New Target Has Legs: Tau PET, Mice, and Antibodies
Quick Links
Coming to the fore as both a disease marker and a therapeutic target, the protein tau drew its share of attention at the 7th Clinical Trials on Alzheimer’s Disease (CTAD) conference, held November 20-22 in Philadelphia. Speakers presented fresh evidence that tau PET imaging reflects the stage of a person’s disease and cognition, suggesting that investigational tracers may allow researchers to track progression in clinical trials. Meanwhile, new animal data strengthened the notion that pathological tau comes in strains that spread through the brain in stereotypical patterns of distinct tauopathies. Would mopping up this extracellular tau halt the march of disease? The idea appears to hold water in animals, with a speaker describing a new monoclonal antibody that restored memory in tau mice. Researchers are trying to learn from past instances where treatment results in mouse models failed to translate to human trials, and one talk introduced a new model that expresses wild-type human tau along with mutant human APP. This mouse develops tangles, memory loss, and neuronal degeneration without amyloid plaques.
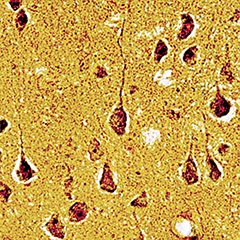
Mouse neurofibrillary tangles without tau mutation.
Tangles came up by introducing wild-type human tau into APP transgenic mice. The tangles were visualized by Gallyas silver staining, and were confirmed to contain both 3R and 4R tau like those in AD brain. [Image courtesy of Takami Tomiyama, Osaka City University.]
With more trials targeting people in prodromal stages of disease, researchers need better ways to monitor progression, said Reisa Sperling of Brigham and Women’s Hospital, Boston. Cerebrospinal fluid (CSF) biomarkers plateau around the time symptoms appear, and barely record ongoing change. “CSF tau is just not a dynamic marker,” agreed Paul Aisen of the University of California, San Diego. Moreover, CSF tau levels do not reflect brain anatomy, and it is not only the concentration of tau but where it spreads that matters, Sperling noted. Tau imaging allows researchers to track how tau creeps through the brain as disease worsens. Sperling presented preliminary data from scans using Eli Lilly’s tracer T807 that offered clues to how tau relates to amyloid deposition and memory loss.
In cognitively normal participants of the Harvard Aging Brain Study, tau began to accumulate around age 40 to 50. By age 65 the majority of people had some tau deposits in their medial temporal lobes, corresponding to a Braak stage of I or II. “As someone who is getting closer to this age, this is frightening,” Sperling said. Older age groups were likelier to have some tau in the neocortex as well, resembling Braak stage III or IV.
How does this relate to CSF biomarkers? In a cohort of about 30 people, high tau PET correlated with high CSF tau. It correlated even more strongly with low CSF Aβ42. What’s more, having low CSF Aβ42 predicted that there would be tau outside of the medial temporal lobe. “Amyloid may accelerate the cortical deposition of tau,” Sperling suggested.
While both amyloid and tau spread from brain region to brain region, Sperling pointed out that the two pathological proteins tend to affect the brain differently. Amyloid disrupts long-range brain networks such as the default mode network, whereas tau tends to gunk up local circuits in the medial temporal lobe. Coordinated activity between the default-mode network and the medial temporal lobe is critical to encode and then retrieve memories. “Perhaps the combination is what harms memory function,” she speculated.
Prior tau imaging studies have found that uptake of tau tracer correlates with early memory problems (see Nov 2013 conference news; Aug 2014 conference news), and more recent data bear this out. Sperling and Keith Johnson at Massachusetts General Hospital, Boston, by now have analyzed data from 93 people who had tau scans and memory tests within six months. Those with neocortical tau performed poorly, even if they had no brain amyloid, Sperling reported. “Tau is the more proximal mediator of cognitive decline,” she said. “Aβ tells you that you’re on the Alzheimer’s train, but tau tells you which stop you’re at.”
If a person has markers of both amyloid and tau, is it too late to intervene? In future prevention trials, Sperling plans to enroll people earlier on the amyloid accumulation curve, who do not yet have widespread tau. At later disease stages, patients might need a combination therapy that attacks both markers, she suggested (see Part 2 of this series).
Other data dovetailed with Sperling’s findings. Adam Schwarz of Eli Lilly in Indianapolis presented findings from a small study that compared T807 tau imaging patterns with Braak stages. The researchers defined eight regions of interest in temporal and occipital lobes based on sections typically used for staging. They scanned seven Alzheimer’s disease patients, five people with mild cognitive impairment, five age-matched controls, and four young controls. The profiles of where tracer bound in these diagnostic groups matched up well with published Braak patterns for these groups, allowing the researchers to categorize participants as stage I/II, III/IV, or V/VI, Schwarz reported. Most healthy controls had at least some tau in the entorhinal cortex, but this was counted as Braak stage zero until it reached a cutoff value. As expected, higher Braak stages correlated with a diagnosis of MCI or AD. Three of the 42 brain hemispheres scanned did not fit into a Braak stage. “We see an encouraging correspondence between typical patterns of Braak progression and T807 binding,” Schwarz said. Autopsy studies will be needed to confirm that the tracer binds pathological tau. Next, Schwarz plans to scan larger cohorts, and to map other tauopathies, such as corticobasal degeneration (CBD), with T807.
The patterns of tau progression suggest that it spreads along neural networks, an idea supported by many animal and cell culture studies (see Jun 2009 news; Mar 2013 conference news; Aug 2013 conference news). At CTAD, John Trojanowski of the University of Pennsylvania, Philadelphia, extended this literature, noting that the injection of small amounts of synthetic preformed tau fibrils into different brain regions of PS19 transgenic mice kicks off tau pathology that spreads to connected regions over time (see Iba et al., 2013). The tau aggregates formed this way resemble the neurofibrillary tangles seen in AD, which would not otherwise develop in these mice, Trojanowski added. Injections into the locus coeruleus, believed to be one of the earliest sites of tau accumulation, resulted in pathological tau spreading both retrogradely to afferent brain regions and anterogradely to efferent regions. In a sense, injection of tau fibrils maps a given area’s anatomical connectome, these studies suggest.
Are all tau aggregates equivalent? Emerging data suggests not. Trojanowski’s group injected tau extracts from postmortem AD and CBD brains into the hippocampus and cortex of young PS19 mice. The injections triggered distinct pathologies reminiscent of their human origin. Mice receiving CBD tau developed tau inclusions mostly in oligodendrocytes near the injection site, from where they spread gradually through white matter to the hippocampal fimbria, external capsule, and stratum radiatum, but not into cortex. CBD is marked by neurofibrillary tangles predominantly in oligodendrocytes, not neurons. Few neurons died in these animals. By contrast, mice injected with AD tau formed tau aggregates in hippocampal neurons, from where they spread to connected brain regions such as the entorhinal cortex and locus coeruleus, and eventually to thalamus and cortex. In these mice, up to 50 percent of neurons died in affected areas (Boluda et al., 2014). The data fit with other recent work hinting that tau can form distinct strains with different pathological characteristics (see Clavaguera et al., 2013; May 2014 news story).
If pathological tau travels between cells, that journey should open a space for antibodies to sweep up the fibrils. Takami Tomiyama of Osaka City University, Japan, told CTAD attendees that both active and passive tau immunotherapy work in mice. Passive approaches are less prone to causing neuroinflammation, he noted, but previous studies have mostly targeted early stages of the disease, before symptoms appear. Tomiyama wanted to develop an antibody to treat advanced disease. First, he studied the phosphorylation patterns of human pathological tau to select the best epitope to target. They picked the phosphorylated Ser413 residue and generated the Ta1505 antibody against it. In human postmortem brain, Ta1505 lit up AD brains but not controls, demonstrating specificity for pathological forms of the protein, Tomiyama said.
The researchers then immunized 14-month-old tau model mice with either Ta1505 or Ta4, which binds to phosphorylated Ser396, another pathological tau residue. Both antibodies recognize only pathological tau in AD brain, not normal tau in control brain, Tomiyama wrote to Alzforum. Mice injected with Ta1505 had less phosphorylated tau, neurofibrillary tangles, and neuron loss. Levels of a synaptic marker rose, and memory returned to wild-type performance, Tomiyama showed. Animals receiving Ta4 improved somewhat on memory tests and phosphorylated tau, but other markers did not change.
Ta1505 may be a promising prototype for humanized antibodies, Tomiyama said. A patent application has been filed, and the paper is in press in Annals of Clinical and Translational Neurology. For previous work along this line, see Aug 2007 news story; Nov 2010 conference story.
Existing tau mouse models have limitations, Tomohiro Umeda, also of Osaka City University, said at CTAD. Animals that overexpress human mutant APP get hyperphosphorylated tau but no neurofibrillary tangles. Double-transgenic mice that express both mutant human tau and APP do get tau tangles, but these animals do not truly model Alzheimer’s, because tau is not mutated in this disease. In human disease, Aβ oligomers are believed to initiate tau pathology by first activating GSK3β, which phosphorylates tau and causes it to aggregate. Why don’t tangles form in APP mice, then? Maybe the process needs human tau, Umeda suggested. He noted that human and mouse tau vary quite a bit at the N-terminal region, which may affect the protein’s ability to aggregate.
To test this idea, Umeda crossed APPOSK mice with tau264 animals. The APPOSK mice express human APP with the Osaka mutation, and have abundant Aβ oligomers but no plaques (see Apr 2010 news story). Tau264 mice carry a human wild-type tau transgene and express equal amounts of the 3R and 4R splice variants (see Umeda et al., 2013). Neither of these mouse models has tangles. The offspring, however, developed tau tangles in cortex and hippocampus at 18 months (see image above). They resembled human tangles and looked filamentous in the electron microscope, Umeda reported. The hybrids also developed more aggressive pathology than their APPOSK parents, with Aβ oligomers, phosphorylated tau, and synapse and neuron loss appearing at younger ages. The results demonstrate not only that tangle formation requires human tau, but also that Aβ oligomers suffice to trigger tangles, Umeda claimed (see Umeda et al., 2014).
Attendees reacted to the model with enthusiasm. Many researchers are trying to move away from overexpression mouse models in favor of more physiological expression levels. At a stem cell symposium held November 6 at Duke University in Durham, North Carolina, Frank LaFerla of the University of California, Irvine, described a mouse model he made that expresses wild-type human APP at native levels and accumulates Aβ plaques by 17 months of age. This animal might resemble sporadic AD more closely than current models do, he suggested.—Madolyn Bowman Rogers
References
News Citations
- Do Tau Tracers Track Cognitive Decline in Disease?
- Scan by Scan, Growing Tau PET Data Picks Up Early Memory Deficits
- Rusty Unleashed: Forget Disease Modification, Go for Big Effect
- Traveling Tau—A New Paradigm for Tau- and Other Proteinopathies?
- Tau, α-Synuclein Spread: Crazy Stuff—How Might It Work?
- Tales of Traveling Tau: Is Transfer Between Neurons Normal?
- Like Prions, Tau Strains Are True to Form
- Tau Vaccine Detangles Mouse Brain
- San Diego: Tau Oligomer Antibodies Relieve Motor Deficits in Mice
- New APP Model: No Plaques—Plenty of Pathology
Research Models Citations
Paper Citations
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci. 2013 Jan 16;33(3):1024-37. PubMed. Correction.
- Hypothesis: a role for macrophages in the pathogenesis of type 1 diabetes. Autoimmunity. 1989;3(2):157-63. PubMed.
- Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013 Jun 4;110(23):9535-40. PubMed.
- Umeda T, Yamashita T, Kimura T, Ohnishi K, Takuma H, Ozeki T, Takashima A, Tomiyama T, Mori H. Neurodegenerative disorder FTDP-17-related tau intron 10 +16C → T mutation increases tau exon 10 splicing and causes tauopathy in transgenic mice. Am J Pathol. 2013 Jul;183(1):211-25. PubMed.
- Umeda T, Maekawa S, Kimura T, Takashima A, Tomiyama T, Mori H. Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice. Acta Neuropathol. 2014 May;127(5):685-98. Epub 2014 Feb 15 PubMed.
Other Citations
External Citations
Further Reading
News
- HAI—Spotlight on Tau Tracers at Human Amyloid Imaging Meeting
- Tau Tracers Shine at Boston Conference
- Tau Tracer May Light Up All Tauopathies
- PET Tracers Enlighten at Neurology Conference
- Mice Tell Tale of Tau Transmission, Alzheimer’s Progression
- Antibodies Stop Toxic Tau in Its Extracellular Tracks
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.