Following up on its October surprise, Biogen showed more data at the Clinical Trials in Alzheimer’s Disease conference, arguing that disparities in drug exposure explained the puzzlingly different outcomes in its one positive and one negative aborted Phase 3 trials. Audience members were put off by the messiness of the data set and not fully convinced by the analysis, but the majority said they believed the general signal that the antibody may slow disease progression. The prevailing mood was one of hopefulness, with most agreeing the results validate the amyloid hypothesis and suggest that effective treatments for AD are within reach.
Exposure, Exposure, Exposure? At CTAD, Aducanumab Scientists Make a Case
There was a new feeling in the air at the 12th Clinical Trials on Alzheimer’s Disease conference. It felt good: After decades of failure, the field might finally be turning a corner. The renewed optimism at the meeting, held December 4–7 in San Diego, was driven largely by Biogen’s shocking announcement last October that one of its two Phase 3 aducanumab trials, after a prior declaration of futility, was looking positive, after all. At CTAD, scientists got a look at some more of the data. It mostly opened a window on how protocol amendments midway through the trial slowly raised exposure among subsets of participants, and tried to link exposure to drug effect.
The audience wanted to see more data still. Even so, they found much to dissect in hallway conversations afterward, with many privately questioning whether the dataset is clean enough to garner marketing approval from the Food and Drug Administration. Nonetheless, the consensus was that the drug probably works. Why? It robustly removes amyloid, possibly clears tau tangles as well, and, at sustained high doses, may modestly slow decline. “All the data suggest this is a disease modification,” Paul Aisen of the University of Southern California, San Diego, noted in a panel discussion, though the panel was criticized as being too one-sided.
It wasn’t only aducanumab that fueled the hopeful mood. Presentations on gantenerumab, donanemab, and BAN2401 all described dramatic amyloid clearance that was sustained over time, although none discussed cognitive outcomes. Other talks also reported positive findings, with researchers getting their first look at detailed Phase 3 data of the anti-agitation drug pimavanserin in AD (Sep 2019 news). Ditto for AMBAR, whose sponsor, Grifols, had previously reported slowing of cognitive and functional decline in AD dementia (Nov 2018 conference news), and in San Diego added cerebrospinal fluid and FDG PET biomarker data, as well as additional statistical analyses.
“For the first time in my 25-year career, we had a conference with not one, but several positive readouts,” said Marwan Sabbagh of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas. “We’re starting to make progress. This has reignited enthusiasm in the field.”
Cynthia Lemere of Brigham and Women’s Hospital in Boston, who investigates anti-amyloid therapies, noted that Biogen’s previous announcement of futility had discouraged her (Mar 2019 news). “These new data have restored my faith in this approach,” she told Alzforum.
So what were the data? In October, Biogen had reported that 10 mg/kg aducanumab slowed cognitive decline by a quarter, and functional decline by 40 percent, in the EMERGE trial. By contrast, the biologic appeared to have no benefit in the identical ENGAGE trial, with placebo and treatment groups performing similarly, and the high-dose group, if anything, trending a tad worse on the primary outcome. The company ascribed the difference to mid-study protocol amendments that raised dosing and disproportionately benefitted EMERGE participants (Oct 2019 news).
In San Diego, Samantha Budd Haeberlein of Biogen elaborated on this point. The key amendment, protocol version 4 (PV4), allowed ApoE4 carriers to be titrated up to the 10 mg/kg dose. About two-thirds of study participants carried an E4 allele, and their dose had been previously capped at 6 mg/kg because of worries over ARIA. To examine the consequences of this dosing change, Budd Haeberlein divided the pooled EMERGE and ENGAGE cohort into people who had enrolled before PV4 was adopted in March 2017, and those who enrolled after. Eighteen percent of pre-PV4 participants received the full possible course of treatment—14 doses of 10 mg/kg—compared with 49 percent of post-PV4 participants. Overall, the amendment boosted the cumulative aducanumab exposure by a third, with the pre-PV4 group reaching a median cumulative dose of 116 mg/kg by the end of the study, and the post-PV4 group, 153 mg/kg. “Protocol 4 had a real impact on dosing,” Budd Haeberlein said.
ENGAGE had begun enrolling before EMERGE, hence fewer of its participants were affected by this protocol change. When PV4 was adopted, ENGAGE had 200 more participants than EMERGE. Each participant had to consent to the protocol change, and PV4 took 18 months to fully implement, Budd Haeberlein noted. As a result, by the end of the trial, 22 percent of ENGAGE patients had received the full course of 14 doses of 10 mg/kg, compared with 29 percent of EMERGE patients. To represent this, Budd Haeberlein showed exposure “heat maps” for ENGAGE and EMERGE of the dose each patient received at each visit. Dark blue indicated maximum dosage, and yellow no dose, providing a visual representation of dosing changes and interruptions. The EMERGE map showed a slightly thicker slice of solid blue than ENGAGE (see image below).
The Blues. More participants achieved 14 treatments at maximum dosage (dark blue) in EMERGE than ENGAGE, with fewer dosing interruptions (yellow). [Courtesy of Biogen.]
Budd Haeberlein showed a slide illustrating marked differences in how fast sites in the countries of these global trials consented their participants to PV4. Biogen issued PV4 in March 2017, and the majority of participants at U.S. sites were on it by December of that year, but participants in Italy and Poland only started signing on by then; in Portugal, signing started in spring of 2018. This implies differences by country, but Budd Haeberlein did not show exposure/efficacy data by country, and declined to answer questions about country or site effects.
Was Exposure the Key? A post hoc subgroup analysis of participants enrolled after maximum dosing was raised suggests a cognitive benefit in both trials (gray, placebo; purple, high dose). [Courtesy of Biogen.]
Biogen contends that participants need to reach this 14 x 10 mg/kg exposure to have a cognitive benefit. Budd Haeberlein showed a comparison of post-PV4 subgroups in ENGAGE and EMERGE, amounting to 886 participants in the former and 790 in the latter, roughly half of each cohort. In this post hoc subgroup analysis, 10 mg/kg aducanumab treatment appeared to slow CDR-SB decline by a similar amount in both trials, 27 percent in ENGAGE and 30 percent in EMERGE. Numerically, CDR-SB worsened by 1.76 points in EMERGE, with treatment reducing this by 0.53. CDR-SB worsened by 1.79 points in ENGAGE, with treatment cutting this by 0.48. In the whole ENGAGE cohort, the placebo group had declined more slowly, by 1.55 points.
Curiously, people who enrolled post-PV4 but were titrated to only 6 mg/kg fared nearly as well as the high-dose group, with a slowing of decline of 20 percent in ENGAGE and 24 percent in EMERGE. On graphs, both treatment groups tracked together (see image above).
The secondary outcome measures followed a similar pattern in this post hoc subgroup, Budd Haeberlein said, but she did not show those data, nor any further characteristics of those subgroups. She declined to address subsequent questions about these subgroups, except to say that their ApoE genotype distribution was as in the ITT population. Biogen believes the findings support the idea that sufficient exposure to drug slowed disease progression in both trials.
Conference attendees had a mixed reaction to this, with most wanting to see more detailed data. Dennis Selkoe at Brigham and Women’s Hospital said that the explanation makes sense to him, but he would like to see the secondary outcomes as well. “If they look similar, it would be compelling,” Selkoe told Alzforum. Henrik Zetterberg, University of Gothenberg, Sweden, essentially concurred, as did other leading researchers at CTAD. At the same time, they noted that other factors could have affected the results. Many wanted to see the percentage of ApoE4 carriers, as well as baseline values and demographic information of each subgroup, and suggested that imbalances there could have skewed the post hoc findings. Howard Feldman of the University of California, San Diego, Bruno Imbimbo of Chiesi Pharmaceuticals in Parma, Italy, and others suggested that a higher incidence of ARIA in treatment groups than controls could have effectively unblinded study participants and some study staff, leading to a placebo effect.
Lon Schneider of the University of Southern California, Los Angeles, said that researchers will need to see sensitivity analyses to know whether the drug effect is real (see full comment below). Others wanted to see spaghetti plots of the dose-response effect. In hallway talk, researchers grumbled that the presentation contained little new data beyond that released in October, and noted that Biogen is holding its cards close to the chest. Maria Teresa Ferretti of the University of Zurich spoke for many when she said, “I’m left with the same questions I had coming in.”
The biomarker data elicited more positive reactions. Budd Haeberlein reiterated amyloid PET findings, reporting consistent plaque removal that correlated with dose. At 6 mg/kg aducanumab, SUVR dropped by 0.17 in both studies. At 10 mg/kg, SUVR dropped by 0.24 in ENGAGE and 0.27 in EMERGE, with group sizes ranging from 74 to 203 people. As previously reported, CSF p-tau and t-tau dropped dose-dependently in EMERGE, though group sizes here were tiny, ranging from 14 to 29 people in a trial that enrolled 1,638 participants total. In ENGAGE, p-tau and t-tau reduction were not dose-dependent, with group sizes ranging from 16 to 21.
Tangle Cleanup? In a small tau PET sub-study, high-dose aducanumab lowered tracer uptake in medial temporal regions over 14 months. [Courtesy of Biogen.]
New in San Diego were tau PET data, using the second-generation tracer MK6240. Examining a medial temporal composite made up of hippocampus, parahippocampus, entorhinal cortex, and other anterior medial and lateral temporal lobe regions, the researchers found that the SUVR increased among people on placebo by about 0.09 over 14 months. For those on drug, the SUVR dropped by about 0.05 (see image above). The PET sub-study comprised 11 participants on placebo, 14 people on 6 mg/kg, and 11 on 10 mg/kg. The change in tracer uptake correlated inversely with dose. No correlation analyses were shown for amyloid removal versus CDR change.
“The tau PET data were impressive,” Eric Siemers of Siemers Integration LLC in Zion, Indiana, told Alzforum in San Diego. Siemers elaborated in writing (see below). Sabbagh, and others, speculated that a reduction of tau pathology might underlie the cognitive benefit.
In trials of aducanumab, as well as other anti-amyloid antibodies like BAN2401 and solanezumab, the cognitive benefit seems to lag behind amyloid removal, hinting it could be due to a downstream effect. Defining the lag time between amyloid removal and a cognitive benefit is a priority for future anti-amyloid trials, researchers agreed. Stephen Salloway of Brown University, Providence, Rhode Island, who was the lead site investigator for the aducanumab trials, noted that although the cognitive benefit seen in the aducanumab trials was small, the data overall suggest an effect on disease progression. “We’re looking for a biological foothold we can build on,” he said.
Across the board, researchers lamented the early termination of these trials, which led to an incomplete, confusing dataset. “The futility decision was highly unfortunate, and puts us in this position of having to analyze a complex dataset,” Aisen said. Budd Haeberlein agreed, but presented no data on the futility analysis. She noted that although Biogen will call patients back to resume dosing, they will have been off drug for varying numbers of months, further complicating long-term efficacy and biomarker data. The dosing changes midway through the trial clouded the picture, but Budd Haeberlein believes amending the protocol was the right decision under the circumstances. “I would not recommend changing the dose in the middle of a Phase 3 trial, but it turned out to be important for this study. Had we not done so, we would not have the results we do today,” she said in San Diego.
Audience members questioned whether the small cognitive benefit, amounting to a 23 percent slowing of decline on the CDR-SB, was meaningful. In the panel discussion, Sharon Cohen of the Toronto Memory Program, a site leader for the aducanumab trials, urged them to focus instead on the 40 percent slowing of decline in activities of daily living (ADL). “Eighty percent of the participants were in the prodromal stage, meaning they are living independently. The ability to continue to work, travel, bank, and enjoy leisure activities matters more to patients than what score they get on a memory test. In this disease, patients lose themselves slice by slice. Anything they can do to hang on is a triumph,” she explained. ADL was a secondary outcome measure. Some in the audience noted that such comments increase public pressure on the FDA to approve the drug, regardless of the unanswered scientific questions, and efficacy data resting largely on one incomplete Phase 3 trial.
Will these data be enough for regulators to allow Biogen to market aducanumab? Many were skeptical. Feldman noted that the FDA might find the risk-benefit ratio insufficient. Although ARIA was manageable and the field in general is less acutely concerned about this phenomenon now than when ARIA first cropped up, its incidence was high. Altogether, 25.8 percent of participants on 6 mg/kg, and 35.1 percent of those on 10 mg/kg, developed ARIA-E, a form of brain swelling. This compared with 2.3 percent of the placebo group. Most ARIA occurred in ApoE4 carriers. For a large majority it was asymptomatic, but about a quarter of those with ARIA complained of headaches, dizziness, or nausea.
In addition, some participants developed ARIA-H, indicating microhemorrhages. These occurred in 17.5 percent of people on drug, versus 6.8 percent of controls. This incidence is higher than in other antibody trials; for example, the Marguerite RoAD extension study of high-dose gantenerumab reported 9 percent (Aug 2018 conference news). ARIA-H has received less attention in AD trials than ARIA-E, with clinicians saying this phenomenon worries them less. Like ARIA-E, it is usually asymptomatic. Seven people on aducanumab and four controls had more extensive brain bleeds. Overall, 31.7 percent of people on 6 mg/kg and 40.7 percent of those on 10 mg/kg experienced some form of ARIA, compared to 10 percent of controls.
Whether aducanumab will be approved is a huge question the FDA and international regulators will decide. That aside, though, most researchers at CTAD saw a deeper significance for the field in these new data. “Everyone I’ve talked to agrees that this validates amyloid as a target,” Selkoe said. Aisen went further. “The lesson here is that the amyloid hypothesis is going to yield effective therapies,” he predicted. Slides of Budd Haeberlein’s presentation are available on Biogen’s website.—Madolyn Bowman Rogers
Fluid AD Biomarkers Link P-Tau to Synapses, Inflammation
CSF Aβ42 and phospho-tau may remain the quintessential fluid Alzheimer’s disease biomarkers for now, but a growing cadre of other markers are also proving their worth. At the Clinical Trials on Alzheimer’s Disease conference, held December 4–7 in San Diego, California, researchers broadened their horizons with data on CSF markers of glial activation, synaptic dysfunction, and neurodegeneration. Particularly in people with amyloid pathology, a bevy of these new markers tracked closely with CSF p-tau, and sharpened predictions of a person’s impending cognitive decline. The expanded set of markers even debuted as outcome measures in clinical trials.
Henrik Zetterberg of the University of Gothenburg in Sweden broached the topic of the burgeoning breadth of biomarkers at CTAD by highlighting upward of 20 emerging biomarkers, ranging from those that track core AD pathology Aβ and tau to markers of synaptic dysfunction, axonal damage, neuroinflammation, vascular function, and neurodegeneration. Plasma biomarkers are increasingly soaking up the limelight (see next story), but Zetterberg said that without validating potential biomarkers in CSF first, the field would have little grasp of what plasma markers portend about goings-on inside the brain. This is especially true for neuroinflammatory markers, many of which are also expressed outside the brain, he said. Therefore, validating all AD biomarkers in CSF is critically important.
Enter NeuroToolKit
With so many new markers coming onto the scene, Zetterberg and his Gothenburg colleagues spearheaded a new effort to validate a curated set of CSF markers across multiple cohorts. The Swedish team selected the markers, and Roche developed a standardized master assay—called NeuroToolKit—to measure all of them in CSF. Besides the core AD biomarkers CSF Aβ42, Aβ40, p-tau-181, and total tau, the markers include α-synuclein as a gauge of synaptic dysfunction; S100b, YKL-40, and glial fibrillary acidic protein (GFAP) as markers of astrocyte activation; soluble TREM2 and IL-6 as markers of microglial activation and inflammation; and neurofilament light (NfL) and neurogranin as markers of axonal injury and synaptic dysfunction. The investigators vote new candidate biomarkers into this jointly evaluated set as individual research labs produce promising evidence on them.
At AAIC earlier this year, scientists presented initial NeuroToolKit measurements in the Wisconsin Registry for Alzheimer’s Prevention (WRAP) cohort (Aug 2019 news).
At CTAD, Carol van Hulle of the University of Wisconsin in Madison presented a deeper analysis of correlations among NeuroToolKit markers in 294 CSF samples taken from 202 participants in the WRAP and Wisconsin Aging and Disability Resource Center (WADRC) cohorts that were sent to Gothenburg for analysis. These samples are a subset from WRAP/WADRC’s larger pool of 1,525 samples; they were picked because they represented the full spectrum of AD stages, including 115 people without cognitive deficits, 40 with MCI due to AD, and 47 with AD dementia. The analysis presented at CTAD is cross-sectional, but the scientists are gathering serial samples and many of the participants now have already donated more than one, van Hulle said.
First, the researchers compared each so-called “exploratory biomarker” in the NeuroToolKit to the core AD markers CSF Aβ42 and p-tau-181. The only marker that correlated significantly with Aβ42 was p-tau-181. In contrast, every biomarker in the tool kit, except IL-6, correlated with p-tau-181—indeed, total tau, neurogranin, and α-synuclein moved in lockstep with p-tau-181. As would be expected, both the Aβ42/40 ratio and p-tau/Aβ42 ratio correlated tightly with amyloid-PET measures in the subset of participants who had been scanned. Several of the neurodegenerative and neuroinflammatory biomarkers were higher in people whose p-tau/Aβ42 ratio indicated they had AD pathology than in those who did not. This was true for NfL, neurogranin, α-synuclein, YKL-40, S100b, and GFAP in people with MCI due to AD, and only true for neurogranin in cognitively normal people. These same biomarkers were also higher among people with MCI who later progressed to dementia.
Do additional biomarkers sharpen predictions of a person’s cognitive performance, over p-tau-181/Aβ42 alone? The answer was yes for the neurodegenerative biomarkers α-synuclein, neurogranin, and NfL, which better distinguished between cognitively normal people and those with MCI/AD, and more tightly correlated with performance on the Preclinical Alzheimer’s Cognitive Composite (PACC) than did the core markers alone. However, the glial activation markers YKL-40, sTREM2, GFAP, and S100b did not improve these cognitive correlations over p-tau-181/Aβ42.
Van Hulle suggested that neuroinflammatory markers might track better with clinical progression at later stages of disease. One audience member countered that the opposite may be true—that inflammation might flare up earlier on, and sharpen predictions about progression from normal cognition to MCI. Van Hulle responded that the NeuroToolKit must be tested in larger cohorts. In this sample, only 11 people progressed from being cognitively normal to MCI.
To Zetterberg, the neuroinflammatory markers don’t improve the power of CSF Aβ42/tau to forecast cognitive decline because inflammation is not specific to AD. Inflammation rises with age, in response to vascular problems, infections and, of course, amyloid and tau pathology. “Glial activation markers are simply noisy,” Zetterberg said. For that reason, he expects that they may track with AD later in disease, as inflammation triggered by tau pathology and neurodegeneration surpasses other age-related triggers.
Will other forms of p-tau—namely p-tau-217, which appears to rise even earlier in disease than p-tau-181—be added to the tool kit? Van Hulle said that while she is aware of the new data on p-tau-217, there are not yet plans to add it.
José Luis Molinuevo of Barcelonaβeta Brain Research Center (BBRC) in Barcelona, Spain, uses the NeuroToolKit to understand the interplay between core AD and neuroinflammation and synaptic dysfunction markers in the earliest stages of AD. Molinuevo presented data from 383 participants from ALFA+, a cohort of cognitively normal people aged 45–65 with a family history of AD (Molinuevo et al., 2016). Though 100 people in this cohort had CSF Aβ42/40 ratios indicative of brain amyloid accumulation, only minimal Aβ deposition showed up on their PET scans, suggesting they were in the earliest stages of the disease.
Like van Hulle, Molinuevo reported strong correlations between p-tau and several other biomarkers, most notably neurogranin, while Aβ42 only correlated with p-tau. Connections between p-tau and other biomarkers were strongest in people with amyloid and tau pathology as per their CSF cutoffs. Plotting biomarker levels against the Aβ42/40 ratio, Molinuevo reported that many of the biomarkers started rising just as the cutoff for amyloid positivity was reached, with neurogranin and p-tau shifting most dramatically. NfL, as well as inflammatory markers sTREM2, YKL-40, and GFAP, rose more gradually after the Aβ42/40 threshold was crossed. The findings placed Aβ aggregation at the beginning of the Alzheimer’s cascade, with p-tau, and markers of synaptic dysfunction, neurodegeneration, and neuroinflammation following suit.
Molinuevo reported that YKL-40 correlated with brain shrinkage, while NFL associated with waning glucose metabolism as assessed by FDG-PET.
Together, the Wisconsin and Barcelona findings suggest that very early in the AD continuum—at a time when Aβ is already abnormal according to CSF but not yet on PET—markers of synaptic dysfunction and neuroinflammation are beginning to change.
The most striking finding from the ALFA+ study, according to Zetterberg, was that it validated neurogranin as being specific for AD. This synaptic protein correlated highly with CSF-p-tau, and only rose in people whose Aβ was abnormal. He thinks neurogranin gets released into fluid when synapses malfunction in response to increasing tau hyperphosphorylation, which are themselves triggered by Aβ. NfL, a more general marker of neurodegeneration, rises later as neurons die, along with the neuroinflammatory markers, he added.
Taking It to Trial
Companies developing therapeutics are starting to implement the expanded set of biomarkers as outcome measures in clinical studies. For example, Lindsay Burns from Cassava Sciences in Austin, Texas, presented biomarker data from a tiny Phase 2a trial of PTI-125, a small molecule that reportedly blocks pathological interactions between Aβ and the α7-nicotinic receptor, and TLR-4 (Jul 2012 conference news; Wang et al., 2017). Aβ binding to α7-nicotinic receptors sets off tau phosphorylation and neurodegeneration, while Aβ binding to TLR-4 unleashes neuroinflammation. PTI-125 blocks Aβ associations with both receptors indirectly, by binding to and changing the conformation of filamin A, a scaffolding protein that facilitates Aβ’s receptor interactions. As such, Cassava proposes that its compound could squelch both neurodegeneration and neuroinflammation, and is using biomarkers to track those phenomena.
Cassava used change in a panel of CSF markers as its primary endpoint for this trial, which evaluated a 28-day course of 200 mg daily dose of PTI-125 in 13 people with mild to moderate AD. The panel included Aβ42, neurogranin, NfL, total tau, p-tau-181, YKL-40, IL-6, IL-1β, and TNF-α. By the end of the month on PTI-125, Burns reported, each biomarker had budged, ranging from a more than 30 percent drop in p-tau and neurogranin to dips of 5 to 14 percent in the inflammatory markers. CSF Aβ42 nudged slightly upward, albeit insignificantly. Cassava measured many of these markers in plasma as well, reporting a 41 percent plummet in neurogranin, a 16 percent drop in total tau, and 11 and 4 percent dips in NfL and YLK-40, respectively.
Cassava scientists also took stock of plasma p-tau. Where previous research gauged “p-tau” based on phosphorylation at threonine 181, nowadays researchers are taking a more granular approach, parsing different forms of tau in plasma and CSF. In San Diego, Burns reported declines in the PTI-125 group in p-tau-T181, p-tau-T202, p-tau-T231, and a nitrated form of tau—n-tau-Y29—in plasma. This small study had no placebo group. Burns claimed that every patient responded to treatment across most markers, and that the drug was safe and well-tolerated.
Burns believes the data support the proposed dual mechanism of action, suggesting a change signal in both neurodegenerative and neuroinflammatory biomarkers, as well as the core AD markers. In September 2019, Cassava started a placebo-controlled Phase 2b trial with 60 participants. For more data on plasma biomarkers, see next story.—Jessica Shugart
Blood Tests of Phospho-Tau, Aβ42, Track With Brain Amyloid
While a suite of new CSF markers has entered a mature stage where they get validated with identical methods in large international cohorts (see previous CTAD story), much newer blood tests are catching up fast. Scientists are pushing the limits of detection of the core AD markers in plasma, and in the process are cracking open what used to be considered the single category of p-tau into a bewildering new spectrum of subspecies. At CTAD, Nicolas Barthélemy of Washington University, St. Louis, showed what this looked like when he presented fresh mass spectrometry data on different forms of plasma phospho-tau. Compared with immunoassays, which rely on antibodies trained against a priori designated phospho-epitopes, mass spectrometry gives an unbiased, and exquisitely specific, view of the landscape of phospho-tau isoforms in a given sample.
Previously, Barthélemy and colleagues put mass spec to use in CSF samples. At AAIC earlier this year, he wowed the crowd by reporting that, in samples collected from the DIAN cohort, CSF p-tau-T217 started rising in CSF just after Aβ started accumulating in the brain—fully 20 years prior to estimated symptom onset (Aug 2019 news). CSF p-tau-181—the epitope most commonly measured in biomarker immunoassays—trailed close behind, at 19 years prior to onset.
At CTAD, Barthélemy presented his mass spectrometry data on plasma tau. This project posed unique challenges. For one, at less than 1 pg/mL, the concentration of p-tau in plasma is vanishingly low, while the complexity of other proteins in this bodily fluid is exorbitant, making p-tau detection there akin to searching for a needle in a haystack. For another, while mass spectrometry is highly specific, it is less sensitive than immunoassays. Compared with the 0.2 mL required to detect p-tau-T181 using the most sensitive immunoassay available, Barthélemy needed 20 mL of plasma to detect it via mass spec. That translates to a 40 mL blood draw, which is not feasible on a large clinical scale, researchers agree. For a typical blood panel in routine medical care, a phlebotomist draws 5–10 mL. This is why mass spec is particularly well suited for discovery research, but will need to become dramatically more sensitive for large-scale use or population screening.
It so happened that Barthélemy had access to large volumes of plasma that had been collected from 35 participants at the Knight Alzheimer’s Research Center during a stable isotope labeling kinetics (SILK) study that tracked the half-life of tau (Mar 2018 news). Twenty of them had tested negative for Aβ accumulation by CSF-Aβ42/40 and amyloid -PET, while 15 were positive. Among the latter, five were cognitively normal, eight were at the stage of MCI due to AD, and two had moderate AD dementia. As was the case in DIAN, too, only the amyloid-positive, cognitively impaired participants had significant accumulation of tangles as assessed by tau PET. In contrast, CSF measurements of p-tau-T181 via mass spec were elevated earlier, in people with amyloid plaques who were still cognitively normal.
P-tauT217 in Plasma: Aβ in Brain. Plasma concentrations of p-tau-T217 were elevated in people harboring Aβ deposits in the brain. [Courtesy of Nicolas Barthélemy, Washington University, St. Louis.]
But what about their plasma? Similar to what Barthélemy observed in CSF, he found tau peptides of 14 different lengths, most of which were truncated at the C-terminus. However, of the 11 unique phosphorylated sites Barthélemy was able to detect in CSF, only four were detectable in plasma: p-tau-T181, p-tau-S202, p-tau-T205, and p-tau-T217. Levels of both p-tau-T181 and p-tau-T217 were significantly higher in plasma from amyloid-positive than -negative participants, including those who were cognitively normal. While p-tau-T181 was 74 percent higher, p-tau-T217 was up by a whopping 365 percent, an exponentially more robust difference than detectable with Aβ42/40 ratio tests. Moreover, p-tau-T217 in plasma correlated closely with p-tau-T217 concentration in CSF, suggesting that its blood level reflects processes in the brain.
In contrast, Barthélemy found that, for both plasma total tau and p-tau-S202, their concentration was about the same in people with and without brain amyloid. He believes p-tau-S202 gets released from cells outside the brain, in which tau is more highly phosphorylated on serine 202 than is tau in the CNS. A high background level of p-tau-S202 in the periphery would mask changes in p-tau-S202 contributed by the CNS. On the other hand, p-tau-181 and p-tau-217 are more specific to the CNS, allowing for detection of subtle differences in the plasma. As for p-tau-T205, Barthélemy only detected it in some plasma samples from the cohort, and its concentration was exceedingly low. He told Alzforum that this phospho-species is likely specific to the CNS, and exists at too low a concentration for consistent detection in plasma even with current mass spec methods.
Will plasma p-tau tests pick up other tauopathies, such as progressive supranuclear palsy or frontotemporal dementia? Barthélemy said that as of now, he has no access to cohorts with sufficient plasma to address this question. Just as is being done in AD, p-tau species that change in other tauopathies will need to be validated in CSF before digging for them in plasma.
Barthélemy acknowledged that the volume of plasma needed to detect p-tau by mass spectrometry precludes implementing the MS-based test on a large scale. MS assays are becoming more sensitive, however. SEABIRD, a St. Louis area study to learn how well MS-based tests pick up CNS Aβ too, is currently validating a protocol that requires only 4 mL of plasma.
For his part, Zetterberg and colleagues are using mass spec as a guide to help them develop sensitive immunoassays, especially for p-tau-T217. At CTAD, Zetterberg talked about measuring p-tau-T181 in plasma using the Simoa platform. In samples from the McGill Translational Biomarkers in Aging and Dementia (TRIAD) cohort, Zetterberg reported that p-tau-181 was two- to threefold higher in people with MCI or AD, compared with cognitively normal elderly people. Yes, fold. Not a few percent higher.
This agrees with findings reported by other groups using different antibodies and platforms, including researchers at Eli Lilly in Indianapolis, the Swedish BioFinder group at Lund University, as well as researchers at the University of California, San Francisco (Mielke et al., 2018, and Aug 2019 conference news). Zetterberg noted that p-tau-T181 has been more extensively validated than p-tau-T217 as an AD biomarker, both in CSF and plasma, and therefore will likely be the first to garner approval as a bona fide AD screening test. If sensitive immunoassays for p-tau-217 are developed and validated in multiple cohorts, it could follow suit.
A test for plasma Aβ is already moving through the paces for regulatory approval, though it has to squeak by with measuring a less than 20 percent difference between groups. At CTAD, Tim West of C2N Diagnostics, a company formed 12 years ago by WashU researchers Randall Bateman and David Holtzman and LifeTech Research, Inc., reported fresh results from C2N’s mass spectrometry-based test for the plasma Aβ42/40 ratio.
Previously, Bateman had reported that among 158 participants in the Knight Alzheimer’s Disease Research Center, the plasma Aβ42/40 ratio correlated tightly with Aβ-PET (Aug 2019 conference news on Schindler et al., 2019). People with elevated plasma Aβ42/40 ratios whose amyloid PET scans were negative were 15 times more likely to be positive on a subsequent scan taken two to nine years later, suggesting the plasma test was predictive of amyloidosis in the near future.
At CTAD, West extended those findings to a more-diverse, less-standardized group of samples that are part of C2N’s clinical validation study. He analyzed 415 plasma samples collected from six independent biobank cohorts, in which blood was collected, handled, and stored in different ways, and Aβ positivity was independently determined using different methods (i.e., CSF versus PET, with a variety of tracers). Thirty-nine percent of the combined cohort harbored brain amyloid, according to these independent methods. Participants averaged 70 years of age, and among 309 people with ethnicity data, 23 percent were Hispanic. Forty percent carried at least one copy of ApoE4. West reported an 81 percent concordance between the Aβ42/40 plasma test and amyloid status. In a receiver operator curve analysis—which measures the probability that a given measure will predict a specific outcome—the results translated to probability of 0.86, which improved to 0.90 once the researchers factored in age and ApoE4 status.
Consistent with Bateman’s previous study, the plasma test produced more false positives than false negatives. West hypothesized that those false positives were not truly false, but rather a reflection of using a binary outcome to classify what is in reality a disease continuum. In other words, these “false positives” might lie just below the threshold of detection via PET. If this is the case, these people, too, would be expected to have a positive scan in the near future.
C2N is currently validating their ratio test in samples from participants in the PARIS study, who had amyloid PET scans as part of the Imaging Dementia—Evidence for Amyloid Scanning (IDEAS) study. Based on data collected from this sample set, C2N will seek approval for the test as an in vitro diagnostic.—Jessica Shugart
Amyloid Clearance: Check. Cognitive Benefit: Um … Maybe.
With aducanumab’s resurrection driving renewed interest in anti-amyloid antibody therapies, researchers at the 12th Clinical Trials on Alzheimer’s Disease conference, held December 4–7 in San Diego, happily provided updates on their own. Presentations on gantenerumab, donanemab, and BAN2401 reinforced previous reports that all three mop up plaques, and added more evidence that this clearance is sustained over time, even after a treatment interruption. There are hints—though that is all they are at this point in time—that amyloid clearance correlates with preserved memory, as well.
Among antibodies still in Phase 2 or 3, Roche’s crenezumab appears to be the outlier. In the terminated Phase 3 CREAD studies, it did not affect cognitive decline, and in San Diego, the company reported that it did not budge amyloid or tau buildup, either. However, crenezumab did lower soluble Aβ plus a marker of synaptic damage, suggesting target engagement. Overall, data at CTAD supported the idea that several antibodies are still in the running as Alzheimer’s treatments.
“I believe amyloid deserves the attention we give it,” Paul Aisen of the University of Southern California, Los Angeles, said in a keynote address. He noted that all evidence suggests the core pathological process of Alzheimer’s disease is closely linked to amyloid accumulation. After the recent signal that aducanumab probably slowed cognitive decline in one Phase 3 trial, researchers in the field are increasingly optimistic that targeting amyloid will be effective (Dec 2019 conference news).
Different Trajectories, Same Result. Three representative patients in the gantenerumab long-term extension study cleared plaque at different rates, but all ended up below zero at year three. [Courtesy of Roche.]
So how well do current antibodies clean up plaque? Gregory Klein of Roche previously reported that two years of treatment with high-dose gantenerumab drove down amyloid load (Dec 2017 news; Aug 2018 news). In San Diego, Klein added three-year data from the same open-label extension study. This OLE originally enrolled 89 patients, and 30 remain in the study at this point. Ten came originally from the Scarlet RoAD trial, 12 from the Marguerite RoAD placebo group, and eight from the Marguerite RoAD treatment group.
Klein noted that the time course of amyloid removal in the OLE varied from patient to patient. He showed three representative examples of people who all started with amyloid loads of 90 to 100 centiloid (see image above). In the first, the plaque burden plummeted by about 90 centiloid in one year, falling to near-zero. Plaque clearance continued at a slower rate for the next two years, with this person now at -30 centiloid. The centiloid scale can go as low as -50. Another participant dropped from 90 centiloid to the positivity threshold of 24 in the first year, then lost plaque more gradually, ending up around -10 centiloid by the third year. The third patient started slow and sped up, clearing about 20 centiloid the first year, 30 the second, and 50 the third, to end below zero in year three. Despite this variation, overall the amyloid load fell continuously in every participant, without hitting plateaus and stalling, Klein said.
After three years, the OLE participants’ average amyloid burden is zero centiloid, representing a drop of 80 or 90 centiloid from baseline. Eighty percent of them are below the amyloid-positivity threshold, compared with 50 percent at the two-year mark. In general, people who start with a higher amyloid burden tend to lose it faster, Klein noted. Scarlet RoAD participants started the OLE with an average centiloid of 53, and ended up losing 57 over three years, while the Marguerite RoAD placebo group started at 91 and lost 90 centiloid on average.
Klein did not discuss cognitive outcomes for these people in San Diego. However, Paul Delmar of Roche had reported two-year cognitive data at AAIC last July, and the data were published on December 12 (Klein et al., 2019). At this timepoint, a regression analysis found that greater amyloid clearance correlated with slower cognitive decline. To look at the data another way, the researchers split the 39 participants remaining in the two-year OLE cohort at that time into those in whom amyloid reduction was either more, or less, than the median of 55 centiloids less than at baseline. Those in the “more-clearance” group had declined 0.8 points less on the CDR-SB, 2.2 points less on the ADAS-Cog11, and 0.9 points less on the MMSE than those in the “less-clearance” group.
These directional trends on cognition were not statistically significant in these small groups, and the authors cautioned against overinterpreting them in a diverse group that included people with prodromal and moderate AD to start with. At OLE baseline, participants had an average MMSE score of 21. When averaged across this OLE group, the cognitive benefit in the high-clearance group amounted to a 20 percent slowing of decline on the CDR-SB, 47 percent on the ADAS-Cog11, and 17 percent on the MMSE, similar in size to the signal reported in the Phase 3 aducanumab trial.
How about Eli Lilly’s donanemab? Previously known as N3pG, this Phase 2 antibody banishes plaque from the brain faster than gantenerumab does. It targets a pyroglutamate form of Aβ found only in amyloid plaques, and reportedly carries a lower risk of ARIA. The Phase 1b trial enrolled 61 participants with prodromal to moderate AD whose average baseline amyloid load was 105 centiloid. Previously, Lilly reported rapid plaque clearance with six months of dosing (Aug 2018 conference news).
In San Diego, Eli Lilly’s Stephen Lowe added 16-month data from this group. At monthly doses of 10 or 20 mg/kg, their amyloid load dropped an average of 90 to 100 centiloid. Curiously, the 10 mg/kg biweekly dose group had a smaller average reduction than either monthly group, amounting to about 50 centiloid, though by this time point, the groups were small. Only nine people remained in the 10 mg/kg biweekly dose group, six in the 10 mg/kg monthly, and five in the 20 mg/kg. In all five in the latter group, their amyloid load was below the threshold of positivity.
As in the six-month data, 26 percent of patients developed ARIA-E. Two of the 12 cases were symptomatic, with participants complaining of headache, confusion, or sleepiness that resolved after dosing was stopped. ApoE genotype did not affect ARIA or the rate of amyloid removal. Lowe noted that this trial has just completed dosing, with the last patient visit on November 5, and will undergo data lock soon. Lilly is currently testing donanemab in an 18-month Phase 2 trial, which enrolls 266 people with prodromal to mild AD.
And Stay Gone! Once removed with high-dose BAN2401 (blue and green lines), amyloid plaque stays low over a gap period as long as four years (black, placebo control). [Courtesy of Eisai.]
And then there is BAN2401. The CTAD presentation on this anti-protofibril antibody discussed what happens when a person takes a break from dosing. In Phase 2, BAN2401 cleared plaque and slowed cognitive decline by a bit after 18 months of treatment (Jul 2018 news; Jul 2018 conference news; Nov 2018 conference news). This led the Eisai/Biogen partners to invite participants back to begin an open-label extension (May 2019 conference news), and in San Diego, Eisai’s Chad Swanson presented baseline data from it.
This OLE enrolled 35 people who had previously been on the 10 mg/kg biweekly dose, 52 people from the 10 mg/kg monthly group, 26 from the 5 mg/kg biweekly, six from the 5 mg/kg monthly, and 37 from the placebo groups. At OLE baseline, they had been off drug for an average of two years but with wide variability, spanning from nine months to 4½ years. Despite this range, their amyloid burden remained almost flat during this gap period. At the end of treatment, participants had a mean SUVR of 1.05, and at OLE baseline, 1.08. About 80 percent of participants were amyloid-negative by visual read at the end of Phase 2, and this remained true for those who enrolled in the OLE. Spaghetti plots of the two 10 mg/kg groups showed similar flat trajectories for nearly all participants, although in a handful of people in the monthly, but not biweekly, dose group, some amyloid returned.
By contrast, their cognition started sliding again during the gap period. There was a dose-dependence to this. For those who had received low doses, their cognitive decline caught up to that of the placebo group by the end of the gap period, with indistinguishable scores. Participants on either of the 10 mg/kg doses, on the other hand, maintained the small numerical improvement over placebo they had gained in the blinded trial. Once they went off drug, however, their cognitive decline resumed at the same rate as in the placebo group. This maintained benefit implies a disease-modifying effect, Swanson noted. Because cognitive decline resumed even while plaque load stayed low, it may be that Aβ protofibrils are doing the damage, Swanson speculated. To prevent this, people might need to remain on treatment even after their amyloid load falls below the plaque threshold, he added.
Eisai updated the CTAD audience on their design of a Phase 3 trial. Called CLARITY, it began enrolling in March 2019 and aims to pull in 1,566 people with MCI or mild AD, the same population targeted in Phase 2. They will be randomized evenly to receive placebo or 10 mg/kg biweekly for 18 months. As in Phase 2, the primary outcome measure will be the CDR-SB, with ADCOMS, ADAS-Cog14, and amyloid PET as secondary measures.
Odd One Out?
Crenezumab stood apart at CTAD, because its data showed little effect on biomarkers of pathology. This antibody recognizes soluble Aβ, with 10-fold selectivity for oligomers over monomers, and was previously reported to lower Aβ oligomers in CSF (Jul 2018 conference news). The two Phase 3 CREAD trials were halted in January, and subsequent analysis of CREAD1 data found no effect on cognitive decline (Jan 2019 news; May 2019 conference news).
In San Diego, Tobias Bittner of Roche presented biomarker findings for CREAD1, which enrolled 813 people with an average MMSE of 24. Of these, 351 took part in the amyloid PET substudy. Over two years, amyloid load increased less in people on drug than those on placebo, but the difference was not statistically significant. CREAD1’s tau PET substudy comprised 22 participants on drug and 24 on placebo. It showed a slight worsening of 0.05 SUVR in those on drug after a year, but this might be noise from the small sample size and short follow-up, Bittner suggested.
The CSF substudy comprised 139 people on drug and 140 on placebo. It showed a slight drop of total tau and p-tau181 with treatment, indicating improvement, although again this fell short of statistical significance. Numerically, total tau dropped by 10 pg/ml, p-tau181 by about two. Volumetric MRI showed no difference at all between groups.
Did anything change substantially with crenezumab? CSF Aβ40 and Aβ42 rose significantly and dose-dependently, suggesting that crenezumab hit its target in the central nervous system. Aβ42 jumped about 300 pg/ml from baseline values, and Aβ40 about 7,000. CSF Aβ falls during the progression of AD pathogenesis. CSF neurogranin, a marker of synaptic dysfunction, dropped significantly, falling by about 100 pg/ml. Four other biomarkers of neuronal injury or inflammation—NfL, α-synuclein, YKL-40, GFAP—nudged in the desired direction. Curiously, sTREM2, which has been associated with protection against decline, dropped (Aug 2019 news). Crenezumab is being tested in the API Colombian trial in a preventative paradigm (Aug 2019 conference news).
Overall, the antibody data fed the enthusiasm sparked by the aducanumab findings also shown in San Diego. Cynthia Lemere of Brigham and Women’s Hospital in Boston noted that the totality of evidence now suggests anti-amyloid antibodies will work as a preventative treatment to delay AD progression. “Now it’s a matter of refining the therapies and trying combinations,” she told Alzforum.—Madolyn Bowman Rogers
Picking Through the Rubble, Field Tries to Salvage BACE Inhibitors
Anti-amyloid antibodies are at long last showing promise for treating Alzheimer’s disease, but for long-term use, researchers are seeking cheaper and easier options than monthly infusions. They want that elusive once-a-day pill. BACE inhibitors have been the leading option, but with all trials of these drugs now halted due to cognitive worsening, isn’t the approach just plain dead? At the 12th Clinical Trials on Alzheimer Disease conference, held December 4–7 in San Diego, California, researchers debated this question at a roundtable. Most were not yet ready to write off BACE inhibitors. As an oral drug that directly suppresses Aβ production, these compounds still represent a practical way to slow plaque accumulation and maybe, just maybe, put the brakes on the disease at an earlier stage.
“It’s hard to look away from BACE as a target,” said Michael Irizarry of Eisai.
To be sure, many questions remain to be answered before researchers will know whether any dose with the current batch of compounds is safe enough for long-term use. Chief among them are: Is the cognitive deficit reversible? Does it relate to neurodegeneration? Does it occur at low doses?
Some of the answers can come from analyzing existing data from the halted trials. This process is currently ongoing at many companies. In San Diego, pharma representatives provided an additional glimpse at this. The data to date suggest that the cognitive side effect does not worsen over time and may perhaps reverse once dosing stops. From the current high-dose trial data, however, researchers cannot tell what might happen to cognition at very low doses. Eric Reiman of Banner Alzheimer’s Institute in Phoenix suggested that lower doses be tested in a new trial that would include extensive safety monitoring. “The benefits of BACE inhibition in amyloid-negative people at genetic risk for AD could outweigh the risks,” Reiman said. Others agreed. Concern about trial participants, particularly people who have found out their ApoE genotype in hopes of prevention trials, pervaded the meeting, and researchers agreed all should be done to keep those cohorts engaged.
Pharmaceutical companies have been sharing their data with each other in hopes of solving the BACE inhibitor puzzle and finding a way to move forward. This cooperative spirit continued in San Diego. “This degree of industry collaboration is unprecedented,” noted Maria Carrillo of the Alzheimer’s Association.
Researchers at Merck first reported finding a small cognitive deficit associated with BACE inhibition in their Phase 3 verubecestat trials, which had already been halted due to lack of efficacy (Dec 2017 conference news; Apr 2019 news). Similar news on the other BACE inhibitors followed in rapid succession, with Janssen’s atabecestat, Novartis’ umibecestat, and Biogen and Eisai’s elenbecestat all now linked to cognitive worsening (Nov 2018 conference news; Jul 2019 conference news; Sep 2019 news). To most researchers, the preponderance of the data imply a drug class effect, rather than something off-target.
In addition to the cognitive deficit, BACE inhibitor treatment also consistently comes with weight loss, skin rashes, and increased neuropsychiatric symptoms. Perhaps most concerning, data from some of the trials show greater brain atrophy on drug, as well, suggestive of accelerated degeneration. Lilly’s lanabecestat has not been tied to a statistically significant cognitive deficit, but does associate with greater volume loss (May 2019 conference news).
Cognitive Deficits Consistent Across the Drug Class
In San Diego, presenters focused most of their attention on the cognitive problems. Ana Graf of Novartis showed more data from the two terminated API Generation trials of umibecestat. The trials enrolled ApoE carriers who were cognitively healthy, with an average MMSE of 29. They took either 15 or 50 mg per day of umibecestat. These doses lowered their Aβ40 in cerebrospinal fluid by 70 and 87 percent, respectively.
On the RBANS cognitive composite, people on either dose of drug performed worse on tests of immediate and delayed memory than the placebo group did. Their deficit appeared at the earliest time point tested, three months, and stayed consistent at six months. The Cohen’s d effect size was 0.2-0.3, which is considered small. In addition, more people in the treatment group than on placebo declined by one standard deviation or more on the RBANS, or experienced any decline on the CDR-SB, Graf said, though she did not provide those numbers. Graf noted that one-third of the cohort was amyloid-negative, demonstrating that plaques are not required for this cognitive effect.
Curiously, however, people taking umibecestat performed better than the placebo group on the RBANS language domain. In the discussion afterward, John Sims of Lilly noted that this is a consistent pattern with BACE inhibitors, seen with lanabecestat and verubecestat as well. It seems people on drug score worse than placebo on memory tests but better on language tests. “This is a huge mystery,” Sims said. Michael Egan of Merck said that these similar effects across many BACE inhibitor trials suggest unknown class effects of these drugs. “We need to understand the biology of BACE better. Are other substrates involved?” Egan asked.
Blinded follow-up visits with Generation participants will continue for the next six months, Graf said. Researchers are currently analyzing cognitive data collected three months after dosing stopped, which will reveal whether the cognitive deficits reversed.
Data from other inhibitors look similar. Irizarry provided a brief update on the elenbecestat Phase 3 trials, which were halted in September after a safety review found an unfavorable risk-benefit ratio. Called MISSION AD1 and 2, these trials tested 50 mg of elenbecestat, which lowers CSF Aβ40 by 70 percent. The last patient visits are finishing now, and the database will be locked early next year, Irizarry said.
Because these trials were halted early, only a few patients completed the full two-year course of treatment. Eisai has six-month data for about 1,700 patients, 12-month data for 900, 18-month data for 300, and two-year data for only 40. A cognitive deficit did not appear at the early time points, unlike with the other inhibitors. At the two-year mark, however, participants on elenbecestat scored worse than those on placebo on the CDR-SB. The difference was not statistically significant, but that may be because of limited power, with only about 20 people in each arm at this time point, Irizarry said.
Participants on elenbecestat also lost more weight and had more skin rashes and neuropsychiatric effects than controls, continuing a common theme with BACE inhibitors. Overall, the negative effects and lack of cognitive benefit tipped the scales against this drug, Irizarry said.
The findings overall echo data from the verubecestat Phase 3 APECS and EPOCH studies, which enrolled people with prodromal or mild AD, respectively. These trials tested doses of 12 or 40 mg verubecestat, suppressing Aβ40 in CSF by 67 to 84 percent, respectively. As with umibecestat and atabecestat, the cognitive deficit on verubecestat appeared at three months, the earliest time point studied, and did not worsen over time, Julie Stone of Merck said in San Diego.
What about lanabecestat? It did not report statistically significant cognitive worsening. Hints of a negative cognitive effect came through here, too, Sims told Alzforum. Researchers saw poorer performance on drug when they looked at subpopulations such as the MCI group, and they also saw memory deficits on highly sensitive exploratory cognitive measures. “My feeling is that the patterns across the inhibitors are pretty similar, although magnitudes may be slightly different,” Sims told Alzforum.
On the important question of whether cognition recovers, researchers thus far have little data to share. David Henley of Janssen presented the only findings on this in San Diego. He reported that participants who had taken five months of atabecestat and developed a cognitive deficit rebounded after three months off drug.
It is also unclear if the cognitive harm varies with disease stage. Eric McDade of Washington University in St. Louis noted that the deficit appears more obvious in preclinical than prodromal cohorts, but this may be because it is easier to measure a subtle cognitive effect in a less-impaired population. Furthermore, researchers do not know whether the memory problems are perceptible to patients. Reiman wondered how the deficit compared with other factors that blunt mental acuity, such as drinking wine.
The mechanism remains a mystery. The idea that has been bandied about the most—that the inhibitors block both isoforms of their target enzyme—seems not to be the reason for the problems with these recent trials. “We don’t think it is a BACE1 versus BACE2 issue,” Sims said. Elenbecestat and umibecestat are about threefold as selective for BACE1, but still triggered this symptom. Henley suggested that BACE1 substrates other than APP are likely involved.
Researchers agreed that animal studies will provide insight here. In San Diego, Riqiang Yan of University of Connecticut Health, Farmington, reminded the audience that BACE-null mice have a learning deficit, suggesting that BACE is required for synapses to function optimally. Knocking out BACE in the 5xFAD model reverses plaque deposition but does not completely restore synaptic function, again hinting that neuronal transmission requires BACE, Yan said (Hu et al., 2018). Other scientists, including Jochen Herms of DZNE in Munich, have been calling attention to this, as well.
Curiously, agonists of the metabotropic glutamate receptor 1 rescue synaptic plasticity in BACE knockouts, Yan said at CTAD. He showed unpublished data that agonists boosted excitatory presynaptic potentials back to wild-type levels and restored memory in the Morris water maze and open-field tests. “BACE inhibitors could be used in combination with a synaptic enhancer for better efficacy,” he suggested. Unlike antagonists, mGluR1 agonists are little studied, and none have been approved by the Food and Drug Administration.
Would a Low Dose Dodge the Bullet?
Many researchers hope that a much lower dose of inhibitor could avoid the cognitive harm, but so far there is little evidence to support this. All trials to date have tested doses that inhibit Aβ production by 50 percent or more. Stone cautioned that the cognitive deficit on verubecestat was not dose-dependent across the range of 70-85 percent inhibition. “We cannot predict from our data what the cognitive response would be at lower doses,” Stone said.
How low a dose would still effectively suppress amyloid accumulation? On this question, Merck researchers have more information. Pharmacokinetic data from their dosing studies allowed them to model the effect of dose on plaque. At the high doses tested, BACE inhibitor treatment nudges down plaque load. Stone and colleagues calculated that a dose of 2 mg verubecestat, corresponding to 27 percent inhibition of Aβ40 production, would stabilize plaque load, preventing further growth. For a person without amyloid accumulation, this dose might be sufficient to halt disease progression.
Sims agreed, noting that Lilly’s internal modeling suggests the same thing. “We should shoot for 25 percent lowering,” he said in San Diego. However, Sims also emphasized there is no guarantee this would prevent cognitive harm. “At a lower dose, would it just take longer for the cognitive deficit to appear?” he wondered. Some animal data do suggest that dose is a key factor for synaptic health. In mouse studies, high doses of BACE inhibitors inhibit dendritic spine growth and synaptic plasticity, while low doses do not (Nov 2014 news). These mouse studies image synapses through a cranial window to get a sense for how these dynamic structures change over time.
What Does Brain Volume Loss Mean?
Researchers at CTAD expressed concern about the cortical shrinkage measured by MRI. This old problem has bedeviled Alzheimer’s trialists since the earliest anti-amyloid drug trials, indeed since the long-defunct AN-1792 vaccine (Jul 2004 conference news). Fifteen years later, scientists are still unsure how to interpret it and, lo and behold, it happens with BACE inhibition too. MRI scans have picked up greater brain volume loss in drug than placebo groups on at least three BACE inhibitors: verubecestat, lanabecestat, and umibecestat. In San Diego, Graf reported that umibecestat shrank whole-brain and hippocampal volume after six months, the earliest time point measured, although she showed no numbers. Eisai researchers have not reported volume loss with elenbecestat, but analysis of this trial is still ongoing. Janssen has not presented MRI data for atabecestat.
It is unclear whether atrophy is linked to the cognitive deficit. It may seem obvious, but some data suggest not. Stone reported that, unlike the cognitive deficit, volume loss was worse on a higher dose of verubecestat. “MRI changes don’t seem to correlate with cognitive effects,” Henley said, as well. Reiman agreed. Others are less certain. McDade noted that some data do suggest a relationship. Intriguingly, volume loss does appear to be linked to amyloid load. In San Diego, Adam Schwarz, who is now at Takeda after 12 years working on brain imaging at Lilly, reported that most of the shrinkage seen on verubecestat occurred in regions with a high plaque burden. Showing data provided by Cyrille Sur at Merck, Schwarz reported that people taking 12 mg verubecestat lost around 0.4 percent more from their baseline volume, compared with the placebo group, in amyloid-positive than in amyloid-negative regions. For people taking 40 mg, this difference was 0.6 percent.
Fluid shifts associated with lessening inflammation are often mentioned as a hopeful potential explanation for the measured volume loss in treatment groups. At CTAD, too, researchers overall sounded cautiously optimistic that the loss may not reflect increased degeneration. Stone noted that the shrinkage appeared early during verubecestat treatment and did not worsen further, with atrophy thereafter tracking at the same rate as in controls. In San Diego, Egan and Matthew Kennedy of Merck reported that there were no differences in several CSF markers of neuronal injury or inflammation in verubecestat Phase 3 substudies. NfL, total tau, GFAP, and UCH-L1 tracked the same in people on verubecestat or placebo for 18 months. Over this time frame, NfL, GFAP, and UCH-L1 bumped up consistently by about 12 percent in all treatment groups. Total tau was noisy, with a high variability from person to person that averaged out to a 20 percent increase. Likewise, Graf reported no worsening of CSF markers of degeneration in the Phase 2a umibecestat trial.
Where Do BACE Inhibitors Go From Here?
At CTAD, academic and company researchers agreed that finishing thorough analyses of the data from the terminated trials is a critically important first step to answer several of the questions they posed. Assuming these data confirm that BACE inhibitors at high doses cause a cognitive deficit that is small, stable, and reversible, without accelerating degeneration, then what would be next? Researchers threw out several ideas. Sims mentioned conducting additional safety studies in a nonhuman primate model, perhaps using volume loss as a proxy for the cognitive deficit, if the two are shown to be linked. Henley suggested testing a lower dose range of BACE inhibitors in healthy volunteers to see if they affect memory. “The dose and target population will be critically important for future trials,” Irizarry agreed.
For his part, Reiman would prefer to start a Phase 3 trial of lower doses, with increased safety monitoring. The Alzheimer’s Prevention Initiative has invested tremendous effort and resources into recruiting the two large Generation cohorts. In San Diego, Jessica Langbaum of Banner announced that in addition to halting umibecestat, researchers have now stopped dosing with the active vaccine CAD106 as well. This decision was made to allow the researchers to conduct unblinded interim analyses to better assess CAD106's efficacy and potential for future trials, Graf said. With all dosing now stopped in these API trials, participants are left at loose ends.
These participants deserve particular respect and thoughtful, ongoing engagement that includes new trial options before too long, because they agreed to find out their ApoE genotype for research and are aware of their high risk of impending Alzheimer’s dementia. “We want to enroll amyloid-negative Generation homozygotes in a new trial before they are lost,” Reiman said. Langbaum noted that Banner researchers are waiting to see whether the memory problems on umibecestat are reversible before deciding whether that new trial will include BACE inhibitors again.
Even in the absence of a new drug to test, Banner researchers, including Pierre Tariot, plan to offer continuing support, follow-ups, and information on future research opportunities to participants to keep them engaged. Novartis is clearly aware of a responsibility toward this cohort. “Banner and partners are committed to finding ways to maintain the cohort,” Graf said.
McDade also thinks future BACE inhibitor trials make sense, particularly for people who are at high genetic risk of AD. “BACE is still a rational target in autosomal-dominant AD. There is a strong mechanistic justification, particularly for prevention at an early time point,” McDade said.—Madolyn Bowman Rogers
Going Indirect: Can Therapies Halt Alzheimer’s from Outside the Brain?
For therapies taking aim at Alzheimer’s disease, exerting an effect in the brain seems critical. And obvious. But according to results on display at 12th Clinical Trials on Alzheimer’s Disease conference, held December 4–7 in San Diego, California, a therapy might also influence the brain without ever entering it. Researchers reported that apabetalone, a drug that tweaks expression of hundreds of genes via an epigenetic mechanism, reduced inflammatory proteins in the blood, including those that make inflammation-stoking leukocytes cling to blood vessel walls. Apabetalone appeared to slow cognitive decline, but only among those who were most impaired at baseline. Separately, a fresh statistical analysis of AMBAR—a trial that evaluated a plasma-exchange protocol—claimed a cognitive benefit in people with moderate AD, and hinted it could work in people with milder disease. Both trials were plagued by the limitation that they did not use biomarkers to select people who truly had AD, muddling signs of efficacy.
Apabetalone exerts its sway by inhibiting Bromodomain Extraterminal Domain (BET) proteins, which bind to the acetylated lysines that mark open, active stretches of chromatin. BET proteins then recruit transcription factors to the scene to switch on gene expression. Apabetalone essentially blocks the association between BET and acetylated lysines, squelching transcription of hundreds of genes. Previously, researchers reported that BET proteins drove the expression of myriad genes involved in inflammation, lipid metabolism, and vascular function, casting apabetalone as a candidate treatment for cardiovascular and other chronic diseases of aging.
At the AD/PD meeting in Lisbon earlier this year, Ewelina Kulikowski of Resverlogix, the Calgary-based company developing the drug, reported that in cell culture, apabetalone blocked expression of pro-inflammatory cytokines and leukocyte adhesion molecules on vascular endothelial cells; this kept monocytes from latching onto them (May 2019 conference news). Apabetalone also spared LPS-ravaged mice from damaging neuroinflammation.
At CTAD, Kulikowski presented data suggesting the drug douses vascular inflammation in people, too. She measured more than 1,300 proteins within plasma samples collected from 94 participants in the ASSURE trial, which evaluated a 26-week course of 200 mg of apabetalone for its effect on atherosclerotic plaques in people with cardiovascular disease (Nicholls et al., 2016). Kulikowski found that between baseline and 26 weeks, samples from the apabetalone group had significant drops in inflammatory mediators, particularly chemokines and cell adhesion receptors known to snag circulating leukocytes and rev up vascular inflammation.
Many of the genes targeted by apabetalone were downstream of tumor necrosis factor α (TNFα), a master regulator of inflammation. Overall, her findings suggested that apabetalone could nip TNFα-driven inflammatory responses in the bud, quell vascular inflammation—and just maybe limit the chronic activation of glial cells on the other side of the blood-brain barrier.
The potential role of TNFα in neuroinflammation and neurodegenerative disease jibes with other findings presented at CTAD. Mark Gurney of Tetra Therapeutics in Grand Rapids, Michigan, and Mengshi Zhou of Case Western Reserve University in Cleveland accessed medical records on more than 56 million people—roughly 20 percent of the U.S. population. They reported that among people with chronic inflammatory conditions such as rheumatoid arthritis, psoriasis, inflammatory bowel disease, and Crohn’s disease, those who took TNFα-blocking drugs had as much as a 60 percent lower risk of AD, depending on the condition and drug used.
Jeffrey Cummings of the Cleveland Clinic’s Lou Ruvo Center for Brain Health in Las Vegas followed Kulikowski’s talk with cognitive data from the BETonMACE trial, a Phase 3 study that tested whether apabetalone could prevent major cardiovascular events in people with high cardiovascular risk. Participants in the trial had Type 2 diabetes, a recent brush with acute coronary syndrome, and low levels of high-density lipoprotein (HDL) at baseline. The trial continued until 250 cardiovascular events had occurred. Though the drug trended toward reducing cardiovascular events, the effect did not meet statistical significance. Of the 2,425 people enrolled in the trial, 469 people over the age of 70 took part in a prespecified cognitive sub-study. It assessed scores on the Montreal Cognitive Assessment (MoCA) at baseline, at one year, and at the last treatment visit, which was at an average of 27 months. Participants were split into three groups based on their baseline MoCA score: 26 or higher (cognitively normal); 22–25 (MCI); or 22 and below (MCI to AD).
In San Diego, Cummings reported that while MoCA scores declined slightly or remained stable in treatment and placebo groups among participants in the two high MoCA groups, those with the lowest baseline MoCA scores appeared to benefit from treatment. Among the 97 participants in this subgroup, those taking apabetalone improved by an average of three points on the test between baseline and 27 months, whereas those on placebo, oddly, improved by 1 point. Breaking the scores down by cognitive domain, Cummings reported that the treatment group posted significant improvements on the abstraction and recall portions of the MoCA. Interestingly, Cummings found that differences between the treatment and placebo groups only emerged between 52 weeks and the final visit. The MoCA is a cognitive screen, which includes 30 questions and takes about 10 minutes to administer.
Based on these results in people with cardiovascular disease, as well as apabetalone’s effects on markers of vascular inflammation, Cummings believes the drug assuages cognitive impairment by targeting the inflamed vasculature. Whether it is more effective at treating people with vascular dementia or AD remains to be determined. BETonMACE was not designed as an AD trial, so the researchers did not measure AD biomarkers. However, given recent advances in plasma biomarker tests, Cummings told the audience that existing plasma samples from the participants will be tested for Aβ42/40 ratio, p-tau-181, neurofilament light (NfL), YKL-40, and other markers.
The idea that an inflamed vascular endothelium—especially within the vessels that comprise the blood brain barrier—might damage brain health and cognition jibes with recent studies from Tony Wyss-Coray’s group at Stanford University. He reported that with age, brain endothelial cells ramp up expression of vascular adhesion molecule 1 (VCAM1), which tethers circulating leukocytes to the endothelium and transmits inflammation-stoking, neurogenesis-dousing signals into the brain (Jul 2018 news; May 2019 news). More recently, the group took stock of the plasma proteome in more than 4,000 people between the ages of 18–95. In this month’s issue of Nature Medicine, the scientists report waves of proteomic changes correlating with specific biological pathways and age-related disease, rising in the plasma in the fourth, seventh, and eighth decades of life (Lehallier et al., 2019).
Similarly, researchers at Alkahest, a San Carlos, California-based company co-founded by Wyss-Coray, recently identified specific proteins in the plasma that helped transmit damaging effects to the brain (Dec 2018 news). As they are hunting for bad actors in aging plasma, Alkahest scientists are also looking for good ones.
At CTAD, Jonas Hannestad presented data from the company’s trial of GRF6019, a plasma fraction that reportedly contains factors that promote a youthful brain. Forty-seven participants with mild to moderate AD were randomized to receive 100 mL or 250 mL doses, infused daily during two five-day inpatient stints at weeks one and 13. There was no placebo. Hannestad reported that participants did not decline on either the ADAS-Cog or MMSE over the six-month trial period, but did slide on the ADCS-ADL over that time. Hannestad used data from placebo groups in published clinical trials as well as in ADNI to argue that GRF6019 slowed cognitive decline. The true test will come from a placebo-controlled trial, which is being planned, Hannestad said.
AMBAR—Elaborate Sham Procedure, or Intensive Alzheimer’s Therapy?
A new look at results from another plasma-based therapy stirred up debate at CTAD. In a roundtable discussion, scientists sliced and diced results from Alzheimer’s Management by Albumin Replacement (AMBAR), a Phase 2b/3 trial performed between 2012 and 2018 that tested a 14-month regimen of repeated plasmapheresis with albumin replacement. In a nutshell, researchers had proposed that removing plasma albumin—which is laden with Aβ and myriad other proteins—and replacing it with fresh albumin could cleanse the blood and act as a peripheral Aβ sink. The trial enrolled 322 participants, of whom 232 completed the trial. It had three treatment arms—one that replaced albumin, and two that added different doses of intravenous immunoglobulin (IVIG) to the replacement fluid. The placebo group received a complicated sham procedure, in which only small volumes of plasma were removed.
Antonio Páez of Grifols, based in Barcelona, presented top-line results of the Phase 2b/3 study last year at CTAD (Nov 2018 news). Essentially, the trial met one of its two co-primary endpoints, and when the three treatment groups were pooled, they declined 52 percent less on the ADCS-ADL than those on the sham procedure, with a p value of 0.03. The combined group declined by 66 percent less on the ADAS-Cog, but this result missed significance, with a p value of 0.06. In a prespecified analysis that split the cohort into two groups based on baseline MMSE scores, people with mild dementia barely declined, while those with moderate dementia did. The treatment thus had no effect on either endpoint in the mild group, though the moderate group had a highly significant treatment effect on the ADCS-ADL and a near-significant one—with a p value of 0.054—on the ADAS-Cog.
In San Diego, Páez presented a more detailed analysis of that data and new findings from secondary outcome measures. Among all participants, the combined treatment groups had 71 percent less decline on the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB) and 100 percent less decline on the Alzheimer Disease Research Center Clinical Global Impression of Change (ADCS-CGIC) over 14 months, he said. Participants in the mild group significantly improved on both these measures in response to treatment, while the treatment appeared to slow decline within the moderate AD group. Across all cognitive outcome measures, participants in all three treatment groups appeared to perform similarly, while the placebo group diverged.
Páez showed some CSF biomarker data, reporting stabilization of Aβ42, tau, and p-tau in the treatment groups relative to placebo. These effects were only significant in the moderate group, for Aβ42 and tau.
Páez also reported that according to CSF Aβ42 measures, 72 percent of people in the trial had brain amyloid accumulation. Amyloid status was not confirmed at screening. Fewer people in the mild AD group—65 percent—had abnormal CSF Aβ42 than in the moderate group, in which 79 percent did. This suggests that 35 and 21 percent of the participants in the mild and moderate groups, respectively, had an underlying reason for their cognitive impairment or dementia other than Alzheimer’s disease. At CTAD, Páez did not present outcome measures broken down by amyloid status.
Suzanne Hendrix of Pentara Corporation followed Páez’s talk with additional statistical analyses of the trial data. She emphasized that her analyses were intended to inform the design of future trials, not “rewrite the story with post hoc analysis.” Hendrix normalized and combined data from the ADAS-Cog, ADCS-ADL, and CDR-SB into one overall score, called a global statistical test. Hendrix considers GST a way to assess consistency between multiple outcome measures. In so doing, she found a statistically significant effect of treatment, with an effect size of 100 percent on the GST at nine, 12, and 14 months.
Analyzing potential consequences of the 28 percent of people who left the trial; Hendrix concluded that, if anything, the dropouts reduced any apparent treatment effect. She also looked at interactions between treatment responses and MMSE scores as a continuum, as opposed to mild versus moderate groups, and concluded that the treatment had an effect on some measures within some people classified as mild. Given the consistency of responses across outcome measures, and large effect sizes, the treatment is likely to be exerting a true effect, and should be further tested in people in both mild and moderate stages of impairment, she believes.
In questions following the talks, and in hallway conversations outside, researchers discussed what, if anything, the new data added to the interpretation. Like many others, Stephen Salloway of Brown University in Rhode Island was unmoved by data from additional outcome measures, noting they were not corrected for multiple comparisons. Paul Aisen at the University of Southern California, San Diego, questioned how “alpha spending” was done, given the myriad analyses and outcomes presented. Hendrix responded that beyond the two co-primary outcome measures analyzed by the MMSE subgroup, all analyses were purely descriptive.
Others questioned whether the sham procedure adequately blinded participants. It used a lower volume than the real procedure, and Salloway noted that plasmapheresis triggers a consistent physiological response, including a lowering of body temperature, so it could have been obvious whether a person was undergoing the real deal or not. Larry Honig of Columbia University in New York pointed out to the panel that the fact that all three treatment groups tracked together across measures, while the placebo group declined, suggests that participants knew whether they were receiving plasma exchange or not. He was unconvinced that the sham worked.
Hendrix and Grifols scientists countered that if the effects had all been due to unblinding, one would have expected larger treatment effects in subjective measures, which are more prone to a placebo effect, than in more objective measures. The one co-primary endpoint that was met—the ADCS-ADL—is a subjective measure; still, Hendrix considers the similar direction of effect across measures compelling. She noted that people in the placebo group became less depressed as the trial went on, suggesting they thought they were receiving a treatment. People in the mild dementia group also would have been more likely affected by knowledge of their treatment group than those in the moderate AD group, Hendrix and Grifols scientists agreed after the talk.
Lon Schneider of the University of Southern California, Los Angeles, commended the AMBAR investigators for responding to questions from the field, and running and presenting further analyses. Even so, he saw problems with the trial beyond the possibility of unblinding. For one, there was no dose response, and there were more infections in members of the treatment group who did not receive IVIG, which he found concerning and confusing. He wondered whether there were differences in outcomes, or infections, between people who received central versus peripheral lines to perform the procedure.
Babak Tousi of the Cleveland Clinic, a participating trial site, believes the sham procedure did blind participants. “If people knew they were receiving placebo, would they have come back to take part in this intensive procedure month after month?” he asked. The placebo group had fewer dropouts than the treatment groups. That all three treatment groups performed similarly across measures suggests removal of pathogenic proteins from the blood confers the treatment effect, rather than what was added back in, Tousi believes.
David Morgan of Michigan State University in Grand Rapids questioned the investigators on the premise of their trial. How, exactly, do they think their treatment works? Páez responded that drawing Aβ from the brain via removal of Aβ-albumin complexes from the blood was the initial premise, but it now appears likely that removal of other proteins from the blood contributed. The researchers did not present a subgroup analysis of people with or without brain amyloid, but Hendrix said such an analysis had been performed. She said even people without brain amyloid responded to treatment, at least on some measures. This suggests the procedure may confer broader benefits, Páez said.
Noting that the procedure was quite intensive, Morgan asked whether the scientists were attempting to zero in on which proteins needed to be removed, which would facilitate specific targeting. Páez responded that to do that, they would first need to figure out the etiology of AD.
Researchers wanted to know how Grifols would move forward, given the lack of difference between the treatment arms. Would they evaluate plasmapheresis with albumin replacement only, or continue to add back IVIG? Páez said that theoretically, any of the treatment regimens would be suitable for a future trial. However, he noted that they are considering the safety data. For example, fewer people in the IVIG groups came down with infections during the study. It is unclear why this was the case, Páez said, but this is something Grifols is investigating moving forward.—Jessica Shugart
At CTAD, Early Failures and Hints of Success, from Small Trials
Though Aβ-targeted therapies hogged the limelight at this year’s Clinical Trials on Alzheimer’s Disease meeting, held December 4–7 in San Diego, California, a large handful of smaller studies had findings to report, as well. These studies evaluated drugs trained against a variety of targets, including telomerase, serotonin receptors, nuclear receptors, and neurotrophic factors. Some clearly flopped, while others posted positive results that, according to their presenters, justify larger trials.
Rasagiline
A Parkinson’s disease drug showed some signs of efficacy in people with AD, according to results from a small proof-of-concept study of rasagiline presented by Dawn Matthews of ADM Diagnostics in Northbrook, Illinois. The drug inhibits monoamine oxidase B (MAOB), an enzyme that promotes the breakdown of dopamine and thus boosts dopaminergic activity in people with PD. Elevated MAOB levels have been detected around Aβ plaques in the AD brain, and the enzyme reportedly generates reactive oxygen species and even promotes amyloidogenic processing of APP, casting it as a candidate target.
Run at the Cleveland Clinics in Las Vegas and Cleveland, the trial enrolled 50 people with diagnoses of mild to moderate AD, who had an AD-like pattern of metabolic dysfunction as indicated by FDG-PET. No Aβ- or tau-based biomarkers were used to confirm their diagnoses at screening. Participants took 0.5 mg rasagiline or placebo for the first four weeks, followed by 1 mg rasagiline, or placebo, for 20 weeks. Forty-three people completed the trial, which used change in FDG-PET from baseline to 24 weeks as its primary endpoint. Exploratory measures included tau PET and several cognitive measures.
The trial met its primary endpoint, Matthews reported. In the placebo group, glucose metabolism waned in prespecified frontal, anterior cingulate, and striatal regions over the 24-week trial. Metabolism slipped significantly less, or hardly at all, in these regions in the rasagiline group. On clinical measures, the rasagiline group posted significant improvements on the Quality of Life-AD (QOL-AD) test. They also trended toward better performance on six out of seven other cognitive tests, including the Controlled Oral Word Association Test (COWAT), the ADAS-Cog, MMSE, CGIC, Digit Span, and NPI.
Matthews reported interactions between some of the outcome measures, saying that quality-of-life improvement correlated with FDG-PET uptake in the anterior cingulate, while improvement on the COWAT and digit span tests correlated with improved metabolism in the middle frontal regions. A higher tau PET burden at baseline correlated with the degree of subsequent slippage on the MMSE for those in the placebo group, and with a larger treatment effect for those in the rasagiline group. Matthews said the findings warrant a larger trial.
NDX-1017 Hans Moebius of Athira Pharma in Seattle presented early stage trial results on the regenerative compound NDX-1017. This small molecule activates the hepatocyte growth factor (HGF)/MET receptor, which is normally expressed in the CNS but less so in AD cortex and hippocampus. Receptor activity promotes LTP and is necessary for learning and memory. It also boosts neuronal survival, neurogenesis, and dendritic outgrowth, and dampens neuroinflammation. In animal models, HGF/MET receptor activation lessened cognitive impairment caused by Aβ and prevented onset of Parkinsonian symptoms (Takeuchi et al., 2008; Koike et al., 2006).
At CTAD, Moebius reported findings from Phase 1a and 1b studies completed in the U.S. and France. A single-ascending-dose (SAD) study enrolled 48 healthy young volunteers and tested doses from 2 to 90 mg. In the multiple-ascending-dose (MAD) study, 29 healthy elderly and 11 AD patients received NDX-1017 for nine days, at doses ranging from 20 to 80 mg. The drug was well-tolerated, with four of the 88 participants experiencing some pain or itching at the injection site. There were no other adverse events deemed related to drug.
Because of the drug’s activity on synapses, the researchers used EEG measures as a readout for target engagement and potential efficacy. The power of gamma waves wanes in people with AD, and in animal studies an increase in gamma power associates with improved learning and memory. Conversely, another EEG measure—the event-related potential latency, aka E300 wave—increases in AD.
In the SAD study, gamma power rose after a single dose of 20 mg or more NDX-1017, indicating target engagement. The increase was dose-dependent and statistically significant in the 90 mg dose group, Moebius reported. In the MAD study, 40 mg NDX-1017 increased gamma power on day four and day eight in all seven AD patients who received the drug. In addition, p300 latency dropped, and this change was significant by day eight. The four AD patients taking placebo had no EEG changes. The trial did not include cognitive tests to determine if these electrophysiological changes would translate into a benefit in thinking or memory.
A Phase 2/3 trial is set to start in 2020. It will enroll 120 people with mild to moderate AD, who will take NDX-1017 for 26 weeks, followed by a six-week washout period to see if any improvement stays or fades. The trial will compare several doses and use EEG as an exploratory outcome measure. The primary outcomes will be the Global Statistical Test and safety; the widely used outcomes ADAS-Cog11, ADCS CGIC, and NPI are secondary outcomes in this trial design.
At CTAD, Jicha presented data from an NIA-funded trial that evaluated a 48-week regimen of 600 mg gemfibrozil versus placebo in 75 people, including 48 who were cognitively normal and 24 with MCI. The former were at increased risk for dementia based on family history, and half of them had abnormal CSF Aβ42, indicative of brain amyloid, Jicha said.
On safety—the primary outcome measure of the trial—the drug passed. Jicha also reported a dip in the plasma concentration of miR-107, although levels of the microRNA in CSF were too minute for detection. The scientists only detected BACE1 in the CSF of about half the participants, and so were unable to conclude whether the drug affected CNS levels of the Aβ-producing enzyme.
Gemfibrozil did appear to reduce change in CSF Aβ42, p-tau, and the p-tau/Aβ42 ratio between baseline and 48 weeks; however, these biomarker trends were not statistically significant. Those in the treatment group had less brain atrophy, better scores on some tests of memory, and reduced plasma levels of the pro-inflammatory cytokine TNF-α. Again, these effects trended in the right direction but none were statistically significant, Jicha reported. He said the findings warrant future trials with adequate power to detect changes on cognitive, MRI, and CSF measures. Finally, Jicha pointed out that at just pennies per dose, gemfibrozil could make a cheap treatment. For this trial, the placebo cost more than the drug, he said.
GV1001 Seong-Ho Koh of Hanyang University Guri Hospital, South Korea, showed topline results from a Phase 2 trial of GV1001 in people with clinical diagnoses of moderate to severe AD. GV1001 is a peptide drug against telomerase reverse transcriptase. Developed by GemVax & KAEL in Daejeon, South Korea, GV1001 kicks up an immune response against a form of telomerase that is known to be over-represented in cancer cells; it also interacts with heat shock protein 90 (Hsp90). GemVax is evaluating this drug for multiple types of cancer. Scientists have reported anti-inflammatory and anti-viral effects as well (Kim et al., 2016; Kim et al., 2018; Moon et al., 2018); and at CTAD, Koh described the first trial to test GV1001 in people with AD. It enrolled 90 participants, who were randomized equally to three groups who received four weekly subcutaneous injections of 0.56 mg or 1.12 mg of GV1001, or placebo, followed by biweekly injections for 24 weeks, making for a total of 14 injections.
Koh said the trial met its primary endpoint, which was improved performance on the Severe Impairment Battery between baseline and week 24. While those in the placebo group dropped by 7.23 points on the SIB, scores stabilized in both treatment groups, ultimately slipping by an average of 0.12 points. To the chagrin of audience members, Koh showed no results on any of the trial’s numerous secondary endpoints, noting that they would be released within the coming month. The trial ended in June 2019, according to clinicaltrials.gov. Others noted a particularly steep decline of the placebo group, wondering how that contributed to the apparent treatment effect.
“It’s not very often that we see positive results like this for an AD trial,” said Mike Weiner of the University of California, San Francisco, who co-chaired the session. He asked if Koh attributed the effect to a symptomatic or disease-modifying mechanism. Koh said that GemVax plans to investigate this question further with a Phase 3 trial of the drug in South Korea. The company has listed a Phase 2 trial in 90 people with moderate AD in the U.S. on clinicaltrials.gov.
Masupirdine Jeffrey Cummings of the Cleveland Clinic’s Lou Ruvo Center for Brain Health in Las Vegas reported that a Phase 2 trial evaluating the 5-HT6 receptor antagonist masupirdine missed its primary outcome. According to a previous report from the drug’s developer, Suven Pharma Inc., the Delaware-based subsidiary of Indian company Suven Life Sciences, this serotonin receptor antagonist boosted acetylcholine levels and improved brain oscillations in rats.
The trial enrolled 543 people with a clinical diagnosis of moderate AD, 60 percent of whom carried an ApoE4 allele. All participants took both donepezil and memantine, and were equally randomized to a 26-week course of 50 mg or 100 mg masupirdine, or placebo. Cummings reported no difference between placebo and treatment groups on ADAS-Cog, the primary outcome measure. Numerous secondary measures told a similar story.
The trial failure marks the latest in a string for this class of drugs (Sep 2017 news; Jan 2018 news). Some researchers at CTAD questioned why the trial was done in the first place. Cummings said previous studies had left open the possibility that dosing could have been higher, noting that this latest study truly tested the maximum dose for a 5HT6 antagonist. “I think we are done testing these drugs for effects on cognition,” Cummings said.
Neflamapimod Philip Scheltens of VU University Medical Center, Amsterdam, presented the results of another negative Phase 2 trial. Unlike the masupirdine trial, however, this one, of the p38a inhibitor neflamapimod, did end with a hint that it might have worked had a higher dose been used.
At CTAD in 2016, Scheltens had reported that in a small pilot study without placebo, the drug had safely lowered amyloid-PET at a twice-daily dose of 40 mg, although, strangely, a higher dose of 125 mg had not budged amyloid deposition. In a subsequent study, researchers reported that higher doses of the drug were associated with anti-inflammatory effects, leading researchers to speculate that higher doses may have hobbled the plaque-clearing prowess of microglia (Dec 2016 news).
This year at CTAD, Scheltens presented the results of a placebo-controlled, Phase 2b clinical study that evaluated a 24-week course of twice-daily 40 mg doses neflamapimod, or placebo, in 161 people with MCI or mild AD. Participants had CSF Aβ42 levels indicative of brain amyloid accumulation.
The drug appeared safe and well-tolerated, but missed its primary endpoint of change on the Hopkins Verbal Memory Test. Participants in the treatment and placebo groups tracked together on this measure, and also across all secondary cognitive endpoints. The drug did appear to budge some CSF biomarkers—reducing an uptick in the placebo group in total tau and p-tau-181 between baseline and 24 weeks, and trending toward reduction of neurogranin.
In a prespecified analysis to explore dose-response relationships of the drug, the researchers found that participants with the highest quartile of plasma neflamapimod at 21 days fared better on the HVLT as well as on the Wechsler Memory Scale than did people whose drug exposure was lower. The apparent benefit among those with highest plasma exposure was most striking among people who were on a background therapy of acetylcholinesterase inhibitors.
Overall, Scheltens said, the findings suggest that neflamapimod might boost cognition at a 50 to 100 percent higher dose. Despite the unmet primary in this study, he believes future studies are warranted.—Jessica Shugart
Tau PET Scans Turn Positive When Amyloid Does; Symptoms Follow
This article was amended on January 10, 2020
Tau PET is prying open a window on when and where neurofibrillary tangles appear as Alzheimer’s progresses. At the 12th Clinical Trials on Alzheimer’s Disease Conference, held December 4–7 in San Diego, California, three presentations on next-generation tau tracers offered some detail on how tangles relate to plaque load and to cognition at different stages of the disease.
One surprise: Some people with MCI who were at the threshold of global amyloid positivity with florbetaben/Neuraceq already have positive PI-2620 tau PET scans. This would contradict scientists’ current sense, seen with flortaucipir/AV-1451, that scans turn positive around the time people become symptomatic, when amyloid is typically well-established. However, these are the early days of tau PET. The data shown at CTAD were cross-sectional, with the caveats that attend to such one-time comparisons. They varied widely from person to person, with a few people remaining tau-negative in the presence of a high global amyloid load. Partly, current data may reflect a lack of consensus on what constitutes a positive tau scan.
Meanwhile, longitudinal studies are starting to show that the more tangles a person has at his or her first scan, the quicker the pathology spreads, suggesting the baseline tau signal might help trialists find fast progressors. Neuropathology data have closely tied tangles to cognitive decline, but intriguingly, new PET data from a small longitudinal cohort hinted that there may be a time lag between the two. As more of these kinds of data roll in, scientists hope it will improve their ability to define the right timepoints for intervening with anti-amyloid or anti-tau drugs, and track their effect in trials.
Tau PET scans with next-generation tracer RO-948. [Courtesy of Gregory Klein at Roche and Oskar Hansson at Lund University, Sweden.]
Compared with the initial tau tracers, such as flortaucipir, the new ones tend to bind more strongly to tangles while ignoring other molecules, yielding a better signal-to-noise ratio (Apr 2017 conference news). This helps with tracking change over time.
One next-generation tracer is Life Molecular Imaging’s PI-2620, which appears to distinguish AD scans from those of healthy controls with little noise (Kroth et al., 2019; Mueller et al., 2019; Bullich et al., 2019). Eisai chose this tracer for its now-terminated Phase 3 MISSION AD program of the BACE inhibitor elenbecestat.
In San Diego, LMI’s Andrew Stephens discussed baseline data from the program’s tau PET sub-study of 77 participants. All were amyloid-positive by a visual read of LMI’s florbetaben PET scans. Visual reads pick up early regional amyloid accumulation, before global amyloid scores turn positive. Participants’ average age was 76, and 43 percent of them carried an ApoE4 allele, a lower rate than is typical in AD trials. At baseline, they had mild cognitive impairment, with a CDR score of 0.5 and an average MMSE of 27.
Based on their tau scans, this population fell into three distinct groups, Stephens said. By visual assessment, 33 of the 77 people were tangle-negative, with barely any PI-2620 retention beyond that seen in healthy controls. In another 18, the tracer was taken up mainly in the hippocampus, parahippocampus, and fusiform gyrus—regions of early tau pathology that correspond to Braak stage I/II. Nearly the same number, 20 people, had more extensive uptake in the neocortex, indicating stage III or later. The remaining seven people did not fit cleanly into any of these categories. “Tau deposition is quite heterogeneous, and we don’t fully understand why yet,” Stephens told Alzforum.
Stephens noted that a person’s age, sex, cognitive reserve, and genetic factors can affect his or her tangle accumulation. For example, at a given level of cognitive decline, older people tend to have fewer tangles than do younger ones, while people with high cognitive reserve have more tangles than do those with less reserve (Apr 2019 conference news). Other data suggest tangles accumulate faster in women, but not men, who carry an ApoE4 allele (Feb 2019 news; Nov 2019 news).
In people who inherit a dominant Alzheimer’s mutation, flortaucipir indicates tangle spread into the cortex around the time of symptom onset. This is a faster tangle progression than in MISSION AD, where most of this MCI cohort had no cortical tangles (Aug 2017 conference news; Feb 2018 news). “We are beginning to define the amount of amyloid that is needed to see tracer accumulation, and we hope to extend these findings to learn what amyloid level is required for neocortical tau deposition,” Stephens wrote to Alzforum.
By quantifying tau uptake, not merely judging it with the naked eye, researchers picked up nine additional positive scans in the MISSION AD cohort. This resulted in a total of 47 positive scans, amounting to 60 percent of this MCI cohort. An audience member expressed the surprise of many when he asked why the percentage wasn’t higher. Previous work had reported 50 percent flortaucipir positivity in amyloid-positive but cognitively normal A4 trial participants—an earlier-stage population measured with a weaker tracer. Stephens said he does not know why the percentages are so similar in these different populations.
How did tau uptake among these people relate to their amyloid burden? For context, healthy young controls average a florbetaben amyloid load of 1.16 SUVR using LMI’s methodology, with cerebellar gray matter as the reference region. Regional accumulation begins two standard deviations above that, at 1.25 SUVR, or 12 centiloids, Stephens said. He will present new florbetaben research at upcoming meetings, showing that 12 centiloids is where amyloid accumulation can first be detected. A previous histopathology study of florbetaben in end-of-life subjects had set a cutoff for global amyloid positivity at 1.48 SUVR, about 35 centiloids. Stephens noted that this study included only people who had moderate dementia or worse, and so this cutoff may not capture everyone at earlier disease stages. The range between 12 and 35 centiloids thus defines a “gray zone,” where regional amyloid has begun to build up but the global score is not yet positive.
In the MISSION AD sub-study, the three people whose global amyloid score was below the gray zone were all tau-negative, Stephens reported at CTAD. “This shows that in Alzheimer’s, there is no PI-2620 accumulation in the absence of detectable amyloid,” he said. Among those in the amyloid gray zone, one in five had a positive tau scan, and they all had amyloid burdens toward the high end of the gray zone. At intermediate levels of amyloid plaque, half the MISSION participants were tau-positive. Among those with high global amyloid loads, 80 percent were tau-positive.
Were there regional patterns? Yes. The higher a person’s global amyloid load, the more tau-tracer uptake he or she had in regions known to succumb to AD early—hippocampus, parahippocampus, fusiform gyrus, amygdala, inferior temporal cortex. In this cohort, as expected, amyloid and tau tracer uptake were less correlated in parietal cortex, a region affected later in disease than the 0.5 CDR stage. The researchers are currently gathering longitudinal tau PET data as MISSION AD participants come for their final visits.
Other talks at CTAD offered a glimpse at longitudinal data. The Swedish BioFinder-2 study, which tracks biomarker change over time, uses Roche’s next-generation tracer RO-948 to follow participants. In San Diego, Gregory Klein and colleagues at Roche presented data from the first 51 participants who were scanned at baseline and one-year follow-up. They spanned the disease spectrum, with nine of them amyloid-negative and cognitively healthy, 14 amyloid-positive and cognitively healthy, 18 amyloid-positive with MCI, and 7 amyloid-positive with AD dementia. There were also three outliers who were amyloid-negative but cognitively impaired.
Among this cohort, tau PET scans diverged sharply according to whether a person was amyloid-negative or -positive. For amyloid-negative participants, baseline tau tracer uptake was low and did not rise over the course of a year. If anything, the RO-948 signal dipped slightly at follow-up. For amyloid-positive participants, the baseline tau signal was higher, as expected, and it rose over the following year. The more tau a person had at baseline, the more it rose, suggesting that high baseline tau could be used as a criteria for selecting trial participants. Klein and colleagues did not quantify exactly how many of the amyloid-positive MCI group were tau-positive, but he noted that roughly half of them fell below a tau PET SUVR of 1.3, considered the threshold for positivity with RO-948. These proportions are consistent with those seen in BioFinder-1 with flortaucipir, he noted (Ossenkoppele et al., 2018). “What truly defines tau positivity is still a subject of discussion in the tau PET community,” he told Alzforum.
Where the tau signal rose depended on disease stage. For cognitively normal people with brain amyloid, tau went up in Braak I/II regions. For the MCI group, it went up in Braak III/IV regions, and for people with AD dementia, in Braak V/VI. Curiously, for people with dementia, tau tracer uptake fell slightly in Braak I/II regions over the course of a year, perhaps due to neuron death there.
Klein said that compared with flortaucipir, RO-948 has less off-target binding in brain structures up close to Braak I/II regions, particularly in the choroid plexus sitting atop the hippocampus. This, he believes, may allow this tracer to better detect longitudinal change in people at the preclinical stage of disease (Smith et al., 2019). It may also explain why the longitudinal effect size for tracer uptake change in this group was greater than that published using AV-1451, Klein told Alzforum (Jack et al., 2018).
He believes the combination of high sensitivity in Braak I/II regions and setting a baseline tau cutoff as an inclusion criteria could enable clinical trials to use tau PET as an outcome measure. “This could be a game-changer for designing clinical trials targeting a preclinical AD population, where prior sample-size estimates have been too large to be practical using a tau PET endpoint,” Klein wrote to Alzforum.
Tangles Before Cognitive Decline? Uptake of tau tracer GTP1 rises early in disease, while memory slips later. [Courtesy of Robby Weimer, Genentech.]
A second longitudinal tau PET study presenting at CTAD tied tangle spread to cognition. Robby Weimer and colleagues at Genentech followed 72 people for 18 months using their in-house next-generation tracer GTP1 (Sanabria-Bohórquez et al., 2019). In a previous cross-sectional study, uptake of this tracer correlated with worse memory (Teng et al., 2019). The new longitudinal study comprised 27 people with prodromal, 19 with mild, and 16 with moderate AD, along with 10 cognitively healthy controls. Everyone with AD had a positive amyloid scan.
The data again tightly linked tangles to cognitive decline, but with a temporal lag, i.e., tau PET changed before cognition slipped. For healthy controls, the tau PET signal fell slightly at 18 months, just as in BioFinder, while cognitive scores on the CDR-SB and ADAS-Cog13 stayed flat. For the prodromal group, tau tracer uptake rose slightly, about 0.05 SUVR, while cognitive scores barely moved. The mild AD group had a similar bump in tracer signal as the prodromals, but their cognition worsened by five points on the ADAS-Cog13 and three on the CDR-SB. For those with moderate AD, GTP1 uptake rose the most, about 0.1 SUVR, while their cognition worsened at about the same rate as in the mild group.
Weimer sees two possible explanations for the apparent delay between rising tau PET and declining cognition. It could reflect a physiological phenomenon, where tangle accumulation precedes synaptic or neuronal damage by some period of time. Or it could be a result of ceiling effects in the cognitive measures used, which may lack the sensitivity to detect early changes. “Evaluation of atrophy from this study, which is ongoing, will help delineate these differences and provide insight into the pathophysiology of disease,” Weimer wrote to Alzforum. He noted that with the current dataset, he cannot quantify the time lag in terms of months or years.
Regionally, data from the longitudinal Genentech cohort resembled that from the BioFinder-2, with Braak stage I/II regions changing the most at the prodromal stage, and V/VI changing the most in moderate AD. People with mild AD gained tau signal in all Braak regions over one year.
Weimer noted that a person’s baseline GTP1signal predicted by how much he or she would worsen on the CDR-SB and ADAS-Cog13 over the following year, whereas his or her rate of tau PET change did not. In other words, tangle pathology at baseline better predicted progression than did the rate of tangle spread. Tau PET could be used for disease staging and as a biomarker of progression, Weimer suggested.
He noted that just as in MISSION AD and BioFinder-2, his group saw a range of tau pathology among prodromal patients. They have not yet defined a positivity cutoff for GTP1. “For all the tracers, we will need larger longitudinal datasets for cognitively normal, MCI, and AD patients before we can fully explore the relationship between baseline tau pathology and disease progression. That information will help define tau positivity,” Weimer wrote.
Regarding other next-generation tau tracers, there is also Merck’s entrant, MK-6240. The only data at CTAD on it on came from the aducanumab sub-study (Dec 2019 conference news). There were no presentations on Janssen’s JNJ-067, currently in a Phase 1 trial.
Meanwhile, researchers led by Alex Whittington and Roger Gunn at the imaging services company Invicro, London, presented a new method for quantifying tau tracer uptake by calculating both global and local tau loads. In cross-sectional analyses, the new measure, TauIQ, arrived at a larger effect size than did SUVR for distinguishing between control and AD scans. For flortaucipir, the effect size was 17 percent larger with TauIQ than with SUVR, while for GTP1, it was 24 to 49 percent larger. In longitudinal scans, TauIQ showed a greater increase over a year than did SUVR. For example, in people with high GTP1 uptake at baseline, TauIQ went up 0.7 standard deviations in a year, compared with 0.45 for SUVR. The use of TauIQ could facilitate clinical trials, the authors suggested.—Madolyn Bowman Rogers
Jack CR Jr, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, Jones DT, Graff-Radford J, Kantarci K, Roberts RO, Mielke MM, Machulda MM, Petersen RC.
Longitudinal tau PET in ageing and Alzheimer's disease.
Brain. 2018 May 1;141(5):1517-1528.
PubMed.
CTAD Lessons for 2020: More Phase 2 Trials, More Diversity
What lies ahead for Alzheimer’s therapy development? While anti-amyloid antibodies are at last signaling some success, researchers agree that these expensive—and, thus far, at best modestly effective—biologic drugs can form only part of the arsenal needed to fight the disease. Researchers at the 12th Clinical Trials on Alzheimer’s Disease conference, held December 4–7 in San Diego, California, broadly agreed that an array of therapeutic approaches will be needed to target symptomatic stages, or to combine with antibodies to boost efficacy. Speakers also discussed how to improve the dismal success rate of Alzheimer’s clinical trials. In particular, there is a push to spend more time in Phase 2 to find the right dose and confirm physiological effects of the drug at hand. Others argued that machine-learning approaches, which have made a slow and steady entrance into Alzheimerology over the past five years or so, are now robust enough to make trials shorter, faster, and more efficient.
The mood overall was upbeat. The crowd in San Diego seemed confident that the field now has the tools it needs to move forward, given recent investments in infrastructure and basic research into the systems biology of AD. That research suggests that therapeutic approaches should target not only Aβ and tau pathologies, but inflammation, vascular dysfunction, and multiple proteinopathies, as well. In addition, research into resilience factors might point toward ways to strengthen function in the aging brain. Eliezer Masliah, who heads the neuroscience division at the National Institute on Aging, struck a particularly bullish note in his keynote address. “We have a blueprint for how to get to a cure for AD by 2025. I feel very confident we’ll make that goal.”
Increased funding for Alzheimer’s research powers his plan. Masliah noted that in 2019 the allocation for AD and related diseases was $2.3 billion, more than four times the 2013 total. Although this still lags behind the $6 billion spent on cancer research, the additional funding has allowed the NIA to invest in major infrastructure initiatives, such as the MODEL-AD program for generating better animal models, and the Alzheimer’s Disease Clinical Trials Consortium to support clinical programs (Jan 2017 news; Dec 2017 news).
Far To Go. The roughly 50 Alzheimer’s trials each year represent a drop in the bucket compared with the 500 for cardiovascular disease and 2,500 for cancer. [Courtesy of NIA.]
The challenge is to convert these resources into actual trials. There are only 50 to 60 AD trials each year, compared with 500 for cardiovascular disease and 2,500 for cancer. Masliah stressed that the AD field needs more shots on goal. NIA believes the key is to diversify and fill up the therapeutic pipeline with a greater variety of targets.
The agency currently funds a dozen different target areas, including tau, ApoE, neuroprotection, and neurotransmitter receptors. Its priorities have changed over time. For example, since 2014, agency funding for amyloid-directed therapies has fallen from 25 percent of the portfolio to less than 10 percent, while funding for inflammation has risen sharply, from about 5 to 20 percent of the portfolio. Is the recent announcement of positive Phase 3 results for aducanumab going to change the agency’s direction? Joe Balintfy, a spokesperson at NIA, said this is unlikely, noting that the agency’ priorities are set in consultation with research leaders at the annual NIH summits mandated by the National Alzheimer’s Project Act (Mar 2018 news; Mar 2019).
The Accelerating Medicines Partnership (AMP-AD) is tasked with discovering additional targets by analyzing systems biology in healthy and diseased brain in search of new pathways relevant to AD (Feb 2014 news). The AMP-AD’s Agora database now includes more than 500 candidate targets, ranked by druggability, Masliah said. Of these, 113 are predicted to be druggable with small molecules. These candidate targets typically are components of regulatory networks within the brain. Researchers can view supporting information on each target, such as how its expression changes in AD and whether that correlates with pathology.
To find further disease targets beyond what AMP turns up, NIA also funds projects to investigate the vascular contribution to AD (M2OVE-AD), neuropsychiatric symptoms (NPS-AD), and resilience mechanisms (Resilience-AD).
To move these candidates toward trials, in October 2019 NIA committed $73 million to launch its newest initiative, the AD Translational Centers for New Medicines. This project brings together researchers at diverse institutions into two multidisciplinary teams charged with developing tools such as antibodies and chemical probes directed against candidate targets, and with performing initial investigations in mouse models (Oct 2019 news). “Having a diversified pipeline will put us on the road to 2025,” Masliah concluded.
Changing Priorities. Since 2014, NIA funding for amyloid-related therapies (blue) has shrunk, while that for inflammatory targets (orange) has ballooned. [Courtesy of NIA.]
Phase 2: Reconnecting With a Long-Lost Friend?
Others dryly noted that more trials may not help much if the success rate doesn’t improve. Currently, most Alzheimer’s drug trials are negative. Alette Wessels of Eli Lilly sees a more deliberate focus on Phase 2 as a solution. “We need to do true dose-exploratory Phase 2 studies,” Wessels said in San Diego. Many scientists at CTAD agreed, noting privately that dose-response data and a promising but unclear efficacy signal is what Biogen got out of its Phase 3 program, which followed from Phase 1 trials. Other programs followed a similar strategy, including Lilly’s solanezumab and lanabecestat, Merck’s verubecestat, and Roche’s gantenerumab. A notable exception is BAN2401, which conducted an 856-person Phase 2 study before starting Phase 3.
At CTAD, Wessels stressed that before being advanced into Phase 3, a drug should have formally demonstrated sufficient concentration in the central nervous system, target engagement, a dose response, and downstream physiological effects on the brain, as well as early indications of clinical effects. She suggested Phase 2 trials incorporate indirect measures of cognition, such as EEG or fMRI scans.
Going forward, Phase 2 trials should include adaptive features, Wessels said. This will allow researchers to hone their approach. If Phase 2 results indicate a partial response to drug, such as biomarker movement without clinical benefit, sponsors should stay in Phase 2, adjusting parameters until they see a strong effect, Wessels said. She emphasized the importance of a large effect size in Phase 2, because there tends to be a regression to the mean in the heterogeneous international populations of Phase 3 trials. “Before we initiate Phase 3, we should have an understanding of efficacy,” Wessels said.
Machines to the Rescue?
If this all sounds like a lot of effort and money spent in Phase 2, could machine learning make trials at this stage more efficient? Nicolai Franzmeier of Ludwig Maximilians University, Germany, believes so. He used artificial intelligence to analyze multiple biomarkers, including structural MRI, FDG PET, amyloid PET, and cerebrospinal fluid Aβ/t-tau/p-tau, from a DIAN cohort of 121 cognitively healthy mutation carriers and 54 noncarriers. The goal was to identify the parameter combination that best predicted a participant’s estimated age of symptom onset, aka EYO. Using data from these markers, Franzmeier developed a computer algorithm that fit the bill, predicting EYO with an R2 value of 0.53.
The researchers tested this model in an ADNI cohort comprising 49 amyloid-negative controls and 216 amyloid-positive people with MCI, who were followed for four years. Model predictions explained about 25 percent of the observed decline on the ADAS-Cog13 and ADNI-MEM, i.e., an R2 of about 0.25.
This algorithm could help trialists select people at high risk of decline, Franzmeier said. He estimated that it could lower the number of participants needed to see a treatment benefit by 60 percent; for example, group sizes needed to detect a 20 percent slowing of cognitive decline would drop from 1,982 to 773. An audience member pointed out that collecting so much biomarker data is expensive in itself; Franzmeier said that even using just two of the four markers could cut enrollment in half.
Aaron Smith of Unlearn.AI, a startup company that uses machine learning to facilitate trials, suggested another way digital technology could cut enrollment. Machines can use data from the control arms of past trials to develop predictions for how disease will progress in a person depending on his or her baseline characteristics, he said. In effect, computers can generate a “digital twin” for each enrollee, describing what would likely happen to them without treatment. These digital twins can supplement the physical placebo group of a trial. This would lower the number of participants needed for a given trial, and allow a greater proportion of participants to receive drug rather than placebo. The chance of ending up in the placebo group deters many a potential participant.
Smith and colleagues used 18 months of data on 1,909 patients from the Coalition Against Major Diseases (CAMD) AD database to develop an algorithm for generating digital twins. This “synthetic data” accurately predicted changes in ADAS-Cog scores, and was indistinguishable from actual data by logistic regression analysis, he reported (Fisher et al., 2019). The researchers have since improved this model, now incorporating data from 5,000 people participating in a variety of clinical trial designs, Smith noted.
Would the Food and Drug Administration accept trial results that use digital twins? Smith said his company is discussing their methodology with the FDA, but at this point, he recommends using the approach for Phase 2.
Overall, machine learning is gaining a toehold in the Alzheimer’s field, with 10 talks and 10 posters on the topic at CTAD. Newman Knowlton of Pentara Corporation said in San Diego that artificial intelligence is particularly adept at pattern recognition, making it ideal for applications such as analyzing speech—or indeed classifying patients by disease stage. Computer models can also adjust individual dosing based on pharmacokinetic data, flag potential medication interactions, and predict disease progression. All these applications require machines to synthesize existing data, which is their forte. However, there are some things artificial intelligence cannot do, such as determine drug efficacy, Knowlton noted. For that, human judgment is required.
Considering the future for the Alzheimer’s field, Stephen Salloway of Brown University in Providence, Rhode Island, sounded hopeful. “Working in Alzheimer’s research, you have to keep at it until you break through to the next level,” Salloway told the CTAD audience. He sees the field as being on the cusp of that next level, noting that research networks, such as AIBL, ADNI, DIAN, BioFinder, EPAD, and others, have delivered major progress in brain imaging, biomarker development, and an understanding of the fundamental biology of AD.
To find effective treatments, Salloway believes, the field will need to move beyond paying lip service to combination therapies and start putting those trials in practice. While employing Aβ-targeted therapies earlier in the course of disease, the field must also doggedly scour other biological pathways, such as inflammation and cellular aging, for targets that will form the basis of future combination therapies. Recruiting young scientists from diverse fields into the Alzheimer’s fold will be key to expanding the range of potential treatments, he said. Finally, Salloway stressed that, to grow cohorts for platform-style trials, public engagement in AD trials must increase exponentially. “It needs to go to a much grander scale, where the public really partners with us. Believe me—they are willing. We just need to figure out how to make that partnership flourish,” Salloway said.
Salloway noted the importance of non-pharmacological approaches. Observational studies suggest that living a healthy lifestyle could shave a third off the population dementia risk, and multimodal intervention trials suggest cognitive benefits (see Aug 2019 news; FINGER clinical trial; Nov 2015 news). The ongoing POINTER trial in the U.S. will compare the interventions in different populations. “We’re sure it’s going to be beneficial—we just need to figure out what the regimen is, how to attract people, and how to maintain adherence,” Salloway said.
Laura Baker of Wake Forest University in North Carolina said that POINTER, and similar trials around the globe, will zero in on the most effective interventions suited for each culture. At the same time, better harmonization between trials in terms of which interventions are used and how their cognitive effects are measured is necessary to enable accurate comparisons between lifestyle and pharmacological trials. Baker believes a healthy lifestyle may prepare the body and brain to better respond to drug therapies, asking, “Can lifestyle interventions be a fertilizer for pharmacological therapies?”—Madolyn Bowman Rogers and Jessica Shugart



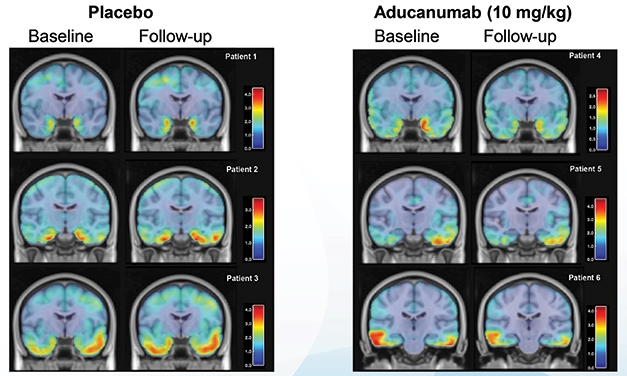






Comments
No Available Comments
Make a Comment
To make a comment you must login or register.