As research on frontotemporal dementia gathers steam, the international conference hosted its largest gathering ever last month in Munich, Germany. More than 750 researchers shared their latest data on basic science, genetics, and clinical outcome measures. Biomarkers dominated the meeting, as the field prepares for therapy trials by seeking better ways to track pathology in living people. The few trials that have been done thus far, without specific biomarkers, have largely been a bust.
Frontotemporal Dementia: The Hard Work of Pushing Toward Trials
In the last 10 years, the science of frontotemporal dementias has exploded, thanks to big openings in the molecular pathologies behind these disorders. Rapidly growing interest among academia and industry was on display at the 10th International Conference on Frontotemporal Dementias, held August 31 to September 2 in Munich. Organizers had to cap attendance at a record 760 participants, who included more junior researchers than previous conferences. Days packed with talks, panel discussions, and poster sessions left attendees little time to enjoy the city’s picturesque streets and legendary outdoor beer gardens; even so, lively hallway and café discussions signaled enthusiasm throughout the conference. While the presentations spanned the range of basic to clinical science, there was a dominant theme: a collective push toward trials. Efforts appeared most intense on two fronts: to delineate the underlying mechanisms of each of this group of diseases in search of the best therapeutic targets for it, and to identify useful biomarkers to enable trials.
“In the last two years, the field has achieved important steps toward deciphering frontotemporal lobar degeneration and laying the groundwork for trials,” conference president Janine Diehl-Schmid of the Technical University, Munich, told the audience.
Frontotemporal lobar degeneration is an umbrella term that covers a wide range of related clinical syndromes, some with signature behavioral symptoms, others language, yet others mostly motor. On the motor end, disorders include amyotrophic lateral sclerosis, progressive supranuclear palsy, and corticobasal degeneration; on the other, behavioral variant frontotemporal dementia and language disorders such as semantic dementia and progressive non-fluent aphasia. The syndromes have atrophy of the front of the brain in common, hence the name, but different molecular pathologies underlie each condition, with the two most common being aggregation of tau and TDP-43 (see Apr 2016 conference news). Genetically, a host of rare mutations can cause FTD. With such heterogeneity, and given the rarity of the diseases, trials have been difficult to run. Only a handful have been completed to date.
Enrollment in the longitudinal GENFI study has climbed steadily since 2012. [Courtesy of Jonathan Rohrer.]
Speakers at ICFTD presented data from three recent trials, with uniformly negative findings (see Part 2 of this series). Undeterred, industry scientists said they were encouraged by the progress in mounting trials in this population. Many other therapeutics are in the pipeline, and speakers updated the audience on several that are preparing to enter human testing. Meanwhile, biomarkers and new clinical measures dominated the meeting, with around two dozen talks and 200 posters devoted to the topic (see Part 3 of this series). The most excitement swirled around the potential of neurofilament light (NfL) as a fluid marker of disease progression, but a plethora of imaging data was also on display. Speakers tied specific behavioral and cognitive symptoms to disruptions of particular brain networks.
On the clinical side, measures of social cognition were a hot topic (see Part 4 of this series). Many speakers believe they will provide more sensitive outcome markers than the cognitive tests currently in use for FTD. On the basic science front, many talks and posters delved into FTD’s genetics and molecular pathology (see Part 5 and Part 6). Scientists urgently need better animal models, and a new TREM2 model, as well as new data on an inducible TDP-43 model, were well-received (see Part 6). Check back in coming days for in-depth stories on all these topics.
Strength in Numbers: Cohort Studies Clarify Disease Progression
FTD researchers are advancing quickly in part because they can follow a path laid down by their Alzheimer’s colleagues. In AD, longitudinal studies such as the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL), Alzheimer's Disease Neuroimaging Initiative (ADNI), the Alzheimer’s Prevention Initiative (API), and the Dominantly Inherited Alzheimer Network (DIAN) have transformed scientists’ understanding of the disease. FTD scientists have launched their own cohort studies, most focusing on familial forms of the disorders, where the molecular pathology is known. By tracking mutation carriers who do not yet have symptoms, researchers can record their preclinical biomarker changes. The idea is to know the natural history of the disease as a basis for trials, as well as build a pool of people interested in enrolling. The FTD cohorts include the Genetic Frontotemporal Dementia Initiative (GENFI) in Europe, as well as the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) and the Advancing Research and Treatment for Frontotemporal Lobar Degeneration Consortium (ARTFL) in the United States (see Jul 2012 conference news; Nov 2014 conference news). GENFI1 has concluded, but GENFI2 is enrolling and gathering baseline data, as are LEFFTDS and ARTFL. At ICFTD, researchers presented interim updates as well as preliminary biomarker data from GENFI1 (see Part 3 of this series).
GENFI, led by Jonathan Rohrer of University College London, is the longest-standing of the cohorts. Researchers previously reported baseline and one-year follow-up findings from GENFI1, which comprised 365 people symptomatic or at risk for genetic forms of FTD (see Nov 2014 conference news). Additional findings from this study were sprinkled throughout the conference. GENFI2, now in its second year, expanded from 13 centers to 27, and currently has 550 people enrolled. Many of them continued onward from GENFI1. Recruitment has been so successful that researchers expect to exceed their original target of 600 participants, perhaps reaching 800, Rohrer told Alzforum. All participants come from families that harbor mutations in either tau, progranulin, or C9ORF72. Participants will return for two follow-ups visits at one-year intervals. About 200 have completed their first follow-up. GENFI2 will institute its first data freeze at the end of 2016, Rohrer said.
Like GENFI, LEFFTDS enrolls only people at autosomal-dominant genetic risk for FTD. In Munich, Brad Boeve of the Mayo Clinic in Rochester, Minnesota, gave an update. LEFFTDS now consists of 209 mostly Caucasian volunteers with a mean age of 50. The cohort comprises about equal parts of each of the three main FTLD genes. About one-third of each group already have disease symptoms. As in GENFI, participants will return for two annual follow-up visits each at the eight participating North American centers.
ARTFL, active at 15 North American centers and led by Adam Boxer of the University of California, San Francisco, casts a broader net. As of August 1, it had enrolled 192 people with sporadic FTLD, plus 240 with a family history, of whom 188 carry a known FTLD gene. Participants have a mean age of 58; 90 percent are Caucasian. ARTFL and LEFFTDS collect biospecimens including DNA, RNA, plasma, serum, monocytes and lymphocytes, as well as CSF from some volunteers. Volunteers with sporadic disease participate in project 1, which seeks to characterize idiopathic FTLD. Researchers are currently investigating blood cytokine profiles of this group. Those with a family history take part in project 2, which tracks clinical and biomarker change over one year.
The three cohorts have joined forces, along with the Australian Dominantly Inherited Non-Alzheimer Dementias (DINAD) study, to form the FTD Prevention Initiative (FPI). DINAD, led by John Hodges and Glenda Halliday at Neuroscience Research Australia, Sydney, has just gotten started and plans to recruit participants with genetic forms of FTD. The FPI umbrella group will establish minimum shared datasets to facilitate the pooling of findings. LEFFTDS and ARTFL already share their database and use the same assessments. Meanwhile, researchers are putting finishing touches on the FTD Disorders Registry in hopes of speeding up recruitment for studies and trials. The registry is expected to go live in October, Boxer said.
Calculating the Cost of FTD
Families are desperately awaiting trials because there are currently no treatments for FTD. Symptoms typically strike in middle age, and can tear families apart. While many anecdotal reports recount the burden of the disease, few studies have tried to quantify its societal and economic cost. James Galvin of Florida Atlantic University, Boca Raton, filled this gap with a Web-based survey administered through the website of the Association for Frontotemporal Degeneration. The survey’s 250 questions take about two hours to complete, which respondents could do over several sessions. To measure the personal toll of the disease, Galvin used the Health Utilities Index 3 (HUI3) and Quality-Adjusted Life Years (QALYs). The HUI3 rates a person’s function on eight attributes pertaining to vision, hearing, speech, ambulation, dexterity, emotion, cognition, and pain. A rating of 1 indicates perfect function, 0 represents death, and a rating below 0 signifies a state perceived to be even worse than death—for example, someone who is bedbound in extreme pain. QALYs are scored similarly, with 1 representing one year of perfect health, 0 death, and negative numbers signifying a state considered worse than death. Scores are multiplied by the number of years spent in that state.
At ICFTD, Galvin reported that 956 people began the survey and 674 completed it. Most were caregivers for someone with behavioral variant FTD. Caregivers reported a very high personal burden, regardless of the stage of their loved one’s disease. More than one-third had quit their jobs to care for their family member, and two-thirds reported declines in their own health. Adult children caring for a parent with the disease reported a worse quality of life than did spouses, with the highest burden falling on daughters caring for mothers. For the person with FTD, on the other hand, quality of life depended on the stage of their disease. At mild stages, caregivers reported an average HUI3 of 0.55 and QALY of 1.2 for their loved one. By moderate disease stages, the HUI3 dropped to 0.24 and the QALY to 0.8. For patients at severe stages, both scores fell into the negative, at -0.09 and -0.64, respectively. The numbers did not depend on what subtype of FTD a patient had; all were scored as equally devastating.
Financial costs were also extremely high. One-third of caregivers reported using paid care several times per week. FTD patients need frequent hospitalizations, more than AD patients do, Galvin said. They can become violent, and often break laws and flout social norms. Reflecting this, caregivers reported spending money on childcare and on moving children to safer locations, as well as legal fees. Caregivers who still worked missed many work days. Overall, they reported a decline in household income after an FTD diagnosis. The survey estimated the yearly cost from an FTD diagnosis to be $135,000 per patient. This is about three times higher than the cost of AD, which has been estimated at $56,000 per year. These figures might help researchers model how much a potential therapy could reduce costs, Galvin suggested.
Several scientists were impressed by how this analysis quantified the social and economic burden of the disease. Boeve called the data “enlightening.” Boxer, noting that researchers need to define what trial outcomes would be clinically meaningful for each FTD patient group, called Galvin’s analysis a starting point.—Madolyn Bowman Rogers
First Round of FTD Therapeutics Fell Short, But Many More Are Up and Running
Even as researchers work to unearth the underlying causes of frontotemporal dementia, they are beginning to develop treatments that target specific mechanisms. Only a handful have come to trial yet, but more are in the pipeline. At the 10th International Conference on Frontotemporal Dementias, held August 31 to September 2 in Munich, speakers presented results from three such trials. All were largely negative, including topline results from a much-anticipated Phase 3 trial of LMTM, a derivative of the dye methylene blue. Though conference attendees were disappointed, their overall outlook was hopeful. “Now that we know the major pathways involved in frontotemporal lobar degeneration, we know what to target and what to look for in trials,” said conference co-organizer Manuela Neumann of the German Center for Neurodegenerative Diseases (DZNE). There was widespread agreement that FTD trials are becoming more rigorous and biomarker-based, improving the odds of seeing a treatment effect.
Better Behavior?
FTD patients taking tolcapone (right bar) improved their scores on the Neuropsychiatric Inventory compared to their baseline measures (left). Statistically, the benefit was not significantly better than that of patients who took placebo (middle). [Courtesy of Edward Huey.]
Both academic and industry groups appear poised to take many shots on goal with immunotherapies, HDAC inhibitors, and antisense agents to suppress expression of harmful genes. In fact, the field’s immediate problem may be finding enough patients with these fairly rare disorders to test multiple therapies at once, noted Adam Boxer of the University of California, San Francisco. FTD is about 200 times less common than Alzheimer’s disease, with an estimated 25,000 people living with the condition in the United States (see Knopman and Roberts, 2011). To help with that, researchers have established observational cohorts and are about to launch an FTD registry to gather a pool of people interested in trials (see Part 1 of this series). Cohort studies are already helping researchers identify biomarkers of disease progression that might change with treatment (see Part 3). “We have made great progress since 2014,” Boxer told the audience.
That First FTD Therapy? It Won’t Be LMTM, aka Methylene Blue
First, the bad news. Claude Wischik of the University of Aberdeen, Scotland, who leads TauRx Pharmaceuticals, described a lack of efficacy for the company’s compound LMTM in patients with behavioral variant FTD (bvFTD). Also known as LMTX or TRx0237, this compound inhibits aggregation of tau and TDP-43, two pathologies in this disorder (see Oct 2012 news). In the largest randomized controlled trial and first Phase 3 study in this population to date, researchers enrolled 221 bvFTD patients who had MRI evidence of frontotemporal atrophy. They were primarily Caucasian, skewed male, and had an average age of 63. Half the cohort took 100 mg of LMTM twice daily, while the other half took a 4 mg LMTM pill twice daily as a placebo. Because LMTM turns urine blue, 4 mg added to placebo was the lowest dose that allowed participants to remain blind to their treatment status, Wischik noted.
Participants underwent clinical, cognitive, and motor assessments at 16, 32, and 52 weeks. The trial used three primary outcome measures: The Addenbrooke’s Cognitive Examination Revised (ACE-R), the Functional Activities Questionnaire, and change in whole-brain volume on MRI. The ACE-R is similar to the MMSE, with the addition of verbal fluency; it can distinguish between FTD and AD. Secondary measures included the ADCS-Clinical Global Impression of Change, the Frontotemporal Dementia Rating Scale, and the Unified Parkinson’s Disease Rating Scale.
On every measure, rates of decline were identical in the treatment and placebo groups. Wischik said the company is still analyzing what he called a “rich data set,” and will publish more results later, including subgroup analyses of patients with different brain atrophy patterns. In addition, he claimed that the whole cohort declined less on the ACE-R than expected based on the literature. ICFTD attendees disputed this, with one audience member pointing out that the rate of decline on the ACE-R matched that seen in the Sydney GENFI cohort, suggesting it falls within the normal range. To see if both groups might have experienced a treatment benefit, TauRx is measuring how much of the drug entered the central nervous system in each group, Wischik said. CNS exposure across a relevant range of doses is typically investigated in Phase 1.
Overall, researchers considered a hidden treatment benefit unlikely and agreed this compound may have reached the end of the road as an FTD drug. “It was disappointing. There was just nothing there,” said Janine Diehl-Schmid, who presided over ICFTD2016 and had been a site leader in the LMTM trial. LMTM previously failed to budge endpoints in a Phase 3 Alzheimer’s trial. The company reported those findings at the Alzheimer’s Association International Conference 2016, where claims of statistical significance of a subgroup effect were broadly challenged (see Jul 2016 conference news).
Two More Strikes Zachary Miller of UCSF presented data from a Phase 1 dose-finding study of nimodipine, an FDA-approved calcium channel blocker that enters the brain well. Because this compound raises progranulin levels in mice, researchers tested it on eight people who carry mutations in the progranulin gene and have half or less of the normal level of the protein in their blood and CSF. Participants had an average age of 57; four had developed symptoms of FTD. They took escalating doses of the drug for four weeks and then remained at their maximum tolerated dose for another four before tapering off. The researchers set the maximum dose at 360 mg/day, based on previous neurological uses of nimodipine.
This regimen appeared safe, with the main side effect being swelling in the hands and feet. Dizziness made one participant withdraw from the trial. However, the researchers saw no change in plasma or CSF progranulin at any dose. Other fluid biomarkers, including CSF Aβ42, tau, and NfL, also remained flat. One industry scientist in attendance asked whether the dose had been too low; but Boxer, who ran the trial, said it would not be feasible to give higher doses. The pills are large, difficult to swallow, and have to be taken every four hours due to the drug’s short half-life in the body. The negative results suggest this drug is finished as an FTD treatment, Boxer told Alzforum.
Data from the third trial were ambiguous. Edward Huey of Columbia University, New York City, described the effects of tolcapone on FTD patients. Tolcapone is approved for the treatment of Parkinson’s symptoms; it raises dopamine levels in the prefrontal cortex by inhibiting an enzyme called COMT. The drug improves executive function in healthy controls, suggesting it might benefit FTD patients, many of whom suffer deficits in this area (see Apud et al., 2007).
The researchers tested tolcapone in a Phase 2b trial of 28 patients with clinically diagnosed, symptomatic bvFTD. Participants either took the drug for two weeks and then switched to placebo for two weeks, or vice versa. While on drug, participants did improve their scores on the Clinical Global Impressions scale, the Neuropsychiatric Inventory, and the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS). Their treatment effect was statistically significant vis-à-vis their baseline measure; however, people on placebo improved somewhat over baseline, as well, and the tolcapone effect missed statistical significance compared to placebo. There was no change in scores on the N-back, a test of working memory, and researchers are still analyzing scans of brain connectivity.
What could this mean? Some people carry a polymorphism in the COMT gene, V158M, that substitutes a methionine for a valine and renders the enzyme less active, raising dopamine levels. People without this polymorphism, i.e., with two copies of the val allele and lower dopamine levels, tend to have worse executive function and smaller brain volumes in the dopamine system than their met/met peers. The val/val participants benefited most from tolcapone treatment, as expected. This difference showed up on the CGI and NPI, but not the RBANS, Huey noted. He conceded the RBANS results were disappointing. Tolcapone is not particularly well-tolerated, either, and better COMT inhibitors are being developed. Some of these might be worth testing, Huey suggested. Others agreed. “There were some measureable effects of tolcapone that might warrant further investigation with new COMT inhibitors that have fewer side effects,” Boxer wrote to Alzforum.
Despite the overall negative findings, researchers said these studies represent progress. “This shows trials can be run in FTD. The targets are there,” said Philipp von Rosenstiel of Biogen Idec in Cambridge, Massachusetts.
In the Pipeline
Many other therapeutics are in trials and expected to release findings soon. A Phase 1 study in 32 progressive supranuclear palsy (PSP) patients of C2N’s 8E12, an antibody to aggregated, extracellular tau, has just finished data collection. Boxer, who ran the trial, said results will be announced at the upcoming Clinical Trials on Alzheimer’s Disease conference December 8-10 in San Diego. Bristol-Myers Squibb has a similar antibody, BMS986168, which is in a Phase 1 study of 48 PSP patients and expected to conclude next year.
Meanwhile, a Phase 2 trial of HDAC inhibitor FRM-0334 in 30 FTD patients with progranulin mutations has also wrapped up. The treatment was designed to boost progranulin levels (see Nov 2014 conference news). Scientists at ICFTD2016 expressed hope that the data will come out this fall; however, the future of the compound hangs in doubt because the sponsor, FORUM Pharmaceuticals, closed its doors in June following the failure of its Phase 3 drug encenicline (see Xconomy story). The company was funded by FMR LLC, as is Alzforum.
Researchers are investigating other ways to boost progranulin. In Munich, Daniel Bittner of the DZNE presented preliminary human data indicating that the malaria medication chloroquine can rescue progranulin levels in people who carry a pathogenic mutation or have low progranulin levels (see Feb 2011 news). In three FTD patients with a progranulin deficiency who received chloroquine for a year, CSF progranulin levels rebounded to control levels, and CSF tau decreased. CSF progranulin and tau remained stable in seven untreated FTD patients and 60 AD patients. Larger studies may be warranted, Bittner concluded.
Ongoing clinical trials not discussed at ICFTD include a Phase 1 of the microtubule-stabilizer TPI 287 in patients with PSP or corticobasal degeneration, and Phase 1 studies in PSP of the NSAID salsalate and of plasma transfusions from young adults.
The conference did feature compounds about to enter human testing, though. Christoph Wiessner of Asceneuron SA, Lausanne, Switzerland, noted that his company will take its O-GlcNAcase inhibitor ASN-561, which lowers tau pathology, into Phase 1 in healthy volunteers in 2017 (see Aug 2014 conference news). If it proves well-tolerated, they will test the therapy in PSP patients. These people are particularly in demand for trials since they have a pure tauopathy and tend to decline quickly. However, the pool of potential participants is small, since most studies report a prevalence of about six PSP cases per 100,000 people, or about 400,000 cases worldwide (see Bower et al., 1997; Nath et al., 2001).
Bucking this trend, Norbert Zilka of Axon Neuroscience, Bratislava, Slovakia, announced that his company’s tau vaccine, AADvac1, currently in Phase 2 for Alzheimer’s, will enter a Phase 1 trial for primary progressive aphasia, a form of FTD (see Aug 2014 conference news). Estimates vary on how common tau pathology is in PPA, but tend to agree it is present in fewer than half the cases (see Knibb et al., 2006; Snowden et al., 2007).
Other treatments target specific disease symptoms. Because many patients with bvFTD lose their ability to empathize, the hormone oxytocin, which mediates compassion, affection, and social bonding, is being tried. A previous study reporting that a single dose of intranasal oxytocin improved recognition of facial emotions in people with FTD suggested it might counteract social deficits (see Jul 2012 conference news; Jesso et al., 2011). Oxytocin appeared safe in a small Phase 1 trial (see Nov 2014 conference news; Finger et al., 2015). At ICFTD, Elizabeth Finger of Western University, London, Canada, described plans for a Phase 2 trial in 100 FTD patients at four Canadian and 11 U.S. sites. It will start with a crossover design of six-week treatment intervals for dose finding; the second phase will advance the best dose. The primary outcome measure will be change on the NPI apathy/indifference score, with secondary measures to include recognition of facial emotions and caregiver distress. The trial will begin enrolling in January 2017, Finger said.
Meanwhile, Don Cleveland of the University of California, San Diego, advocated for directly shutting down pathogenic genes by targeting their RNA with antisense oligonucleotides (ASOs). ASOs bind to RNA messages and trigger their degradation (see Oct 2013 news; Nov 2015 conference news). Cleveland noted that this strategy appears to work in the childhood disorder spinal muscular atrophy, where a Phase 3 study in 110 infants recently met its endpoints at the halfway point (see press release). Biogen has now licensed the treatment from Ionis Pharmaceuticals and has applied for FDA approval. Ionis has initiated a new Phase 1 trial for ALS caused by mutations in the SOD gene (see Miller et al., 2013, for previous Phase 1 results).
Next year, Ionis plans to take an ASO targeting the C9ORF72 expansion to trial, Cleveland said. In mice with the expansion, a single injection at six months of age squelched production of the abnormal RNA and peptide, and delayed a cognition phenotype for six months (see Apr 2016 news). The expansion accounts for about one-quarter of familial FTD cases and half of familial ALS.
Many groups are gearing up to test therapies in people with the C9ORF72 expansion, and the first trials are expected to begin next year, Neumann told Alzforum.—Madolyn Bowman Rogers
Fluid NfL Shines, Tau PET Dims, in the Hunt for FTD Biomarkers
Biomarkers are the obsession du jour in frontotemporal dementia research. Over and over at the 10th International Conference on Frontotemporal Dementias, held August 31-Septemper 2 in Munich, speakers emphasized the need for new markers to enable research and trials. In a set of diseases bedeviled by both tremendous heterogeneity and small patient populations, biomarkers are must-haves for tracking how disease gets worse and selecting the right participants for trials. In Munich, the consensus was that the field has advanced more on the first goal than the second. Several presentations bolstered the case for the protein neurofilament light (NfL) in blood and cerebrospinal fluid to mark disease stage; NfL also appears to predict future decline. Growing evidence from longitudinal studies suggests that atrophy in key brain areas can flag how close a person is to symptoms. Such biomarkers may help researchers monitor trial participants and perhaps indicate drug effects, although that still has to be tested. “It’s critical to have biomarkers for rational dose finding in Phase 1,” noted Christoph Wiessner of Asceneuron SA, Lausanne, Switzerland.
On the other hand, diagnostic markers that reveal a person’s underlying protein pathology remain elusive. Without them, researchers cannot select the right patients for sporadic FTD trials and instead have to rely on a clinical diagnosis that includes people with similar symptoms but different underlying causes. For example, a person with behavioral variant FTD is about equally likely to harbor tau or TDP-43 pathology, but their clinical symptoms provide no clue which. “We urgently need to develop in vivo biomarkers,” said Ian Mackenzie of the University of British Columbia, Canada. Researchers hoped tau PET might fit the bill, but data at ICFTD made clear that existing tracers cannot discriminate tau and TDP-43 and give but a weak signal from FTD brains. Preliminary data hint that some fluid biomarkers might distinguish between forms of FTD.
Altogether, the research is opening a window into how frontotemporal dementia affects living brains, said Jason Warren of University College London. “The interest in biomarkers represents a paradigm shift for [studying] these diseases. We stand on the verge of a revolution in the treatment of FTD.”
NfL Tracks Degeneration.
CSF levels run high in people with FTD (top) and correlate with brain shrinkage (bottom) in presymptomatic carriers (blue) and FTD patients (orange). [Courtesy of John van Swieten.]
NfL Up and Coming as Progression Marker
Researchers at ICFTD hailed NfL as the most promising FTD biomarker to emerge to date. The protein leaks out of injured axons, and high levels in cerebrospinal fluid and blood appear to reflect the intensity of neurodegeneration in several disorders, including FTD, ALS, and Alzheimer’s disease (see Jun 2015 news; Jun 2016 news). While not specific for disease type, the marker tracks severity quite well, researchers said.
At ICFTD, speakers added new evidence that NfL can also predict progression. John van Swieten of Erasmus Medical Center, Rotterdam, The Netherlands, described findings from 174 Rotterdam participants, most of them in the Genetic Frontotemporal Dementia Initiative 1 (GENFI1). About half of those had symptoms of FTD, with the remainder split between asymptomatic gene carriers and healthy controls. CSF NfL levels in asymptomatic carriers resembled those in controls, while the marker ran eightfold higher in symptomatic participants, van Swieten said. NfL levels correlated with cortical atrophy and a loss of white-matter integrity in the brain (see Meeter et al., 2016). Moreover, in two people who developed symptoms over the course of the study, CSF NfL shot up fourfold over 18 months, suggesting the marker could herald imminent decline. More longitudinal data may help pin down exactly when NfL begins to crest, van Swieten said. In transgenic mice, NfL rises before symptoms appear, he noted. Likewise, data from the Advancing Research and Treatment for Frontotemporal Lobar Degeneration Consortium (ARTFL) also finds differences in NfL levels between asymptomatic carriers and controls, implying the marker precedes symptoms, Brad Boeve of the Mayo Clinic in Rochester, Minnesota, told Alzforum.
While CSF data are informative, clinicians would much prefer a marker they can measure in blood. Here, too, NfL performs well. As other groups had previously, van Swieten reported that serum NfL correlated closely with CSF levels. Julio Rojas of the University of California, San Francisco, added yet more evidence supporting blood measurements. He reported findings from a cohort of 15 people with progressive supranuclear palsy (PSP) and 12 controls. The researchers measured NfL in plasma using a single-molecule array (Simoa), an ultrasensitive technique that employs a digital readout to detect minute quantities (see Apr 2016 conference news). Plasma NfL ran twice as high in cases as controls, with a cutoff of 20 pg/ml providing 80 percent sensitivity and specificity for discriminating the groups, Rojas said (see Rojas et al., 2016).
Rojas and colleagues validated the findings in an independent cohort of 147 PSP patients. Half of this group had very high plasma NfL and half lower, although all were above control levels. Higher NfL predicted worse cognitive, functional, and motor decline one year later, as well as more brain atrophy. Plasma NfL remained stable over that year, and correlated closely with CSF levels. “Tracking blood NfL is feasible, and may support prognosis,” Rojas concluded.
Others at ICFTD reported similar findings for blood measurements. Ione Woollacott of University College London presented data from 74 FTD patients and 28 healthy controls. Serum NfL ran about four times higher in patients than controls. Levels varied widely among individuals, however, and correlated with how quickly their frontal lobe was shrinking (see Rohrer et al., 2016). “NfL is the first good blood biomarker in FTD,” first author Jonathan Rohrer of UCL told Alzforum.
Beyond NfL, researchers are searching for additional fluid markers of progression. MicroRNAs have been found to change in frontotemporal lobar degeneration, and the pattern can vary based on the subtype of disease or the underlying genetics (see Kocerha et al., 2011; Freischmidt et al., 2014). In Munich, Maria Carmela Tartaglia of the University of Toronto reported finding seven microRNAs that were reduced in CSF in 16 people with familial FTLD compared with 19 presymptomatic carriers and seven controls in the GENFI1 cohort. Most carried progranulin mutations. The microRNAs appear to regulate molecular pathways that previously have been implicated in this form of the disease, she told the audience. In a similar vein, van Swieten described a mass spectrometry study of CSF from two Dutch progranulin families. He turned up multiple proteins that distinguished between symptomatic and asymptomatic carriers, including four complement factors and two synaptic proteins. All of these preliminary markers remain to be validated.
Brain Atrophy Marks Progression. In the GENFI genetic cohort, people who carry a C9ORF72 expansion show accelerated atrophy (yellow) in some brain regions up to 10 years before developing symptoms. [Courtesy of Jonathan Rohrer.]
Structural Imaging May Flag Progression Too
What about brain volumetric imaging? Could it pick out the people most at risk of progressing to FTD? Early indications at ICFTD suggest yes. Martina Bocchetta of UCL presented MRI data from 56 asymptomatic mutation carriers in the GENFI1 cohort. Those who were more than 10 years away from their expected age of onset resembled age-matched controls. However, the brain began to shrink in people within 10 years of their expected onset age. In particular, the researchers saw rates of about 1 percent decline per year in the temporal lobe, cingulate, amygdala, and striatum in this group (see image above). In controls of the same age, these regions remained stable. The findings suggest there might be an atrophy signature that could pick out the people closest to progressing, who might be the best candidates for a clinical trial. “We may be able to use structural imaging [as a biomarker] in trials at that late preclinical period,” Rohrer, who led the study, told Alzforum.
A different progression marker came from the work of Lize Jiskoot, also of Erasmus Medical Center. She analyzed four years of MRI and clinical data from 25 progranulin mutation carriers, nine MAPT mutation carriers, and 29 controls in the Rotterdam Risk Cohort, which is part of GENFI. All were asymptomatic at the start, and thus far three of each genotype have developed FTD symptoms over the course of the study. The researchers saw the cortex shrink faster in MAPT than in GRN carriers. In addition, the four people who developed bvFTD, but not the two who were diagnosed with progressive non-fluent aphasia, lost white matter in the right uncinate fasciculus before symptoms developed, regardless of genotype. This tract connects frontal portions of the salience network, which becomes disrupted in FTD (see Nov 2014 conference news). The findings suggest there may be mutation- and diagnosis-specific atrophy patterns, Jiskoot said.
Howard Rosen of UCSF took a different tack. Instead of presymptomatic carriers, he analyzed MRI images obtained over two years from 44 people with clinical diagnoses of bvFTD, 30 with semantic-variant primary progessive aphasia, and 26 with non-fluent variant primary progressive aphasia, as well as 97 age-matched controls. He asked which brain regions changed the most over time in each disorder and found a different pattern in each. The bvFTD group showed shrinkage in the anterior caudate, insula, and medial frontal and parietal cortex, people with nfPPA lost volume in the perirolandic and periinsular cortices, and those with svPPA in the inferior temporal lobe (see Pankov et al., 2016). An analysis found that these regions detected longitudinal change better than either more commonly used brain regions or clinical measures, Rosen said. If the findings hold up in independent samples, researchers using these regions might need fewer participants in drug trials to see a significant effect, he believes.
Tau PET Varies by Diagnosis.
PET tracer AV1451 lights up different brain regions in a person with bvFTD caused by a MAPT mutation (right) than in a person with corticobasal degeneration (left), matching expected patterns of tau deposition. [Courtesy of Richard Tsai.]
Fading Hope for Tau PET Leaves Differential Diagnosis Difficult as Ever
While progression markers show promise, the field appears stalled for the moment on its goal of biomarkers that reveal an FTD patient’s underlying pathology. Without that, researchers will remain unable to run effective clinical trials in sporadic bvFTD, said conference co-organizer Manuela Neumann of the German Center for Neurodegenerative Diseases (DZNE). She noted that a clinical diagnosis of bvFTD is wrong half of the time. Even when correct, people with this diagnosis are equally likely to have tau or TDP-43 deposits, and some can have yet other pathologies. FTD researchers had hoped tau PET would distinguish these groups, but numerous presentations at ICFTD emphasized that existing tau tracers work poorly for FTLD. “The tau PET data was the biggest disappointment of the conference,” said Christian Haass of Ludwig-Maximilians University, Munich, expressing a common sentiment.
Existing tracers bind well to the type of tau deposits found in Alzheimer’s disease, which are composed of paired helical filaments made up of 3R and 4R tau strands. Most tau pathology in FTLD, however, consists of either pure 4R or pure 3R tau and, for unknown reasons, these don’t bind the current tracers well. In Munich, Boeve reported that five people with the N279K MAPT mutation, who are known to make 4R tau, weakly bound the tracer AV1451, with intensities falling within the range of binding in healthy control brains. By contrast, two people with the S305N MAPT mutation, who make 3R + 4R tau tangles, took up tracer at levels comparable to AD patients. In an overview talk, Brad Dickerson of Massachusetts General Hospital noted that the tracer THK5351 gives a slightly higher signal in 4R tauopathies than does AV1451, but still far below that seen in AD.
Moreover, AV1451 and THK5351 bind to things other than tau. They recognize still-unknown targets in the midbrain and basal ganglia in particular, confounding the signal from some types of FTLD that feature neurofibrillary pathology in those regions, such as PSP and corticobasal syndrome (CBS). Even more problematic, existing ligands light up brains that contain TDP-43 deposits. Thus, they cannot discriminate these two forms of the disease, Boeve noted. In addition, in autoradiography studies on postmortem tissue samples from FTLD brain, current tracers bind even more poorly than they do in living tissue. This puzzles researchers. Dickerson called the autoradiography data “underwhelming.”
Are the existing tracers useful at all in FTD? Richard Tsai of the UCSF Memory and Aging Center reported that AV1451 uptake matched the expected patterns of tau or TDP-43 pathology in three bvFTD, 10 PSP, nine CBS, and six PPA patients, as well as six FTD mutation carriers and 24 controls (see image above). Corey McMillan of the University of Pennsylvania, Philadelphia, analyzed data from an AV1451 imaging study sponsored by AV1451’s maker, Avid Radiopharmaceuticals. He reported that an AV1451 uptake cutoff of 1.71 SUVR discriminated between 20 PSP patients and 20 controls with a sensitivity and specificity of 80 percent. However, binding was too weak to allow individual diagnosis, McMillan said, and CBS patients did not take up the tracer at all. Jennifer Whitwell of the Rochester Mayo Clinic reported a different pattern of AV1451 binding in logopenic PPA compared with semantic and agrammatic PPA, suggesting PET scans could help differentiate among the different aphasias. In one of the few studies examining THK5351, Sonja Schönecker of Ludwig-Maximilians University reported that this tracer differentiated 10 PSP patients from healthy controls and correlated with clinical severity.
Overall, the data suggest that AV1451 and THK5351 might be useful in clinical trials to show target engagement and determine dosing, but are probably not helpful diagnostically, concluded Adam Boxer of UCSF during a panel discussion. Other companies such as Merck, Genentech, and Roche are developing tau tracers, but did not present at ICFTD (see Feb 2015 conference news; Aug 2016 conference news).
Better Luck with Fluid Markers?
Could biomarkers in CSF or blood become diagnostic? At ICFTD, presentations hinted at some discriminatory power, but so far no marker has broken through as a leading candidate for the non-genetic forms of FTLD. David Irwin of UPenn probed whether CSF p-tau might do the job. He used data from people with FTLD who had undergone a lumbar puncture during life and donated their brains after death. Among 28 people with tau deposits, CSF p-tau ran about 50 percent higher than it did in 45 people with TDP-43 pathology. The difference became even more significant when Irwin screened out 14 cases with comorbid Alzheimer’s pathology. When combined with an Alzheimer’s screen, CSF p-tau might help distinguish these forms of FTLD during life, Irwin suggested.
Rojas presented similar data, reporting that FTLD patients with autopsy-confirmed TDP-43 deposits had about half the CSF p-tau/total tau ratio, and more than twice as much CSF NfL, as did patients with tau pathology. An NfL readout above 6,757 pg/ml and a tau ratio below 0.28 discriminated the two disorders with more than 80 percent sensitivity and specificity in a cohort of 37 patients, Rojas found.
Others focused on new markers. Charlotte Teunissen of VU University Medical Center, Amsterdam, had previously reported that a combination of the inflammatory markers YKL-40, MFGE8, and catalase discriminated 12 patients with FTLD-TDP from eight patients with FTLD-tau (see Teunissen et al., 2016). In Munich, Marta del Campo in Teunissen's group presented the results of replication in two cohorts, one from Emory University and one from Milan University, containing a total of 30 FTLD-TDP patients, 20 FTLD-tau, 29 controls, and 66 with other dementias. The combined biomarkers distinguished the FTLD-TDP cohort from controls with better than 80 percent sensitivity and specificity, suggesting this might become useful to diagnose this form of the disease.—Madolyn Bowman Rogers
Freischmidt A, Müller K, Zondler L, Weydt P, Mayer B, von Arnim CA, Hübers A, Dorst J, Otto M, Holzmann K, Ludolph AC, Danzer KM, Weishaupt JH.
Serum microRNAs in sporadic amyotrophic lateral sclerosis.
Neurobiol Aging. 2015 Sep;36(9):2660.e15-20. Epub 2015 Jun 9
PubMed.
Tests of Social Cognition Hold Potential as FTD Outcome Measures
As frontotemporal dementia researchers gear up for trials, high up on their wish lists are better clinical outcome measures to detect change over time more sensitively than standard cognitive tests do. At the 10th International Conference on Frontotemporal Dementias, held August 31-September 2 in Munich, researchers discussed possibilities, including an old informant-based test of social cognition and a new social cognition battery. Both are being evaluated in ongoing cohort studies. Other presenters discussed innovations in measuring the functional deficits that are specific to FTD, often done in combination with structural or functional imaging that ties a deficit to degeneration and connectivity loss in particular brain regions. Attendees were excited about the potential of these tests to flag common behavioral deficits of FTD.
“Hopefully we can start to use social cognition measures. Right now we’re missing key aspects of the disease,” said Edward Huey of Columbia University, New York City.
Currently, most FTD trials use cognitive or functional measures such as the MMSE, CDR, and the ADCS-CGIC. However, these were designed for Alzheimer’s disease, with its primary features of impaired executive function and memory. That is inadequate because the primary symptoms of FTD are behavioral or affect different cognitive domains than AD does. In fact, Everard Vijverberg of VU University Medical Center, Amsterdam, reported in Munich that bvFTD patients perform better than psychiatric patients in tests of executive function and verbal and working memory.
Specific for FTD.
On the SEA social cognition battery, individual FTD patients (red) score lower than most AD patients (blue) and all controls (green). Each dot represents a one-time measurement of an individual person. [Courtesy of Bruno Dubois and Aurélie Funkiewiez-Guignebert.]
Many researchers are exploring measures of social cognition as more specific outcome markers for FTD. In early FTD, personality changes above all else. FTD patients typically lose social skills and become insensitive to other people’s emotions. They can become unfiltered, speak rudely to strangers, or miss social cues and the meaning of jokes.
Katherine Rankin of UCSF reminded the audience of one measure to quantify changes of this sort, the Revised Self-Monitoring Scale (RSMS) developed more than 30 years ago (see Lennox and Wolfe, 1984). On this 13-item questionnaire, an informant rates a person’s ability to modulate his or her behavior in various social situations. Rankin said this questionnaire performs well, with a normal distribution and a wide range of scores suggesting it is unlikely to be hobbled by floor or ceiling effects. Scores on the RSMS start to dip in asymptomatic mutation carriers and drop steadily throughout the course of FTD, Rankin said.
Worsening scores also predict a loss of connectivity in the salience network. The longitudinal studies GENFI, ARTFL, LEFFTDS, and the National Alzheimer’s Coordinating Center’s FTLD Module are currently evaluating the RSMS using their respective data sets. Other researchers were cautiously optimistic about it, but said it needs further validation.
In the long run, a battery of tests may perform better than any single one, several researchers said. “It’s unlikely there will be one magic bullet,” Jonathan Rohrer of University College London told Alzforum. Bruno Dubois of Pierre and Marie Curie University, Paris, developed a battery of five subtests to evaluate prefrontal functions. Called Social Cognition and Emotional Assessment, or SEA, this battery in initial testing separated FTD patients from AD patients and controls with a specificity of 89 percent (see Funkiewiez et al., 2011). GENFI, ARTFL, and LEFFDTS use a shorter version of it, the mini-SEA, which includes only the tests of facial emotion recognition and the ability to recognize a social faux pas (see Bertoux et al., 2012). The cohort studies will gather data on how well the battery performs longitudinally, said Brad Boeve of the Mayo Clinic in Rochester, Minnesota.
Numerous other talks discussed exploratory clinical measures. David Perry of UCSF reported that people with bvFTD show less aversion to unpleasant smells than controls do. This behavioral deficit reflects atrophy in the insula and amygdala, not the anosmia known to occur early in AD, PD, and DLB. People with FTD do smell the garbage left out in the heat—they are simply less bothered by it. Charles Marshall of University College London described dulled reaction to facial emotions in people with bvFTD compared with controls, as measured by facial electromyography. This characteristic correlated with atrophy of several brain regions, particularly the supplementary motor cortex. Other speakers presented evidence that tests of social cognition can distinguish between bvFTD patients and those with semantic dementia, or between people with MAPT mutations and GRN mutations.
Some researchers are thinking further afield in their search for ways to measure FTD. Jason Warren of UCL reviewed measures that can flag disruption of specific processes (see Nov 2014 conference news). For example, people with bvFTD can seem quite impervious to pain, heat, and cold, while people with semantic dementia appear more sensitive (see Fletcher et al., 2015). People with bvFTD respond less than controls to pictures evoking emotion, as measured by skin conductance and heart rate (see Balconi et al., 2015).
Electrophysiological measures, such as transcranial magnetic stimulation, reveal impaired cortical inhibition and plasticity in people with FTD, in some cases before symptoms develop (see Burrell et al., 2011; Benussi et al., 2016). Task fMRI uncovers differences in how FTD patients process music and think about moral dilemmas (see Agustus et al., 2015; Chiong et al., 2013).
All these data are preliminary. While researchers sounded excited about the potential of these measures, they emphasized that work remains to develop and validate them. Warren noted that these potential markers should be evaluated in large longitudinal cohorts to figure out which ones are both useful and robust for widespread use.—Madolyn Bowman Rogers
Researchers know the strongest genetic factors that cause frontotemporal dementia and amyotrophic lateral sclerosis: the genes for tau, progranulin, and the C9ORF72 repeat expansion. However, just as in the Alzheimer’s field, a majority of the population genetic risk remains unexplained and may be due to rare genes or ones with small effect sizes. To root them out, geneticists are analyzing affected families and using new statistical techniques. At the 10th International Conference on Frontotemporal Dementias, held August 31-September 2 in Munich, researchers chronicled their incremental progress toward unraveling the remaining genetic signals. There were no big “finds,” but speakers did identify a few new rare genes, including one on chromosome 16 that generated some buzz. Many presentations focused on modifying factors rather than causal ones, discussing genes that affect the onset age or clinical symptoms of the disease. In panel discussions, researchers agreed that the next step is to figure out how genetic factors contribute to disease, so that better therapies targeting these mechanisms can be developed.
Latest FTD Gene Highlights Autophagy.
Geneticists linked a variant in CYLD, a deubiquitinase that slows autophagy, to familial frontotemporal dementia. [Courtesy of Molecular Cell, Komander et al., 2008]
New Genetic Signals
Linkage analysis of affected families helps researchers pinpoint genes. Carol Dobson-Stone of Neuroscience Research Australia, Sydney, described one such study in an Australian family with a history of dementia and motor neuron disease that spanned four generations. Affected family members had both TDP-43 deposits and tangles of 4R tau in their brains. Linkage analysis revealed a region on chromosome 16 that segregated with the disease. To narrow in on the gene involved, researchers performed whole-exome sequencing on five family members. In the region of interest they found a single variant in the CYLD gene that always segregated with the disease and never appeared in unaffected family members. Variants in CYLD appear to be quite rare, as analysis of an additional 442 people with FTD or motor neuron disease turned up only one additional person with a functional CYLD variant.
CYLD encodes a protein that cleaves ubiquitin chains at their lysine 63 residue. This site promotes a given substrate protein’s degradation by autophagy, so by removing the chains, CYLD puts the brakes on disposal (see Komander et al., 2008; Ferreira et al., 2015). A loss-of-function mutation in CYLD, D681G, leads to more protein degradation and causes the skin disorder cylindromatosis (see Almeida et al., 2008). However, the new FTD variant, M719V, boosts CYLD’s deubiquitinase activity fourfold, Dobson-Stone said. Heightened activity of CYLD in bladder cancer cells has been associated with weakened autophagy (see Yin et al., 2016). That suggests the M719V variant might lead to a buildup of aggregated protein, she speculated, adding, “This reinforces the importance of the autophagy pathway in FTD.”
In addition to its role in autophagy, CYLD also inhibits the activation and nuclear translocation of the transcription factor NFkB. The variant M719V shuts down NFkB more effectively than does its wild-type counterpart, Dobson-Stone noted. Other researchers found this a particularly intriguing potential mechanism for FTD. Finally, Dobson-Stone found that CYLD physically interacts with the protein products of several other FTD risk genes, including OPTN, TBK1, and SQSTM1.
A different linkage site came out of the work of Rita Cacace of the VIB at University of Antwerp, Belgium. A previous study by the group, led by Christine Van Broeckhoven, pointed to a region on chromosome 7q36 in a Dutch family with Alzheimer’s disease (see Rademakers et al., 2005). To pin the gene there, Cacace sequenced the whole genome of four affected family members. She found a four megabase inversion that segregated with disease and disrupted the gene dipeptidyl peptidase 6 (DPP6). To validate the association, the researchers screened 453 FTD, 124 ALS, 335 AD patients, and 755 controls. They found significantly more DPP6 rare and loss-of-function variants among the FTD group than in controls. Alzheimer’s patients tended to have more DPP6 variants as well, but the finding missed significance.
DPP6 increases expression and regulates the function of the potassium channel Kv4.2, which controls the excitability of hippocampal neurons. FTD patients carrying the variants made less DPP6 and less Kv4.2. “Loss of function variants in DPP6 may lead to synaptic hyperexcitability,” Cacace suggested. DPP6 was previously linked to sporadic ALS, but failed to replicate in a large GWAS (see Dec 2007 news; Feb 2009 news; Sep 2009 news).
Researchers are also using new genetic techniques to turn up fresh associations. Aniket Mishra of VU University, Amsterdam, re-analyzed GWAS data from 3,526 FTD patients and 9,402 controls (see Ferrari et al., 2014), but instead of looking for linkage with single SNPs, he tested the set of all markers within a given gene for association with FTD. This method pumps up the statistical power to find associations (see de Leeuw et al., 2015).
In this way, Mishra and colleagues identified variants in two genes—ARHGAP35 and SERPINA1—that associated with progressive non-fluent aphasia (PNFA). SERPINA1 is a secreted protease inhibitor found only in peripheral blood and liver, not the central nervous system. However, it enables circulating glucocorticoids to enter the brain, Mishra noted. The risk allele boosts SERPINA1 protein levels, perhaps increasing stress signaling in the brain, he suggested. Meanwhile, ARHGAP35, which is expressed in the brain, binds to glucocorticoid receptor genes and represses their transcription, thus dampening glucocorticoid signaling. Together, these variants suggest that stress signaling may play a role in PNFA, Mishra said.
Tracking Down Factors That Influence Disease
Many genes with weak effects do not trigger disease by themselves, but instead may hasten or delay its onset, or affect how fast it moves. Cumulatively, such genes may wield a large effect. For example, carriers of GRN mutations and C9ORF72 expansions display great variability in when and whether disease develops. Several researchers at ICFTD shed light on this by reporting numerous modifying factors. Protective factors may be particularly helpful in highlighting potential therapeutic targets, researchers noted.
Cyril Pottier of the Mayo Clinic in Jacksonville, Florida, focused on GRN mutation carriers. Pottier and colleagues analyzed DNA from 491 carriers of 113 different variants, as well as 1,173 matched controls. All were Caucasian, unrelated to each other, and had FTD symptoms whose onset ranged from 39 to 87 years. Two SNPs appeared connected with onset age. One was near TNIK, aka TRAF2 and NCK-interacting kinase, a gene that regulates dendrite growth and synaptic transmission. The other was near RRBP, ribosome-binding protein 1, which regulates the transport and secretion of intracellular proteins.
The researchers also identified an SNP in TMEM106B that affected how likely someone with a GRN mutation was to get the disease. Other SNPs around this transmembrane protein have been previously associated with FTLD-TDP due to GRN and C9ORF72 mutations, although the functional variant is unknown (see Feb 2010 news; Aug 2012 news; Feb 2015 news). The new variant, rs6966915, seems to protect people against the disease. About 14 percent of controls carried two copies of the protective T allele, but only 1 percent of GRN carriers did, Pottier reported. In them, the allele delayed onset age by a decade, with TT carriers typically developing symptoms in their 70s instead of their 60s. Pottier is attempting to replicate the association in a larger cohort, including sporadic FTLD patients with TDP-43 pathology and familial patients without known mutations.
New information on a different protective allele of TMEM106B came from Nina Rostgaard of the Danish Dementia Research Centre, Copenhagen. She analyzed DNA from 80 members of a large Danish family with inherited FTD due to a CHMP2B mutation. Of this group, 19 had symptoms, 14 were asymptomatic carriers, and 47 were unaffected family members. CHMP2B carriers who had the minor C allele at SNP rs3173615 of TMEM106B developed symptoms about five years later on average than those without the SNP. This variant was previously reported to protect GRN and C9ORF72 carriers from disease. The new findings imply that TMEM106B modifies FTD across its multiple causative genotypes, Rostgaard said.
In a similar vein, Elisa Rubino of the University of Turin, Italy, dug into the effect of repeat expansions in the ataxin-2 gene. Healthy people have 23 or fewer repeats, while 34 or more repeats cause spinocerebellar ataxia type 2. Between these extremes, intermediate expansions are known to be a risk factor for ALS and FTD/ALS, but have not been associated with pure FTD (see Aug 2010 news; Sep 2014 news). Rubino analyzed 243 sporadic FTD patients with a mean age of 65, as well as 176 controls. She found no difference between patients and controls in how common ataxin-2 expansions were. However, FTD patients who had intermediate expansions of 27 to 33 repeats developed disease earlier than repeat non-carriers, and were four times more likely to display symptoms of parkinsonism. Ataxin-2 repeats may affect the clinical features of FTD, Rubino concluded.
Researchers also highlighted new genes that influence when disease strikes. Raffaele Ferrari of University College London and Mario Grassi of the University of Pavia, Italy, described an association analysis of 411 FTD patients from the Italian Network on FTD. The analysis found six SNPs that, when they were summed in a single genetic score, lowered the average age of onset in patients by about four years. The SNPs were found near the genes NRXN, HPCAL1, PTPRD, and GPR137B, which are involved in processes such as neuronal development, myelination, and synaptic transmission. The effect of the genes was independent of which ApoE allele they carried. Furthermore, the researchers’ analysis suggested that about 14 percent of the variability in age of onset of FTD might be accounted for by genetic factors. That figure is much higher than what has been calculated for Alzheimer’s disease, where ApoE alone accounts for almost four percent of the variability, and other genes together about 2 percent, and Parkinson’s disease, where genes are estimated to account for only 0.6 percent of variability in onset age, he noted (see Nov 2014 news; Lill et al., 2015).
With this steady stream of tidbits of information on an ever-growing list of genes, researchers hope to make inroads in understanding the processes that affect disease. “Genetics provides the first entry to new biology,” noted Stephan Züchner of the University of Miami. However, scientists at ICFTD agreed that finding causative and modifying variants is but a first step. To emphasize the importance of subsequent functional studies, Christian Haass of Ludwig-Maximilians University, Munich, referenced a line from Sydney Brenner’s 2002 Nobel Prize lecture, where Brenner noted that genomics discoveries can generate a collective state of “drowning in a sea of data and starving for knowledge.” Haass suggested that geneticists go after the big fish in this sea by prioritizing genes with the strongest effects for follow-up. Those are the ones most likely to have cellular effects, he said.—Madolyn Bowman Rogers
Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB, Dobson-Stone C, Brooks WS, Schofield PR, Halliday GM, Hodges JR, Piguet O, Bartley L, Thompson E, Haan E, Hernández I, Ruiz A, Boada M, Borroni B, Padovani A, Cruchaga C, Cairns NJ, Benussi L, Binetti G, Ghidoni R, Forloni G, Galimberti D, Fenoglio C, Serpente M, Scarpini E, Clarimón J, Lleó A, Blesa R, Waldö ML, Nilsson K, Nilsson C, Mackenzie IR, Hsiung GY, Mann DM, Grafman J, Morris CM, Attems J, Griffiths TD, McKeith IG, Thomas AJ, Pietrini P, Huey ED, Wassermann EM, Baborie A, Jaros E, Tierney MC, Pastor P, Razquin C, Ortega-Cubero S, Alonso E, Perneczky R, Diehl-Schmid J, Alexopoulos P, Kurz A, Rainero I, Rubino E, Pinessi L, Rogaeva E, St George-Hyslop P, Rossi G, Tagliavini F, Giaccone G, Rowe JB, Schlachetzki JC, Uphill J, Collinge J, Mead S, Danek A, Van Deerlin VM, Grossman M, Trojanowski JQ, van der Zee J, Deschamps W, Van Langenhove T, Cruts M, Van Broeckhoven C, Cappa SF, Le Ber I, Hannequin D, Golfier V, Vercelletto M, Brice A, Nacmias B, Sorbi S, Bagnoli S, Piaceri I, Nielsen JE, Hjermind LE, Riemenschneider M, Mayhaus M, Ibach B, Gasparoni G, Pichler S, Gu W, Rossor MN, Fox NC, Warren JD, Spillantini MG, Morris HR, Rizzu P, Heutink P, Snowden JS, Rollinson S, Richardson A, Gerhard A, Bruni AC, Maletta R, Frangipane F, Cupidi C, Bernardi L, Anfossi M, Gallo M, Conidi ME, Smirne N, Rademakers R, Baker M, Dickson DW, Graff-Radford NR, Petersen RC, Knopman D, Josephs KA, Boeve BF, Parisi JE, Seeley WW, Miller BL, Karydas AM, Rosen H, van Swieten JC, Dopper EG, Seelaar H, Pijnenburg YA, Scheltens P, Logroscino G, Capozzo R, Novelli V, Puca AA, Franceschi M, Postiglione A, Milan G, Sorrentino P, Kristiansen M, Chiang HH, Graff C, Pasquier F, Rollin A, Deramecourt V, Lebert F, Kapogiannis D, Ferrucci L, Pickering-Brown S, Singleton AB, Hardy J, Momeni P.
Frontotemporal dementia and its subtypes: a genome-wide association study.
Lancet Neurol. 2014 Jul;13(7):686-99.
PubMed.
New Data Reinforces Concept of Protein Propagation
Research into frontotemporal dementias has powered forward in the last few years, propelled by a new understanding of the underlying biology of this group of diseases. Nonetheless, one major roadblock is the lack of good animal models. At the 10th International Conference on Frontotemporal Dementias, held August 31-September 2 in Munich, Virginia Lee of the University of Pennsylvania, Philadelphia, presented new data on an inducible model of TDP-43 pathology. It provided more clues to how the brain becomes less resilient to FTD with age, and generated excitement among attendees. Meanwhile, researchers at Ludwig-Maximilians University, Munich, debuted a new model of TREM2 deficiency. Other speakers focused on deciphering when disease starts and how it progresses, in particular on how pathologic proteins spread through the animal brain. Data from the UPenn group support the idea that misfolded proteins propagate, corrupting native protein along the way. Other scientists added human data on a vulnerable cell type in behavioral variant FTD, and highlighted different patterns of progression in bvFTD and amyotrophic lateral sclerosis (ALS). Researchers were hopeful that a deeper understanding of molecular mechanisms will one day lead to better treatments.
Building a Better Mouse Model
TDP-43 proteinopathy, seen in half of FTD and nearly all ALS cases, has been difficult to model in mice. Knockouts die because the protein plays an essential role in development, and overexpression models poorly reflect human disease (see Sep 2012 news). Lee and colleagues at UPenn addressed these problems by generating mice with an inducible TDP-43 transgene in neurons only. Lacking a nuclear localization signal, the transgenic TDP-43 aggregates in the cytoplasm. After induction of this transgene at five weeks of age, the mice rapidly formed TDP-43 deposits and lost endogenous nuclear TDP-43.
They developed a selective loss of certain motor neurons, and died at 13 to 15 weeks old. That said, when researchers switched off the transgene after six weeks, the mice recovered. The effect was dramatic. Remaining motor neurons re-innervated previously wasting muscle, and the mice lived to a normal age. This work is published (see Nov 2015 conference news; Walker et al., 2015; Spiller et al., 2016).
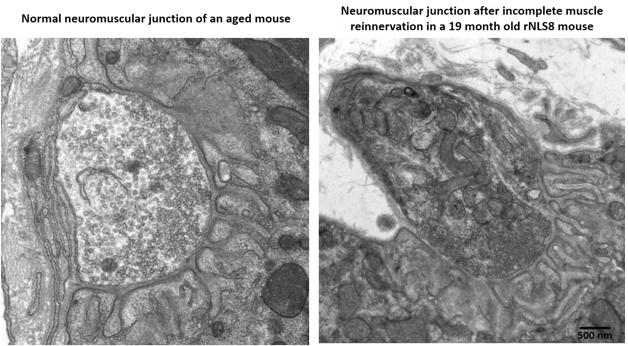
Aged mice recover poorly after a TDP-43 transgene switches off, with partially innervated, disorganized neuromuscular junctions (right) compared to age-matched controls (left). For pictures of recovering young transgenic mice, see upcoming paper. [Courtesy of Krista Spiller and Virginia Lee.]
In Munich, Lee fleshed out this picture with fresh data. She reported that young mice still recovered well even after eight weeks of transgene expression, when they were at death’s door. Her group stressed mice by cycling the transgene on and off every two weeks for six cycles before turning it on permanently. Mice remained healthy during the gene cycling, and followed a normal disease course after the final induction. However, these mice started as adolescents, whereas people do not develop this disease until late in life. To model this, the researchers induced the transgene in middle-aged mice more than a year old. The older mice developed the disease as rapidly as their younger littermates, but they declined faster and died sooner, at four to six weeks after induction. Middle-aged mice tolerated the gene cycling as well as the young ones. But when the TDP-43 transgene was switched off after six weeks, the older mice did not recover as well as young mice did. Surviving motor neurons only partially re-innervated muscle, and their limbs remained weak. Exactly why the older mice recovered poorly is unclear, but Lee noted that their neuromuscular junctions appeared less organized than did those in young, healthy animals after re-innervation (see image above; paper in press).
Researchers were intrigued that TDP-43 had to be permanently active in order to trigger neurodegeneration and death. They said this may help illuminate how the protein damages aging brain. “This model looks promising,” said Manuela Neumann of the German Center for Neurodegenerative Diseases (DZNE). Brad Boeve of the Mayo Clinic in Rochester, Minnesota, called the talk “one of the hottest things” he heard at the conference.
Researchers also want knock-in mouse models of TREM2, the microglial receptor whose variants inflate the risk of FTD, ALS, and AD (see Oct 2012 news; Oct 2013 news; Feb 2014 news). In Munich, Gernot Kleinberger of Ludwig-Maximilians University described the generation of knock-in T66M mice. In people, this mutation lowers levels of the protein; it causes an autosomal recessive early onset dementia when homozygous and predisposes to FTD when heterozygous. Previous studies by senior author Christian Haass at LMU found that this variant becomes stuck inside cells rather than trafficking to the surface membrane (see Apr 2015 conference news).
In keeping with this, Kleinberger reported that the knock-in T66M mice make almost no soluble TREM2, which is produced by cleavage at the cell surface, but do accumulate immature TREM2 inside microglia and bone marrow-derived macrophages. The mice live and make healthy bones, he noted. However, their midbrain develops with a delay and, starting at six months, less blood flow reaches their brains. After one year of age, the mice’s microglia are less activated than those in controls, as seen by PET imaging. In addition, bone marrow-derived macrophages isolated from the knock-in brains poorly phagocytose bacteria and fibrillary Aβ in vitro. Several studies report that TREM2 aids microglial activation and phagocytosis (see Feb 2015 news; Jul 2016 news). Kleinberger is currently collecting behavioral data. Because this model includes many features seen in people with TREM2 mutations, it might be valuable for testing drugs targeting the receptor, he suggested.
Identifying Disease Beginnings
Having better mouse models will enable researchers to investigate how disease begins and progresses. Many scientists now believe that misfolded, pathologic proteins spread along anatomic connections in numerous neurodegenerative diseases, though the concept has also been challenged (see Apr 2015 conference news; Jan 2016 news; Apr 2016 webinar). At ICFTD, speakers added new evidence for this process in FTD, some from animal models and some from postmortem studies.
John Trojanowski described data from his and Lee’s labs at UPenn. Previously, they had reported that injecting either synthetic tau fibrils or pathological tau isolated from AD brain into transgenic tau PS19 mice jump-started tau pathology, with greater verisimilitude to AD-like tangles than seen in aged, uninjected PS19s (see Dec 2014 conference news; Peeraer et al., 2015; Iba et al., 2015). In Munich, Trojanowski reported that injecting tau extracts from AD brain into wild-type mice also gave rise to AD-like tau pathology, although a milder one than in the transgenics. The new tangles were composed of endogenous mouse tau, and spread from the injection site to anatomically connected brain regions. Importantly, injecting AD tau extracts into a tau knockout mouse did not produce pathology. The data confirm that misfolded tau proteins can act as proteopathic seeds and recruit native versions, even at endogenous levels, and they strengthen the idea that tau pathology could propagate in this way through human brain, Trojanowski said. Christian Haass of Ludwig-Maximilians University, Munich, who was not involved in the work, was impressed by this evidence of propagation in a wild-type mouse.
TDP Exodus.
In an FTD patient, TDP-43 (brown) flees the nucleus and gathers in cytoplasmic speckles in VEN, aka spindle cells (long vertical cells). Top and leftmost one show speckles, bottom has normal nuclear TDP. [Courtesy of Bill Seeley.]
But how does disease begin in a person? Bill Seeley of the University of California, San Francisco, described a working model that posits a tug of war between natively folded and misfolded forms of proteins. People have intrinsic regional vulnerabilities to protein misfolding based on their genetics, lifestyle, and other factors. Pathology expresses itself first in the most vulnerable neurons, then leaks out of this epicenter into neighboring cells along network connections, Seeley believes. In behavioral variant FTD, the most vulnerable neurons appear to be von Economo neurons (VENs, aka spindle cells) and fork cells found in the anterior cingulate cortex and frontal insula, Seeley said. He had previously reported that fork cells and VENs selectively die in bvFTD (see Kim et al., 2012).
These cells are large layer 5 projection neurons, similar to the corticospinal neurons that degenerate in ALS, he noted. Regions containing the VENs and fork cells form part of the salience network, which sputters in people with bvFTD. To investigate their vulnerability, Seeley and colleagues analyzed postmortem tissue from 17 people who had had bvFTD and/or ALS, and 10 age-matched controls. They found that more than one-quarter of VENs and fork cells in FTD patients harbored TDP-43 inclusions, while only 8 percent of neighboring layer 5 cells did. Deposits were present in people who had just developed symptoms, and their extent correlated with clinical severity, but were absent in controls. By the time of death, FTD patients had lost up to half of their VENs and fork cells. In surviving cells, dendrites and the cell body had shrunk by a third in cells with TDP-43 inclusions compared to those without. In future work, Seeley wants to find out what renders these neurons especially susceptible to TDP-43.
Other investigators focused on propagation itself. Glenda Halliday of the University of New South Wales, Randwick, Australia, noted that data from human postmortem studies support the concept of protein spread, with distinct patterns in different diseases. Scientists led by Lee and Trojanowski recently proposed four neuropathological stages of TDP-43 pathology in ALS patients, based on the regions affected in people with an increasing pathological load (see Brettschneider et al., 2013). Meanwhile, Halliday and colleagues developed a scheme for differentiating neuropathological bvFTD-TDP from ALS based on deposits in the anterior cingulate cortex versus the hypoglossal nucleus (see Tan et al., 2015). These studies highlight the varied propagation patterns of each disease, Halliday said. In bvFTD, lesions start in frontal regions and move back through the brain, whereas in ALS, inclusions first appear in the motor cortex and then descend into anterior and posterior brain regions. This staging has been replicated by other groups as well (see Fatima et al., 2015; Takeuchi et al., 2016). The data support the idea that misfolded proteins propagate along axons, she noted.
Ultimately, researchers believe that understanding onset and progression will enable them to develop strategies to slow or prevent disease, Seeley said. “I have hope [for treatments] that is born out of the progress made in the last 10 years,” he told the audience.—Madolyn Bowman Rogers









Comments
No Available Comments
Make a Comment
To make a comment you must login or register.