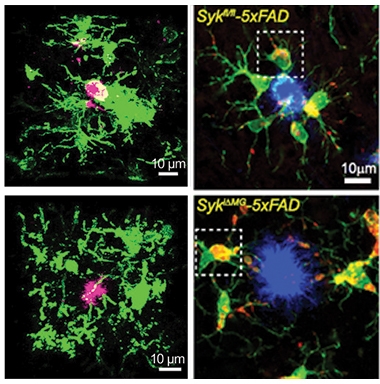
“Syk”ened Microglia. Compared to 5xFAD mice (top), SYK knockouts (bottom) had fewer microglia (green) surrounding amyloid plaques (pink or blue) at both 5 (left) and 9 months old (right). [Courtesy of Ennerfelt et al., 2022 (left column) and Wang et al., Cell, 2022 (right column).]
Notably, feeding tamoxifen to 1-month-old animals before AD pathology was present had the same effect as knocking out Syk in 4-month-old mice that already had extensive plaque pathology. Both sets of authors concluded that SYK drove microglial responses to amyloid during both disease onset and progression.
Knockout effects were not limited to AD models. Ennerfelt and colleagues found that it stymied microglial activation in two mouse models of multiple sclerosis, as well. Compared to control mice, the knockouts lost more spinal cord myelin, accumulated more toxic myelin fragments, and had worse hindlimb paralysis.
How does SYK regulate microglial activation? Scientists in both labs suspected this was through PI3K and AKT, which activates mTOR. This signaling trio controls many processes associated with microglial activation in AD (Chu et al., 2021). Indeed, compared to those from 5xFAD mice, SYK-negative microglia contained less phosphorylated AKT and less phosphorylated GSK3β, an AKT substrate. Wang and colleagues concluded that SYK deficiency ultimately dampens mTOR signaling, suppressing gene expression patterns (see image below).
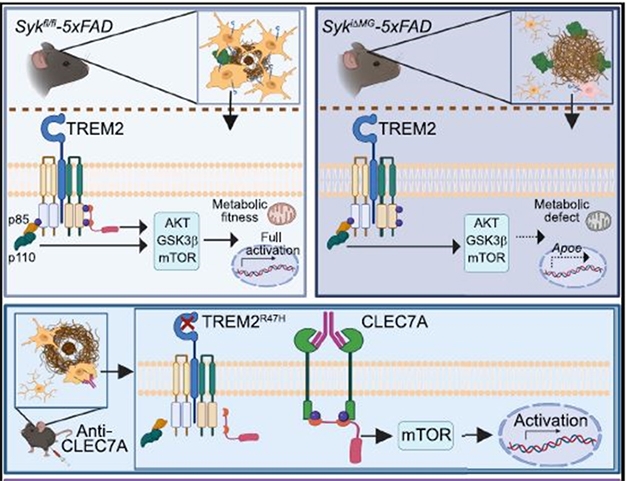
With and Without SYK. Microglia (orange) expressing SYK (pink) surround and compact amyloid plaques (top left), driven by activation of genes through TREM2 signaling and the mTOR pathway. Without SYK (top right), this signaling wanes and microglia struggle to mobilize, leading to filamentous plaques (brown) and more dystrophic neurites (green). Giving mice an antibody that activates CLEC7A (bottom) skirts defective TREM2 to activate SYK and mTOR. [Courtesy of Wang et al., Cell, 2022.]
Beyond Syk
How does TREM2 signaling factor in? This receptor binds to its co-receptor DAP12 to activate SYK. While knocking out TREM2 suppresses DAMs, Wang and colleagues noticed that SYK-negative microglia adopted an intermediate state, expressing both homeostatic and DAM genes. They think that TREM2 partially activates the microglia through SYK-independent signaling.
Wang suspected that DAP10, another TREM2 binding partner, may be involved. Indeed, just like microglia from SYK knockouts, those lacking DAP10 contained less phosphorylated AKT and GSK3β and phagocytosed less Aβ. It turns out that DAP10 activates PI3K directly, thus bypassing SYK, to activate AKT and mTOR (see image below). The scientists concluded that DAP10 signaling also plays a role in microglial responses to amyloidosis.

Bypassing SYK. In microglia, TREM2 and its binding partner DAP12 (left) activate SYK. However, TREM2 may also tango with DAP10 (right) to activate PI3K, which lies directly downstream of SYK. Therefore, TREM2/DAP10 skirts SYK yet activates the same pathway. [Courtesy of Marco Colonna.]
Moreover, both groups found a way to enhance microglial phagocytosis by skirting TREM2 and both its co-receptors. They activated CLEC7A. Expressed in DAM microglia, this cell surface receptor can directly signal through Syk without binding DAP10 or DAP12 (see graphical abstract above). Ennerfelt boosted this signaling by injecting 5xFAD hippocampus with pustulan, a β-D-glucan and natural ligand of CLEC7A. Wang targeted CLEC7A with an antibody delivered by systemic injection into 5xFAD mice that carried the R47H hypofunctional variant of TREM2. In both experiments, the mice had fewer filamentous plaques and more of the compacted type of plaques in their cortices than did untreated animals.
“Targeting the intracellular pathway downstream of TREM2 produces a potent effect not only in microglial response but also by changing the course of disease,” wrote Renzo Mancuso, University of Antwerp, Belgium. Tyler Ulland of the University of Wisconsin, Madison, a co-author on the Luken manuscript, agreed. “These results show that Syk is an essential downstream regulator of the microglial response to [amyloid] and, excitingly, identifies Syk as a new target for possible therapeutic intervention,” he wrote (comments below).
All told, both papers suggest that SYK is needed to spur microglial responses to toxic debris, such as amyloid and damaged myelin, ultimately protecting neurons from degeneration. Boosting SYK signaling, such as through immunotherapies that activate CLEC7A, may be an effective therapeutic strategy for multiple neurodegenerative diseases, say the authors.—Chelsea Weidman Burke
References
Mutation Interactive Images Citations
Research Models Citations
News Citations
Paper Citations
- Chu E, Mychasiuk R, Hibbs ML, Semple BD. Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic neuroinflammation. J Neuroinflammation. 2021 Nov 27;18(1):276. PubMed.
Further Reading
Papers
- Schweig JE, Yao H, Coppola K, Jin C, Crawford F, Mullan M, Paris D. Spleen tyrosine kinase (SYK) blocks autophagic Tau degradation in vitro and in vivo. J Biol Chem. 2019 Sep 6;294(36):13378-13395. Epub 2019 Jul 19 PubMed.
- Schweig JE, Yao H, Jin C, Crawford F, Mullan M, Paris D. Neuronal Spleen tyrosine kinase (SYK) mediates cytokine release in Transgenic Tau P301S mice organotypic brain slice cultures. Neurosci Lett. 2020 Jun 11;729:134992. Epub 2020 Apr 22 PubMed.
- Schweig JE, Yao H, Beaulieu-Abdelahad D, Ait-Ghezala G, Mouzon B, Crawford F, Mullan M, Paris D. Alzheimer's disease pathological lesions activate the spleen tyrosine kinase. Acta Neuropathol Commun. 2017 Sep 6;5(1):69. PubMed.
News
- Nilvadipine Fails to Slow Cognitive Decline in AD Patients
- Deleting CD33 Benefits Mice—If Their Microglia Express TREM2
- Potent TREM2 Antibody Stirs Microglia to Prune Plaques in Mice
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
Primary Papers
- Ennerfelt H, Frost EL, Shapiro DA, Holliday C, Zengeler KE, Voithofer G, Bolte AC, Lammert CR, Kulas JA, Ulland TK, Lukens JR. SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell. 2022 Oct 27;185(22):4135-4152.e22. Epub 2022 Oct 17 PubMed.
- Wang S, Sudan R, Peng V, Zhou Y, Du S, Yuede CM, Lei T, Hou J, Cai Z, Cella M, Nguyen K, Poliani PL, Beatty WL, Chen Y, Cao S, Lin K, Rodrigues C, Ellebedy AH, Gilfillan S, Brown GD, Holtzman DM, Brioschi S, Colonna M. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell. 2022 Oct 27;185(22):4153-4169.e19. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Kentucky
In this new manuscript by Ennerfelt et al., the authors mechanistically assess the contributions of microglial spleen tyrosine kinase (SYK) across multiple mouse models that recapitulate aspects of drivers associated with neurodegeneration. Multiple complementary approaches were taken to examine known pathobiological responses in light of a loss-of-function conditional removal of Syk from microglia to ultimately determine whether this kinase is a critical regulatory checkpoint of multiple cell-surface receptors (i.e., Clec7a, CD33, and TREM2), which have been previously implicated in neurodegenerative conditions. Excitingly, the authors demonstrate a consistently worsened outcome in both the 5xFAD and cuprizone/experimental autoimmune encephalomyelitis models if they lack microglial Syk. Further, removal of Syk from microglia reduced the proportion of disease-associated microglia (DAM) transcriptional phenotypes in favor of both homeostatic and a newly described high Cd36-expressing population. These findings add further support to the notion that the maintenance of certain microglial subsets (i.e., DAM) is neuroprotective in the chronic stages of neurodegenerative disease models.
Critically, the authors demonstrate that SYK is an indispensable regulatory molecule for maintaining these subsets in the degenerative milieu. Given the signal transduction role of SYK relative to TREM2, these findings are in line with several prior works demonstrating that removal of TREM2 exacerbates neuropathological sequelae in similar animal models (Piccio et al., 2007; Wang et al., 2015). Excitingly, the findings by Ennerfelt et al. suggest that new therapies with agonistic targeting of SYK may offer beneficial outcomes for neurodegenerative disease, similar to work targeting TREM2 directly (Zhao et al., 2022).
Complementary to these findings, Wang et al. demonstrate the TREM2-SYK signaling axis as a primary effector driving microglial responses to amyloid-induced neuropathology, implicating TREM2-SYK signaling as a key regulatory checkpoint for microglial transcriptional phenotype progression into the “DAM” staging. What is interesting in this study is that the authors demonstrate potential alternative mechanisms to engage SYK functionality if/when TREM2 signaling may be defective. They demonstrate that engaging the cell-surface receptor DECTIN1 (CLEC7A) via monoclonal antibody treatment was able to alter some pathological burden.
Collectively, both of these manuscripts highlight potential therapeutic targets associated with driving increased SYK signaling as a means to ramp up microglial function in the face of amyloid-induced neuropathology.
References:
Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, Panina-Bordignon P. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007 May;37(5):1290-301. PubMed.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015 Mar 12;160(6):1061-71. Epub 2015 Feb 26 PubMed.
Zhao P, Xu Y, Fan X, Li L, Li X, Arase H, Tong Q, Zhang N, An Z. Discovery and engineering of an anti-TREM2 antibody to promote amyloid plaque clearance by microglia in 5XFAD mice. MAbs. 2022 Jan-Dec;14(1):2107971. PubMed.
Massachusetts General Hospital and Harvard Medical School
Massachusetts General Hospital
Wang et al. elegantly described new mechanistic insights into how TREM2 induces microglial cell responses to Aβ in mouse models of Alzheimer’s disease (AD) through SYK-dependent and SYK-independent pathways.
Recent studies have shown characteristic expression changes in microglia around Aβ plaques, labeling them as disease-associated microglia (Keren-Shaul et al., 2017). DAM microglia have been characterized by TREM2-dependent upregulation of phagocytic and lipid metabolism genes (Keren-Shaul et al., 2017; Krasermann et al., 2017). Several innate immune receptors that are upregulated in DAM signal via SYK, including TREM2 and CLEC7A. Here, Wang et al. showed that Syk-deficient microglia impaired the formation of a protective microglial barrier around Aβ plaques and enhanced Aβ accumulation and behavioral deficits in in the 5xFAD mouse model of AD. Targeted deletion of SYK in microglia disrupted the PI3K-AKT-GSK3β-mTOR signaling pathway, thereby leading to metabolic defects. In line with the new and interesting study by Ennerfelt and colleagues, showing SYK modulates microglial activation and that SYK loss of function exacerbates Alzheimer’s disease neuropathology, Wang et al. likewise found that SYK was required for microglial clustering around Aβ plaques during both disease onset and AD progression in 5xFAD mice.
While the articles by Ennerfelt et al. and Wang et al. showed that SYK deficiency impaired generation of the DAM response, Wang and colleagues also uncovered that SYK deletion led to accumulation of an Apoe-expressing microglial cell population that is prodromal to DAM. Furthermore, comparison of single-cell RNA-Seq datasets from 5xFAD;Syk-deficient and 5xFAD;Trem2-deficient microglia by Wang et al. suggested that Aβ pathology induced a TREM2-dependent but SYK-independent pathway in microglia that was sufficient to advance these cells to a prodromal stage of DAM. Biochemical analysis using bone-marrow-derived macrophage cultures from mice deficient for Syk, Trem2, or Dap10 showed that DAP10 (which also binds TREM2) sustained microglial cell proliferation, metabolic fitness, and Aβ phagocytosis independently of SYK. However, both non-redundant SYK and DAP10 signaling pathways were required for the implementation of full DAM response. Thus, TREM2 mediates microglial cell response to Aβ accumulation via SYK-dependent and -independent (e.g., DAP10) signaling pathways.
Our recent observations suggest a critical role for the innate immune receptor TREM2 in modulating microglial pathology in AD (Griciuc at al., 2019). Although previous studies showed that SYK acts downstream of TREM2 (Yao et al., 2019), additional studies are required to investigate whether TREM2 relies partially or exclusively on SYK to control AD pathogenesis. To this end, Ennerfelt et al. compared their RNA-Seq dataset with our previously published dataset analyzing 5xFAD;Trem2-deficient microglia (Griciuc et al., 2019). They found that 25 percent of genes upregulated and 60 percent of genes downregulated in 5xFAD;Trem2-deficient microglia were shared with 5xFAD;Syk-deficient microglia. However, 5xFAD;Syk-deficient microglia also showed uniquely expressed genes not observed in 5xFAD;Trem2-deficient microglia. These important findings suggest that TREM2 signaling through SYK partially mediates the DAM response, and that SYK acts downstream of other receptors in addition to TREM2 in the 5xFAD mouse model.
One such receptor, CLEC7A, which was originally defined as a fungal pathogen receptor, directly activates SYK. Ennerfelt and colleagues found that SYK was required for the CLEC7A-mediated phagocytic response in microglia, following injection of pustulan, a β-D-glucan that acts as a ligand for CLEC7A. These data suggest that CLEC7A signals through SYK to promote microglial phagocytosis in 5xFAD mice. Moreover, Wang et al. showed that antibody-mediated cross-linking and activation of CLEC7A in microglia expressing a TREM2 hypofunctional variant (TREM2R47H) on a 5xFAD background improved microglial activation.
In summary, both papers suggest the exciting prospect that in addition to TREM2 signaling, the CLEC7A-SYK signaling pathway in microglia may represent a novel therapeutic target for AD.
References:
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017 Jun 15;169(7):1276-1290.e17. Epub 2017 Jun 8 PubMed.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017 Sep 19;47(3):566-581.e9. PubMed.
Griciuc A, Patel S, Federico AN, Choi SH, Innes BJ, Oram MK, Cereghetti G, McGinty D, Anselmo A, Sadreyev RI, Hickman SE, El Khoury J, Colonna M, Tanzi RE. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer's Disease. Neuron. 2019 Sep 4;103(5):820-835.e7. Epub 2019 Jul 10 PubMed.
Yao H, Coppola K, Schweig JE, Crawford F, Mullan M, Paris D. Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFκB Antagonistic Activities. Front Cell Neurosci. 2019;13:457. Epub 2019 Oct 10 PubMed.
University of Wisconsin
SYK is an essential regulator of several cellular responses and, in microglia, serves as downstream signaling molecule for many important receptors with innate immune function, including TREM2, CD37, Clec7A, as well as several members of the Fc receptor family. Mutations in several of these genes have been linked to increased risk for developing Alzheimer’s disease (AD) and/or multiple sclerosis (MS). In particular, TREM2 has been a focus of a great deal of recent research because it dramatically impacts the ability of microglia to interact with amyloid plaques in AD, and clear cellular debris and enhance recovery in models of MS. With that said, a role for SYK in the microglia response to AD or MS has not been established.
These two new papers by Ennerfelt et al. and Wang et al. have described an essential role for SYK signaling in the normal microglial response to Aβ plaques and oligodendrocyte loss. Ennerfelt et al. utilized the 5xFAD mouse model of AD and the experimental autoimmune encephalomyelitis (EAE) model of MS, to show that SYK is an essential regulator of the microglial response to neurodegeneration. In 5xFAD mice, SYK-negative microglia were less likely to cluster around plaques, were less proliferative, less activated, and less phagocytic. The plaques in these mice were more prominent and neurotoxic.
Meanwhile, in EAE, silencing Syk in microglia resulted in mice that were less able to recover post-challenge and were unable to mount a normal disease-associated microglial (DAM) response.
These results show that SYK is an essential downstream regulator of the microglial response to challenge, and excitingly identifies SYK as a new target for possible therapeutic intervention. Wang et al. confirmed that SYK signaling was necessary for the normal DAM response in 5XFAD mice. Wang et al. also confirmed that loss of SYK signaling recapitulated the autophagy phenotype observed in microglia from Trem2-/- 5XFAD mice (Ulland et al., 2017). Finally, Wang et al. showed that in a mouse model with decreased TREM2 signaling—the Trem2R47H 5xFAD mice—engaging Clec7A with an activating antibody could increase SYK signaling, leading to activation of microglia independently of TREM2 signaling.
The findings in these two new papers are exciting and could lead to the development of new therapeutics targeting the SYK pathway.
References:
Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell. 2017 Aug 10;170(4):649-663.e13. PubMed.
VIB-Center for Molecular Neurology
These are very relevant papers given the central role of microglia in Alzheimer’s disease and other neurodegenerative disease. It is also very nice to see that research performed independently and in parallel yields the same results.
From a scientific point of view, the role of Syk in governing macrophage activation via ITAM receptors is not new—you can find literature on that over more than a decade ago. So, to me it is perhaps not surprising that this mechanism is conserved in microglia and TREM2 signaling. Having said that, it is very nice and relevant to see how targeting the intracellular pathway downstream of TREM2 produces a potent effect, not only in microglial responses, but also by changing the course of disease.
Whereas these two papers focus on TREM2, Syk also acts downstream of other ITAM receptors, such as AXL and MERTK, that play a crucial role in interactions with Ab plaques, so it would be very interesting to investigate beyond TREM2 in follow up studies.
Along the same lines, it would be important to determine how other microglial functions are altered—I see for example that microglial numbers and proliferation show changes—and to explore whether there could be any potential therapeutic window in modulating microglial intracellular signaling pathways. Whereas genetics clearly point to a role for microglia in AD, we see that manipulating microglial function has a strong impact in other models of disease, as well, such as multiple sclerosis.
Regarding TREM2, it seems that whereas activation of microglia helps remove amyloid deposits, there is evidence that it may exacerbate tau pathology. While this may be related to timing, disease stages, etc., I think it is crucial to understand those fundamental differences before we can use these strategies in the clinic.
Make a Comment
To make a comment you must login or register.