Vancouver Frontotemporal Dementia Conference Shows Awakening Field
Quick Links
It can start with insensitive comments at the dinner table. It can start with theft by a previously law-abiding citizen, or sexual misconduct by a hitherto faithful spouse. It can start with halting speech or a blank stare at a simple sentence. No matter the beginning, frontotemporal lobar degeneration (FTLD) usually ends in the mute, bed-bound misery of advanced dementia, and death.
This mysterious set of neurodegenerative diseases was the topic when 590 researchers from 30 countries met in the rainy city of Vancouver, Canada, on October 23 and descended into a hotel basement ballroom for 2½ days of exchange about the latest research findings. When they re-emerged, into a still-rainy city, they expressed awe at how their branch of study has hurtled forward in the eight years since the rapid-fire discoveries of progranulin, TDP-43, C9ORF72 hexanucleotide repeats, and tau PET tracers essentially broke it open. “The field has built on these discoveries so rapidly and with such volume, it’s remarkable,” said Ian Mackenzie of the University of British Columbia.
Co-hosted by Mackenzie and Howard Feldman of UBC, the 9th International Conference on Frontotemporal Dementias (ICFTD) featured 56 talks, 319 posters, and 13 satellite meetings—a far cry from its humble beginnings in 1986 in Lund, Sweden. FTD research languished for years in the shadow of its larger cousin, Alzheimer’s research, but now the pieces have moved into place for it to draw broad-based interest among scientists, funders, and even some drug-development companies. In fact, many players from the Alzheimer’s field are pushing in, sensing action and the chance to find something big. Clinical, neuropathological, and molecular genetics studies are each advancing. A detailed classification system is gradually being worked out; it is heterogeneous and therefore complex, but serves as a basis from which to work, scientists agree. Cellular pathways from lysosomal storage to RNA regulation are coming into view. Therapeutic trials are starting to target underlying mechanisms.
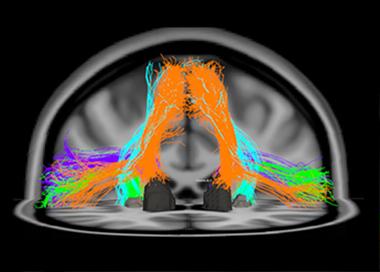
This diffusion tensor image shows fiber tracts of the speech-production network that degenerate in primary progressive aphasia, a language form of FTLD. The orange tracks, for example, connect the supplementary motor area to the Caudate nucleus. [Image courtesy of Maria Luisa Mandelli and Marilu Gorno-Tempini, UCSF. For more, see Mandelli et al., 2014.]
Perhaps the most important innovation at this moment lies in newly forged international collaborative relationships to comprehensively examine patients and their families in longitudinal cohort studies. Inspired by the success the Alzheimer’s field has had with natural history cohorts such as the Alzheimer's Disease Neuroimaging Initiative (ADNI), the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL), and the Dominantly Inherited Alzheimer’s Network (DIAN), every FTD center participating in such a joint venture agrees to use the same methods of deep phenotyping, genotyping, and imaging, and to pool their findings into shared databases. At ICFTD, two such initiatives presented baseline data, one on 336 presymptomatic mutation carriers in Europe and Canada (see Part 2 of this series) and one on 700 symptomatic patients in Germany (see Part 3). Two large North American initiatives announced that they had just received $5.1 million to get going on their own cohorts (see Part 4). “Working across the progranulin, C9ORF72, and tau forms of these diseases is an accessible problem, but it requires global effort,” said Feldman. “There is tremendous power when we work together.”
Therapeutic studies are beginning, as well. Here, researchers work amid two cross-currents. They talk about avoiding the mistakes that have marked the history of Alzheimer’s trials, where some drugs were rushed into Phase 3 without sufficient evidence that they entered the human brain and adequately engaged their molecular target there to have a chance at making a dent in the disease. At the same time, a sense of urgency to relieve the profound suffering FTD causes pervaded this conference. This awareness was made more acute by a simultaneous meeting of 200 FTD caregivers hosted by the Association for Frontotemporal Degeneration (AFTD). Most of the carers had traveled to ICFTD from outside British Columbia. “In FTLD there is a faithful network of caregivers that spans the globe,” Feldman said. “Caregivers hold the key to what matters; we need to listen to them.”
No one disputes that new therapies that target central FTLD molecules are sorely needed. No drugs approved specifically for FTLD exist. Rather, some patients with behavioral symptoms take antidepressants with modest success; patients with parkinsonian symptoms often are prescribed levodopa, but typically do not respond to it.
As scientists tried to advance the understanding of frontotemporal dementia from basic science to clinical trials, their discussions reflected the realization that they are working together through the early days of translational medicine, rather than entrenched in competing interests. “We heard people from different centers and backgrounds stating similar viewpoints about how to go about those trials. We have not just random individual strategies; there really seems to be a community approach to this,” said Mackenzie. “That gives me optimism that the translational process will be effective.” Throughout the conference, scientists showed unpublished data on work in progress. They asked colleagues from other labs to pressure-test a new assay, or offered to teach them new analysis algorithms. “There is a spirit of generosity right now where people are forthcoming with their assets to work toward a common goal. That touches us all as a community,” Feldman said.
The Clinical Picture
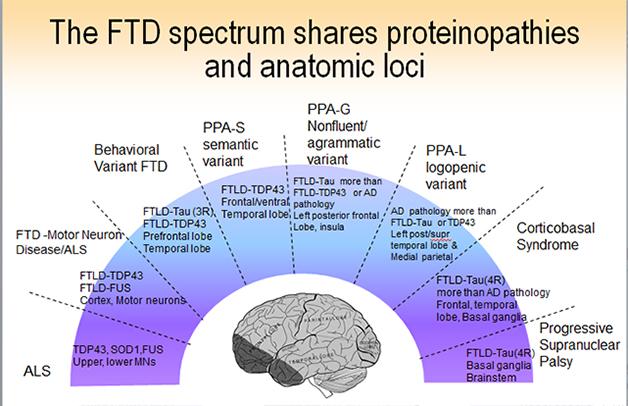
Some 50,000 people in the United States have some form of FTD. The pathological name, frontotemporal lobar degeneration (FTLD), is an umbrella term for a diverse set of diseases that are all marked by atrophy starting somewhere in the frontal or temporal lobe of the brain, often on one side. Beyond this common feature, however, FTLD is heterogeneous at every level—the clinical presentations, the underlying neuropathology, the neural networks that become dysfunctional, and the genes that cause the havoc. Symptoms, proteins, and genes do not map neatly onto separate pathways, but into overlapping groups. Researchers now agree that FTLD represents a spectrum of diseases that stretches toward parkinsonian symptoms one on end and amyotrophic lateral sclerosis (ALS) on the other.
©2014 The Association for Frontotemporal Degeneration.
Outwardly, frontotemporal dementia comes in two main flavors, though subtypes and atypical variants abound and not every research center groups them in exactly the same way. In defining the expressions of FTLD, clinicians are redrawing the line between psychiatry and neurodegeneration. The signature disease is behavioral variant FTD, or bvFTD. This starts primarily as a disease of social cognition, emotion, and executive control, said Bruce Miller of the University of California, San Francisco. A formerly warm, interested, disciplined person can become selfish, apathetic, and gluttonous. He may ignore distress in others, or do embarrassing things in public. She may abuse drugs or alcohol, or become compulsively engaged in the arts or business. Misdiagnosis happens in both directions: bvFTD is frequently mistaken for depression or schizophrenia, but people are also referred to UCSF with a diagnosis of bvFTD when in fact they have a psychiatric illness. “Apathy, change in eating habits, and disinhibition are great distinguishing factors between psychiatric disease and bvFTD that are not yet being applied in the diagnostic community,” Miller said.
The other main form of FTLD, primary progressive aphasia (PPA), starts with problems understanding speech (the semantic variant) and generating speech (the nonfluent or agrammatic form). PPA starts with specific deficits, such as confusion about the meaning of words and concepts, or halting, laborious speech. The underlying functions map to frontal cortical and subcortical areas and the left posterior insula. They are connected into language networks by fiber bundles, such as the aslant tract, that are the subject of study with diffusing tensor imaging and tractography (see image above). At ICFTD, Marilu Gorno-Tempini of UCSF discussed emerging evidence to support a suggestion proffered originally by Marsel Mesulam at Northwestern University, Chicago, that certain developmental language disabilities may render the brain’s language network less resilient. Childhood dyslexia, for example, may make the brain more prone to a particular, logopenic subtype of PPA later in life.
In addition, neurologists record a long and growing list of atypical frontotemporal dementias. The motor symptom variant corticobasal degeneration (CBD) renders the upper body rigid and the arms increasingly unusable until the hand becomes permanently clenched in a fist, said Bradley Boeve of the Mayo Clinic in Rochester, Minnesota. Less well known is a language variant that starts with what is called prosopagnosia, a curious inability to recognize the faces of familiar people. Patients with this disease will view a photo of Barack Obama and say “I like him” or “I do not like him,” but will not be able to name the president. Boeve told the story of a man who played bingo with his second wife. The man stepped away, chatted with someone, and when he returned his wife asked, ”Why were you talking to her?” He replied that he just had a talk with someone—he did not realize the person was his first wife. Another rare variant is primary progressive apraxia of speech. Disease in these people starts with reversals of the correct response. “Are you married?” “No! .... Yes.” Clinicians agreed that the heterogeneity of FTLD subtypes urgently demands robust biomarkers to help them diagnose what causes a given patient’s disease.
At ICFTD, John Hodges of Neuroscience Research Australia in Sydney discussed the puzzling phenomenon of FTLD phenocopies. A minority of bvFTD and aphasia patients meet diagnostic criteria when they come in, but then stay stable for many years (Rascovsky et al., 2011; Khan et al., 2012). This runs counter to the classic view of neurodegenerative disease as inexorably worsening. It is good news for the individual patient, because it means not everyone who receives a bvFTD diagnosis will head toward profound suffering and death in only a few years. These patients can be bad news for clinical trials, however. If researchers unwittingly enroll a “phenocopy” soon after diagnosis, they might mistake his lack of progression for a drug benefit.
At ICFTD, Emma Devenney in Hodges’ group noted that six-year observational data tipped them off to certain clues that predict a suspected bvFTD patient is not a phenocopy, but will indeed progress. The clues include a family history, low baseline performance on memory, visuospatial- and language-based tests such as the RAVLT, poor self-care or other activities of daily living, and definite atrophy on MRI. “It makes sense to think that if a person has at baseline a deficit in a domain we know is part of the spectrum, such as memory, then it is more likely to be an organic disease and not just an isolated personality problem that may phenocopy bvFTD,” commented Mackenzie. Some slow progressors who did eventually get worse after an initial stable phase turned out to have C9ORF72 mutations, Hodges said.
The Genes and Proteins
Beneath the panoply of outward expressions of disease lie three core RNA/protein pathologies, but they do not map in one-on-one relationships to a given clinical type. A few subtypes of FTLD do have a consistent underlying pathology; for example, semantic dementia patients typically have TDP-43 pathology in their brain, and progressive supranuclear palsy patients have tau pathology. But the majority of FTLD cases could be driven by any of a number of different underlying neuropathologies; “guestimating” the correct one has become part research, part art form. This is true of bvFTD, as Bill Seeley at University of San Francisco and collaborators documented on a poster at ICFTD. At least six different protein pathologies have been described for this clinical syndrome. Likewise, tau, TDP-43 and even Alzheimer’s pathology underlie the primary progressive aphasia syndromes in about equal measure, said Mesulam.
©2014 The Association for Frontotemporal Degeneration.
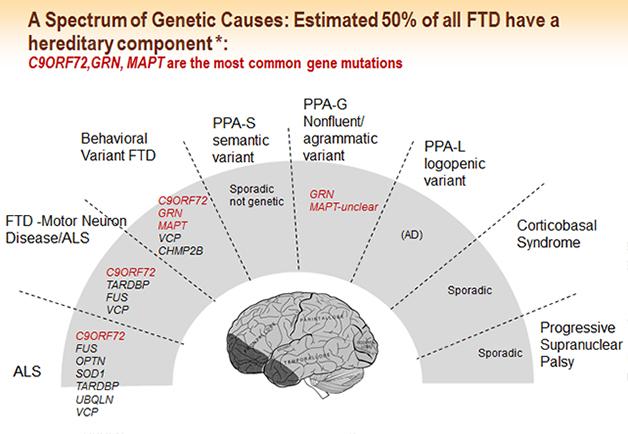
FTLD is highly genetic. Up to 50 percent of all cases have a family history, and 10 to 30 percent are autosomal-dominant. Mutations in the genes for tau, progranulin, and C9ORF72 account for a majority of familial cases, but more genes continue to be implicated in FTLD overall. Neurogeneticists disagree about whether additional highly penetrant genes remain to be found. At ICFTD, geneticists proposed several new genes that may alter FTD risk or change the course of the disease in people who already harbor one of the dominant mutations. Christine van Broeckhoven of the University of Antwerp, Belgium, proposed VPS13C as a new FTD gene. Rosa Rademakers of the Mayo Clinic in Jacksonville, Florida, found that expression of hox genes was boosted in C9orf72 carriers. Hox (aka homeobox) genes were identified as master regulators of patterning during development, but they also play a role in neuronal repair. Studying a family with FTLD in whom she had excluded all the known dominant mutations, Rademakers presented to the audience a list of potential genetic variants she thought might be responsible for this family’s disease, and asked the scientists to dig through this list of genes in their own unexplained familial samples in hopes of homing in on a new genetic culprit (see Part 8 of this series).
Mapping from gene to neuropathology is more straightforward than mapping from pathology to clinical syndrome. Mutations in the gene MAPT give rise to tauopathy; mutations in progranulin, C9ORF72, and VCP give rise to TDP-43 pathology. Alas, tau and TDP-43 pathologies each can lead to a range of different and overlapping clinical pictures, even in a single family. Mutations in the gene FUS cause a different neuropathology and lead to bvFTD or ALS (for review, see Riedl et al., 2014). In Vancouver, a key focus was on presentations devoted to parsing the molecular mechanisms of how the pathogenic hexanuceotide repeats in C9ORF72—in the form of RNA foci, peptide inclusions, or an unknown soluble entity—relate to aggregates of TDP-43, and which part of this axis of evil ends up killing neurons (see Part 5 of this series).
Even as this picture was being filled in, researchers at ICFTD saw a problem in the wide span of symptoms that can develop from each gene or protein a given therapy might target. This range will make it difficult for symptom-based outcome measures to capture whether the drug works. Early stage trials can start out by addressing just the question of whether the drug changed levels of its immediate target, e.g. progranulin, as their efficacy outcome (see Part 6 of this series). But where to go from there, asked Feldman? How to tell whether elevating progranulin levels benefits a treatment group?
On this question, the conference showcased budding research on physiological markers. They could, if they hold up in further research, be developed into “intermediate” outcome markers that lie downstream of target engagement, but remain short of classical clinical symptoms of FTLD. For example, it appears that people with FTLD have abnormal perceptions of pain, temperature, or even music. Researchers working with Jason Warren at University College London, U.K., are characterizing brain-network signatures that could explain why a bvFTD patient can wear a fur coat on a hot day, yet remain unaware of her overheating body and the beads of sweat standing on her forehead. The conference also featured news on social cognition. Researchers are fingering some of the same brain networks that have been implicated in the personality changes seen as disease progresses and formerly warm personalities become emotionally blunted (see Part 7 of this series). Readers used to the entorhinal cortex, perforant path, and hippocampal pyramidal neurons: Get ready to hear about the insula, thalamus, uncinate fasciculus, and von Economo cells.—Gabrielle Strobel
References
News Citations
- First Data from GENFI1: Brain’s Insula Region Shrinks A Decade Before FTD
- German Network of 700 FTLD Patients Presents Baseline Data
- Meet the Artful Leftie: NIH Jump-Starts U.S.-Canadian FTLD Cohorts
- Stream of Genetics Pushes FTD Research Forward
- Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
Paper Citations
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011 Sep;134(Pt 9):2456-77. Epub 2011 Aug 2 PubMed.
- Khan BK, Yokoyama JS, Takada LT, Sha SJ, Rutherford NJ, Fong JC, Karydas AM, Wu T, Ketelle RS, Baker MC, Hernandez MD, Coppola G, Geschwind DH, Rademakers R, Lee SE, Rosen HJ, Rabinovici GD, Seeley WW, Rankin KP, Boxer AL, Miller BL. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012 Apr;83(4):358-64. PubMed.
- Riedl L, Mackenzie IR, Förstl H, Kurz A, Diehl-Schmid J. Frontotemporal lobar degeneration: current perspectives. Neuropsychiatr Dis Treat. 2014;10:297-310. Epub 2014 Feb 13 PubMed.
Other Citations
Further Reading
Papers
- Mandelli ML, Caverzasi E, Binney RJ, Henry ML, Lobach I, Block N, Amirbekian B, Dronkers N, Miller BL, Henry RG, Gorno-Tempini ML. Frontal white matter tracts sustaining speech production in primary progressive aphasia. J Neurosci. 2014 Jul 16;34(29):9754-67. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.