Serial PET Nails It: Preclinical AD Means Amyloid, Tau, then Cognitive Decline
Quick Links
Amyloid deposits without causing symptoms for years, but once tau tangles accumulate, cognition craters. That’s the prevailing model for the progression of Alzheimer’s disease. The newest support for it comes from the first longitudinal study to repeatedly scan older adults for both amyloid and tau over years, and correlate the PET results with cognitive changes. In the June 3 JAMA Neurology, investigators in the lab of Keith Johnson at Massachusetts General Hospital in Boston reported that the rate of change of amyloid and tau was key—people who accumulated amyloid fastest experienced the most rapid ramp-up of tau, which in turn drove the biggest declines in cognition. Researchers saw it as an affirmation of a wealth of evidence gathered mostly from cross-sectional and pathological studies. “While it is not exactly surprising, it is exciting to see it demonstrated that it is not just cross-sectional amyloid or tau levels that are most closely linked to cognitive function … it is amyloid and tau change that are most critical to final cognition seven years later,” wrote Samuel Lockhart, Wake Forest School of Medicine, Winston-Salem, North Carolina, to Alzforum (see comment below).
- Serial PET scans evaluate amyloid and tau, compare to cognition.
- Amyloid PET is most useful for detecting early pathology.
- Tau PET looks promising for tracking disease progression.
First author Bernard Hanseeuw had presented this longitudinal data, hot off the scanner, at CTAD in 2017 (Dec 2017 conference news). The paper adds a deeper analysis and modeling of the relationships between amyloid, tau, and cognition.
The data fits with previous PET studies but paints a fuller picture of the sequential changes occurring in otherwise cognitively healthy older adults, researchers agreed. “This helps to put the pieces in order,” said Beau Ances, Washington University, St. Louis.
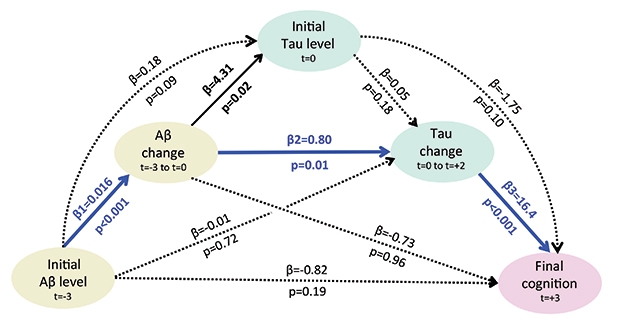
Road to Ruin. Statistical modeling of serial amyloid and tau PET suggests a sequential model of decline (blue lines). Baseline Aβ three years before a tau PET scan (t=-3) drives Aβ change and tau change after the scan (t=0 to t=+2). In turn, tau change best correlates with cognitive decline six years after the initial Aβ scan (t=+3). Other scenarios explaining cognitive decline (black and dotted lines) are less likely. [©2019 Hanseeuw BJ et al., JAMA Neurology.]
“This is all consistent with the view that Alzheimer’s disease is an Aβ-facilitated tauopathy leading to neurodegeneration, cognitive decline, MCI, and dementia,” said Michael Weiner, University of California, San Francisco. He called it a pioneering study, but noted the small number of subjects. “This work needs to be replicated,” he said.
Hanseeuw analyzed scans from 60 people in the Harvard Aging Brain Study, a longitudinal cohort of cognitively healthy men and women between 65 and 85. The participants had multiple Pittsburgh compound B (PiB)-PET amyloid scans starting in 2010, and repeated flortaucipir-PET scans starting in 2013. Each year participants took the Preclinical Alzheimer’s Clinical Composite (PACC), a battery designed to detect cognitive decline too subtle to qualify as cognitive impairment or prodromal AD.
Based on the first PiB scan, Hanseeuw divided the sample into groups of high and low cortical amyloid, using a global standardized uptake volume ratio of 0.724 as the cut point. At the time of the first tau scan, the high-amyloid group had more tau tracer uptake in the inferior temporal region, a site of early tau spread that the investigators focused on in their analysis. In subsequent years, amyloid and tau continued to rise in both groups, and cognitive scores decreased, but on average, the process moved faster in people whose initial amyloid load was higher.
The rates of change were even more telling than baseline values. The change in amyloid correlated most strongly with subsequent tau accumulation. In turn, the speed of tau accumulation tracked closely with cognitive decline, and better predicted it than initial measures of amyloid or tau. Cognitive decline weakly correlated with baseline amyloid, but not with the change in amyloid. Additional statistical modeling of the data supported the idea that Aβ weakens cognition mostly by boosting tau (see image above). In other words, Aβ by itself only weakly affects cognition, but when it’s rising, it triggers a burst of tau that appears to cause to cognitive loss.
At the time of analysis, six of 17 people who had had high amyloid on their first scan, but none with low amyloid at that time, had declined cognitively to the point of mild cognitive impairment. All six had been accumulating tau at a significantly faster clip than the other 11 high-amyloid participants who were cognitively stable. These 11 saw their tau rise more slowly, comparable to the low-amyloid group. Likewise, some of the people with low amyloid at baseline had high rates of subsequent Aβ accumulation, and they also were increasing tau at a faster clip. This suggests that they, too, were on track for cognitive decline. The results highlight the value of serial tau measures to predict progression, Hanseeuw said.
Because how fast a person’s brain accumulated tau pathology most strongly predicted that their cognition would decline, the authors propose serial tau PET as a surrogate marker for disease progression in clinical trials. “I would be very confident that a drug that targets and reduces tangles seen on PET would preserve cognition, and I believe tau PET could be useful as an outcome measure,” said Hanseeuw.
Regulatory authorities require cognitive outcomes in AD trials (Nov 2018 conference news). Hanseeuw noted that cognition is highly variable in prodromal AD and that cognitive measures take a long time to show significant drug response effects. “Tau PET might shorten the wait,” he said. His data suggest that scans two years apart would detect change in prodromal AD. In people with active disease, he predicts a quantifiable reduction could show up in six months or less.
Michel Grothe, German Center for Neurodegenerative Diseases, Rostock, wonders at what point amyloid accumulation causes tau pathology to accelerate. “What are the critical levels of amyloid that are sufficient to have this effect on progressing tau pathology?” he asked. Grothe pointed out that even in the low-amyloid group, tau clearly increases over time. Some of these people may have no amyloid, others may have subthreshold or regional amyloid that might be associated with tau deposition and memory decline (Sakr et al., 2019; May 2018 news). Grothe would like to see stratification of subjects according to their regional Aβ deposition, to see how that tracks with tau changes (Oct 2017 news). That type of study will require a larger sample, he said.
Tau spreads in a characteristic pattern. In people who don’t have amyloid, tangles arise in the medial temporal lobe in the course of normal aging, but only there. In people who do have amyloid, tangles fan out into surrounding areas, starting from the entorhinal cortex and hippocampus and continuing to the adjacent limbic cortex and inferior temporal cortex, and finally invading the association cortex and primary sensory cortex.
Hanseeuw’s main analysis was limited to the inferior temporal lobe, but he found similar relationships with Aβ and cognition when he measured tau in other neocortical regions such as the temporal neocortex and precuneus. Hanseeuw said the team was unable to study early tau deposition in the medial temporal lobe, because participants were past the age when that process begins.
Tau’s spread depends not only on the presence of Aβ, but also on functional and structural connections between brain regions. In a manuscript on BioRxiv, Jacob Vogel and Alan Evans of the Montreal Neurological Institute in Canada, along with Oskar Hansson, Lund University, Sweden, virtually modeled the spread of tau over neuronal connections in the human brain, then compared those patterns to tau PET scans of 312 people on the AD spectrum. Their results support the idea that tau spreads through interconnected neurons, rather than simply diffusing through the interstitial milieu.
Interestingly, the pattern of spread was similar in amyloid-negative people, suggesting that tau uses the same pathway to propagate in normal aging, and Aβ simply speeds up that process. In light of this, Hansson told Alzforum he would like to see more sophisticated analyses of tau spread in longitudinal studies like Hanseeuw’s. “It’s likely not the same in all patients, and that will be very important to study,” he said.—Pat McCaffrey
References
News Citations
- At CTAD, Tau PET Emerges as Favored Outcome Biomarker for Trials
- Which Are the Right Tests to Satisfy New FDA Guidance?
- A Little Amyloid, A Lot of Trouble?
- PET Staging Charts Gradual Course of Amyloid Deposition in Alzheimer’s
Paper Citations
- Sakr FA, Grothe MJ, Cavedo E, Jelistratova I, Habert MO, Dyrba M, Gonzalez-Escamilla G, Bertin H, Locatelli M, Lehericy S, Teipel S, Dubois B, Hampel H, for the INSIGHT-preAD study group, Alzheimer Precision Medicine Initiative (APMI). Applicability of in vivo staging of regional amyloid burden in a cognitively normal cohort with subjective memory complaints: the INSIGHT-preAD study. Alzheimers Res Ther. 2019 Jan 31;11(1):15. PubMed. Correction.
External Citations
Further Reading
Primary Papers
- Hanseeuw BJ, Betensky RA, Jacobs HI, Schultz AP, Sepulcre J, Becker JA, Cosio DM, Farrell M, Quiroz YT, Mormino EC, Buckley RF, Papp KV, Amariglio RA, Dewachter I, Ivanoiu A, Huijbers W, Hedden T, Marshall GA, Chhatwal JP, Rentz DM, Sperling RA, Johnson K. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019 Jun 3; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Perceptive/Wake Forest School of Medicine
This JAMA Neurology paper by Hanseeuw et al. is a true tau-PET tour de force. With a sample of just 60 people who started with normal cognition and had seven years of follow-up data, the authors were able to provide some of the first in vivo longitudinal confirmation of the leading research hypotheses that we have in the field today about amyloid, tau, and cognitive function—“that Aβ precedes and accelerates neocortical tau pathology, which together precipitate cognitive decline.”
The results are a reaffirmation of recent research efforts, implying that the growing trends in AD studies—to focus earlier on preclinical disease, and to amplify use of imaging biomarkers—may prove critical to successfully measuring, predicting, and preventing cognitive decline in clinical trials of disease-modifying drugs.
While it is not exactly surprising, it is exciting to see it demonstrated that it is not just cross-sectional amyloid or tau levels that are most closely linked to cognitive function. Rather, as the mediation model shows, it is processes of amyloid and tau change that are most critical to final cognition seven years later.
This paper did a solid job of supporting the argument that serial tau-PET biomarkers will be critical to understanding whether neurodegenerative disease processes are truly being slowed by treatments, regardless of whether enough follow-up has occurred over the length of a standard clinical trial to see a slackening of cognitive decline. In other words, the take-home for clinical trialists is that these findings may bring us closer to using in vivo imaging biomarkers to measure the impacts of interventions in AD clinical trials, as neuroimaging may allow us to detect significant group differences over a shorter time and in an earlier disease state, even without observing differences in clinical and cognitive function.
Lund University
I think the paper is very well performed and the results indicate that tau pathology is more closely related to cognitive decline than Aβ pathology. More importantly, the study indicates that it is the rate in increase in tau pathology (as measured with change in FTP PET) that is strongly related to cognitive decline.
The results support investigating therapies aimed at halting spread of tau pathology. However, causal relationships cannot be proven until a therapy directed against an AD-relevant pathology has been shown to halt cognitive decline. Further, larger studies are needed evaluating this and other tau PET tracers before we can make more firm conclusions, because the study is quite limited in size and in the regions evaluated for tau pathology.
This paper provides a crucial window into the pathology of AD. The results appear to imply that tau is more directly related to cognitive decline than Aβ, and that developing therapeutics directed at abrogating tau might more likely to impede dementia than developing therapeutics directed at impeding Aβ. Perhaps this is so, but a crucial gap remains between Aβ, tau, and the onset of dementia symptomology, and this involves smaller, and potentially more toxic forms of Aβ that are difficult to measure using PiB. This means that missing from the cascade described by Hanseeuw et al. was the influence of smaller forms of Aβ on the symptomatology of AD.
As the authors of this paper highlight, PiB is not particularly sensitive to prefibrillar or low levels of fibrillar Aβ (also see Vlassenko et al., 2012), and an inverse correlation between the size of Aβ assemblies and the potency of toxicity has been implicated (Sengupta et al., 2016).
Thus, lacking data on this important subgroup of Aβ in the milieu of AD pathology and in the context of the sequential model by Hanseeuw et al. makes it more challenging to attempt to infer from these results that a therapeutic strategy directed at impeding tau will be more or less viable than a strategy directed at impeding Aβ.
References:
Vlassenko AG, Benzinger TL, Morris JC. PET amyloid-beta imaging in preclinical Alzheimer's disease. Biochim Biophys Acta. 2012 Mar;1822(3):370-9. PubMed.
Sengupta U, Nilson AN, Kayed R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine. 2016 Apr;6:42-9. Epub 2016 Apr 5 PubMed.
Make a Comment
To make a comment you must login or register.