What went down in London town during the AAIC meeting? Whether you were there or could not go this year, Alzforum reporters captured the highlights. Read about a promising plasma Aβ assay that flags people who have brain amyloid plaques, raising hope for faster, cheaper, less-invasive screens for therapy studies. Witness a bidding push for primary prevention trials, as well as an official call to be ambitious on modifiable risk factors. New variants of microglial genes emerged, as did basic science proposing that the AD brain is filled with a “cloud” of different Aβ strains.
Finally, a Blood Test for Alzheimer’s?
In the quest for the Holy Grail of Alzheimer’s disease biomarkers—a blood test for brain amyloid accumulation—researchers have been stymied. Over and over, studies have failed to find a robust and reproducible relationship between Aβ42 levels in brain and blood, suggesting the latter might not reliably indicate disease. Lo and behold, new research unveiled at the Alzheimer’s Association International Conference, held July 16-20 in London, upends the conventional wisdom. Randall Bateman of Washington University School of Medicine in St. Louis presented a new method for measuring plasma Aβ that relies on mass spectrometry. It greatly increases accuracy and precision over previous antibody-based measures. The method lowers background noise enough to reveal an average 15 percent drop in the plasma ratio of Aβ42/Aβ40 in people with brain amyloid compared to those without, Bateman said. The findings have been replicated in an independent cohort, but the method requires validation in multicenter studies and clinical settings, he added. The data were published in the July 11 Alzheimer’s and Dementia.
The presentation excited researchers in London. “This was by far the biggest news at the meeting. It’s a game-changer with huge implications for prevention trials,” Paul Aisen of the University of Southern California in San Diego told Alzforum. He noted that the high negative rate on amyloid PET scans presents a big problem as prevention studies try to enroll, driving up cost and thereby limiting how large the trial can be. If researchers could screen potential participants with a blood test instead of a scan, costs would drop and trials could screen many more people. Pharma researchers flocked to pick Bateman’s brain after the talk.
Blood Test for Brain Amyloid.
A representative “area under the curve” graph, showing the specificity and sensitivity of the plasma Aβ42/40 ratio for identifying people with amyloid plaques. [Courtesy of Randall Bateman.]
While previous studies have clashed on whether blood and CSF Aβ correlate (for review see Toledo et al., 2013), Bateman saw encouraging indications that these measures might indeed be linked while studying Aβ production and clearance using stable isotope labeling kinetics (SILK). In this technique, people ingest 13C-leucine, which incorporates into newly made Aβ, allowing researchers to track how quickly the peptide appears and disappears and to follow where it goes (Jun 2006 news). In a separate study, Bateman was surprised to find that between 30 to 50 percent of brain Aβ flowed into the bloodstream (Roberts et al., 2014). Since CSF Aβ levels are 50 times higher than peripheral levels, this suggests the bulk of blood Aβ comes from brain, suggesting to him that blood Aβ levels should reflect those in brain, after all, and that a blood test was worth pursuing.
In his AAIC talk, Bateman described how he applied SILK to study the 24-hour kinetics of Aβ in the blood of 41 cognitively normal older adults, about half of whom had amyloid plaques, as determined by CSF Aβ levels or an amyloid PET scan. In the plaque-positive group, the researchers saw a more rapid appearance and clearance of newly synthesized soluble Aβ42 in plasma that mirrored kinetics in the CSF, strengthening the idea that much of the plasma Aβ comes from brain.
In previous CSF studies, overall clearance of Aβ from the brain has been found to slow in aging and Alzheimer’s disease (Jul 2010 conference news; Dec 2010 news). However, the turnover of the soluble Aβ42 relative to soluble Aβ40 speeds up in people with brain amyloid accumulation, likely reflecting the deposition of Aβ42 into plaques (Patterson et al., 2015; Dec 2016 news). The finding that newly made Aβ42 also appears and disappears faster from plasma in people with plaques confirmed the idea that blood levels indicate amyloid status, and suggested that plaques might lead to low steady-state levels of plasma Aβ42.
Bateman and colleagues then directly measured total Aβ blood levels. They immunoprecipitated the peptides from a 1.7 ml sample of plasma, then digested them with proteases and analyzed the fragments by high-resolution liquid chromatography mass spectrometry. Since their method measures the C-terminal ends of Aβ, it can distinguish between common isoforms such as Aβ42, Aβ40, and Aβ38, but cannot detect N-terminal truncations, Bateman noted. Mass spec analysis does not require isotopic labeling of the peptide.
Just as for Aβ42/Aβ40 in the CSF, this ratio in plasma was lower in the 18 people with brain amyloid than in the 23 without, Bateman reported in London. Plasma and CSF levels correlated with a regression coefficient of 0.70. Samples taken at any time of the day produced similar results. In plasma, an Aβ42/Aβ40 ratio above 0.124 distinguished amyloid-negative from -positive participants with 88 percent accuracy, Bateman found.
To validate these findings, the researchers tested stored plasma samples from a separate cohort of 164 people seen at the Knight Alzheimer’s Disease Research Center at WashU. Again they saw a consistently lower Aβ42/Aβ40 ratio in people with brain amyloid, with a staggering statistical significance of p = 0.00000006.
These samples had been handled extensively, transferred between tubes and frozen. Even so, a cutoff Aβ42/Aβ40 ratio of 0.103 was 76 percent accurate in detecting amyloid accumulation. The researchers determined this cutoff using an external standard curve, so it cannot be directly compared to the value obtained in the smaller study, Bateman noted. In future studies, following standardized protocols could probably improve accuracy, he added. The researchers are also working on lowering the amount of plasma needed for the test.
Eric Reiman of Banner Alzheimer’s Institute, Phoenix, called the work extraordinary. He asked why previous studies failed to find a consistent correlation between Aβ42 levels in CSF and blood. Bateman noted that blood constitutes a much “noisier” environment, chock-full of proteins and ions. Aβ levels in blood are 50-fold lower than in CSF, while other proteins are 1,000-fold higher, making Aβ 50,000 times harder to detect. In addition, because the background solutes are unique for each person, Aβ measurements are affected differently from one individual to the next. For this reason, the coefficient of variation for ELISAs or other antibody-based assays of plasma Aβ averages around 20 percent, which would mask the 15 percent difference in the Aβ42/Aβ40 ratios, Bateman said. By immunoprecipitating Aβ to isolate it from blood, and analyzing it by very-high-resolution mass spec, the background noise falls essentially to zero, he claimed.
Plasma Predicts Brain?
Blood Aβ levels may be specific enough to serve as a potential screening test. In the first study of 41 participants, there was one false negative (triangle above dotted line), but eight false positives (circles below), suggesting such a test could prescreen trial candidates before a PET scan. [Courtesy of Randall Bateman.]
How would such a blood test be used? Bateman noted that the assay produces more false positives than false negatives, with the initial 41-person cohort having eight of the former and only one of the latter. Thus, a negative result could be considered definitive enough to avoid further testing, but a positive result would need to be confirmed by CSF testing or PET scans. Based on the initial data, about two-thirds of people without brain amyloid would test negative on this assay and would not need follow-up PET scans or lumbar punctures.
Bateman suggested that this blood test would serve best as a quick initial screen for people in preclinical or prodromal disease phases. With the costs of mass spec analysis running about one-tenth the price of an amyloid PET scan, that could create savings for large trials, which need to screen thousands of potential participants.
C2N Diagnostics, a company founded by Bateman and others that develops AD assays, is considering how to implement this assay in trials, Bateman noted (Apr 2009 news). Meanwhile, clinical diagnostic use of the test may be possible within a few years, Bateman said.—Madolyn Bowman Rogers
Roberts KF, Elbert DL, Kasten TP, Patterson BW, Sigurdson WC, Connors RE, Ovod V, Munsell LY, Mawuenyega KG, Miller-Thomas MM, Moran CJ, Cross DT 3rd, Derdeyn CP, Bateman RJ.
Amyloid-β efflux from the central nervous system into the plasma.
Ann Neurol. 2014 Dec;76(6):837-44. Epub 2014 Oct 24
PubMed.
In Clinical Use, Amyloid Scans Change Two-Thirds of Treatment Plans
Brain amyloid imaging has become an essential tool for Alzheimer’s research, but the technology has not yet proved its value in clinical practice. Several studies are underway to investigate this. Chief among these is the massive Imaging Dementia—Evidence for Amyloid Scanning (IDEAS) study in the United States, which examines how having an amyloid PET scan changes treatment plans and health outcomes in 18,500 Medicare beneficiaries. At the Alzheimer’s Association International Conference held July 16-20 in London, principal investigator Gil Rabinovici of the University of California in San Francisco reported interim results from the first 4,000 people scanned. The bottom line: The scans had a much greater impact than expected. After seeing scan data, physicians changed medications or recommendations for patients in two-thirds of cases, many more than previous smaller studies had reported. Secondly, and perhaps as importantly, diagnoses shifted dramatically in accordance with the scans, particularly for people without amyloid plaques who had been wrongly diagnosed with AD. The researchers are still collecting data to find out if these treatment changes made a difference in how well patients fared one year later.
Clinicians hailed the findings. “These are very encouraging preliminary results … showing that amyloid imaging has a major impact on clinical diagnosis and treatment,” Stephen Salloway of Butler Hospital at Brown University, Providence, Rhode Island, wrote to Alzforum. “The foundation of medical care rests on an accurate diagnosis,” he added, with regard to the study’s second finding (see full comment below). Kejal Kantarci of the Mayo Clinic in Rochester, Minnesota, who chaired the AAIC session, agreed. “The findings will likely have a significant impact on clinical practice, and perhaps set the stage for similar studies involving other upcoming AD biomarkers,” she wrote.
Across the pond in Europe, other ongoing studies are also strengthening the case that amyloid scanning provides clinical benefits. In the same AAIC session, Arno de Wilde of VU University Medical Center, Amsterdam, reported on Alzheimer’s Biomarkers in Daily Practice (ABIDE). This study of about 500 people took a different approach from IDEAS, enrolling a distinct population that included people with subjective cognitive complaints as well as people with clinically unambiguous AD diagnoses. In this group, amyloid scans changed physicians’ diagnoses or treatment plans about one-fourth of the time. Meanwhile, the European AMYPAD study, which will analyze about 6,000 brain scans, is still enrolling.
The IDEAS study stands out for its size, as well as its potential impact on whether insurers will cover amyloid PET. The Centers for Medicare & Medicaid Services (CMS) pays for the IDEAS scans as part of its “coverage with evidence development” process to find out if the technology helps patients. Positive findings may lead CMS to cover the scans for its beneficiaries, after which other insurers are likely to follow suit (see Apr 2015 news). The study, which was organized by the Alzheimer’s Association, now includes 674 clinical practices and 1,124 dementia experts across the United States, Rabinovici said in London. These experts enroll Medicare beneficiaries who meet appropriate-use criteria defined previously. In a nutshell, the criteria specify that patients must have either mild cognitive impairment or dementia with an uncertain cause before undergoing scanning (Jan 2013 conference news). IDEAS started enrolling in February 2016 and is on track to finish before the end of this year, with almost 12,000 people already scanned, Rabinovici noted.
For the interim analysis, the researchers included data from the first 3,979 participants. Two-thirds of them had MCI and one-third had dementia, and their average age was 75. At baseline, about three-fourths of the cohort had been diagnosed with prodromal or full-blown AD. Importantly, they were almost equally likely to have this diagnosis whether or not they turned out to actually have amyloid plaques, Rabinovici found. In other words, in a community setting, diagnosing challenging cases was only 50 percent accurate. About 40 percent of people labeled as having AD did not have amyloid plaques, while more than half of those with other diagnoses did have them. Unsurprisingly, clinicians changed many diagnoses after seeing these data, with the percentage of AD cases rising from 78 to 95 in the amyloid-positive group, and falling from 73 to 15 in the amyloid-negative group. Thus, scans seemed to have the largest effect in ruling out Alzheimer’s disease in uncertain cases.
The IDEAS study probably saw such a large effect on diagnoses because the appropriate-use criteria singled out uncertain cases, selecting for those who are most likely to benefit from amyloid scanning, Rabinovici noted. This contrasts with early trials that chose participants based on criteria for probable AD, where one-fourth to one-third turned out to be amyloid-negative (Mar 2012 conference news; Jan 2014 news; Apr 2013 conference news).
Treatment plans in IDEAS changed in synch with diagnoses. Dementia specialists wrote up an initial care plan before seeing the scan data, then revised their plan based on the results and shared their recommendations with patients. The specialists started or stopped acetylcholinesterase inhibitors or memantine for about half the cohort as a result of the scan.
Clinicians were likelier to write new prescriptions than to discontinue drugs, with about one-third of amyloid-negative cases remaining on the medications. This last point engendered discussion at AAIC, with researchers debating whether this represents inappropriate medication use. Kantarci noted that acetylcholinesterase inhibitors can be helpful for some other conditions, such as dementia with Lewy bodies, and so may still benefit some amyloid-negative patients.
Meanwhile, use of non-AD drugs, such as antidepressants, antipsychotics, and other neurologic drugs, changed in about one-third of the cohort. In one-fifth of cases, the specialists changed their recommendations for counseling about safety and long-term care. Referrals to clinical trials dropped from 20 to 12 percent, mostly due to clinicians pulling referrals for amyloid-negative people. Recommendations for follow-up MRI and FDG-PET scans changed in about 10 percent of cases. Overall, care plans shifted in some way for 67.6 percent of participants.
This number is far higher than in several previous small studies, where scans typically led to new treatment recommendations for one-fourth to one-third of patients (Aug 2015 conference news; Nov 2016 news). Rabinovici noted that many of those studies occurred in academic settings. He suggested that the current findings might more accurately represent what would happen in general medical practice.
Data from the ABIDE study complements the IDEAS findings. This three-year project analyzes how amyloid PET, MRI, and CSF measurements affect patient care (de Wilde et al., 2017). The researchers invited all patients seen at their memory clinic during 2015 and 2016 to participate, and about half agreed. The final cohort consisted of 507 people, almost 50 percent of whom had dementia, with the remainder having MCI or subjective cognitive decline. Thus, the population was much broader than that seen in IDEAS, including cases where the initial diagnosis was clear-cut and cases with no clinical diagnosis. Participants had a standard neuropsychological workup and MRI scans, but did not have lumbar punctures.
Amyloid PET scanning showed that around half the cohort had plaques. After scanning, doctors changed the diagnoses of one-quarter of the patients; they altered treatment recommendations for the same percentage, mostly the amyloid-negative patients, de Wilde said. Treatment changes included referrals for further testing or clinical trials as well as starting or stopping medications (de Wilde et al., 2016).
Wiesje van der Flier of VU University, who leads ABIDE, noted that even among people with subjective cognitive decline, amyloid imaging resulted in new treatment recommendations 10 percent of the time. “This is a very important group of patients; they constitute 25 percent of the memory clinic population, and ask for an explanation of their [cognitive] complaints,” van der Flier wrote to Alzforum. Current appropriate-use criteria in the United States exclude this group, but van der Flier believes they can benefit from scanning. She suggested that the field develop guidelines for how best to disclose scan results to this population.
Clinicians still have little information on how amyloid scanning affects a patient’s state of mind. The ABIDE study asked some participants to fill out questionnaires before and after amyloid scanning, and found that they reported more certainty about their diagnosis afterward, but no change in their anxiety regardless of the results. Studies show that patients want more information about what they can expect from diagnostic tests, and what the results mean for their own situation, van der Flier said (Kunneman et al., 2016; van der Flier et al., 2017). Rabinovici agrees that the issue of how best to disclose scan results needs more study, while noting that most patients find value in seeing the data. “People want to know, even if it’s bad news,” Rabinovici said at a AAIC press conference.—Madolyn Bowman Rogers
de Wilde A, van Maurik IS, Kunneman M, Bouwman F, Zwan M, Willemse EA, Biessels GJ, Minkman M, Pel R, Schoonenboom NS, Smets EM, Wattjes MP, Barkhof F, Stephens A, van Lier EJ, Batrla-Utermann R, Scheltens P, Teunissen CE, van Berckel BN, van der Flier WM.
Alzheimer's biomarkers in daily practice (ABIDE) project: Rationale and design.
Alzheimers Dement (Amst). 2017;6:143-151. Epub 2017 Jan 23
PubMed.
Searching for New AD Risk Variants? Move Beyond GWAS
Geneticists continue to plumb the genome for clues to AD risk. New variants presented at this year’s Alzheimer’s Association International Conference, held in London July 16-20, reinforce the central role of microglia in AD and link new genes to traits such as Aβ and tau pathology or metabolic dysfunction early in the process.
Large genome-wide association studies (GWAS) have identified about 27 susceptibility loci for late-onset AD (LOAD), implicating diverse biological functions, such as immune responses, cholesterol transport, endocytosis, ubiquitination, and protein folding pathways (see Apr 2011 news; Jul 2013 conference news; International Genomics of Alzheimer's Disease Consortium (IGAP), 2015; June 2017 news). Those variants account for 30–40 percent of the estimated 58–76 percent heritability of LOAD, claimed Julie Williams from Cardiff University in Wales. Geneticists have since adopted other methods to identify the rest. In London, Williams described how she, with eight other principal investigators and more than 450 scientists, used exome genotyping to find rare variants associated with LOAD. They went undetected in GWAS, which generally identify only common variants.
Finding rare variants presents challenges. “In an ideal world, one would sequence the complete genomes of maybe hundreds of thousands of individuals,” Williams told Alzforum. Since that would require more time and money than genetics has at the moment, Rebecca Sims, also at Cardiff, Sven van der Lee of the Erasmus Medical Center in Rotterdam, the Netherlands, Adam Naj of the University of Pennsylvania in Philadelphia, and Céline Bellenguez of the INSERM in Lille, France, decided to focus on the exome. Using a microarray enriched in rare coding variants, they genotyped samples from the approximately 85,000 people covered in the International Genomics of Alzheimer's Project (IGAP). “The approach is notable for its very large sample size,” wrote Johnathan Cooper-Knock from the University of Sheffield in England (see full comment below). The results were recently published in Nature Genetics (Sims et al., 2017).
The researchers used the Illumina HumanExome BeadChip, which includes nearly 250,000 variants of which roughly 75 percent have minimum allele frequencies of less than 0.5 percent. Co-author Rita Guerreiro from University College London told Alzforum that this was the first time this type of chip had been used in AD studies. Sims and colleagues identified 43 candidate AD variants, excluding known risk loci, after genotyping about 16,000 LOAD cases and 18,000 cognitively normal, elderly controls from IGAP. The researchers validated these initial hits in two additional cohorts, also from IGAP. One had 14,000 cases and 22,000 controls, the other 6,000 cases and 8,000 controls. Sims and colleagues tracked the 43 candidates found in the first analysis and used imputation to look for the variants in samples that had been previously genotyped for common variants. Because DNA is inherited in blocks, imputation allows geneticists to predict the presence of alleles based on co-inheritance of other variants nearby. In this case, they imputed based on reference genotypes from the Haplotype Reference Consortium, which includes nearly 65,000 haplotypes covering close to 40 million single nucleotide polymorphisms (SNPs).
The results revealed three new non-synonymous coding substitutions associated with LOAD in the phospholipase C γ2 gene (PLCG2), the ABI family 3 gene (ABI3), and TREM2, a known susceptibility gene for AD. All genes are highly expressed in microglia. While the PLCG2 variant, an arginine for proline at position 522, was protective, the ABI3 Ser209Phe and TREM Arg62His variants were associated with increased AD risk.
“This is exciting news—these researchers provide more evidence for a causative role of microglial dysfunction in AD,” commented Christian Haass and Gernot Kleinberger of the German Center for Neurodegenerative Diseases in Munich. They noted TREM2 variants are risk factors for other neurodegenerative diseases as well. “Microglia are thus clearly not simple bystanders or only secondary troublemakers,” they wrote (see full comment below).
All three genes have a similar expression profile in the human cortex, said Williams: high in microglia, low in neurons, oligodendrocytes, astrocytes, and endothelial cells. The findings align well with a raft of recent studies implicating microglia in AD. Indeed, a GWAS published last month found that among the 112 genes lying within AD-associated loci, 60 were expressed in human microglia and contained binding sites for the master regulator of microglial function and identity, PU.1 (Jun 2017 news).
Williams’ lab is exploring how the new PLCγ2 variant might protect. PLCγ2 is a transmembrane signaling enzyme that generates two second messengers. Myoinositol 1,4,5-trisphosphate (IP3) regulates cytoplasmic calcium levels, and diacyl glycerol initiates the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways. To dissect how the Pro522Arg substitution alters its functions, Georgina Menzies in Williams’ lab modeled the molecular structure of three parts of the protein. In her poster, she showed that the overall structure and flexibility of the protein appears to remain unchanged, despite the removal of a proline, which often introduces kinks into protein backbones. Nevertheless, Menzies showed how the positively-charged arginine resides in a loop on the edge of the enzyme’s active site. Because it can attract surrounding negatively charged amino acids, the arginine likely changes the structure of the loop. Menzies said the loop could partially cover the entrance for the substrate and affect catalysis. The researchers will track how the variant affects calcium dynamics in cultured cells. PLCγ2 might also be linked to TREM2. Gary Landreth of Indiana University School of Medicine in Indianapolis noted that the TREM2 binding partner DAP12 activates the phospholipase in osteoclasts (Mao et al., 2006).
ABI3 is an adaptor protein. It forms part of a complex that regulates actin polymerization. Although mostly studied in T cells, in the brain it is predominantly expressed in microglia. How the AD risk variant alters ABI3 function is unclear. Haass said he wanted to investigate this. “ABI3 activities may be related to cytoskeletal rearrangement and the formation of membrane protrusions,” he and Kleinberger wrote. “It is therefore tempting to speculate that its dysfunction may interfere with a central function of TREM2 in chemotaxis and phagocytosis (Mazaheri et al., 2017; Kleinberger et al., 2014).”
TREM2 is a microglial transmembrane receptor. Previous studies have shown that loss-of-function variants reduce microglial phagocytosis, impair lipid sensing, prevent binding of lipoproteins, and disrupt microglial chemotaxis (Apr 2017 conference news). Sims and colleagues revealed not only the new Arg62His variant, but also confirmed the previously reported Arg47His variant. The exome analysis hinted at the existence of additional risk variants in this gene.
Connecting the Dots
While TREM2, ABI3, and PLCG2 might at first glance seem unrelated, Peter Holmans, also at Cardiff University, found they are part of a single protein-protein interaction network. Holmans discovered the network by looking for interactions between proteins related to GWAS hits. He used data from a previous study that mapped GWAS risk variants to clusters of co-expressed genes found in the brains of healthy individuals (International Genomics of Alzheimer's Disease Consortium (IGAP), 2015). Four of the 117 clusters found were enriched with AD GWAS genes. A set of 151 genes captured this GWAS signal.
Microglial AD Network? An interaction network of 56 proteins is enriched by genes harboring common and rare variants associated with AD (in boldface). It includes the three new exome hits. [Courtesy of Sims et al., Nature Genetics.]
Holmans then used high-confidence human protein-protein interaction data to see if proteins encoded by the 151 genes formed a network. In step-wise fashion, he used one protein as a start and looked to see how each of the other 150 might link to it, then how each of the remaining 149 might link to that, and so on. He reiterated this exercise 151 times, using each protein as a hub. At the end of this analysis, one network stood out for its size. It contained 56 proteins—more than would occur by chance. Surprisingly, it included TREM2, PLCG2, and ABI3 (see image above). It also contained a list of microglia-related genes that have been genetically linked to AD. They are two master regulators of microglial function, SPI1, aka PU.1, and TYROBP, aka DAP12 (Zhang et al., 2013; May 2013 Alzforum webinar); SYK, a signaling protein downstream of TREM2/DAP12 that controls PLCγ2 activity (Xing et al., 2015; Paris et al., 2014); INPP5D, which forms a complex that regulates SYK; and CD33, which interacts with TREM2 and promotes microglial phagocytosis of Aβ processing (Aug 2013 news; Oct 2015 news). The network also includes CSF1R, a master regulator of microglial proliferation. Curiously, pathway databases, such as Gene Ontology or the Kyoto Encyclopedia of Genes and Genomes, don’t link ABI3 to these genes. Furthermore, because the co-expression and protein interaction data are derived from healthy controls, the clusters of correlated genes and the protein network cannot have arisen as consequences of neurodegeneration, noted Holmans. This, plus its enrichment with AD risk genes, indicates that the network is consistent with microglial responses in LOAD being directly involved in disease, rather than simply a downstream consequence of neurodegeneration, he said.
Williams thinks the new TREM2, AIB3, and PCLG2 variants found in the exome search account for a small portion of AD’s missing heritability. The authors speculate that the remainder may reside anywhere in the genome. There could be common variants of small effect size, rare variants found in other exons, even rarer variants that may only be identified by analysis of larger cohorts, and variants within introns and intergenic sites. Researchers at AAIC asked about additional variants in other populations. Williams agreed this was important, as IGAP includes mostly Caucasians of European descent. “Looking at other populations will help us understand AD mechanisms and help refine risk predictions,” she said.
Exactly how much of AD’s heritability remains to be found is subject to debate. A recent study from John Hardy and colleagues at University College London, concluded that a polygenic score based on known AD variants predicts 84 percent of the risk for AD, which comes close to the concordance seen in studies of twins, said Hardy (Escott-Price et al., 2017). These authors concluded that, though rare variants are still likely to be found, studies would be well advised to focus on targeted sequencing of known AD pathways and on cell and animal experiments to further delineate those pathways.
Other Methods to Find AD Variants
Geneticists are inventing new ways to hunt AD variants that went undetected in GWAS and might shed light on AD pathogenesis. Many labs are searching for polymorphisms tied to specific quantitative traits. As Yuetiva Deming from Carlos Cruchaga’s group at Washington University, St. Louis, pointed out, GWAS identify risk variants but say nothing about how that risk manifests. Finding variants that associate with specific endophenotypes could be extremely informative, she said. Leigh Christopher, who works with Michael Greicius at Stanford University, California, agreed. “We are not only interested in causative genes, but in those that modify the course of the disease or the rate of decline, she told Alzforum. “We can’t necessarily find those genes in case-control GWAS,” she said.
Deming and colleagues ran a GWAS to find variants linked to CSF Aβ, tau, or phosphorylated tau, using data from more than 3,000 people in nine different cohorts. About half of them were women, the average age was 72. Deming and colleagues found four variants for CSF tau or p-tau and two for Aβ. The former lie near the genes GMNC, GLIS3, PCDH8, and NFATC1, while the latter lie near the GLIS1 and SERPINB1 genes (see Deming et al., 2017).
Deming used independent data sets to test whether these variants also associated with AD, age of disease onset, or progression. GLIS1 popped out in the AD risk and progression analysis, while SERPINB1 associated with age at onset. Deming said that the GLIS1 locus might alter expression of the gene SLCA17, while the SERPINB1 variant, which lies in an intron, seems to alter its own expression. SERPINB1 encodes an elastase found in immune cells; Deming thinks the variant might control levels of the enzyme in macrophages, perhaps explaining its association with amyloid accumulation.
Also trying to tease apart genetic links to tau and Aβ, Jaeyoon Chung from Lindsay Farrer’s lab at Boston University took a so-called bivariant approach, asking if genetic polymorphisms associated with any two of plaques, tangles, or cerebral amyloid angiopathy. “We thought we might find variants with pleiotropic effects,” he said.
Chung built on a recent univariant analysis that used data from the Alzheimer's Disease Genetics Consortium to find SNPs that associated with AD neuropathology among 3,135 people, 463 of whom were healthy controls (Beecham et al., 2014). Chung found a variant upstream of the ECRG4 gene that associated with plaques and tangles, and another in the HDAC9 gene that associated with tangles and CAA.
How the ECRG4 gene might be involved in AD pathology is unclear, but Chung noted that RNAseq analysis of brain samples from the Mayo Clinic hints that the risk allele reduces expression of the gene. It has also been reported to be suppressed in brain injury, he said.
An AD link to HDAC9 seemed more plausible, since the gene associated with other neurological conditions, as well, including stroke and schizophrenia. Prior data suggest it protects against neuronal apoptosis, said Chung. HDAC9 inhibits expression of MEF2C, another AD risk gene (Jul 2013 conference news). Chung presented expression quantitative trait loci analysis that suggests the HDAC9 risk allele reduces expression of the gene in the brain. Analyzing postmortem brain samples, he found lower HDAC9 expression in the prefrontal and visual cortices of AD patients than controls, and lower expression to correlate with Braak stages.
Emrin Horgusluoglu in Andrew Saykin’s lab at Indiana University School of Medicine, Indianapolis, took a different tack. She ran GWAS of ADNI samples, then applied a gene set enrichment analysis called GSA-SNP to identify functional pathways associated with tau accumulation (Nam et al. 2010). From 1,200 volunteers with either CSF tau/p-tau or tau PET data, she identified 39 pathways. Fifteen were related to neurogenesis. Horgusluoglu then used gene-based association analysis to pull out specific genes in the pathways that correlated with tau levels. Four genes fit the bill: APOE, PVRL2, APOC4, and MAP3K10.
Tweaking Tau. Gene set enrichment analysis points to pathways that influence accumulation of tau or p-tau in the CSF. [Image courtesy of Ermin Horgusluoglu and AAIC 2017.]
In her presentation, Stanford’s Christopher reported a variant that associated with glucose hypometabolism in the posterior cingulate cortex, an early marker of AD. She found a single SNP, rs2273647, among 606 participants in the ADNI study. The SNP lies in the SMEK1 gene, which encodes regulatory subunit 3a of protein phosphatase 4 (PP4R3a). The minor T allele seems protective, associating with less hypometabolism than seen in FDG PET scans. People with two T alleles metabolized glucose normally.
Looking further, the researchers found that the polymorphism protected people with mild cognitive impairment from progressing to Alzheimer’s dementia. Carriers of the C/T or T/T genotype were at lower risk of progressing. The T allele also slowed cognitive decline in those with AD. Carriers performed better over time than noncarriers on the MMSE and in the Boston naming task.
How does this variant protect? It may alter expression of the phosphatase subunit, Christopher said. She reported that healthy controls make less of one mRNA isoform than AD patients do. Among healthy controls, T allele carriers also make less of this isoform than noncarriers. However, among people with AD, carriers and noncarriers appear to express the gene equally well. “I think this suggests that something turns on the gene in the disease state,” suggested Christopher.
Given that glucose hypometabolism is not unique to AD, others at the meeting asked if the variant might protect against other neurodegenerative diseases as well. Christopher considers this plausible. Some scientists found it curious that the effect of a second T allele was additive in the FDG PET analysis but not the cognitive testing, and Christopher agreed. “There is definitely an additive effect in imaging and a dominant effect for cognitive traits, but we are not sure why,” she said.
Chloe Sarnowski from Boston University focused on different imaging endophenotypes. Working with Sudha Seshadri and colleagues at BU and at the University of California, Davis, Sarnowski used whole-genome sequence analysis to look for variants and genes that associate with total cerebral volume, hippocampal volume, or white-matter hyperintensities (WMH). She mined data from Trans-Omics for Precision Medicine (TOPMed), an NHLBI program, to identify genetic variants and other factors that associated with vascular disorders.
Variants and Volume. WGS analysis uncovers variants in chromosomes 1 and 16 that associate with total cerebral volume (top) and hippocampal volume (bottom), respectively.
Sarnowski found previously reported variants at chromosome 12q24 and 17q25 that associated with hippocampal volume and WMHs, respectively. She also detected two new variants. From 2,180 TOPMed samples, she found a 1p21 variant that associated with total cerebral volume, and from 2,170 samples a 16q23 variant that associated with hippocampal volume (see image above). Sarnowski found about 10 other genes that almost reached statistical significance, including the GBA3 gene associated with Parkinson’s and the UNC5D gene that has been linked to AD. Those genes appeared to be enriched in immunity, inflammation, and related AD and PD pathways.—Marina Chicurel and Tom Fagan
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C.
TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis.
Sci Transl Med. 2014 Jul 2;6(243):243ra86.
PubMed.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V.
Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease.
Cell. 2013 Apr 25;153(3):707-20.
PubMed.
Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, Carrell D, Cai Y, Fernandez MV, Budde J, Ma S, Saef B, Howells B, Huang KL, Bertelsen S, Fagan AM, Holtzman DM, Morris JC, Kim S, Saykin AJ, De Jager PL, Albert M, Moghekar A, O'Brien R, Riemenschneider M, Petersen RC, Blennow K, Zetterberg H, Minthon L, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Schellenberg G, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Peskind ER, Li G, Di Narzo AF, Alzheimer’s Disease Neuroimaging Initiative (ADNI), Alzheimer Disease Genetic Consortium (ADGC), Kauwe JS, Goate AM, Cruchaga C.
Genome-wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers.
Acta Neuropathol. 2017 May;133(5):839-856. Epub 2017 Feb 28
PubMed.
Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, Corneveaux JJ, Hardy J, Vonsattel JP, Younkin SG, Bennett DA, De Jager PL, Larson EB, Crane PK, Kamboh MI, Kofler JK, Mash DC, Duque L, Gilbert JR, Gwirtsman H, Buxbaum JD, Kramer P, Dickson DW, Farrer LA, Frosch MP, Ghetti B, Haines JL, Hyman BT, Kukull WA, Mayeux RP, Pericak-Vance MA, Schneider JA, Trojanowski JQ, Reiman EM, Alzheimer's Disease Genetics Consortium (ADGC), Schellenberg GD, Montine TJ.
Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias.
PLoS Genet. 2014 Sep;10(9):e1004606. Epub 2014 Sep 4
PubMed.
Monomeric Seeds and Oligomeric Clouds—Proteopathy News from AAIC
Researchers studying protein aggregation have long debated two questions: What is the smallest structure than can seed different protein aggregates, aka strains, and how many different strains form within a brain? Researchers answered both at this year’s Alzheimer’s Association International Conference, held July 16-20 in London. Marc Diamond, University of Texas Southwestern Medical Center, Dallas, reported that single molecules of monomeric tau have the wherewithal to seed if certain motifs are exposed, while Mathias Jucker, University of Tübingen, Germany, presented evidence for “clouds” of different Aβ strains in the human brain. Other researchers thought the former offered a solid mechanistic explanation for seeding, and that the latter could have profound implications for amyloid imaging and therapy.
Fluorescent Probes.
Detection of different conformers of Aβ is possible with fluorescent dyes—conjugated oligothiophenes such as heptamer formyl thiophene acetic acid (h-FTAA) and heptamer hydrogen thiophene acetic acid. [Courtesy of Choong et al., Biofilms and Microbiomes 2016.]
Researchers in Jucker's lab spotted these clouds while using a type of dye to probe the structure of Aβ aggregates. Developed by Peter Nilsson and Per Hammarström at Linköping University, Sweden, luminescent conjugated oligothiophenes (LCOs) have a flexible backbone that conforms to protein structures that they bind. This molecular contortion changes the emission spectra of the LCOs. The dyes can theoretically identify dozens of β-sheet conformations because each will fluoresce differently. In effect, said Jucker, the spectrum is indicative of the amyloid structure.
Nilsson, Jucker, and colleagues have used these dyes to detect different forms of Aβ plaque in transgenic mouse models of AD and to distinguish them from fibrils of tau (see Aslund et al., 2009). In London, Jucker showed how two of these dyes detected a myriad of Aβ structures in human brain samples, as well.
Using a combination of two LCO dyes, Jay Rasmussen, Jasmin Mahler, and Nathalie Beschorner from the Jucker lab probed postmortem tissue from the frontal, temporal, and occipital cortices of 40 people who had had different types of AD. Twenty-one samples came from sporadic cases, three each from familial AD patients who carried APP V717I or PS A431E mutations, five from PS E280A FAD carriers, two from PS F105L patients, and six from patients with posterior cortical atrophy (PCA).
Spectral Plaque.
An LCO labels an amyloid plaque in a tissue section from an AD brain. Different colors represent different conformations of the plaque’s Aβ components. [Courtesy of Natalie Beschorner.]
While the mean spectral characteristics of plaques from different regions of a given brain looked similar, plaques from different mutation carriers had unique spectral features, which in turn differed from the spectra of plaques from sporadic AD and from PCA. Spectra of plaques from AD and PCA also differed. The heterogeneity did not stop there. When Jucker zoomed in on small areas within each plaque, many different individual spectra opened up before his eyes, indicative of many different conformations of Aβ. To quantify this, he measured the spectra from each of the two dyes at different regions in each plaque, using the ratio of the peak emission intensities as a measure of heterogeneity. He found dozens of different ratios across each plaque. “The data indicate that there are clouds of conformations of Aβ that are similar in regions of the same brain, but that differ between brains,” said Jucker.
Jucker does not yet know the exact structures to which these dyes bind. The spectra did not seem to correlate with the amount of Aβ in a plaque, or with whether the Aβ was resistant to proteases. “We need further analysis to explore the putative links between conformations detected by the dyes and phenotypes,” he said.
However, it does seem clear that these conformational clouds can be transmitted. The researchers injected extracts from PS A431E, APP V717I, and sporadic AD patients into APP transgenic mice. Months later LCOs detected clouds of different Aβ conformers in induced Aβ plaques in the brains of the mice, and these were similar overall to the LCO spectra of the source plaques in the human brain.
Researchers in the audience were intrigued. “This is really, really great work,” noted Diamond. He asked how it gibes with findings from Robert Tycko at the NIH, who reported that one type of Aβ structure predominantly forms when extracts from many regions of a single brain are used to seed amyloid fibrils in vitro (Sep 2013 news). “Does your result suggest that it may be very difficult to accurately amplify, in a single reaction, the diverse structures from the brain?” asked Diamond. Jucker agreed it might. “You could argue that Rob amplified the most important structure,” he suggested. Tycko was not at AAIC, but told Alzforum that his most recent data, which indicate heterogeneity among Aβ42 fibrils from various forms of AD, seems consistent with Jucker’s findings (Jan 2017 news).
Others wondered what Jucker’s finding means for amyloid PET scanning or for immunotherapy, particularly in a prevention paradigm where one might need an exquisitely specific antibody to prevent seeding and propagation of amyloid. Jucker said the heterogeneity could complicate both, further cautioning that the heterogeneity might be dynamic. “The seeds you see at end-stage disease may be very different from those you see early on,” he said.
Some of those concerns may not apply in cases where the seeds are monomers, and in the case of tau, that now seems more likely. Previously, researchers had concluded by extrapolating from the concentration of tau in solution that a monomer might be the smallest structure that could seed fibrillization (Chirita et al., 2005). In London, Diamond presented physical evidence that this is the case.
Diamond and colleagues have previously identified many different strains of tau in tissues samples from different tauopathies. These faithfully seed the propagation of identical strains in vitro and in vivo (May 2014 news). To determine the smallest seed that can pull off such a feat, Hilda Mirbaha in Diamond’s lab made tau fibrils in vitro, sonicated them, fractionated them by exclusion chromatography, and then tested the fragments in a cellular biosensor assay developed in Diamond’s lab (Oct 2014 news).
Mirbaha found that monomers derived from fibrillized tau seeded aggregation of tau in the cell assay. Curiously, monomers that had never been fibrillized could not. Mirbaha and co-workers ran extensive biophysical tests to ensure that these seeds were indeed monomers, including fluorescence correlation spectroscopy, which measures fluctuations in fluorescence intensity due to diffusion of molecules in solution to determine the size and number of particles present as they pass through an incident light beam.
Satisfied that monomers from fibrils made in vitro can act as seeds, the researchers then asked whether monomers from brain extracts do, as well. They captured tau aggregates from AD brain extracts using immunoprecipitation, gently homogenized them, and then separated tau species by exclusion chromatography. Again, fractions from AD brain containing only monomers seeded tau aggregation in cells. In contrast, monomer fractions from control brains did not. Interestingly, after sitting at room temperature for 24 hours, monomers from the AD but not control brain formed larger assemblies. “It appears we have two different types of monomer, one that likes to seed and self-assemble, and another inert monomer that does not,” said Diamond. The researchers have dubbed the recombinant seed competent and inert monomers Ms and Mi, respectively.
How do these monomers differ? Diamond and colleagues used cross-linking with mass spectrometry (XL-MS) to probe their structure. This technique identifies which parts of a structure lie adjacent to each other. The data indicated that Ms and Mi had distinct cross-linking patterns. In fact, one region around amino acids 150-275 seemed to cross-link solely in Ms. Based on the XL-MS data and the known structures of tau, Diamond worked with Lukasz Joachimiak, also at UT Southwestern, to model the structures of tau. These suggest that two motifs—VQIINK and VQIVYK, near the beginning of the second and third repeat domains, respectively—are accessible in Ms but buried in Mi.
Again, the audience was impressed. “I think the cross-link mass spec work is important, because it appears to identify the core element of the tau seed,” noted Benjamin Wolozin, Boston University. “VQIINK/VQIVYK were previously known to be important for tau aggregation. That in seed-competent tau these are solvent-accessible is very interesting, because it presents a concrete mechanism through which seeding becomes enabled,” Wolozin added (von Bergen at al., 2000; Apr 2017 news).
How do these monomer structures relate to strains? Diamond will investigate this. He believes the relative exposure of the VQIINK and VQIVYK motifs might be germane. For example, VQIVYK forms part of the core of the recently derived tau structure, whereas VQIINK does not (Jul 2017 news).
Are specific monomers responsible for different strains in different tauopathies? Here it gets complicated. Diamond has isolated monomers and larger seeds from AD and corticobasal dementia brain samples. All the seeds from the AD sample yielded one morphogenic strain in cells. Whole extracts from CBD brains yielded two strains, but Ms from CBD yielded those two strains plus a third. Diamond went on to show that monomers from CBD could apparently morph to give rise to at least three different strains. “A single monomer in CBD appears to account for the different strains we observe,” Diamond told Alzforum. “The critical idea is that there is a dominant superstructure of the monomer, but that it has some 'flexibility' to sample other shapes that, when assembled, form unique strains,” he added. By contrast, the “superstructure” of the AD monomer is relatively rigid, and give rise to just one strain.
Diamond believes there are hierarchies of tau conformation based on seeding competency. He showed a representative “family tree” of tau strains, with seed-competent strains branching into AD and CBD, and CBD strains branching further into at least three specific species. “This concept helps us pick apart how we can have the different types of tauopathy, and has implications for imaging, diagnosis, and treatment,” Diamond said.—Tom Fagan
Planning the First Primary Prevention Trial for Alzheimer’s Disease
Is it time for the first primary prevention trial for Alzheimer’s pathology? Observational studies have made clear that amyloid accumulation begins more than two decades before symptoms appear, creating a huge window for intervention. Armed with better biomarkers for detecting early accumulation, researchers have pushed treatment to ever-earlier stages of the disease, and secondary prevention trials are underway to see if anti-amyloid drugs can stave off cognitive decline in preclinical disease populations. But what about stopping the disease process before it even gets a foothold?
Such was the goal discussed at a working meeting among leading clinician-researchers in the field, held in conjunction with the Alzheimer’s Association International Conference that took place July 16-20 in London. The assembled researchers agreed that all necessary pieces have finally fallen into place to allow them to tackle primary prevention of the central Alzheimer’s pathogenic pathway. These pieces include a trial-ready population of people certain to develop Alzheimer’s, detailed knowledge of how the disease progresses in this group, biomarkers that can track early amyloid accumulation, and drugs that prevent accumulation and are safe and cheap enough for long-term use. In a July 13 Nature commentary, Eric McDade and Randall Bateman of Washington University in St. Louis argued that now is the right time to begin this endeavor.
“We are at a point where a primary prevention trial makes sense. It’s time to move forward and truly test the amyloid hypothesis,” McDade told the working group. McDade helps direct the Dominantly Inherited Alzheimer Network trials unit (DIAN-TU). He described to the assembled scientists plans for a prevention trial in this population. In a lively discussion, the working group debated the issues surrounding how to implement such a trial.
DIAN participants come from families who carry pathogenic mutations in APP, PS1, or PS2 (see Nov 2008 news series; Aug 2016 news). All of these mutations cause the brain to produce more Aβ42 than normal, leading to early amyloid accumulation and plaque formation. Mutation carriers develop symptoms at around the same age their affected parent did. In many cases, this occurs in the mid-40s, meaning that the first insidious buildup of plaques in their brains probably begins in their 20s.
Because the disease starts so early in this group, DIAN researchers will enroll amyloid-negative carriers and noncarriers as young as 18. Noncarriers will be included so that participants will not have to find out their mutation status, although they will have the option to do so, McDade told Alzforum. On average, participants will be 22 years away from their estimated age of symptom onset.
The trial will test whether BACE inhibitors can slow the growth of plaques, as seen by amyloid PET, over the course of four to five years. Either one-third or one-half of participants will receive placebo.
The researchers are focusing on BACE inhibitors because these drugs can suppress production of Aβ42 and Aβ40 by 80 to 90 percent (Mar 2013 conference news; Oct 2014 news; Apr 2015 conference news). About half a dozen BACE inhibitors are currently in Phase 2 or 3 trials. So far, no serious side effects have cropped up in these multiyear studies, suggesting the drugs might be safe enough for long-term use. Because they are small molecules taken orally, they would be cheaper and more feasible for long-term use than antibody infusions, the scientists noted.
The working group largely supported the choice of BACE inhibitors, but voiced some cautions. Some noted that these drugs, unlike antibody therapies, have yet to demonstrate any slowing of cognitive decline in later AD. Mathias Jucker of the University of Tübingen, Germany, saw potential for antibodies to benefit young mutation carriers. Animal studies indicate that very few pathogenic seeds are present at early disease stages, hence mopping up those seeds could delay disease for a long time, Jucker suggested. But most researchers thought BACE inhibitors represented the most practical option for people in the early stages of familial AD.
“The case for BACE inhibitor treatment in this population is extremely strong. It would be unethical not to try it,” said Paul Aisen of the University of Southern California, San Diego.
To determine if the inhibitors work, the researchers will examine changes in the trajectory of plaque growth by PiB PET, McDade said. The goal is to slow amyloid accumulation compared to the placebo group, ideally keeping plaques below the threshold for amyloid positivity. The working group endorsed amyloid PET as the only feasible outcome measure for people at this early stage of AD pathogenesis. Reisa Sperling of Brigham and Women’s Hospital, Boston, noted that the brains of young mutation carriers do not harbor tau pathology yet. Nor have synapses begun to degenerate at this early stage, she added. Aβ in cerebrospinal fluid, while it detects very early signs of amyloidosis, does not track progression as well as amyloid PET, according to biomarker experts.
The greatest debate at the meeting centered around the size of this trial. DIAN researchers calculate that a cohort of 80 to 100 mutation carriers will provide 90 percent power to detect a 50 to 80 percent slowing of amyloid accumulation. Bateman noted that this calculation is based on analyses of extensive observational data that delineate the rates of change in the PiB PET signal for people carrying various mutations. The researchers will exclude people carrying the Arctic or Dutch mutations from the trial, due to the unique effects of these variants on amyloid, Bateman said.
Other scientists, such as Aisen and Sperling, urged the DIAN leaders to go bigger. Sperling pointed out that the calculation assumes a best-case scenario for BACE inhibitor performance, and makes no allowances for unexpected variability. Although researchers know something about how different mutations affect accumulation, it remains unknown how slashing the levels of Aβ monomer would affect the amyloid accumulation curve for each mutation, Sperling said. Moreover, other genes may influence the rates of amyloid buildup among different individuals, potentially muddying the results. A larger starting population would also help buffer against the expected attrition over a long trial, Sperling added. She recommended doubling the size to 100 people per arm to increase the odds of seeing a positive outcome. Cost, and caution about how many young adults to expose to an investigational drug were the counterpoints on this question. In the end, the discussion persuaded DIAN leaders to raise their enrollment goal to 140–180 mutation carriers.
Maintaining the trial population will be crucial, because the researchers have long-term plans for them. If the initial four- to five-year study does show a slowing of plaque growth, then the researchers will switch all participants to drug and follow them for several more years to try to detect a cognitive benefit. This phase of the trial will not include non-carriers. Researchers have not yet settled on a cognitive outcome measure, but several composites that detect change in preclinical populations have now been developed. These include the Preclinical Alzheimer Cognitive Composite (PACC), the Cognitive Function Instrument (CFI), ADCOMS, and a cognitive composite used in DIAN (Jun 2014 news; Mar 2015 news; Wang et al., 2016; Aug 2016 conference news). The researchers will compare cognitive change in this cohort to historical data from DIAN to look for any slowing of the trajectory. They will also analyze whether people initially on drug maintain their abilities better than those who started on placebo.
Exposing young, outwardly healthy people for so many years to experimental drugs raises ethical issues. Participants will be asked not to get pregnant during the trial, because what BACE inhibitors might do to a developing fetus is not yet known. Animal studies demonstrate a much bigger role for BACE in the developing than in the adult brain (Oct 2016 news). This requirement might represent a significant sacrifice for people in their prime child-bearing years, especially for noncarriers who run no risk of passing on the disease to their child, some researchers argued. McDade noted, however, that participants might be able to take a “drug holiday” for a year to have a child, and then resume dosing. Others noted that these issues must be spelled out clearly during the consent process.
A final conundrum concerns whether findings in the autosomal-dominant AD population will translate to sporadic disease. The latter involves impaired clearance, not overproduction, suggesting the findings might not be directly applicable to late-onset disease, noted José Luis Molinuevo of Barcelonaβeta Brain Research Center in Barcelona, Spain. Sperling agreed that if the study is successful, it will need to be repeated in the sporadic AD population. Such a trial would have to be larger, perhaps 250 people per arm, she suggested. A previous, negative, trial of ginkgo biloba pills demonstrated that a large prevention study can be done in sporadic AD, with the majority of the 3,000 elderly participants staying with the regimen for six years (Nov 2008 news). But for a mechanism-based, anti-amyloid drug such as a BACE inhibitor, an ADAD trial is a crucial first step to show efficacy, the researchers concurred.
McDade noted that the families involved in DIAN are eager to participate in this. At the DIAN Family Conference held in conjunction with AAIC, they expressed strong support for the trial, with some pressing to start treatment earlier than age 18. “The stakes are extraordinarily high. If successful, a primary prevention treatment could avert the loss of memories, thoughts, and independence for a significant proportion of the world’s older population,” wrote McDade and Bateman in their Nature commentary.—Madolyn Bowman Rogers
New Dementia Trials to Test Lifestyle Interventions
Hoping to stem the growing worldwide burden of dementia, scientists at the Alzheimer’s Association International Conference 2017 held in London July 16–20 described new studies to test whether multidomain lifestyle interventions can slow cognitive decline. Laura Baker of Wake Forest School of Medicine, Winston-Salem, North Carolina, outlined Protect through a Lifestyle Intervention to Reduce Risk (U.S. POINTER), a two-year study that will test the combined effects of physical and mental exercises, a healthful diet, and careful management of heart health. The trial is part of a larger, international effort, aka Worldwide FINGERS, that comprises studies in the U.K., Singapore, and China. Those are in the early planning stages. A similar trial will enroll soon in Australia, as well.
Two years ago the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability, or FINGER study, reported that a multimodal lifestyle intervention improved cognitive scores in older adults at risk for Alzheimer’s disease (see July 2014 news; Nov 2015 news). While encouraging, the findings were limited to one study of a single population. Researchers have since called for replication, most recently in a report from the U.S. National Academies of Sciences, Engineering, and Medicine (NAS) (Jun 2017 news), which noted that “multiple, independent studies testing the same combination of component elements will be necessary before strong conclusions can be drawn regarding the effectiveness of any specific multimodal intervention.” Ron Petersen of the Mayo Clinic in Rochester, Minnesota, who is a member of the NAS committee, told Alzforum that U.S. POINTER fulfills that recommendation and should provide confirmation, or not, of the FINGER results.
Baker will co-lead U.S. POINTER with FINGER lead investigator Miia Kivipelto of the National Institute for Health and Welfare, Helsinki, and Rachel Whitmer of the Kaiser Permanente Northern California Division of Research in Oakland. Like the other Worldwide FINGERS trials, POINTER will be tailored to fit the populations’ cultures and will build on lessons learned from FINGER and other recent lifestyle intervention studies, such as the French Multidomain Alzheimer’s Prevention Trial (MAPT) and the Dutch Prevention of Dementia by Intensive Vascular Care (PreDIVA). Diets will be adjusted to local tastes and cognitive and physical interventions adapted to suit common practices.
Funded by $20 million from the Alzheimer’s Association, POINTER will recruit 2,500 participants aged 60–79 years from five–seven healthcare networks across the United States, including Wake Forest’s large Accountable Care Organization for treatment of Medicare patients, which has more than 80 locations across North Carolina, and the Kaiser Permanente managed care consortium, which has more than 10 million members. Enrollment will start in 2018. In line with a trend in the dementia field to intervene early in disease progression, U.S. POINTER will recruit healthy people at risk for dementia, or people in early stages of mild cognitive impairment (MCI). Researchers suspect that a key to FINGER’s success was focusing on clinically asymptomatic people who performed at, or slightly below, average on neuropsychological tests.
Baker said U.S. POINTER is creating an algorithm to screen medical records for recruits. It will flag those with hypertension or elevated blood sugar, people who have siblings or a parent with memory impairment, and those who engage in less than 20 minutes of aerobic activity per week. It will exclude people with dementia and late-stage MCI or who perform above average on cognitive tests. By reaching out to these candidates, the researchers hope to avoid recruiting people who actively seek out studies, since they tend to be highly motivated and not representative of the population as a whole.
Baker hopes to include people with diverse backgrounds and ethnicities. A poster presented by Anna Rosenberg, a student of Kivipelto at the University of Eastern Finland in Kuopio, reported that FINGER participants benefited regardless of sex, age, education, household income, baseline cognition (MMSE score), cardiovascular risk factors, and cardiovascular comorbidity, but Finland is a more homogeneous society than the United States, for example.
U.S. POINTER’s interventions will be similar but not identical to FINGER’s. Instead of following the Nordic diet used in Finland, recruits will follow the MIND diet, a hybrid of the Mediterranean and the Dietary Approaches to Stop Hypertension (DASH) diets, both of which reduce risk for hypertension, diabetes, heart attack, and stroke by limiting red meat, butter and margarine, cheese, pastries, and sweets, and fried or fast foods, and by incorporating vegetables, especially leafy greens, along with nuts, berries, beans, whole grains, fish, poultry, olive oil, and wine. Scientists led by Martha Clare Morris at Rush University, Chicago, are already testing if the MIND diet can slow cognitive decline and neurodegeneration in a three-year, randomized controlled Phase 3 trial. As in FINGER, participants in U.S. POINTER will receive regular medical checks as well as advice and interventions to manage hypertension, metabolic problems, and weight. The control group will attend group meetings on health and aging topics, and receive annual feedback on laboratory tests.
U.S. POINTER’s cognitive training and physical exercise program, which will include mostly aerobic workouts four times a week, will be very similar to FINGER’s but will include more group sessions. Nicola Coley from Sandrine Andrieu’s lab at the University of Toulouse in France found that participants in FINGER struggled to adhere to unsupervised tasks. Less than a quarter carried out at least 66 percent of their at-home cognitive training, for example. Baker expects follow-through to be better in a group setting. Also, social isolation is common among elderly people, she noted, so these group activities are likely to improve participants’ mental health. Overall, U.S. POINTER will include more contact and communication with patients than FINGER. In fact, each participant will be assigned a “navigator” to help coordinate with exercise specialists, nutritionists, and health educators.
Baker emphasized the importance of meeting the psychological needs of the participants. U.S. POINTER will employ cognitive behavioral psychologists, who will try to facilitate participants’ transition to a healthier lifestyle by helping them realize the benefits for themselves and their families. POINTER will ramp up exercise programs slowly to give participants time to adjust, and is creating phone health apps to provide feedback on performance.
To make interventions accessible, standardized, and sustainable after the trial, U.S. POINTER will partner with national community-based organizations, including YMCAs and Alzheimer’s Association local offices. YMCAs have a nationwide network of gyms, and some of these are being used in the EXERT study run by the Alzheimer’s Disease Cooperative Study. This Phase 3 randomized, controlled trial tests whether aerobic exercise can slow cognitive decline in adults aged 65–89 who have memory complaints or mild MCI and who do not exercise regularly. Baker co-leads the study.
As in FINGER, POINTER’s primary outcome measure will be a composite score from the standard Neuropsychological Test Battery (NTB). The researchers also intend to track secondary outcomes, but it is unclear if it will track AD biomarkers, which could shed light on how these interventions work. FINGER tracked no biomarkers and no lifestyle intervention study has been shown to have a clear effect on AD-specific markers. As such, these studies cannot distinguish interventions that work for AD and non-AD causes of dementia.
The architecture of U.S. POINTER’s large database facilitates data sharing, said Baker, and complies with Kivipelto’s ongoing efforts to develop a platform for joint analysis of multidomain data from thousands of patients.
In Asia, Christopher Chen of the National University of Singapore and Edward Koo from the University of California, San Diego, co-lead the Singapore Intervention Study to Prevent Cognitive Impairment and Disability (SINGER). Chen, who presented SINGER at AAIC, noted that they will first develop pilot studies to test FINGER interventions modified to suit Singaporeans. For example, they may adapt computer-based cognitive tasks back to paper and pen because many older Singaporeans resist using electronics, and they are designing a diet matched to Asian tastes.
In Australia, Henry Brodaty of the University of New South Wales in Sydney will coordinate the Maintain Your Brain (MYB) study, a four-year randomized controlled trial that will give lifestyle advice via the internet to people at risk of dementia. Although not part of the Worldwide FINGERs consortium, this trial is modeled on the FINGER study, and Kivipelto is associate investigator. Brodaty hopes this trial will be cheaper and more easily deployed on a large scale than FINGER. MYB has started recruiting 16,000 participants aged 55–75 from the 45 and Up Study, an ongoing survey of roughly a quarter-million Australians who were 45 or older when recruited between 2006 and 2009 from lists held by Australian Medicare, a publicly funded health care system.
Participants must have a home computer with internet access, and at least two of these dementia risk factors: type 2 diabetes, hypercholesterolemia, hypertension, depression, obesity, or low levels of physical or cognitive activity. In the first year, they will receive, two to four times a week, interactive programs tailored to their risk factors, including physical and mental exercise programs, diet plans, and guidelines for managing depression, stress, and sleep problems. They will get advice on managing high blood pressure, hyperlipidemia, alcohol consumption, and smoking. After the first year, monthly booster programs will be sent out. The control group will receive less interactive, non-individualized information about exercise, diet, and depression, as well as National Geographic videos and questionnaires on health.
Researchers at AAIC were excited about the new trials, but cautioned that questions remain about multidomain interventions, most notably whether their effects will last, and how to determine which of the multiple interventions is responsible for any positive outcome. Kivipelto hopes a seven-year extended follow-up of FINGER will answer the first question. The follow-up will also test whether booster interventions on cognition, dementia/AD incidence, and secondary outcomes help, she said. As for parsing cause and effect, Baker acknowledged this limitation, but defended the appeal of fast-tracking a package of preventive strategies that has the potential of being more effective than single interventions on their own.—Marina Chicurel
Lancet Commission Claims a Third of Dementia Cases Are Preventable
Spurred by the 2013 G8 Dementia Summit in London and by the First WHO Ministerial Conference on Global Action Against Dementia in March 2015, The Lancet commissioned an expert project to review available evidence and recommend how best to manage and prevent dementia. The group’s report, “Dementia Prevention, Intervention, and Care,” was released to coincide with a symposium on the topic at this year’s Alzheimer’ Association International Conference, held in London July 16-20. The 65-page document, authored by 24 leading dementia researchers from Europe, North America, and Australia, delivers 10 key messages (see box below). Most address patient treatment and care. The one that captured the most attention, especially in the general media, was to “be ambitious about prevention,” since it concluded that a third of dementia cases might be delayed or prevented.
As outlined by Gill Livingston, a psychiatrist at University College London who led the effort, the commission arrived at that number by estimating the population-attributable fraction for a variety of risk factors, even those at play from an early age. “This is the very first life-course analysis of risk factors for dementia,” Livingston stressed in an AAIC press briefing. Livingston captured that life-course in a figure that many at AAIC, including commission member Eric Larson, Kaiser Foundation Health Plan of Washington, found compelling (see figure below). “Without sounding hyperbolic, I think this is just brilliant,” Larson told Alzforum. “It shows that the [dementia] condition has its roots throughout the life, which is true for other chronic conditions, such as vascular disease and even cancer,” he said.
Taken from the literature, poor childhood education; midlife hearing loss, hypertension, and obesity; and smoking, depression, physical inactivity, social isolation, and diabetes in late life emerged as modifiable risk factors for dementia. Added together, they accounted for 35 percent of cases, the authors concluded. That compares to only 7 percent of cases attributable to ApoE, the strongest genetic risk factor for late-onset AD. The report stresses treating midlife hypertension as an immediately actionable priority for physicians and patients, and claims that controlling the other risk factors would help reduce the number of dementia cases.
Life Course of Dementia Risk.
Starting from childhood, modifiable risk factors add up to 35 percent of dementia cases, according to the report. [Courtesy of Livingston et al., The Lancet 2017.]
In an accompanying comment in The Lancet, Martin Prince, King’s College London, calls the report “a timely evidence-driven contribution to global efforts to improve the lives of people with dementia and their carers, and limit the future impact on societies.” Lancet editors Helen Frankish and Richard Horton called on governments to use the report to help them update action plans for dementia care. Researchers at AAIC welcomed the report but asked what practical effect it will have without a public information campaign to go along with it. Livingston didn’t dismiss such an effort, but noted that evidence is still insufficient to recommend specific interventions to reduce most of the risk.
For her part, Deborah Barnes, University of California, San Francisco, questioned whether the evidence was strong enough to draw causal connections between the risks and dementia. Livingston said that the evidence for causality was stronger for some factors, such as hearing loss, than others. Experts on the commission conceded that randomized, placebo-controlled, clinical trials remain the gold standard to prove causality, but for some risk factors, RCTs would be unethical or impractical, e.g., to prove childhood education influences risk for dementia. In the absence of clinical trial data, the commission relied on epidemiological criteria for causality established by Bradford Hill (Hill, 1965). “The standard of evidence is as high as possible for something like childhood education wherein we can’t do randomized controlled trials,” committee member Lon Schneider, University of Southern California, Los Angeles, told Alzforum. He added that treating midlife hypertension in a RCT with cognitive impairment as an end point might be as impractical as doing RCTs of cigarette smoking with lung cancer end points. The report also acknowledged that the incidence of dementia in the developed world has fallen in the last few decades, a trend that researchers have attributed to modifiable risk factors identified in the report, such as healthier lifestyles and reduction in cardiovascular disease (Nov 2016 news; Feb 2016 news).
Beyond prevention, the commission devoted much of the report to recommendations for treatment and care of people who have dementia, and for their caregivers. Led by Livingston, Schneider, and Andrew Sommerlad at UCL, commission members constructed flow charts detailing approaches for managing psychosis, agitation, and depression that clinicians and caregivers may find useful. They offer guidance on dealing with sleep disorders and apathy, as well. In emphasizing the need to protect people with dementia from abuse, the report explains how it likely occurs and outlines approaches to counter it. The report recognizes the difficulties of managing people with end-stage dementia and recommends approaches for palliative care. It also discusses the value of technological advances for care management.
The Lancet report follows on the heels of a similar review from the National Academy of Science Engineering and Medicine that stopped short of issuing guidelines for the general public (Jun 2017 news). Are the two reports at odds? Experts don’t see it that way. The NASEM report also identified physical exercise, cognitive training, and blood pressure management as potential interventions to prevent dementia; however, according to Larson that committee was tasked to review the available evidence, mostly from RCTs, and make recommendations that would satisfy the U.S. Preventive Services Task Force. “As such, the standards of evidence the NASEM committee considered had to cross a much higher bar,” he said. Larson served on both the NASEM committee and the Lancet Commission. Ron Petersen, Mayo Clinic, Rochester, Minnesota, agreed. “The NASEM recommendations are consistent with what the Lancet commission says,” he told Alzforum. He also served on the NASEM committee.
Larson, Petersen, and Schneider all emphasized that the Lancet commission report was much broader in scope, tackling everything from prevention to care to public policy. “The NASEM committee just looked at intervention,” noted Petersen, “and while the level of data, and how you interpret it, may be different [between the two reports], the overall message is similar.”—Tom Fagan
CSF and Brain Markers Highlight Different Facets of Dementia
Part 1 of a two-part story. Click here for Part 2.
While biomarkers have opened a window onto Alzheimer’s pathogenesis, the view has remained cloudy. At the Alzheimer’s Association International Conference 2017 held July 16-20 in London, the number of biomarker talks was noteworthy, with nearly one-quarter of all sessions devoted to this topic. So inescapable was the subject that John Hardy of University College London quipped, “This is the biomarkers meeting.” Researchers discussed new data that clarify how various biomarkers relate to each other, and what they say about the underlying disease. Speakers solidified a recent trend showing that cerebrospinal fluid proteins and PET scans, while correlated, provide different types of information, with the former best suited for diagnosis, and the latter better for tracking progression. Several presentations reinforced the idea that tau pathology, rather than amyloid plaques or atrophy, drives cognitive decline, while others discussed how the presence of some of these pathologies can be used to predict others (see Part 2 of this story). Overall, researchers are painting an increasingly panoramic picture of what biomarker data means. Keep an eye on Alzforum for upcoming stories on longitudinal biomarkers and neuroimaging correlates of cognitive reserve.
CSF vs PET. CSF Aβ42, phospho-tau, and total tau correlate with tau PET signals in a mixed AD and non-AD population (top row), but not within the AD group alone (bottom row). [Image courtesy of Renaud La Joie.]
Markers Tell Different Stories
Many clinical studies use either CSF or PET measures of amyloid as inclusion criteria, on the assumption that they are equivalent. In London, researchers asked if they truly are. The answer was a clear no. Susan Landau of the University of California, Berkeley, noted that PET signals from amyloid and tau tracers measure the accumulation of fibrils in the brain, whereas CSF markers of Aβ and tau reflect the balance between production and clearance of soluble forms. Nonetheless, both modalities can indicate disease, and abnormal CSF and PET values typically correlate with each other across populations.
In a recent study from Landau’s group, diagnoses based on predetermined CSF Aβ and amyloid PET cutoff values agreed in 82 percent of participants. Landau noted that other studies have found even higher concordance, in the range of 85–90 percent. In her study, CSF values were determined using new automated assays that are more precise than previous ELISA-based methods (Aug 2015 conference news; Apr 2017 conference news). Still, some of the discordance may be due to remaining technical issues with assays, Landau told Alzforum. There may also be floor effects that occur with CSF Aβ42 because these values drop so low that they are difficult to measure. However, much of the discordance likely reflects real differences between how CSF and PET measures behave, she said.
Notably, among patients who tested positive on both measures, the researchers found no correlation between absolute CSF Aβ levels and PET binding values. Partly, this occurs because PET and CSF markers follow distinct trajectories throughout the course of disease, Landau said (Toledo et al., 2015). CSF Aβ drops early in preclinical AD and then stays low, whereas the amyloid PET signal rises steadily for a longer period of time before falling later, in the symptomatic phase of the disease, forming an inverted U curve. This means the two measures will not change precisely in tandem.
Supporting this, longitudinal changes in CSF Aβ and amyloid PET were correlated at four years of follow-up, but not at two. “That is not encouraging in terms of their potential use as secondary endpoints in a clinical trial,” Landau wrote to Alzforum. Trials often span just one or two years. An underlying problem is that researchers do not yet understand how longitudinal changes in CSF Aβ versus amyloid PET relate to pathology, which can only be directly measured at autopsy, Landau added.
What bearing do these differences between CSF and PET have on diagnosis and prognosis? Clifford Jack of the Mayo Clinic in Rochester, Minnesota, summed up the issue as a matter of state versus stage. CSF analytes reflect a pathophysiological state, he said, whereas imaging stages the disease. Thus, either measure can work for diagnosis, but PET scans will perform better in measuring progression and trial outcomes, Jack said.
Other talks supported this conclusion and extended the concept to tau imaging. Renaud La Joie of the University of California, San Francisco, compared 24 amyloid-positive AD patients to 29 amyloid-negative people diagnosed with other neurodegenerative diseases, such as frontotemporal dementia or progressive supranuclear palsy. Across the whole cohort, the tau PET signal correlated positively with CSF tau and negatively with CSF Aβ. This shows either biomarker modality works for diagnosis, La Joie said. Intriguingly, he saw the strongest correlation between CSF phospho-tau and tau PET, particularly in the cingulate, precuneus, and lateral parietal regions (see image above). Previous work suggests that CSF p-tau reflects a process specific to AD, whereas total tau indicates general neurodegeneration. If so, that might explain why p-tau and tau PET correspond closely. The findings also support the proposed A/T/N classification scheme, where CSF p-tau and tau PET are grouped together as markers of tau pathology, while CSF t-tau is considered a neurodegeneration marker (Aug 2016 conference news; Chhatwal et al., 2016).
Despite the overall correlation between tau PET and CSF markers in the whole UCSF cohort, the tau PET signal correlated poorly with CSF Aβ or even CSF tau within the AD group. In other words, although everyone in the AD group had low CSF Aβ, there was no relationship between how low Aβ was and how high tau PET was. Likewise, there was almost no relationship between how high CSF tau was and how high tau PET was in AD patients (see image). The finding emphasizes that CSF and PET measures are not interchangeable for scoring severity of AD, La Joie said.
Notably, the strength of the tau PET signal correlated with symptom severity better than CSF tau did. Many studies have tied tau tangles to brain atrophy and cognitive decline (May 2016 news; Aug 2016 conference news; Mar 2017 news). In years past, researchers believed that CSF tau would continue to rise in AD and would therefore constitute a progression marker, but La Joie’s new research strengthens a more recent realization that CSF tau does not actually track progression through the symptomatic stage of disease. Researchers are hoping that tau PET will provide a more useful progression marker.
“In the next few years, it will be crucial to analyze longitudinal multimodal imaging data to help us disentangle the respective dynamics of each modality,” La Joie wrote to Alzforum.—Madolyn Bowman Rogers
All Signs Point to Tau Tangles as the Culprit in Fading Memory
Part 2 of a two-part story. Click here for Part 1.
The two hallmark pathologies of Alzheimer’s disease, amyloid plaques and tau tangles, play different roles in disease progression. Accumulating research suggests that amyloid plaques kick off the disease, precipitating the spread of tau, which then does the actual dirty work of harming neurons. At the Alzheimer’s Association International Conference 2017 held July 16–20 in London, researchers bolstered this theory with new data tying tangles to most types of cognitive decline. However, one talk bucked this trend by proposing a specific role for amyloid in semantic memory. In addition, researchers said that because tau, cortical atrophy, and amyloid all mingle together in Alzheimer’s disease, the presence of one biomarker can be used to predict the simultaneous presence of others. Some groups fashioned a preclinical AD signature from plaque-induced cortical thinning.
Tau and Cognitive Deficits. When AD patients (top row) voice concerns about their own cognitive abilities, they accumulate tau in frontal areas of the brain. When family members notice a memory change, tangles appear more posteriorly (bottom row). [Courtesy of Shannon Risacher, AAIC2017.]
Tau Drives Decline
Many talks at AAIC emphasized the connection between tangles and clinical decline. For example, Andrew Aschenbrenner of Washington University in St. Louis noted that markers of amyloid, tau tangles, and brain atrophy all predict future cognitive decline, though this does not prove that any of these pathologies cause decline. To pinpoint causation, Aschenbrenner and colleagues analyzed 104 older adults with an average age of 70, who ranged from cognitively healthy to mildly symptomatic. Participants were drawn from various aging studies at WashU, including the Adult Children and the Healthy Aging and Senile Dementia studies, who had undergone an average of five cognitive assessments over the course of six years. During this period, they underwent one florbetapir amyloid scan, one AV1451 tau scan, and one structural MRI to measure hippocampal volume.
As expected, abnormal values on any of these three imaging modalities correlated with later cognitive decline. However, when the researchers performed regression analyses to look for causal factors, they found that the tau signal alone accounted for all the cognitive decline in this cohort. Parsing tau’s effect on different cognitive domains in a cross-sectional analysis, the scientists found that tau as measured by PET most strongly affected episodic memory, then processing speed, executive function, and lastly semantic memory, Aschenbrenner said. A longitudinal analysis suggested a slightly different sequence of effects; over time, tau pathology appeared to hit processing speed the most, followed by executive function, episodic memory, and lastly semantic memory.
Taking this a step further, Shannon Risacher of the Indiana Alzheimer Disease Center in Indianapolis reported that tangles in different regions may exert distinct effects on cognition. She analyzed data from 36 amyloid-positive older adults in ADNI-2. Fifteen appeared cognitively healthy, four had memory concerns, and 17 mild cognitive impairment. All underwent an AV1451 tau scan. To assess cognitive function, the researchers analyzed the memory subscale from the Measurement of Everyday Cognition (ECog) test, which participants and informants separately completed (Farias et al., 2008).
The researchers saw a surprising pattern. When participants themselves reported concerns about their cognition, they tended to have tau tangles in the frontal cortex, but not in posterior regions. When informants reported concerns but the participant appeared to have no insight, participants harbored tangles in the precuneus and parietal cortex but not in frontal regions.
Although the reason for the difference is unclear, Risacher speculated that people are most likely to notice when they have problems with executive function, which taxes the frontal cortex. On the other hand, informants might be more likely to notice memory problems caused by posterior tangles, she said. Other studies have reported that informant and self-concerns act as independent, additive risk factors for Alzheimer’s progression, reinforcing the idea that these two measures pick up different types of pathology.
In Risacher’s study, all participants had plaques in the medial temporal lobe (MTL). In another study, Anne Maass, working with Bill Jagust at the University of California, Berkeley, focused on what tau pathology in this region does to cognition. The MTL accumulates tangles early during normal aging; in fact, two-thirds of people over 60 have tangles in their MTL. While these tangles associate with poor episodic memory, it is unclear how that happens. Maass analyzed data from 83 cognitively normal people at an average age of 77 who participated in the Berkeley Aging Cohort Study. They underwent amyloid PiB and tau AV1451 PET, structural MRI, and took tests of episodic memory. Fifty-seven of the participants had at least one additional MRI and subsequent memory tests one to two years later. Tau scans were mostly acquired after the other data was collected.
In this cohort, tau tracer uptake in the MTL was the best predictor of baseline memory performance, accounting for 20 percent of the variance in these scores, Maass said. Notably, no other measure explained memory variance. Within the MTL, tangles in the entorhinal cortex and parahippocampal cortex most associated with poor baseline memory. Forty percent of the participants were amyloid-positive, but amyloid had no relationship to memory in this group, Maass noted. Moreover, MTL tau measures associated with memory decline and entorhinal atrophy over time. The data suggest that tau tangles underlie faltering memory even in normal aging, Maass concluded (Maass et al., 2017).
Then What’s Amyloid Doing?
Does amyloid play any role in the progression of symptoms? Several imaging studies have suggested that cortical amyloid seems to unleash tau, allowing it to spread from the medial temporal lobe into surrounding regions and kicking off neurodegeneration (Jul 2016 news; Aug 2016 news; Jun 2017 news). At AAIC, Keith Johnson of Massachusetts General Hospital, Boston, underscored this finding. He noted that in multiple cohorts of healthy elderly people, including those in the Harvard Aging Brain Study, the Mayo Clinic Study of Aging, and the National Alzheimer’s Coordinating Center database, neocortical tau appears to interact with amyloid plaques to bring on hypometabolism and bring down memory. In particular, temporal lobe tau associates with waning metabolism in the posterior cingulate, but only when cortical amyloid is also there, Johnson said.
Beyond amyloid’s role in precipitating tau pathology, does it do anything to harm memory by itself? Perhaps, according to Kate Papp at Brigham and Women’s Hospital, Boston. Papp previously found that semantic memory, which stores factual knowledge, declines in cognitively normal older adults who have amyloid plaques (Papp et al., 2015). This was notable because unlike episodic memory, which wanes with age, semantic memory tends to increase with healthy aging. It was unclear what type of pathology caused this form of memory to drop in preclinical AD. To investigate, Papp compared episodic and semantic memory in 74 cognitively healthy participants in the Harvard Aging Brain Study whose average age was 78. All had PiB and AV1451 PET scans.
Papp found that the extent of amyloid plaques, but not of tau tangles, correlated with worse semantic memory. Something about how semantic memory is stored could render it more susceptible to amyloid, Papp speculated. Some researchers believe that semantic memory is processed in a distributed neocortical network, which could leave it vulnerable to the effects of widespread neocortical Aβ, she suggested. Next she plans to examine longitudinal and brain network data to see if the plaque-semantic memory effect holds up, and what might explain it.
Because of the close relationship between plaques, tangles, and cortical neurodegeneration, the latter can be used as a surrogate for the former, other speakers noted. Aylin Dincer, working with Tammie Benzinger at WashU, analyzed structural MRI data from participants with and without amyloid plaques in the Dominantly Inherited Alzheimer Network (DIAN) to derive a specific signature of AD-related cortical thinning. She found thinning in amyloid-positive people, who were also the mutation carriers, primarily in the posterior regions of the brain, such as the precuneus and lateral parietal lobes. Because DIAN participants are young, this signature was not confounded by age-related brain volume loss. When Dincer looked for this AD signature in a cohort of cognitively normal elderly seen at the Knight ADRC at WashU, it only turned up in those with brain amyloid and/or cognitive impairment. In fact, this thinning signature better predicted amyloid status than did hippocampal volume, Dincer said, noting that hippocampal shrinkage is not specific to AD.
Paula Petrone of Barcelonaβeta Brain Research Center, Spain, described a complementary approach. She used structural MRI and diffusion tensor imaging, which measures axonal integrity, to derive an AD signature from 96 ADNI participants and 87 elderly patients seen at the Hospital Clinic of Barcelona. Both groups included cognitively healthy controls as well as people with preclinical, prodromal, and full-blown AD. Using machine-learning techniques, the researchers found a set of 45 structural features, 43 from DTI scans and two from MRI, that best predicted whether people had amyloid plaques. This AD signature was 84 percent accurate, suggesting it could serve as a prescreening tool in place of expensive PET scans, Petrone suggested. She calculated DTI could save about 75 percent of screening costs over PET for clinical trials. Petrone noted, however, that the signature performs best in younger cohorts with fewer age-related changes. In future work, she plans to include cognitive and genetic biomarkers to try to improve its performance. —Madolyn Bowman Rogers
New Ties between AD and the Stages, Waves, and Molecules of Sleep
At the Alzheimer’s Association International Conference 2017 held in London July 16-20, the relationship between disturbed sleep and AD came into sharper focus. Scientists revealed how sleep-disordered breathing boosts the risk for cognitive impairment. They also implicated disturbances in specific phases of sleep as risk factors for Aβ pathology and dementia. They pinpointed regions in the brain most affected by this sleep/neuropathology axis and shed light on molecular mechanisms that likely support it. Treating sleep disorders more widely may substantially reduce cognitive decline, and even AD, said scientists at the meeting.
People with sleep-disordered breathing (SDB) experience episodes of abnormally slow or shallow breathing (hypopnea) during sleep, or brief periods of no breathing at all (apnea). Several studies have reported a link between SDB and cognitive impairment in the elderly, but the results have been inconsistent. To better understand this relationship, Yue Leng, working in Kristine Yaffe’slab at the University of California in San Francisco, sifted through a massive dataset. She pooled results from eight cross-sectional and six longitudinal studies, which together had medical data, including records of sleep patterns and cognition, on more than 4 million people. She selected those studies because each included at least 200 individuals who were older than 40, had used a standard apnea-hypoapnea index or a clinical diagnosis to ascertain SDB, and had assessed cognitive function using standard tests. To account for the different cognitive tests, SDB diagnostic methods, and populations recruited in each investigation, Leng and colleagues used statistical modeling to normalize the data.
Timing is Everything
Driven by sleep (black) and arousal (blue) circadian rhythms, production and clearance of Aβ in the parenchyma can complicate interpretation of its levels in the CSF (red). See below. [Image courtesy Cedernaes et al., 2017]
Analyses of the eight cross-sectional studies revealed that people with SDB had normal global cognitive function and memory, but slightly worse executive function. Might this increase risk for dementia? Data from the six longitudinal surveys suggested yes. From the records of more than 200,000 people, the researchers found that over three-15 years of follow-up, the roughly 11,000 people with SDB were 26 percent more likely to develop mild cognitive impairment (MCI) or dementia than were those who slept soundly. Given well-established interventions to treat SDB, such as continuous positive airway pressure devices and dental appliances, Leng and Yaffe emphasized the importance of improving SDB diagnosis and setting up studies to determine if these treatments can delay cognitive impairment. “It’s quite convincing that there’s a causal link. SDB could be a really important modifiable risk factor,” said Yaffe.
Disordered breathing conditions are quite common, affecting 13 percent of men and 6 percent of women aged 30-70 (Peppard et al., 2013). Obstructive sleep apnea (OSA), caused by partial or complete closing of the upper airway, strikes 9-24 percent of men and 4-9 percent of women aged 30-60. Obesity nearly doubles the risk (Romero-Corral et al., 2010). Also, the prevalence rises sharply with age, increasing two- to threefold in people over 65 (e.g., Young et al., 2002).
To investigate how OSA might increase risk for dementia, researchers led by Omonigho Bubu at Wheaton College in Illinois tested if it altered the rate of change of Aβ42, tau, and phosphorylated tau (p-tau) in the CSF, as well as Aβ burden in the brain, as assessed by positron emission tomography (PET) using the amyloid marker florbetapir. They obtained the data from the Alzheimer’s Disease Neuroimaging Initiative study (ADNI) database and included 1,639 individuals with an average age of 72-75 years who had reported as being either OSA-positive or OSA-negative. Of these, 516 were cognitively healthy, 798 had mild cognitive impairment (MCI), and 325 had been diagnosed with AD.
The researchers found that over approximately 2.5 years, Aβ built up more rapidly in the brains of OSA-positive individuals than in OSA-negatives, while Aβ42 levels dropped and tau and p-tau levels rose in the CSF. Interestingly, these associations emerged in the cognitively healthy and MCI groups, but not the AD group. “OSA might act as a modulator, accelerating amyloid deposition in those at risk for AD,” said Bubu. He cautioned that the sample size was small and that ceiling and floor effects may have limited biomarker changes in the AD group, since they already have substantial biomarker signals.
Other groups drilled down into the what, where, and how of sleep’s effects on dementia. In London, David Holtzman of Washington University in St. Louis showed how disrupting a particular stage of sleep called slow-wave sleep (SWS) promotes the buildup of Aβ in the CSF in healthy volunteers. During SWS, also known as deep sleep because it is harder to awaken from, the brain spends over 20 percent of its time generating slow (0.5 to 4.0 Hz), synchronized oscillations that can be recorded by electroencephalography (EEG). This slow-wave activity (SWA) contrasts with that during wakefulness and during rapid eye movement (REM) sleep, when electrical oscillations are mostly fast and desynchronized.
Holtzman and colleagues previously reported that in mice, neuronal activity, which drops during SWS, caused the release of soluble Aβ into the interstitial space. Others have found, in people, that clearance of Aβ from the brain accelerates during SWS (Oct 2013 news). In keeping with this, Holtzman and others found that in cross-sectional studies of healthy middle-aged and older people, stronger SWA correlated with lower CSF Aβ40 (Ju et al., 2016; Varga et al., 2016).
To more definitively prove that SWA modulates Aβ, Yo-El Ju at WashU and Sharon Ooms at Radboud University in Nijmegen, Netherlands, monitored CSF Aβ levels in 17 healthy volunteers, aged 35-65, who were prevented from slipping into SWS. Right before bed, the researchers hooked up the volunteers to an EEG recorder to capture snapshots of their brain waves every 10 seconds, and fitted them with earphones. As soon as the tell-tale slow waves of SWS surfaced in the EEG, some participants heard a tone through the earphones that increased in volume until their slow waves subsided, indicating they were no longer in SWS. Others were allowed to sleep at will. The following morning between 9:30 and 10 am, the scientists collected CSF from the volunteers, and measured Aβ40 levels. As expected, for the most part, the volunteers who missed SWS the night before had higher levels of CSF Aβ than did controls. Also, the weaker the SWA, the greater the increases in CSF Aβ40. Why did CSF Aβ increase when its clearance from the parenchyma would be expected to decrease if sleep is disrupted? This may be because they measured close to the nadir of the daily CSF Aβ rhythm (9:30-10 a.m.). If SWS disturbance increased neural activity and delayed clearance, the nadir would come at a later time, and CSF Aβ at 9:30-10 a.m. be higher than in control groups (see figure above).
Interestingly, levels of no other protein the scientists measured in the CSF, including tau, changed in these overnight experiments, suggesting that Aβ production and clearance mechanisms are specifically related to sleep. However, when sleep disruptions occurred over a longer time, CSF tau rose as well. Ju and Ooms found that volunteers who slept poorly at home, as indicated by actigraphy wristbands that recorded how much they tossed and turned during six nights prior to CSF collection, had higher CSF tau levels. The findings appear in the Jul 10 Brain online (Ju et al., 2017).
“This is an important study and very nicely done,” said Barbara Bendlin of the University of Wisconsin in Madison. “It is consistent with prior work and shows a direct relationship between SWA and Aβ.”
In an accompanying commentary in Brain, Matthew Walker and colleagues from the University of California, Berkeley, noted the study extends the mostly correlational human studies to date (Mander et al., 2017). They cautioned, however, that the sample size is small and that other factors, such as the stress induced by the tones, changes in other sleep stages, or number of arousals, could contribute to the observed changes in CSF Aβ. Looking ahead, Walker and colleagues suggested questions for future studies: Which properties of SWS are critical for its effect on Aβ? Are slow waves produced in particular brain regions more likely to contribute to changes in Aβ or, conversely, more likely to be affected by AD pathology?
At AAIC, Kate Sprecher from Bendlin’s lab, addressed the last question. She described a preliminary study to probe the relationship between AD neuropathology and deficits in SWA in specific brain regions. The investigators recruited 19 middle-aged, cognitively healthy women and one man with a parental history of AD from the Wisconsin Alzheimer’s Disease Research Center. Their mean age was 57. The scientists measured CSF Aβ42 and t-tau, and monitored sleep with standard polysomnography, measuring global brain waves, blood oxygen levels, heart rate, breathing, as well as eye and leg movements overnight. They also recorded electrical activity in specific regions of the brain with high-density EEG using 256 electrodes. To measure amyloid burden in the brain, the scientists used florbetapir PET. The PET and EEG data were collected within days or weeks of each other, and CSF measurements were separated by a longer time interval. Unlike Holtzman’s study, Bendlin focused on chronic, rather than acute, levels of CSF Aβ and tau.
Sprecher looked for correlations between the t-tau/Aβ42 ratio and the power of the EEG recordings in the slow-wave range of 1-4.5 Hz across the cortex. The data revealed a strong link between CSF t-tau/Aβ42 and SWA specifically in the centro-parietal cortex: Higher CSF t-tau/Aβ42 levels correlated with reduced SWA in this area. And when they examined brain amyloid in the same volunteers using florbetapir PET, they found that reduced SWA significantly associated with greater amyloid burden in a subregion of the parietal lobe, the precuneus, which is affected very early in the course of AD pathology.
Might localized disruptions in SWA precede early AD neuropathology? Sprecher and Bendlin think so, but emphasize their study is preliminary. “We are collecting more EEG and PET data to extend these studies,” said Bendlin. Interestingly, obstructive sleep apnea also seems to correlate with disrupted SWA in posterior areas of the brain, including the precuneus, noted Sprecher (Jones et al., 2014). This could be a factor contributing to the observed links between OSA and dementia. An earlier study showed a correlation between amyloid deposition in the medial prefrontal cortex and fewer slow waves generated in this area during sleep in 75-year-old healthy controls (Jun 2015 news). Bendlin told Alzforum that she does not know how this relates to her findings, noting the work is preliminary and that the two studies had important differences, including the participants’ ages.
Making Brain Waves
Sleep spindles and K-complexes occur during a light sleep stage known as non-REM stage 2. Sleep spindles dwindle when tau rises in cerebrospinal fluid.
Others at AAIC claimed slow waves are not the only components of sleep linked to dementia. Ram Sharma, working with Ricardo Osorio at New York University School of Medicine, associated sleep spindles with biomarkers of tau pathology. These bursts of 10-16 Hz oscillations (see image above) appear mostly during non-REM stage 2 sleep, a stage of light sleep that accounts for about half of our sleeping time. Sharma suspected a connection between spindles and dementia because p-tau accumulates in the thalamus, where sleep spindles are generated. Others have reported fewer spindles per minute in AD and MCI (Gorgoni et al., 2016).
Sharma tracked sleep spindles by EEG and measured t-tau and p-tau in the CSF from 50 cognitively healthy adults aged 53 to 84. He found that shorter spindle duration ,as well as less dense spindles, correlated with higher levels of tau and even more tightly with p-tau. No correlations were found with Aβ42. Neither the spindles nor tau correlated with sleep quality, suggesting a poor night’s sleep cannot explain the correlation. Sprecher was intrigued by the findings. “The involvement of sleep spindles is really interesting,” she told Alzforum. “This is a very important wave form that plays a role in memory and cognition, but it has not been explored much in AD.” Holtzman’s group also found a relationship between tau and sleep in the P310S transgenic mouse, which expresses a mutant form of human tau. Jerrah Holth and colleagues found that decreased REM and non-REM sleep in these animals associated with an increase in tau pathology in the brainstem (Holth et al., 2017).
Additional evidence supporting a link between REM and dementia came from the studies of Matthew Pase of Swinburne University, Australia. While at Sudha Seshadri’s lab at Boston University, Pase found a link between this dream state and dementia in a subset of participants in the Framingham Heart Study Offspring cohort. The Offspring cohort is composed of more than 5,000 sons, daughters, and their spouses, of the original participants in the original Framingham Heart Study. Most of them were in their 20s-50s between 1971 and 1975, when the Offspring study started. The researchers pored over data from 321 cognitively healthy individuals who had gone through sleep assessments at a mean age of 67 using home-based polysomnography between 1995 and 1998. Pase then examined the incidence of dementia after a mean follow-up of 12 years. By then, 32 of the 321 had developed dementia, 24 due to AD. Pase was surprised to find that individuals who spent more time in REM during the night, and fell into REM sleep more quickly, had a reduced risk of all forms of dementia, including AD. Each percentage increase in REM sleep was associated with a 9 percent reduction in dementia risk. Interestingly, the team found no association between non-REM sleep and dementia risk. Although some studies have hinted at a link between REM and AD (e.g., Liguori et al., 2014), said Pase, most current observations point to a connection between SWS and dementia pathology. Pase offered a few hypotheses to explain his findings: a drop in REM may be a marker of a degenerating cholinergic system; REM might be curtailed by stress, which may also increase the risk of dementia; or a drop in REM could facilitate the development of dementia by reducing synaptic consolidation.
Molecular mechanisms to explain these multiple sleep-dementia connections are still lacking. However, researchers, including John Cirrito at WashU, are beginning to tackle the issue. Cirrito studies the signaling pathways that underlie the fluctuations in Aβ secretion that occur during the sleep-wake cycle. Previous studies had shown that more Aβ releases into the interstitial fluid (ISF) during wakefulness than during sleep. To delve into the molecular signaling that controls these fluctuations, Cirrito uses in vivo microdialysis to monitor Aβ levels in the ISF of living APP transgenic mice, while manipulating signaling cascades pharmacologically. At AAIC, he reported that inhibiting extracellular regulated kinase (ERK) increased ISF Aβ levels by 50 percent and blocked the sleep/wake fluctuation in ISF Aβ, suggesting ERK plays a role in the diurnal rhythm of Aβ production. Cirrito’s group found that orexin signaling, which normally promotes wakefulness, regulates the phosphorylation/activation of ERK, which in turn contributes to the diurnal fluctuations in ISF Aβ during the sleep-wake cycle. “It’s an interesting finding. It shows that ERK signaling is important in Aβ production and connects it with sleep,” said Holtzman.—Marina Chicurel
Data from DIAN Revise Familiar Biomarker Trajectories
Part 1 of a two-part story.
As researchers analyze data in longitudinal biomarker studies, they find that some previously estimated biomarker trajectories do not hold up. At the Alzheimer’s Association International Conference 2017, held July 16–20 in London, one tenet to tumble was the idea that biomarkers change along the neatly stacked sigmoidal curves drawn in progression models of AD that have become so familiar to the field. In reality, in-person serial data now suggest that biomarkers change in a range of different patterns, and vary by brain region and disease stage. Rates of change accelerate and decelerate at different points in time; some markers, for example cerebrospinal fluid p-tau181, even reverse course from rising to falling as disease worsens.
These revelations not only add nuance to disease diagnosis, progression, and prognosis, they also complicate interpretation of biomarker outcome measures in clinical trials, speakers said. “In order to plan interventions for a chronic disease, researchers need good estimates for when a biomarker starts changing and how it behaves,” noted Eric McDade of Washington University in St. Louis.
Much of the longitudinal data presented at AAIC came from the Dominantly Inherited Alzheimer Network (DIAN), which enrolls adults from families with autosomal-dominant AD. Because they are younger than people with sporadic AD, early pathological signs in their brains are not confounded by age-related changes, allowing researchers a glimpse into a pure AD process. In this population, some changes, such as hippocampal atrophy and tau tangles, occurred later than researchers had expected based on prior cross-sectional and sporadic AD data. The findings discussed at AAIC are preliminary as, in some cases, they are based on but two or three time points. More data will refine estimates of biomarker change, researchers say.
This story sums up several DIAN presentations. Part 2 of this story covers other AAIC talks presenting longitudinal data from sporadic AD that added insight into age-related changes in ApoE4 carriers, and from people who maintain excellent memory into old age.
Changing Trajectories. Rates of amyloid accumulation (left column), neuronal hypometabolism (center), and tissue atrophy (right) vary by time and brain region. Top, inferior temporal (orange), precuneus (blue), and insula (green); bottom, caudate (purple), putamen (red), and hippocampus (turquoise). [Courtesy of Brian Gordon, AAIC 2017.]
Longitudinal Data Spring Surprises
The DIAN observational study started enrolling in January 2009 (Nov 2008 news series). Longitudinal data remain limited, in part because DIAN participants who were many years away from their expected age at onset initially came in for comprehensive testing only once every three years. Moreover, many participants left the observational study to join DIAN-TU treatment trials, and others left because they became too ill to travel.
At AAIC, McDade reported findings based on data collected through July 2016. This covered 251 carriers of pathological mutations in APP, PS1, or PS2, and 160 noncarriers. The cohort’s average age was 39 and a little more than half had made two or more clinic visits, which take three to four days each and encompass the full suite of imaging and fluid biomarkers and cognitive and clinical tests. From this initial dataset, the researchers were surprised to learn that some biomarkers changed more rapidly than they had expected based on DIAN cross-sectional results.
To be sure, some biomarkers did behave as anticipated. Rates of change for amyloid PET and cerebrospinal fluid Aβ42 in mutation carriers diverged from those in noncarriers about 25 years before the expected onset of symptoms (Jul 2012 news). Whereas CSF Aβ42 levels fell most rapidly early in the disease, with the rate of change slowing as people approached symptom onset, brain amyloid deposition as per PET continued accelerating even after symptoms appeared. Other researchers at AAIC noted that CSF and PET measures followed their own trajectories in sporadic disease as well (Aug 2017 conference news).
Likewise, glucose metabolism in the precuneus started to wane at 15 years before expected symptom onset, just as researchers anticipated from the cross-sectional data. Decline was gradual until symptoms began, at which point it accelerated sharply.
That said, other biomarkers tracked differently than expected. Hippocampal atrophy and cognitive decline both appeared later than in the cross-sectional study (Bateman et al., 2012). There, hippocampi in mutation carriers appeared shrunken as early as 10 to 15 years before symptom onset, while cognition dipped five years before. Longitudinal data showed, however, that hippocampal atrophy accelerated only a year before symptoms appeared. Scores on a cognitive composite only began to worsen two years prior to symptoms, while the CDR-SOB did not budge until symptoms emerged. “The longitudinal changes [in hippocampal atrophy] correlate closely with cognition, which is what we might expect,” McDade noted. McDade emphasized that this young population has no age-related hippocampal atrophy, which might explain why shrinkage shows up only as symptoms begin. A separate analysis of DIAN data recently pegged hippocampal atrophy to symptom onset as well (Kinnunen et al., 2017).
The biggest surprise in the data was the behavior of CSF total tau and phosphorylated tau. In cross-sectional data, these markers are first elevated about 15 years before symptom onset and steadily climb as people approach and even pass diagnosis. In longitudinal data, however, total tau levels were already high in mutation carriers when they entered the study, and the researchers saw little difference in a person over time, with the rate of change close to zero. Probably the rise occurs so early in the disease that it did not show up in these data, McDade said. Phospho-tau followed a different course. Its level stayed high in mutation carriers until symptom onset, at which point it dropped dramatically. Previous longitudinal data from DIAN had hinted at a drop in CSF total tau and p-tau once symptoms emerged (Mar 2014 news). The findings emphasize that CSF total tau and p-tau measure different things, McDade said. He noted, however, that these proteins were measured using older ELISA-based assays, rather than newer automated systems that are more accurate (Aug 2015 conference news; Apr 2017 conference news). He plans to repeat the CSF analysis using high-performance systems.
Tau PET Distinct in Familial Disease
Could tau PET clarify how tau pathology evolves in familial AD? This marker was recently added to DIAN, and longitudinal data are not yet available. However, initial cross-sectional results already have surprised researchers. At AAIC, Tammie Benzinger of WashU presented tau PET data from 50 DIAN participants. Fourteen were symptomatic carriers with an average age of 50, 20 were asymptomatic carriers averaging 39 years old, and 16 were non-carriers with an average age of 38. The asymptomatic carriers were about 11 years shy of their estimated age of onset on average, while the symptomatic carriers averaged two years past it.
To Benzinger’s surprise, tau PET was negative in asymptomatic carriers up until the age of onset. This contrasts with late-onset AD, where researchers have found positive tau PET signals in amyloid-positive people who have no clinical symptoms (Mar 2016 news; Aug 2016 conference news). Moreover, DIAN mutation carriers at onset age or beyond had intense uptake of tau tracer, two to three times higher than that seen in symptomatic late-onset AD. Benzinger had reported similar preliminary findings from growing numbers of DIAN participants last year, noting that in familial disease, tau tangles seem to storm onto the scene late (Feb 2016 conference news; Aug 2016 conference news). Those hints are reproduced in this larger dataset.
In addition, the pattern of deposition in the brain varied from that seen in LOAD. In DIAN, tau deposition appears concentrated in posterior and precuneus regions, which also become hypometabolic in DIAN participants, Benzinger noted. Furthermore, the tau PET signal correlated with atrophy in these regions.
What do these findings of a late tau PET signal mean? Intriguingly, the signal shows up around the same time as CSF p-tau drops in longitudinal samples. McDade noted that existing tau tracers bind to hyperphosphorylated paired helical filaments of tau. Thus, tau tracers may be detecting the same form of tau as CSF p-tau assays. Possibly, as tau tangles spread across the brain, they absorb soluble p-tau, leading to its drop in CSF, much as amyloid plaques are believed to soak up soluble Aβ42, McDade speculated.
CSF total tau, by contrast, increases long before PET detects tangles in the brain. Total tau likely reflects neuronal injury and the lysing of cells, and bears little relationship to tangle deposition, McDade said.
Some researchers at AAIC speculated that differences in tau pathology in young and old brains may explain the late but rapid rise of the tau PET signal in young mutation carriers. Older brains accumulate some age-related tangles without developing notable cognitive decline. In the young DIAN brains, however, tau tangles seem tightly linked to clinical symptoms.
At AAIC, Keith Johnson of Massachusetts General Hospital, Boston, reported that in the Harvard Aging Brain Study, the older the person, the weaker the link between tangles and cognitive decline. HABS tracks cognitive change in older adults who start out cognitively healthy. William Jagust of the University of California, Berkeley, said he has seen a related phenomenon in ADNI data: Older people with AD take up less tau tracer than younger AD patients do. Overall, tau seems less informative about disease in older brains. The reason is unclear, although some researchers have previously suggested that tau may no longer track decline at the end stage of the disease because too many neurons have already died. “These findings may be telling us something fundamental about the biology of tau,” Benzinger agreed. However, she also noted that the AV1451 tau tracer used in DIAN binds off-target to the glial inflammatory marker monoamine oxidase. Thus, some of the intense signal in young mutation carriers could be a result of neuroinflammation, Benzinger suggested.
Changing Biomarker Trajectories
The longitudinal findings highlight a dilemma for researchers planning to use biomarkers in clinical trials: They cannot yet predict how a biomarker might change with treatment. For example, if an intervention raised CSF p-tau, would that indicate improvement or worsening of disease? It might depend on exactly where in the disease trajectory the person was when treatment started, researchers agreed. The same applies to CSF Aβ, where researchers have wondered for years what a treatment benefit would look like. They also do not know how a therapy that shifts one marker might affect others. Would a treatment that lowers brain amyloid below the threshold for PET positivity stop the spread of tau or improve cognition? If a runaway process has already taken hold in the brain, perhaps not, McDade noted. For these reasons, cognitive impairment will remain the key outcome measure for trials, he predicted.
Other data from DIAN may help researchers pick the best biomarker for a particular disease stage by showing when and where each marker changes most rapidly. Brian Gordon at WashU presented longitudinal imaging findings from 88 symptomatic carriers, 141 presymptomatic carriers, and 148 non-carriers in DIAN. Most participants had made two clinic visits. Gordon compared amyloid PET, FDG PET, and structural MRI scans for 34 cortical and seven subcortical regions. He found tremendous variety in rates of change by brain region and disease stage (see image above).
For example, amyloid plaque load climbed fastest in the precuneus up until about 10 years before symptom onset. At that point, precuneus plaque accumulation slowed down, and after symptoms appeared, began to fall. From –10 years onward, plaques grew the fastest in the inferior temporal lobe. In the hippocampus, on the other hand, virtually no plaque deposited at any point in disease, belying data from LOAD. Thus, a trial in a very early preclinical familial population might best track amyloid PET in the precuneus, while the inferior temporal lobe signal would be more telling for later disease stages. “These would be the best regions to look at to judge the potential effectiveness of an anti-amyloid drug,” Gordon wrote to Alzforum.
Intriguingly, the precuneus also showed the most dramatic changes in FDG PET signal and brain volume throughout most of the course of disease. This region seems particularly susceptible to AD pathology, Gordon noted. The hippocampus appeared to shrink steadily throughout the whole disease course, from –30 to +10 years. “Rather than considering aggregate, summary measures of pathology, clinical trials should take into account these regional patterns and look at areas of the brain that have the most optimal signal properties,” Gordon wrote.
Mutation-Specific Effects
Another complication for DIAN studies regards how particular mutations may affect the results. At AAIC, Jasmeer Chhatwal at Massachusetts General Hospital, Boston, presented new data suggesting that specific mutations cause unique patterns of amyloid deposition. Early studies had reported that plaques form first in the striatum in familial disease, although later research noted tremendous variability in DIAN (Jun 2007 news; Mar 2012 conference news).
From 129 DIAN participants, who between them had made 181 clinic visits, Chhatwal stratified amyloid PET data by mutation. He found that only those people who carried a presenilin 1 mutation in either transmembrane domain 2 or 8 developed early striatal plaque. People with presenilin 2 transmembrane domain 2 mutations, by contrast, accumulated cortical plaque first. For all other mutations, accumulation occurred at about the same time in the two regions. Chhatwal did not analyze other regions such as the precuneus. It is still unclear why mutations have these distinct effects, he noted, suggesting that animal studies may be able to parse out the mechanisms. Intriguingly, a previous study found that presenilin 2 mutations churn out more intracellular Aβ42 than presenilin 1 mutations do (Jun 2016 news).—Madolyn Bowman Rogers
Longitudinal Data Say: Nope, CSF Markers Do Not Track Progression
Part 2 of a two-part story.
As longitudinal biomarker data begin to roll in, they challenge some previous assumptions about Alzheimer’s disease progression. Researchers at the Alzheimer’s Association International Conference 2017, held July 16–20 in London, presented data from studies of familial and sporadic AD that, on the face of it, contradict previous cross-sectional findings. While progression models based on the latter propose that most biomarkers change gradually during the preclinical phase of the disease, in-person serial data from familial AD show instead that several biomarkers change abruptly around the time symptoms begin (see Part 1 of this story). Serial studies of sporadic AD painted yet a different picture. Researchers reported no difference in how quickly CSF biomarkers changed between controls and AD patients, confirming the idea that these markers do not track progression. Imaging showed more promise for this. Amyloid PET suggested that plaques appeared first in frontal and only later in posterior brain regions. FDG PET and structural MRI pinned down the earliest neurodegeneration in ApoE4 carriers to about 10 years before age at onset. MRI also indicated that so-called “superagers,” older adults who maintain the memory skills of youngsters, preserve cortical thickness. But don’t take all this to the bank. Speakers stressed that some of these findings are based on only two or three time points, and that more data may change the picture. Even so, these preliminary data may help researchers pick biomarkers to use in clinical trials.
Frontal First. Amyloid PET of cognitively healthy people finds the earliest amyloid accumulation (red lines) in frontal (left) rather than posterior regions (right). Gray lines represent those with stable/declining amyloid. [Courtesy of Michelle Farrell, AAIC 2017.]
Imaging Pins Down Early Changes
In London, several scientists focused on early imaging changes in AD. Previous neuropathology and imaging studies suggested that amyloid plaques first begin to deposit in the frontal cortex (see Braak and Braak, 1991; Villemagne et al., 2011). Specifically, the earliest accumulation may occur in the orbitofrontal cortex, claimed Michelle Farrell of the University of Texas at Dallas. She analyzed longitudinal data from 83 cognitively healthy participants in the Dallas Lifespan Brain Study. Participants ranged from 30 to 89 years old, and only 15 of them were amyloid-positive at baseline. They underwent amyloid PET and structural MRI and took cognitive tests at baseline and four years later.
Farrell examined the PET signal in frontal and posterior brain regions and distinguished people who accumulated amyloid from those who remained stable (see image above). Looking just at the accumulators, she found that those who started out as amyloid-negative had their first sign of accumulation in frontal regions, namely the lateral and medial orbitofrontal cortex and the pars orbitalis. That suggests these are the first regions to lay down plaques, and that an amyloid PET signal in the OFC might serve as a very early biomarker of AD, Farrell said. Intriguingly, accumulation in the lateral OFC predicted a decline in reasoning on follow-up testing, but no effect on memory or processing speed. This makes sense because the pars orbitalis plays a role in reasoning, Farrell noted. Others at AAIC suggested that the finding of early OFC accumulation could help explain the presence of olfactory dysfunction in the very early stages of AD (Jan 2010 news).
On the other hand, people who were already amyloid-positive at baseline accumulated further amyloid in both frontal and posterior regions, namely the precuneus, posterior cingulate, and isthmus cingulate. This group tended to be older than the frontal accumulators. Amyloid deposition in the precuneus predicted a drop in episodic memory upon follow-up, but did not affect reasoning or processing speed. This is in keeping with the role of the precuneus in memory, Farrell said. The SUVR changes in this study were small and often in the amyloid-negative range, but others at AAIC noted that accumulation can still be measured in this way. “It is quite reasonable to look for rising SUVRs even at levels below threshold,” William Jagust of the University of California at Berkeley told Alzforum.
Richard Caselli of the Mayo Clinic in Scottsdale, Arizona, reported on longitudinal changes in ApoE4 carriers. They tend to accumulate amyloid in the brain faster than noncarriers, and some studies have shown that they can have subtle metabolic or functional deficits in their 30s or even younger (Apr 2009 news; Oct 2015 news). Despite these early signs, neurodegeneration in ApoE4 carriers begins late in life, just as in noncarriers, Caselli said.
He selected 36 cognitively healthy ApoE4 carriers and 10 noncarriers from a larger cohort who attended clinics in the Phoenix area. All were over 50 and had a first-degree relative with dementia. Although the study was not specifically designed to study ApoE4 carriers, it was highly enriched for people with this risk factor, Caselli noted. To spot early brain changes, Caselli tracked changes in cognition as well as in brain volume and metabolism every two years for an average 14-year follow-up time. During this time, 12 of the carriers and three of the noncarriers progressed to MCI.
Caselli found that rates of hippocampal atrophy accelerated in ApoE4 carriers about 10 years before an MCI diagnosis. Deficits in verbal memory appeared seven years prior to diagnosis, and hypometabolism in posterior regions at six. On average, these declines started when people were in their 60s, suggesting that the deficits reported in the literature for young ApoE4 carriers may reflect differences in brain development and are not progressive.
Researchers also believe superagers can help them understand what goes on in the aging brain (Rogalski et al., 2013). Studies have reported greater cortical thickness in these lucky folks than in peers with age-related decline, but it was unclear if superagers start out with bigger brains, or simply maintain their brain volume (Oct 2016 news). Hamid Sohrabi of Edith Cowan University in Perth, Australia, investigated this by following 21 superagers and 24 typical agers older than 60 enrolled in the Australian Imaging, Biomarkers & Lifestyle (AIBL) study. Superagers were defined as having a baseline memory performances on the second edition of the California Verbal Learning Test that matched those of 30- to 44-year-olds. Participants underwent five MRI scans and took four cognitive assessments over six years.
Surprisingly, the baseline cortical thicknesses of superagers resembled those of the typical agers. Super- and typical agers also had the same degree of amyloid pathology as seen by PET.
Over time, however, the superagers maintained their cortical thickness, particularly in the median cingulate, paracingulate gyri, and superior occipital gyrus, while their peers with age-related cognitive decline lost brain volume at typical aging rates (Salat et al., 2004). Superagers kept more volume than typical agers in 25 of 34 brain regions examined. This study represents some of the first longitudinal data on volume changes in superagers, and the data need to be replicated, Sohrabi said. He speculated that genetic factors allow some people to resist the losses associated with aging. In future work, Sohrabi will look for genetic differences that might support this idea.
Nothing Much to See in CSF
Many researchers investigate the utility of CSF biomarkers for trials. CSF analysis costs less than brain imaging and allows researchers to monitor numerous biological processes. In London, Alberto Lleó of the Hospital de Sant Pau, Barcelona, Spain, reported CSF findings from the longitudinal BioMark APD study, which tracks volunteers at 45 centers across 19 European countries and Canada. Lleó analyzed data from a subset comprising 154 cognitively healthy people, 75 with subjective cognitive decline, 128 with mild cognitive impairment, and 111 people with clinical late-onset AD. Most were in their 60s. Participants took the MMSE and donated CSF at baseline, and gave CSF again during at least one follow-up visit about two years later.
At baseline, the findings matched other studies: People with mild cognitive impairment (MCI) or AD had lower CSF Aβ42, and higher total tau and p-tau, than the other groups. Inflammatory and injury markers such as YKL-40 and NfL also ran high in CSF of people with clinical symptoms.
Over time, however, no differences emerged between diagnostic groups in how fast biomarker levels changed. CSF Aβ42 levels rose slightly, but at the same rate, in all four groups. Inflammatory markers also climbed similarly in all groups. By contrast, CSF tau markers remained stable over time, with one exception: In people with AD who did not carry an ApoE4 allele, CSF total tau and p-tau dropped. The reason for this is still not clear. Possibly, the patterns of AD biomarker changes in people without an ApoE4 allele are shifted compared to those with ApoE4, so that they represent different time frames despite similar clinical status, the researchers said. Intriguingly, at AAIC researchers reported that CSF total tau levels stayed flat in people with familial AD in the Dominantly Inherited Alzheimer Network (DIAN), too, but CSF p-tau dropped close to disease onset. Researchers saw this drop as a sign p-tau was getting swept into neurofibrillary tangles in the brain (see Part 1 of this story).
What explains the static biomarker signatures in sporadic AD? Daniel Alcolea at Sant Pau’s noted that the study’s two-year time span may be too short to capture true long-term change. “Longer follow-up periods, and a special focus on preclinical stages might give us more information about the relevance of these changes,” Alcolea wrote to Alzforum.
Meanwhile, the lack of longitudinal change over the short term casts fresh doubt on the ability of CSF markers to serve as outcome measures for clinical trials, as others have noted (Aug 2017 conference news). Lleó suggested that CSF markers might best track progression early in disease. In preliminary analyses of people with preclinical AD, he does see a rise in p-tau in this group, he told Alzforum. “The main changes in core CSF biomarkers occur during the asymptomatic phase of AD. Our study supports the idea that these biomarkers are not very dynamic during the symptomatic phase of the disease,” Lleó wrote.—Madolyn Bowman Rogers
Salat DH, Buckner RL, Snyder AZ, Greve DN, Desikan RS, Busa E, Morris JC, Dale AM, Fischl B.
Thinning of the cerebral cortex in aging.
Cereb Cortex. 2004 Jul;14(7):721-30. Epub 2004 Mar 28
PubMed.
Teasing Out the Brain Features Behind Cognitive Reserve
Some older adults maintain good memory and cognitive skills even in the face of extensive amyloid and tau pathology in the brain. This resilience has been dubbed “cognitive reserve,” but researchers still do not know exactly what explains it or how to measure it directly. At the Alzheimer’s Association International Conference 2017, held July 16–20 in London, hints emerged that specific organizational features of the brain underlie this phenomenon. Several researchers highlighted functional brain networks that correlate with preserved cognition. One reported that more efficient neuronal networks help people resist the effects of AD pathology. Another suggested using brain volume to devise a simple num erical measure of cognitive reserve, which scientists still cannot quantify. Such a measure could allow researchers to predict how quickly AD will progress in a given person. Though preliminary, these initial studies are beginning to paint a better picture of brain resilience.
Memory Network.
Brain regions connected to the frontoparietal control hub activated (yellow and red) when people successfully memorized face-name pairs. [Courtesy of Michael Ewers, AAIC 2017.]
Reserve has been loosely defined as the variation in functional brain characteristics that provides resilience against the cognitive decline brought on by aging, injury, or disease (Apr 2017 conference news). Researchers detect it by identifying people who maintain better cognition than expected given their age or disease stage. Because years of education and IQ score correlate with reserve, these measures are often used as proxies for it.
Frontoparietal Control Network Crucial for Reserve Michael Ewers of Ludwig-Maximilians University, Munich, wondered if he could pinpoint a more specific measure of cognitive reserve. In previous work, he found that highly educated people with preclinical AD had worse brain glucose metabolism in the posterior cingulate and angular gyrus than less-educated people at the same level of cognitive performance (Ewers et al., 2013). This suggested to him that cognitive reserve was helping the more educated people resist declines due to hypometabolism.
What brain feature might explain this resistance? One candidate is a hub of the frontoparietal control network found in the left frontal cortex (LFC). This LFC hub has been associated with IQ in young people, and tends to be spared in AD, Ewers said. It couples with various other networks to perform different tasks. To find out what role this hub played in cognitive reserve, Ewers analyzed ADNI resting-state fMRI data on 44 amyloid-positive people with MCI. He found that MCI patients whose LFC hub was more tightly connected with the rest of the brain maintained sharper episodic memory than those with low connectivity for the same degree of hypometabolism. High connectivity also associated with more education, as would be expected for a measure of cognitive reserve (Franzmeier et al., 2017).
Was this link between resting-state connectivity and memory just a correlation, or does greater LFC connectivity actually help people perform better while doing a task? Ewers investigated this question in a cohort of 37 healthy controls and 17 MCI patients who completed a face-name association task that measured episodic memory while being scanned (see image above). Higher connectivity of the LFC hub to regions such as the medial temporal and parietal lobe during the task correlated with better performance, indicating that this network played a direct role in episodic memory. High connectivity again correlated with years of education, supporting the idea that LFC hub connectivity measures cognitive reserve (Franzmeier et al., 2017).
Ewers next wondered if high LFC connectivity might delay cognitive decline. He analyzed data from the Dominantly Inherited Alzheimer Network (DIAN) and from DELCODE, a German study of sporadic AD. In both cohorts, people with higher LFC connectivity had better cognitive abilities than those with low connectivity at any given level of pathology. Those with higher connectivity declined more slowly as well. Strengthening this network might be a therapeutic strategy, Ewers suggested.
Another talk at AAIC complemented these findings. Rachel Buckley of Massachusetts General Hospital in Charlestown investigated the relationships of five functional brain networks to several proxies of cognitive reserve, including education, IQ, occupational attainment, and self-reported cognitive activities. She analyzed resting-state fMRI data from 243 cognitively healthy participants in the Harvard Aging Brain Study; 182 were amyloid-negative, while 61 were amyloid-positive.
She found that connectivity in the right frontoparietal control network was most associated with cognitive reserve. While her data implicate the right rather than the left frontal cortex as Ewers found, the two studies both point to the importance of the frontoparietal network. The default mode network (DMN), dorsal attention, and salience networks had a weaker relationship to proxies of cognitive reserve, and the motor network had none, as might be expected. Connectivity of these networks correlated least with occupational attainment and most strongly with IQ score, suggesting this was the best proxy for cognitive reserve. Amyloid burden did not affect the relationship between connectivity and reserve.
A Cognitive Reserve Network? Parts of the DMN and task-positive networks either activate (red), or deactivate (blue), respectively, during all cognitive tasks, and also correlate with IQ. [Courtesy of Yaakov Stern, AAIC 2017.]
Identifying a Cognitive Reserve Network for All Cognitive Domains
Another talk at AAIC identified a different brain network associated with IQ that may play an important role in cognitive reserve. Yaakov Stern of Columbia University, New York, used task-related fMRI rather than resting-state to find a network involved with preserving cognitive abilities. He noted that many previous studies of cognitive reserve examined the functional brain networks that activated when participants perform only a single cognitive task (e.g., Steffener et al., 2011). However, cognitive reserve preserves numerous cognitive abilities, such as memory, language, and executive function, not just single cognitive domains, Stern said. To identify a single brain network that correlated with reserve across a variety of mental challenges, Stern analyzed data from the Reference Ability Neural Network study. This aging study employs fMRI to identify brain networks responsible for the mental abilities that change the most with age: reasoning, episodic memory, processing speed, and vocabulary. About 300 cognitively healthy adults from age 20 to 80 completed 12 cognitive tasks that called on these skills while under the scanner (Stern et al., 2014; Habeck et al., 2016).
Stern analyzed data from 220 participants, using IQ scores as a proxy for cognitive reserve. He searched for a pattern of brain areas that activated during all 12 tasks, with the degree of activation correlating with IQ. He found a network that encompassed multiple brain areas, including parts of the DMN and parts of the task-positive network (see image above). The former, which includes the medial temporal lobe, medial prefrontal cortex, and posterior cingulate cortex, turns off during tasks, while the latter, which includes the superior parietal lobe, lateral prefrontal regions, and insular cortex, activates. To find out if this combination network responded during any mental challenge, Stern tested participants on a different type of activation task. Again, the same network lit up, with its degree of activation correlating with IQ score. Thus, this pattern of activation may point to a cognitive reserve (CR) network, Stern suggested.
To test this idea, Stern correlated the CR network activation with cognitive ability and brain volume. Normally, older people who have thinned cortices perform worse cognitively than those with more preserved thickness. Stern found that people who highly activated the putative cognitive reserve network executed better than expected on cognitive tests given their cortical thickness. This supports the idea that this network embodies cognitive reserve. Stern suggested that measuring this network could help predict the rate of cognitive decline in AD, and help researchers account for differences in cognitive reserve among clinical trial populations. In addition, interventions meant to raise cognitive reserve might measure activation of this network as an outcome, Stern said.
Efficient Brain Networks Lead to Better Performance Marina Weiler, now at the National Institutes of Health, Bethesda, Maryland, took yet a different approach to identifying what underlies cognitive reserve. She noted that neuronal networks organize themselves according to psychologist Stanley Milgram’s “small-world problem,” a.k.a. six degrees of separation (Travers and Milgram, 1969). This is the idea that any two people in the world can be connected to each other by a chain of no more than six acquaintances. In the case of neurons, they achieve this high connectivity by making many local connections and only a few long-range connections. This allows them to perform complex local processing but also send messages across the brain quickly, reaching any other neuron through few intermediaries. Previous research demonstrated that such small-world networks are highly efficient, with a high benefit and low cost to the brain, Weiler said.
Does this type of organization help brains resist pathology? Weiler, who at the time worked at the University of Campinas, São Paulo, Brazil, examined a cohort of 70 people, which consisted of 28 controls, 28 people with amnestic MCI, and 14 with mild AD. Weiler noted that the participants represented a wide range of education levels and IQs. She measured pathology using cerebrospinal fluid markers of Aβ, t-tau, and p-tau, and brain efficiency by applying a graph theory approach to resting-state fMRI data. By analyzing functional connectivity data with the GraphVar toolbox, which allows users to deduce the brain’s topographic organization based on fMRI data, Weiler obtained multiple measures of network efficiency (Kruschwitz et al., 2015).
Across all diagnostic groups, the people who had the most cognitive reserve, as determined by education and IQ score, also had the greatest small-world connectivity, Weiler found, suggesting that this could be a measure of reserve. As some other studies have reported, higher levels of education and IQ did not protect people from developing the pathophysiological features of AD. Notably, however, education level did seem to modify the effects of pathology on network efficiency in aMCI and AD patients. AD patients normally lose the small-world organization of neuronal networks, but those with more education better maintained this. The finding suggests that AD patients with more reserve are better able to cope with the effects of pathology, Weiler said.
Late Onset, Faster Decline. People with high cognitive reserve better maintain abilities as pathology advances, but at late stages decline more quickly. [Courtesy of Anita Van Loenhoud, AAIC 2017.]
A Dynamic Measure of Reserve
One drawback to these functional connectivity measures of cognitive reserve is that they are not easily quantifiable. Anita Van Loenhoud of VU University Medical Center, Amsterdam, derived a simpler, numerical measure of cognitive reserve. She used data from amyloid-positive participants in the Amsterdam Dementia Cohort, 56 of whom had subjective cognitive decline, 108 MCI, and 347 AD. She compared their brain volumes, as determined by structural MRI, to their scores on a cognitive composite. Because the brain shrinks with aging and with AD, people with lower volume typically have poorer cognition. Van Loenhoud calculated a statistical W-score for each person based on how much their cortical volume differed from what she expected given their cognitive score. W-scores are similar to z-scores, but adjusted for specific covariates like age and sex. Those who had a low W-score, meaning less brain volume than their cognitive scores would suggest, were considered to have high cognitive reserve.
Van Loenhoud then examined how W-scores related to disease progression. Unexpectedly, she found that MCI patients with low W-scores, and thus high cognitive reserve, were more likely to progress to AD than were their peers with high W-scores (van Loenhoud et al., 2017). Though initially puzzling, the data fit with some previous observations. Although people with high reserve have a lower risk of dementia, after an AD diagnosis, they actually progress more rapidly than others do, likely because they do not show symptoms until their pathological burden is large and their disease quite advanced. These MCI patients were probably already at the point in disease where decline accelerates, Van Loenhoud suggested (see image above).
W-scores represent a dynamic measure of cognitive reserve, since they depend on current brain volume, which changes over the course of the disease, van Loenhoud noted. This may make the measure more useful for prognosis than static measures. Supporting this, education level, a static measure, did not predict disease progression in this cohort. In future work, van Loenhoud plans to incorporate other measures of pathology into the score to refine its prognostic use.—Madolyn Bowman Rogers
At AAIC, Yet Another Phase 3 Flop While Phase 1 Trials Forge Ahead
Part 1 of a three-part story.
At this year’s Alzheimer's Association International Conference, held July 16–20 in London, researchers reported negative data from three Phase 3 trials of the 5HT6 antagonist idalopirdine. It came as no surprise, since topline data released last year had already deemed one of the trials to have fallen short. In contrast, Phase 2 data for VX-745, a kinase inhibitor, and leucine, a recombinant human cytokine, hint that they may slow cognitive decline. Safety and tolerability data from Phase 1 trials appeared to give a green light thus far for everything from small molecules such as BACE inhibitors to passive immunotherapies for Aβ and tau (see Part 2 and Part 3 of this story). Read on for a summary of the latest clinical data.
Once Again, Phase 3 Brings Down a Rising “Star” Alireza Atri from the California Pacific Medical Center, San Francisco, reviewed three Phase 3 clinical trials of H. Lundbeck’s 5HT6 antagonist idalopirdine. This drug is thought to enhance neurotransmission in several neuronal subtypes in the brain, including cholinergic and glutamatergic. Phase 2 data had suggested to its sponsors that idalopirdine and the acetylcholinesterase inhibitor donepezil together slowed cognitive decline in people with moderate Alzheimer’s disease better than donepezil alone did (Jun 2012 news), and the company launched a Phase 3 program of three trials all dubbed STAR.
Alas, already last September, topline data from the STARSHINE study dimmed idalopirdine’s luster. Among 932 patients taking 10 mg donepezil daily, those who also took 30 or 60 mg of this study drug performed no better on the ADAS-Cog over 24 weeks (Sep 2016 news). At AAIC, Atri presented data from STARSHINE as well as from its sister trials STARBRIGHT and STARBEAM. The latter tested 10 and 30 mg of the drug versus placebo in combination with 10 mg donepezil among 858 patients, while STARBRIGHT tested a combination of 60 mg idalopirdine or placebo with any approved AD treatment in 734 patients. Atri did not report data from an ongoing extension trial.
Overall, the 5HT6 antagonist had no effect. Of 4,000 patients who were screened for inclusion and exclusion criteria, the three trials enrolled about 2,500. On average, they were around 74 years old and had been diagnosed with AD 2.2 years prior. They had a mix of mild and moderate AD; 40 percent scored between 19 and 22 on the MMSE, while 60 percent scored between 12 and 18.
Atri showed plots of treatment and placebo data for the 24 weeks of each trial. In all three trials, scores for the ADAS-Cog, the ADCS-Activities of Daily Living, and the Clinician Global Impression of Change (CGIC) followed the same trajectory in both the treatment and placebo groups. In STARSHINE there was a possible trend toward a signal in the CGIC, said Atri.
Breaking down the data by disease severity at baseline exposed hints of an effect in people with moderate AD, Atri said. This prespecified analysis covered patients with MMSEs of 12 to18, which resembled the baseline scores in the positive Phase 2 proof-of-concept study. In STARBRIGHT, moderate AD patients in the treatment group did better on the ADAS-Cog, with a p value of 0.03. There was also a trend for efficacy on the ADCS-ADL. In STARSHINE, a trend for efficacy on the CGIC also emerged. Patients with MMSEs of 19–22 did no better than placebo on any outcome measure in any of the trials.
Atri said the drug seemed safe and well-tolerated. Patients on drug and placebo reported similar adverse events, such as diarrhea, nausea, vomiting, falls, and headaches. They were a tad more common in the 30 and 60 mg doses.
Researchers at AAIC were disappointed but congratulated Atri, calling the trials a considerable effort. Some questioned if the Phase 2 trial had been robust enough to warrant a Phase 3 program, given the lack of a benefit on the ADCS-ADL. Others wondered if the dosing was high enough. Atri said dose occupancy tests had been done, though not in AD patients but in young healthy controls. Nonetheless, Atri said he expected 80 percent target occupancy over 24 hours with the doses used. He did acknowledge that having no biomarker selection was a limitation of the program. He said some of the patients likely had no brain amyloid and therefore were misdiagnosed as having AD.
Atri did not review data from an ongoing extension trial that monitors 1,500 patients on 60 mg of the drug for an additional 28 weeks.
A Neuroinflammation Target? Kinase Inhibitor and Cytokine in Phase 2 Niels Prins, VU University Medical Center, Amsterdam, outlined cognitive results of two small trials of the p38 MAP kinase inhibitor neflamapimod, a.k.a. VX-745. It is purported to reduce harmful inflammation and improve neural plasticity while stimulating microglia to phagocytose amyloid. Two tiny AD trials on this compound have been done. In one, 16 patients took either 40 mg or 125 mg of it twice daily for 12 weeks. In a second, nine-week trial, five people took 40 mg, and one person took 125 mg. The primary outcome measure, a change in plaque burden as judged by quantitative PiB PET, suggested that the drug stimulated plaque removal. Philip Scheltens from VU Amsterdam outlined those results at CTAD last December (Dec 2016 conference news).
In London, Prins said that when patients in the 12-week trial sat for the Wechsler Memory Scale, their average scores for its immediate recall composite increased by 5.7 points at four weeks and by 10 points at the end of the trial, a significant improvement. Delayed recall scores also improved, from 13.2 at baseline to 18.2 and to 22 at four and 12 weeks, respectively. The nine-week trial used the Hopkins Verbal Learning Test-Revised. It also indicated significant improvement in delayed recall, with participants scoring on average 5.4 at baseline and 7.5 at day 40.
Researchers at AAIC expressed concern that there was no placebo group for comparison in these studies. They wondered why Prins assumed there would be no practice effect in the memory test, which could confound interpretation of the data. Prins said that prior literature has shown no strong practice effects with these tests. He also said pharmacokinetic data bolstered the idea that the improvements were drug-related. This analysis indicated a correlation of 0.7 between the drug’s concentration in plasma and combined immediate and delayed recall tests in the 12-week trial.
Prins said they were planning a six-month double-blind, placebo-controlled trial.
In their poster, researchers led by Huntington Potter, Jonathan Woodcock, and Tim Boyd at the University of Colorado Anschutz Medical Campus, Aurora, and Ashok Raj at the University of South Florida, Tampa, showed preliminary analysis of a Phase 2 trial to test the safety, tolerability, and efficacy of leukine in patients with mild or moderate AD. This is a recombinant form of human granulocyte-macrophage colony stimulating factor (GM-CSF) made by Genzyme/Sanofi. The FDA has approved this drug for treating bone marrow transplant (BMT) patients to beef up their blood cell counts. Potter had reported that in a BMT clinical trial, GM-CSF improved neuropsychological test scores, indicating that the cytokine improved cognition (Jim et al., 2012). Other hints that the cytokine might benefit AD come from patients with rheumatoid arthritis. They have elevated levels of GM-CSF in their blood, explained Potter, and they also have lower risk of developing Alzheimer’s. To investigate this correlation, the researchers gave injections of GM-CSF daily to transgenic mouse models of AD for 20 days. The treatment halved the amyloid burden in the mice and improved their navigational skills (Boyd et al., 2010).
In the Phase 2 trial, volunteers were injected subcutaneously with placebo or 250 μg/m2 leukine five days a week for three weeks. Patients were examined at baseline, at the end of the trial, and again 45 and 90 days later for safety and with a battery of cognitive and functional tests. Potter reported data for13 people on drug (six men) and 19 on placebo (10 men). Those in the randomized treatment arm had an average MMSE score of 16.46 at baseline, versus 20.63 for those on placebo, a significant difference. ADL scores were also lower—54.61 for treatment group and 63.16 for placebo.
The drug seemed well-tolerated. The poster listed no serious adverse events and reported no signs of ARIA, or amyloid-related imaging abnormalities. Plots of MMSE and ADL scores suggested that patients on the drug benefitted at the end of the three-week treatment period. At this point the treatment group scored about 1.5 points higher in MMSE than at baseline, while the placebo scores had not changed. Similarly, the ADL score rose about 1.5 points at the end of treatment but then fell by about the same margin in treatment and placebo arms, respectively. No differences emerged between the two groups at later time points. No significant difference emerged at any time between treatment and placebo arms on the ADAS-Cog, CDR-sb or MOHS tests. Given these results, the authors wrote that the “Part the Cloud” 24-week trial of leukine in patients with mild to moderate AD is warranted. Clinicaltrials.gov list a Sanofi-sponsored Phase 2 trial of leukine, a.k.a. sargramostim, as “withdrawn.”—Tom Fagan
At AAIC, Encouraging Safety Data on a Variety of Small-Molecule Candidates
Part 2 of a three-part story.
At AAIC 2017 in London last month, researchers introduced data from ongoing Phase 1 trials of a plethora of small-molecule drugs, including BACE inhibitors, a γ-secretase modulator, and a phosphodiesterase inhibitor. Though early days, the candidates looked safe for further testing.
Researchers from Merck, Pfizer, and Novartis outlined safety data from their ongoing BACE inhibitor trials. This enzyme cleaves the extracellular domain from the amyloid precursor protein, leaving a C-terminal fragment that γ-secretase processes further to release the Aβ peptide. Researchers had raised concerns with targeting BACE because it comes in two isoforms and each cleaves multiple proteins. Fears that blocking cleavage of any one of these substrates might cause serious side effects had led to calls for caution (Dec 2013 conference news). At the doses used and with new inhibitors that are more specific for BACE1 than for BACE2, so far the strategy appears to have avoided any safety pitfalls.
RuolunQiu from Pfizer in Cambridge, Massachusetts, reported on safety, tolerability, and pharmacokinetics from two small trials of PF-06751979. Qiu said this compound is a selective inhibitor of BACE1, blocking this CNS isoform with 30-fold more potency than the more peripherally expressed BACE2. In keeping with this, Qiu reported that in mice, a dose of PF-06751979 that reduces Aβ by 55 percent caused no hypopigmentation of their fur, unlike a similar dose of a non-selective BACE inhibitor. BACE2 plays a critical role in the generation of melanin in the skin and hair follicles.
Reviewing the human data thus far, Qiu said the drug seems well tolerated. Placebo or single ascending doses from 3 to 540 mg were given to 16 healthy young men in their 30s; multiple ascending doses of 5 to 275 mg or placebo were administered over 14 days to 50 more men and women around the same age. Ten Japanese volunteers received 125 or 275 mg, or placebo, over the same time frame. Lastly, 24 volunteers in their 60s and 70s were given 50 or 125 mg, or placebo, also for 14 days.
In these trials, this BACE2 inhibitor seemed safe, Qiu said. Two volunteers discontinued due to adverse events; one of them was on placebo, the other developed a skin rash. Treatment-related adverse events were mild, said Qiu, and included fatigue, headache, insomnia, and acne. Levels of the enzyme plasma transaminase temporarily increased in one person on placebo, one on 125 and one on 275 mg of PF-drug, but this was asymptomatic. There were no serious adverse events, Qiu said.
Pharmacokinetic data suggested that the drug is suitable for once per day dosing, said Qiu. Both the maximum concentration and total amount in the blood increased with dose, and on average the maximum concentration was reached in three to four hours. Half-life in the blood was about 30 to 40 hours. Qiu claimed the drug entered the brain well but showed no CSF data for inhibitor levels. She did show that 50, 125, and 275 mg of drug reduced CSF Aβ42 by 70, 85, and 95 percent, respectively. Qiu did not show data for sAPPβ or sAPPα, but said they tracked in the expected direction.
Cristina Lopez-Lopez from Novartis, Basel, Switzerland, reviewed a three-month dose ranging and tolerability study for the BACE inhibitor CNP520, which is being tested in the ongoing Generation secondary prevention trials conducted by the Alzheimer’s Prevention Initiative (Jul 2014 conference news). Lopez-Lopez said that Novartis now has data on five studies that include 422 subjects, 355 on drug. She said that up to now, testing has revealed no eye, skin, cardiovascular, or cognitive problems. She said across these trials, adverse events were similar among placebo and drug groups, except for one case of pruritus.
A three-month dose ranging trial administered placebo or up to 85 mg CNP520 daily to healthy volunteers over 60. Novartis researchers use pharmacometric modeling to predict doses that would lead to 80 and 50 percent reduction in CSF Aβ in 90 percent of people on the given dose. This turned out to be 15 and 50 mg/day for the high and low reduction, respectively. On this base, Generation 1, which will test CNP520 and Novartis’ Aβ immunotherapy CAD106 in homozygous ApoE4 carriers, will use 50 mg of the BACE inhibitor, and Generation 2, 15 and 50 mg doses. The latter is recruiting both heterozygous and homozygous ApoE4 carriers. Lopez-Lopez noted that in clinical and preclinical studies, ApoE genotype does not alter reduction in CSF Aβ by the drug.
Describing a new line of attack on BACE, JuliyaKalinina from Merck & Co., West Point, Pennsylvania, described how a new inhibitor, MBi-10, reduced CSF levels of Aβ oligomers (AβOs). Merck researchers led by Mary Savage have developed an immunoassay to detect these ephemeral Aβ species (Savage et al., 2014). Kalinina gave no details on this compound save to say it potently inhibited APP processing in vitro and in vivo, and that it is highly selective for BACE1 over BACE2. In monkeys, a related BACE inhibitor, MBi-5, reduced Aβ monomers, AβOs, and sAPPβ in the CSF but not sAPPα, said Kalinina.
What about in people? Kalinina and colleagues administered a single dose of placebo, 10, 60, or 800 mg MBi-10 to each of 24 healthy men. The researchers then collected serial CSF samples over 36 hours and assayed for Aβ monomers and oligomers species. Levels of Aβ40 and AβOs gradually fell from the time the drug was taken. The reduction was dose-dependent, reaching 10 percent relative to baseline. Kalinina noted that the kinetics and extent of the changes were similar for the two forms of Aβ, suggesting that monomers and oligomers rapidly reach equilibrium.
Other researchers at the meeting were intrigued by the findings but wondered what they mean. Jeff Cummings, Cleveland Clinic at the Lou Ruvo Center, Las Vegas, noted that there are many different species of oligomers and asked Kalinina if she knew exactly what was being detected in the assay. She acknowledged that they don’t know for sure, but that this is something they are studying at Merck.
Green Light for γ-Secretase Modulator and Phosphodiesterase Inhibitor
Researchers from Pfizer showed data from three Phase 1 trials of their γ-secretase modulator PF-06648671. As is the case for BACE, γ-secretase cleaves many substrates besides the amyloid precursor protein (APP), most notably Notch, and in clinical trials inhibitors led to serious adverse reaction in patients (Oct 2015 news). Researchers are now focused on modulators. These have no effect on γ-secretase endopeptidase activity, thus allowing it to cleave Notch and other substrates as usual. Instead, these drugs modulate subsequent carboxypeptidase activity of the enzyme that sequentially cleaves three-amino acid peptides from the Aβ peptide before releasing it. The idea is to coax the enzyme to make shorter, less amyloidogenic peptides. Jae Eun Ahn reported how he used pharmacokinetic modeling to determine how plasma concentration of the PF-06648671 affected CSF concentrations of various Aβ species, including Aβ42, Aβ40, Aβ38, and Aβ37.
Ahn said that the GSM blocked production of Aβ42 more than 40, and boosted production of Aβ37 more than 38, showing that the modulator shifted the cleavage of APP toward generating shorter Aβ peptides. He concluded that 100 mg/day and 350 mg/day doses would reduce Aβ42 by 50 and 66 percent, respectively, while increasing Aβ37 two- and fourfold. He said these results will be used to guide dosing in future Phase 2 or 3 trials.
Reporting on a totally different approach, Robert Lai, Eisai, Hatfield, U.K., described results of a Phase 1 trial of a phosphodiesterase-9 inhibitor, E2027. Blocking PDE9, a strategy that other companies have tried before to no avail, boosts levels of cyclic GMP in the brain. This purportedly strengthens synaptic activity and may attenuate the toxic effect of Aβ on neurons (Jan 2010 conference news).
Eisai tested E2027 in a four-part Phase 1 proof-of-target-engagement study. Part A gave single doses of 10 to 1,200 mg to non-elderly healthy controls, while part B looked at taking this drug with food. Parts C and D tested single ascending doses in healthy elderly and in non-elderly Japanese volunteers. To measure cGMP levels, the researchers collected serial CSF samples from volunteers in parts B through D.
Lai reported that the drug was readily absorbed, reaching maximum concentration in the blood within two to four hours. The maximum concentration and the total amount seen in the blood, a.k.a. area under the curve in pharma parlance, increased with the dose but plateaued at 800 mg. The AUC ticked up 20 percent when the drug was taken with food, and the maximum blood concentration jumped about 50 percent. The pharmacokinetic data was similar in Japanese and other ethnicities.
E2027 increased cGMP in the CSF, with single doses of 100 and 400 mg showing a three- and fourfold increase before a slow decline. Levels remained above baseline three hours after dosing. Lai said the drug seemed well tolerated.—Tom Fagan
High-Dose Aβ and Tau Immunotherapies Complete Initial Safety Tests
Part 3 of a three-part story.
Since Aβ passive immunotherapy emerged as a potential treatment for Alzheimer’s disease, drug developers have struggled with a major challenge—how to get enough antibody into the brain to have the desired effect without unleashing potentially dangerous inflammatory responses. Many antibodies tested in AD trials have caused amyloid-related imaging abnormalities on MRI scans. ARIAs reflect blood vessel damage, likely caused by microglial responses to the therapy. Reducing effector function, or the antibody’s ability to activate microglia, has emerged as a viable strategy, as was evident by two presentations at this year’s Alzheimer’s Association International Conference, held July 16–20 in London. Two other talks outlined safety data from two novel tau immunotherapies, which have also been tested at much higher doses than some of the early Aβ antibodies, such as gantenerumab.
Genentech’s Helen Lin reviewed long-term safety data from crenezumab, which is currently undergoing two Phase 3 trials called CREAD1 and CREAD2. Lin reviewed a Phase 1 trial designed to test the safety and tolerability of up to 120 mg/Kg crenezumab given intravenously. That’s higher than the 15 mg/Kg intravenous doses used in a prior Phase 2. This Phase 1 trial was a bridging study, said Lin, to help the researchers to determine how high they can dose in Phase 3 trials. Being an IgG4, a class of antibody with reduced effector function, crenezumab was expected to elicit less ARIA.
This high-dose Phase 1 recruited 50- to 90-year-old people with a diagnosis of mild to moderate AD. They had an MMSE of 18 to 28 or a CDR of 0.5 or 1.0., and a positive amyloid PET scan to ascertain AD was the reason for the cognitive symptoms. The volunteers were split into three cohorts, each of whom received placebo or drug once a week for four weeks. Crenezumab doses of 30 or 45 mg/Kg were given to 26 people in cohort 1. The 26 and 23 patients in the second and third cohorts each received either 60 or 120 mg/Kg, respectively, if on drug. At week 13, patients had the option of rolling over into an active extension trial at the same dose, except for those originally taking 120 mg/Kg, who continued with 60 mg/Kg. The high dose works out to 7,800 mg/week for a 65 Kg patient, or much higher than the 225 mg per month of gantenerumab given to patients in the failed Scarlet Road trials (Dec 2014 news).
The cohort studies are completed, and the active extension has begun, said Lin. Genentech designed the trial to test the lowest dose first. If no safety or toxicity concerns emerged, the next cohort was enrolled, and so on until the highest dose was tested. Lin said the majority of adverse events recorded during the 13-week blinded phase were unrelated to the drug. So far in the active extension there have been five adverse events that turned up in three or more individuals, but seem unrelated to drug dose, said Lin. These included anxiety, falls, and headache. Out of the 75 patients there have been nine serious adverse events. In general, the SAEs were consistent with those in the Phase 2 clinical trials, said Lin. One patient in each of cohorts 2 and 3 developed pneumonia, were treated, and continued in the trial. Some patients developed infusion site reaction, but these were mild and tended not to recur, said Lin. Again, these appeared unrelated to dose.
On MRI there were no cases of imaging abnormalities due to leaky blood vessels, called ARIA-E. ARIA-H, which are due to microhemorrhages, occurred in patients in cohorts 1 and 2, but they were asymptomatic and required no treatment said Lin.
Lin concluded that no dose-limiting toxicity was apparent from this trial. “This Phase 1 supports this idea at doses four times greater than used in the Phase 2 trial,” she said.
Thor Ostenfeld, AstraZeneca, Cambridge, U.K., outlined data from a Phase 1 trial of MEDI1814. This monoclonal antibody developed at MedImmune, which is now part of AstraZeneca, was designed to target Aβ42 and not Aβ40. An IgG1, its effector function has been reduced with a triple mutation in its Fc tail. In rats and monkeys, MEDI1814 increased total Aβ42 and decreased free Aβ42 in the CSF, without changing Aβ40 levels, said Ostenfeld.
The two-part Phase 1 trial tested the antibody in single and multiple ascending doses in patients with probable AD and an MMSE of 16 to 26. An MRI scan during screening had to be consistent with an AD diagnosis. In the first part, 45 volunteers received placebo or 25, 100, 300, 900, and 1,800 mg of MEDI1814 intravenously, or 100 mg subcutaneously, and were then followed for 16 weeks. In the multiple ascending dose part, 32 volunteers received either placebo, 300, 900, or 1,800 mg IV or 200 mg subcutaneously. Three doses were given at four-week intervals. Safety evaluations included an MRI at day 36 for the single dose, and day 92 for those on multiple doses.
Ostenfeld reported no serious adverse events. None of the 57 people on drug in either the SAD or MAD part of the trial had signs of either ARIA-H or ARIA-E. Ditto for changes in vital signs. About half the people on drug and 40 percent of those on placebo had adverse events, which included headache, dizziness, and diarrhea. “The overwhelming majority of adverse events were mild and resolved during the trial,” said Ostenfeld.
The plasma concentration of the antibody increased with dose in both the single and multiple dosing regimens, said Ostenfeld. The antibody reduced free Aβ42 in the CSF, to the point of almost completely eliminating this peptide at higher antibody doses. Total CSF Aβ42 increased, but there was no change in levels of Aβ40 in the CSF, in keeping with the drug’s proposed mechanism of action. Ostenfeld showed that subcutaneous administration also reduced free Aβ42 in the CSF.
Tau Antibodies Look Promising
Researchers from AbbVie/C2N and from Genentech outlined results from their Phase 1 trials tau antibodies. AbbVie 8E12 recognizes extracellular, aggregated tau; Genentech humanized RO7105705, a mouse monoclonal antibody, to also target tau in the extracellular space, hoping to block the spread of toxic forms.
The mouse version of AbbVie 8E12 reduced disease progression in tauopathy models, slowed brain volume loss, and improved performance in behavioral tests, noted Kumar Budur from AbbVie in North Chicago. He reviewed Phase 1 results in people with progressive supranuclear palsy and gave a brief overview of a Phase 2 AD trial.
AbbVie conducted the PSP study at 12 clinical centers in the United States. Single doses of 2.5 to 50 mg/Kg were injected intravenously and volunteers were tracked for three months for signs of clinical change. Volunteers had to have a diagnosis of possible or probable PSP with a PSP rating scale score between 20 and 50. They had to have an MRI scan consistent with the disease, and be able to walk at least five steps with minimal assistance, said Budur. People who had had symptoms for more than five years, or signs of another neurodegenerative disorder, were excluded. Of 38 patients screened, 30 enrolled and were randomized three to one drug to placebo, which worked out to seven on placebo and 23 on drug. Sixteen were men, the average age was 69, and PSP ratings score was 36. Nine people received 50 mg, while three received each of the other doses.
Adverse events occurred in 21 volunteers, including seven people on placebo. Five people each had dermatitis or a fall, and three each had headache or skin abrasions. Other AEs included fatigue and bacteria in the urine, which was asymptomatic. Budur described three serious adverse events. A patient on 15 mg/Kg suffered a subdural hematoma following a fall; however, this person had a history of falls, which are common in PSP patients, and this patient’s latest fall was likely unrelated to the treatment, Budur said. One person on 50 mg/Kg developed cardiovascular disease due to high blood pressure, which was also deemed unrelated to the drug. The third SAE, a case of increased agitation, was in a person who got 25 mg/Kg. Investigators thought this might be related to the treatment, though this person, too, had a history of anxiety and agitation during times of stress, including while undergoing medical procedures.
The antibody’s plasma half-life is 27 to 37 days, said Budur. Its maximal blood concentration increased with dose, and the CSF-to-plasma ratio was .18 to .35, said Budur.
In the ongoing Phase 2 trial for AD, Budur reported no evidence of dose-related adverse events. He said the pharmacokinetics and were similar to other antibodies. This trial is testing three doses of AbbVie 8E12, each in 100 people who have mild or probable AD against placebo in another 100 patients. Patients will be treated over 96 weeks with a 16-week follow-up period. Budur also said that AbbVie is recruiting for a Phase 2 trial in PSP. This multiple dose trial will recruit 180 people to evaluate safety and efficacy over 52 weeks, he said. Primary outcome measure will be change in PSP rating scale.
Geoffrey Kerchner from Genentech, South San Francisco, reviewed data from a Phase 1 trial of RO7105705. He said no adverse events had been detected in rats or monkeys in preclinical testing. The Phase 1 trial recruited healthy volunteers between ages 18 and 80, and patients with mild to moderate AD who were between 50 and 80. The AD patients had an MMSE of 16 to 28 and a positive brain amyloid scan. The trial tested escalating single doses in seven cohorts of healthy volunteers, then four weekly doses in healthy volunteers and AD patients. The single doses in healthy volunteers went from 225 to a whopping 16,800 mg. Kerchner noted a 15-day window between doses for safety evaluations. The drug was given intravenously or subcutaneously.
The last patient has completed the final clinic visit, said Kerchner, but as of his talk in London, results were still blinded. All Kerchner could say was that the antibody had generated no serious adverse events, and minor AEs unrelated to the drug included headache, respiratory tract infection, nausea, and vomiting. Nine volunteers experienced AEs attributed to the drug, including bruising and pain at the injection site.
Kerchner said the antibody’s half-life in plasma was 32 days, which is on par with other antibodies. Both plasma and CSF concentration increased with dose.—Tom Fagan

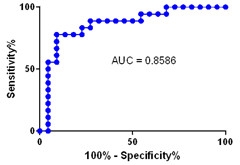




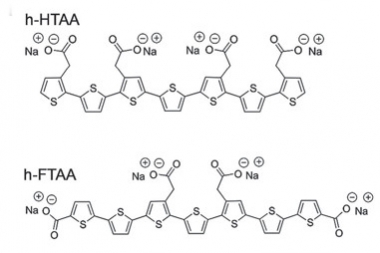








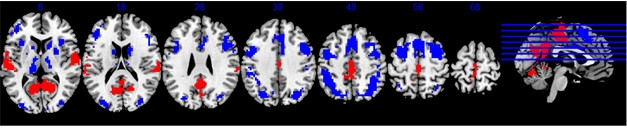
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.