The port city of Lisbon, the launch point of many a voyage of exploration, seemed a fitting site to host the 18th International Conference on Alzheimer’s and Parkinson’s Diseases and related neurological disorders. Disease-modifying therapies for amyloid plaques now approved, researchers are searching for similar treatments for tau, synuclein, and other potential drivers of neurodegeneration. With more than 4,700 attendees navigating 600+ presentations during five days, often spread across six parallel sessions, the conference was bustling, yet imbued with a sense of discovery. Speakers discussed new small-molecule and antibody therapies, combination approaches, new plasma biomarkers for tau and TDP43, and a good smattering of basic biology, from cellular resilience to microglial diversity. Follow along with Alzforum’s conference coverage.
Fast Plaque Clearance with Little ARIA? So Teases Trontinemab at AD/PD 2024
A record 4,700 people from 70 countries attended the 18th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 5 to 9 in Lisbon, Portugal. Those who attended this hybrid meeting in person sometimes packed rooms to capacity to hear 692 onsite talks or forums—another record. Scientists expressed a new sense of confidence that Alzheimer’s disease can be slowed or halted. With amyloid immunotherapies in hand, much of the discussion centered around how they can be improved and where the field goes next.
A consensus emerged that researchers need to target additional disease mechanisms and work toward combination therapies. Multiple sessions discussed the latest in tau biology, inflammation, and vascular research. Symposia focused on resilience mechanisms, prevention such as lifestyle changes, and advances in biomarkers. For Parkinson’s and other neurodegenerative proteinopathies, speakers showcased promising preclinical and biomarker data.
“Optimism is taking over. We have momentum,” Philip Scheltens of EQT Life Sciences Dementia Fund said in a forum sponsored by the Alzheimer’s Drug Discovery Foundation. Many noted that the recent successes have inspired biotech to make new investments in the field. “More deals are happening,” said Laurence Barker of the Dementia Discovery Fund. Susan Kohlhaas of Alzheimer’s Research U.K. has seen a change in patient attitudes as well, with people more likely to seek treatment for AD.
While the range of therapeutic approaches is broad, several sessions kept a spotlight on amyloid immunotherapy. Here, the buzziest data came from Roche, which reported that at the highest dose yet tested, its new antibody trontinemab virtually abolished plaque in three months in a small dose-finding study, while causing no ARIA in the eight participants who had reached this timepoint. Trontinemab combines the Fab fragment from gantenerumab with a transferrin-based “brain shuttle,” allowing the molecule to slip past the blood-brain barrier and perhaps avoid the bulk of the vascular amyloid that triggers ARIA. The data raised hopes that scientists can skirt this most troubling side effect. Meanwhile, Lilly and Eisai researchers shared new analyses from their immunotherapy programs that elucidated links between amyloid removal, tangle accumulation, and cognition.
During the conference, the U.S. Food and Drug Administration announced it would convene an advisory committee to consider Lilly’s application for traditional approval for its plaque-targeting antibody donanemab. Many had expected the antibody to be approved without this step, but Howard Fillit of the ADDF noted that this is the same procedure that was followed for Aduhelm and Leqembi. “[The] FDA decision is not a setback, but another step forward in the drug approval process, with the regulatory agency doing its due diligence before the distribution of the drug to patients,” Fillit said in a statement. Lawrence Honig of Columbia University, New York, told Alzforum he was not surprised by the decision.
Vanishing Plaque. Trontinemab clears plaque at lower doses than did previous antibodies. Participants taking 3.6 mg/kg (purple) fell below the positivity threshold (dotted line) by three months. [Courtesy of Roche.]
Better Efficacy, and Safety?
Trontinemab is the first anti-amyloid antibody in trials to use brain-shuttle technology, which Roche and other companies have been developing for years. In Phase 1, the shuttle approach delivered eight times more antibody into the brains of healthy volunteers than did conventional delivery (Mar 2021 conference news). At last fall’s Clinical Trials on Alzheimer’s Disease conference in Boston, Roche’s Luka Kulic reported six-month data from three dose cohorts in an AD Phase 1b/2a dose-finding study. The highest dose, 1.8 mg/kg, dropped plaque load by 62 centiloids at three months and by 84 centiloids at six. One person developed ARIA-E and one ARIA-H, for a 7 percent incidence of each (Nov 2023 conference news).
In Lisbon, Kulic added preliminary data from the highest planned dose, 3.6 mg/kg. As with the previous dose cohorts, the group comprised 15 people, of whom three received placebo. They averaged 72 years old and had either mild cognitive impairment or mild dementia due to AD, with an MMSE of 21. Kulic showed data from the eight participants on trontinemab who had reached the three-month timepoint as of last October, when the data were analyzed. Combined, the study enrolled a total of 59 people treated with four different doses.
People in the high-dose cohort started with an average amyloid PET score of 119 centiloids, a higher plaque load than in most immunotherapy trials. For comparison, participants in the negative Phase 3 gantenerumab studies started with 95. In three months, trontinemab mopped up 91 centiloids, plunging plaques below the amyloid positivity threshold of 24.1 centiloids in five participants, and below 11 centiloids in four of those (see image above). In other words, five of eight participants on trontinemab became amyloid-negative, with an average load of 21 centiloids at three months.
Preliminary safety findings looked good, with fewer concerns than at lower doses. There were no deaths, serious adverse events, or study withdrawals due to adverse events. In previous trontinemab cohorts, participants frequently developed infusion-related reactions, such as flushing and chills, after their first intravenous dose. These reactions were more frequent at higher doses, with three-fourths of people in the 1.8 mg/kg group experiencing them. However, in the fourth, 3.6 mg/kg dose cohort, clinicians pretreated participants with anti-inflammatories, such as acetaminophen, to head this off. As a result, less than half had this reaction. Kulic believes this side effect could be lowered further, or nearly eliminated, by pretreating with corticosteroids before the first dose.
Another issue in earlier cohorts was anemia, cropping up in a third of people in the 1.8 mg/kg cohort, for example, but also in people on lower doses and on placebo. Kulic believes frequent blood draws in the study may be partly to blame. The protocol was changed to stipulate iron supplements for participants who have low blood iron at baseline. So far, in the fourth cohort, one person developed anemia.
Likewise, anti-drug antibodies (ADAs), which lowered drug exposure by 70 percent in the lowest dose cohort but were less troublesome at higher doses, were more muted yet at 3.6 mg/kg. Only one person at this dosage developed ADAs, and this did not affect drug pharmacokinetics, Kulic reported.
Finally, the big question—what about ARIA? Despite the speed of amyloid removal, ARIA remained low at the 3.6 mg/kg dose, with no cases in the first eight people to reach three months of treatment. Though the numbers are small, Kulic called the data encouraging. Other work has linked ARIA to antibody interactions with vascular amyloid, which trontinemab’s delivery route may largely bypass (Aug 2023 conference news).
Kulic said Roche will select the most promising dose to take into part two of the study. That extension will expand the number of participants to 210 in order to gather more safety data before moving to Phase 3. The decision on which dose to take forward has not yet been made, Kulic said.
“These early results with trontinemab are exciting, and suggest that a brain-shuttle delivery has the potential to clear fibrillar amyloid rapidly and extensively without increasing circulating antibody levels,” Christopher van Dyck of Yale School of Medicine in New Haven, Connecticut, who co-chaired the session, told Alzforum. He noted that circulating antibody levels have been linked to the risk of ARIA. However, he cautioned that more data will be needed to understand the potential risks of the brain shuttle mechanism.
Dueling Antibodies. In a head-to-head study, donanemab (green) cleared plaque faster than aducanumab (gray). [Courtesy of Eli Lilly.]
Donanemab Data Highlight Benefits of Starting Early
Data from donanemab, too, suggest that fast plaque clearance does not hike the risk of ARIA. Lilly had previously reported 12-month data from its Trailblazer-Alz4 trial. This pitted donanemab head-to-head against aducanumab, with no placebo arm. In the first year, donanemab banished 80 centiloids, compared with aducanumab’s 56 (Jul 2023 conference news).
In Lisbon, Stephen Salloway of Butler Hospital in Providence, Rhode Island, reported on the final timepoint of 18 months. Aducanumab had almost caught up, removing an average of 72 centiloids to donanemab’s 84 (see image above). Partly, this was because plaque levels in the donanemab group had nearly bottomed out at the one-year timepoint, with 71 percent of people already amyloid-negative, compared with 22 percent of those on aducanumab. At the final timepoint of 18 months, 78 percent of those on donanemab were amyloid-negative, versus 43 percent of those on aducanumab. To put it another way, it took a person on donanemab an average of 359 days, or about a year, to completely clear amyloid. On aducanumab, this took 568 days, or seven months longer. Donanemab has not yet been tested head-to-head against lecanemab.
Despite the faster plaque removal, ARIA-E remained lower on donanemab, at 24 percent versus aducanumab’s 35. “We can lower plaque quickly without [worsening] safety issues,” Salloway concluded, noting that ARIA incidence does not directly relate to the speed or amount of plaque clearance.
Early Start Crucial. A model based on Phase 3 donanemab data predicts that treatment effects will be greater in people who start at an earlier disease stage, with trajectories (green) suggesting delayed progression to loss of independence, i.e., CDR-sb score of 11 (dotted line), compared with untreated AD (gray). [Courtesy of Eli Lilly.]
Other analyses in Lisbon focused on the antibody’s cognitive effects. Lilly’s Mark Mintun had previously modeled how donanemab changed the trajectory of cognitive decline, using data from the Phase 3 Trailblazer-Alz2 trial. He found the drug’s benefits were dramatically better for those at an earlier stage of disease, slowing decline by 88 percent, compared with 6 percent for those later in disease (Nov 2023 conference news).
In Lisbon, Mintun used the model to predict how much this slowing might delay progression to advanced disease stages. A person’s baseline tangle load determined where that person started in the disease trajectory. People with a baseline tau PET below 1.10 SUVR were defined as “low tau,” those between 1.10 and 1.46 as intermediate, and those above 1.46 as high. Because AD patients value keeping their independence, naming this as one of the most meaningful treatment outcomes, Mintun calculated the time to probable loss of independence, using a CDR-SB of 11 as a proxy.
In his model, a high-tau participant taking donanemab would stay independent about five months longer than if he or she were on placebo. At an intermediate to low tangle load, the person would keep their independence for an extra 11 months. For those in the intermediate-to-low tau group who were less cognitively impaired, at the MCI stage, this delay jumped to 37 months, or more than three years (see image above). Mintun cautioned that this is simply a model, and its assumptions need to be validated with additional data. However, he believes the findings emphasize the importance of starting plaque clearance early, before tangles have spread.
John Morris of Washington University, St. Louis, came to a similar conclusion using different methodology. To derive average rates of cognitive decline for people at different disease stages, Morris analyzed data from AD patients being seen at the Knight Alzheimer’s Disease Research Center in St. Louis. He determined disease stage by CDR-SB score, rather than by pathology, but, like Mintun, used a CDR-SB of 11 as the threshold for loss of independence. Then he calculated how much independent time a person would gain from a hypothetical therapy that slowed decline by 30 percent, which is about the same efficacy as lecanemab and donanemab. For someone who started at a CDR-SB of 1, the least impairment, it would take an additional 2.2 years to lose their independence. Someone who started at a CDR-SB of 4.5, or mild dementia, would gain only 0.9 years of independent life, Morris said.
Tangles, Interrupted. On placebo (blue, left), tangle load in the medial temporal lobe (x axis) determines how fast tangles spread in the parietal lobe (y axis); on lecanemab (pink), this relationship disappears. For cognition (right), lecanemab does not change the effect of MTL tangle load on the rate of decline, but it slightly reduces the amount. (CFB=change from baseline.) [Courtesy of Eisai.]
Lecanemab Disrupts Tangle Growth
Data from the lecanemab program reinforces this. Previous analyses had divided the Phase 3 Clarity population into a low-tau group, with baseline SUVRs below 1.06, and an intermediate-to-high tau group above that threshold. The low tau group had the greatest cognitive benefit, with 60 percent of them actually improving on their baseline CDR-SB scores over 18 months (Nov 2023 conference news).
In Lisbon, Eisai’s Arnaud Charil tied baseline amyloid and baseline tangle load to tangle growth. He divided the brain into seven composite regions, and noted that within each, the amyloid PET signal at baseline was associated with the baseline tau PET signal. If the amyloid PET signal was less than 1.2 SUVR, the tau PET signal in that region was typically negative. Above that threshold, baseline tau PET was positive. However, future tangle accumulation depended on baseline tau PET, rather than baseline amyloid. Charil showed that the higher the baseline tau signal, the more tangles accumulated over the next 18 months on placebo. On lecanemab, however, this relationship was broken. Tangle accumulation became negligible, and was unrelated to the baseline tangle load (see image above left). “Treatment with lecanemab disrupts tau accumulation,” he said.
How does this affect cognition? Here, lecanemab did not break the relationship between tangles and cognitive decline, but it did lessen the effect. For the placebo group, higher baseline tangles in any brain region correlated with steeper decline on the CDR-SB. On lecanemab, higher tangles still correlated with steeper decline, but this decline was slightly less pronounced than on placebo. Graphically, the slope was the same on lecanemab or placebo, but the line was shifted downward (see image above right). As with donanemab, benefits were higher in people who started with lower tangle loads. Because this analysis compares baseline tangle load in each brain region to the rate of cognitive decline, it is not equivalent to the common analyses that track the rate of decline over time and show an altered slope on lecanemab. This analysis indicates that even on drug, people who start with more tangles will have faster decline than those with fewer tangles. This again stresses the importance of early treatment.
Data from these programs have convinced many researchers that anti-amyloid antibodies will hold a key role in treating AD. “Immunotherapies will be widely used,” Barker of the DDF predicted, adding, “We’re already thinking about how to position new drugs in combination with them.” At the same time, some people will not be able to take these drugs, due to having high vascular amyloid or other contraindications. “Amyloid immunotherapy is not for every patient,” Scheltens cautioned.—Madolyn Bowman Rogers
TauRx Parses Subgroups to Make the Case for Methylene Blue Derivative, Again
Old drugs die hard. Despite a string of negative Phase 3 trials, HMTM, a derivative of the malaria drug methylene blue, resurfaced again at AD/PD 2024, held March 5-9 in Lisbon, Portugal. TauRx CEO Claude Wischik reported results from exploratory and post hoc analyses, as well as from an open-label extension, of the Lucidity trial, which evaluated HMTM in people with MCI and mild to moderate AD. As had been reported previously, the trial failed to meet its co-primary endpoints. At the meeting, Wischik reported that over the yearlong trial, participants with MCI who took the highest dose of the drug were half as likely to progress to AD as were volunteers in the control group. Among a subset of participants for whom blood samples were available, the drug also appeared to stem a small rise in plasma neurofilament light—a marker of neurodegeneration, he said. Further, in the open-label extension, participants with MCI who received the drug all along declined more slowly than those who had previously been in the control group, the company claimed. Given the troubled past of TauRx trials, many scientists who spoke with Alzforum were reticent to weigh in on the latest presentations of the data. Those who did were not convinced by the various subgroup analyses or, in a new twist for methylene blue trials, comparisons to external, historical controls. “Overall, there continues to be no evidence that these methylene blue derivatives have biomarker or clinical efficacy in Alzheimer’s disease,” wrote Lawrence Honig of Columbia University in New York (comment below).
Lucidity and its predecessors have been marred with complications, mostly wrought by the lack of a true control group. Because the compound gives urine a greenish-blue tinge, TauRx gave control volunteers just enough of the drug, or of a related compound, called MTC, to pee blue, to maintain blinding. Issues with this emerged in 2016, which saw HMTM, also known as LMTM, fail to slow cognitive decline in three Phase 3 trials—two in AD and one in FTD. Much to the chagrin of clinicians at the time, TauRx identified glimmers of hope within controversial subgroup analyses. They claimed that the treatment and placebo arms declined equally because the supposedly inactive placebo—8 mg/day of the drug versus 75 mg or 125 mg twice per day in treatment arms—was actually active. They also used subgroup analyses to claim that results were skewed by participants taking acetylcholine esterase inhibitors or memantine, and that in the 15 percent of people not taking those medication, LMTM slowed cognitive decline (Jul 2016 conference news; Dec 2016 conference news). They subsequently reported that HMTM boosts acetylcholine in the brains of mice (Kondak et al., 2022). Other scientists noted that people who are not taking cholinesterase inhibitors (AChEIs) typically have less-advanced dementia and therefore decline more slowly than people taking these drugs, explaining the differences in rates of cognitive decline between the LMTM monotherapy group and placebos who were mostly taking AChEIs.
Undeterred, TauRx opted for a new trial, excluding people taking AChEIs, and using much lower doses of HMTM in the active treatment group. It began in 2018, and, after several major changes, including to inclusion criteria, outcome measures, and duration, the final version of the trial protocol was published in 2022 (Wischik et al., 2022). Lucidity enrolled 598 people with MCI due to AD, mild AD, or moderate AD, all of whom were amyloid-positive according to PET scans. The 266 participants randomized to the control group took 4 mg MTC—a compound that Wischik said has the same activity as HMTM—twice weekly to maintain urine discoloration, while 252 received 16 mg/day of HMTM. A group receiving 8 mg/day was included for comparison to past trials, but these 80 volunteers were not included in analysis of the co-primary endpoints—change in ADAS-Cog11 and ADCS-ADL23 over 52 weeks. Secondary endpoints included change in whole-brain volume, while exploratory endpoints included change in clinical dementia rating scale (CDR) analyzed by disease severity.
At AD/PD, Wischik said that plasma NfL was measured as a prespecified biomarker endpoint; however, this is not listed within the published protocol or on clinicaltrials.gov.
After completing the 12-month randomized portion of the trial, all participants were invited to join an open-label extension, in which they received 16 mg HMTM per day for an additional 12 months. Wischik reported that 21 percent of participants dropped out of the blinded portion of the trial. Of those who completed it, 95 percent joined the extension. The drug was safe and well-tolerated. As determined previously, there were no significant differences between treatment and control groups for either co-primary outcome over the first 12 months of the trial (image below).
No Benefit. On co-primary outcomes (two left graphs) and secondary outcome of whole-brain volume (right), there were no differences between controls and the 16 mg/day group during the 12-month randomized portion of the trial, nor during the 12-month open label extension. [Courtesy of TauRx.]
In Lisbon, Wischik presented findings from a post hoc analysis of only those participants with MCI, who made up just under half of the total enrollees. In these participants, both the control and 16 mg/day groups improved over baseline in the first six months, this time on the ADAS-Cog13 (image below). Wischik claimed that because it is more sensitive, the ADAS-Cog13 better suits MCI patients than does the ADAS-Cog11 that was stipulated as the primary endpoint. He attributed the improvement on the ADAS-Cog13 to a tau-independent, symptomatic effect, purportedly mediated by a rise in acetylcholine in the hippocampus.
At 12 months, among those with MCI, scores in the control group had started to worsen while those in the treatment group held steady. In the open-label extension, MCI participants who had previously received 16 mg/day held steady on the ADAS-Cog13 out to 18 months, before dropping back to their baseline scores at 24 months. Scores continued to worsen between 18 and 24 months in people who had previously been part of the control group (image below). Wischik interpreted this finding to suggest that the placebo dose conferred a brief symptomatic effect, and that by the time the participants switched to the 16mg/day dose, it was too late to change the course of disease.
MCI Alone. A post hoc subgroup analysis of participants with MCI over the randomized and open-label portions of the trial suggest a benefit on the ADAS-Cog13 (left) among those taking 16 mg/day HMTM the whole time. [Courtesy of TauRx.]
Curiously, per the ADCS-ADL23, only the control MCI group experienced an apparent functional boost, doing slightly better than baseline at six months, but at no other time point. For the 16 mg/day group there was no statistically significant change from baseline during the two years to the ed of the extension. No difference emerged at any time point between the placebo and treatment groups.
This post hoc MCI subgroup analysis also teased out an effect on brain atrophy, said Wischik. At the 18- and 24-month timepoints in the extension, less brain shrinkage occurred among those with the treatment group relative to placebo.
As part of an exploratory analysis of CDR scores broken down by AD severity at baseline, Wischik claimed a 48 percent reduction in transition from MCI to AD at 12 months. What is this based on? Essentially, of 79 controls with MCI, 20 progressed from a CDR 0.5 to 1. By comparison, of 63 people with MCI in the treatment group, eight progressed from MCI to AD over 12 months. Most AD trials now eschew the CDR for the CDR sum of boxes, a much more refined test, especially for people in early stages of dementia.
HMTM Halts NfL’s Rise?
Plasma NfL, widely used as a marker of neurodegeneration in the brain, was measured in approximately 70 percent of the trial cohort “with available samples suitable for analysis,” according to Wischik. He did not explain why some samples were unavailable. He did report that the biomarker’s rise, not its concentration, over the 12-month trial was curbed by 95 percent among 136 people in the 16 mg/day group relative to 157 controls. Essentially, plasma NfL had inched up by 3 pg/mL in controls, but stayed put among people in the treatment group. Separating by disease severity, the effect was only significant among people with MCI. Among these milder cases, NfL held steady in 55 people in 16 mg/day groups and among 22 people in the 8mg/day group, while it rose by 3pg/mL among 68 controls.
NfL Stabilized? Over the 12-month randomized portion of Lucidity, plasma NfL rose by about 3 pg/mL in controls (left bar), but not among the 16 mg HMTM per day group (right bar).
Henrik Zetterberg, University of Gothenburg in Sweden, told Alzforum that the NfL findings were interesting, and may indicate that neurodegeneration became less intense in the treatment group. He has reported plasma NfL levels of around 40 pg/mL in MCI, increasing by about 2.7 pg/mL per year (Mar 2017 news; May 2019 news). Wischik did not show the absolute NfL levels. He also reported that among people whose NfL increased least, fewer transitioned from MCI to AD.
Finally, he presented comparisons between the pooled, three arms of Lucidity (including the placebo group) with two cohorts of external, historical controls. When compared to ADNI volunteers who were matched by sex, age, baseline MMSE, ApoE genotype, and who were not taking acetylcholinesterase inhibitors or memantine, Lucidity participants declined less on the ADAS-Cog11 and preserved more brain volume between baseline and 24 months. Those with MCI who received 16 mg/day were 75 percent less likely to transition to AD over 12 months than matched ADNI MCI cases. Similar effects were found when “meta-analytic” MCI controls from multiple trials were used as the external comparator.
Honig was not convinced by the trial data or the post hoc historical comparisons. “This duality thus involved both declaring the per protocol ‘placebo control’ group to actually be an active ‘non-placebo’ group, and using historical controls to imply efficacy in what was otherwise a negative randomized controlled trial,” he wrote.
Lon Schneider, University of Southern California, Los Angeles, was similarly incredulous. “Many pharmas—not having significant clinical outcomes from their randomized trials—just go on to find subsets and compare their outcomes to old data or ADNI instead of proper controls,” he wrote. “It’s a can’t-miss strategy that is sure to hack a p-value. We should do better in reporting trials results, and am confident we will in the future,” he wrote.
Although all of the apparent benefits of HMTM were found among exploratory endpoints, subgroup, or post hoc analyses, Wischik announced that TauRx will apply for marketing authorization for HMTM from the U.K.’s Medicines and Healthcare Products Regulatory Agency, and that discussions with the European Medicines Agency and Chinese regulators are moving forward as well.—Jessica Shugart
Therapeutic Contenders Target Hard-to-Reach Pockets of Tau
At this year’s AD/PD meeting, held March 5-9 in Lisbon, Portugal, no splashy Phase 3, or even Phase 2, data on tau-targeted therapies wowed attendees. Still, Phase 1 and preclinical data showcased a variety of therapeutic approaches the field has latched onto to find treatments for tauopathies. Biogen reported that BIIB113, a small-molecule O-GlcNAcase inhibitor designed to prevent tau from transforming into a pathological form bound to its target in the brain and was safe in healthy volunteers. Similarly, Eli Lilly reported promising Phase 1 findings from its OGA inhibitor, which is being evaluated in a fully enrolled Phase 2 trial in people with AD. On the vaccine front, AC Immune’s phospho-tau vaccine seems to spawn antibodies that thwart tau seeding. Meanwhile, preclinical data on nanobodies and anti-oligomer antibodies encased in slippery micelles hinted that engagement of tau inside of cells might be possible.
This new crop of tau-targeted therapies speaks to a shift away from the N-terminally trained antibodies that failed in previous trials, toward approaches that seek to ferret out other, possibly more pathogenic, reservoirs of tau. This includes tau traveling between cells in extracellular vesicles or within tunneling nanotubes, as well as tau pathology brewing within the cells, noted Luc Buée of the University of Lille in France.
In a forum on tau targeted therapies in Lisbon, Bradley Hyman of Massachusetts General Hospital in Boston said that when it comes to figuring out which species of tau to take down and how to do it, the field is still in an exploratory phase. The kinetics of tau release, uptake, and processing are still largely uncertain, he said. “Each of these kinetic steps could be explored, but we have to make a best guess. Sometimes that means just doing the experiment.”
Sugarcoating It
A glycoside hydrolase, O-GlcNAcase, aka OGA, removes N-acetylglucosamine moieties post-translationally bound to the hydroxyl groups of serine and threonine residues. Stripped of these sugars, tau is more likely to form filaments, hence some believe that by bolstering tau glycosylation, OGA inhibitors will prevent neurofibrillary tangles (Liu et al., 2004; Mar 2012 news).
Sugar-Coating Tau. OGT adds N-acetylglucosamine sugars (green) to unbound tau, i.e., free in solution and not attached to microtubules. OGA, on the other hand, removes said sugars. Without them, tau is likelier to become phosphorylated and misfold, heading down the path toward tangles. [Courtesy of Dustin Mergott, Eli Lilly.]
Flavia Nery from Biogen presented the first in-human data on that company’s OGA inhibitor, BIIB113. The compound has been tested for safety, and for target occupancy using a PET tracer for OGA activity that Biogen developed in-house (Cook et al., 2023). A drop in PET signal indicates that the OGA active site is occupied.
In a single-ascending-dose trial, 35 healthy volunteers aged 18-64 who took between 0.5 mg and 50 mg BIIB113 once orally, were monitored. Subsequently, 27 participants, up to age 75, took placebo, 15 mg, or 50 mg BIIB113 daily for 14 days. Results of both the single- and multiple-ascending-dose regimens indicated only mild to moderate adverse events, mostly deemed unrelated to the drug. Headache was the most common complaint among people in the treatment arms, and one person who received the 50 mg dose during the multidose regimen withdrew due to tremor. There were no other serious adverse events.
For target analysis, 10 healthy adults underwent OGA PET scans before and after receiving the drug. Nery reported that 48 hours after receiving 3 mg BIIB113, the PET signal in the brain had dropped by 90 percent, suggesting the drug had broadly engaged the enzyme. In a multidose study, 0.5 mg daily maintained this target occupancy over the two-week interval, Nery reported. Based on these Phase 1 findings, Biogen is planning a Phase 2 trial.
Attendees in Lisbon asked how OGA target occupancy might translate into reduction of tau pathology. Nery said that prior studies in mice indicated a target occupancy of 85 percent would be needed, meaning BIIB113 would pass muster at the 3 mg dose. Seiko Ikezu of the Mayo Clinic in Jacksonville, Florida, wondered about potential safety concerns of inhibiting OGA this much, given the enzyme’s role in stripping sugars from proteins across the body. Nery said that Biogen’s initial dosing studies, as well as the broader body of trials that tested other OGA inhibitors, have generally found this class of drugs to be safe. Participants will be closely monitored for any side effects of longer treatment in the upcoming Phase 2, she said.
Of the handful of OGA inhibitors in clinical development, Lilly’s LY3372689 is furthest along. At AD/PD, Lilly’s Dustin Mergott presented preclinical data and findings from Phase 1 studies of the compound in healthy volunteers. He reported that single oral doses ranging from 0.16 to 16 mg of the drug appeared safe, in that adverse events were mild, and did not relate to drug. Pharmacokinetics indicated a plasma half-life of six hours. To analyze target engagement, the scientists used an OGA PET tracer, [18F]LSN3316612, they had developed in collaboration with researchers at the National Institutes of Health (Lu et al., 2020; Shcherbinin et al., 2020). In Lisbon, Mergott reported that in four volunteers, the OGA PET tracer signal plummeted by 97 percent two hours after receiving 1 mg of LY3372689, and the signal was still down by 81 percent 22 hours later. In a subsequent multidose PET study, this target occupancy was sustained over a 14-day period. Finally, in a multiple-ascending-dose study, 40 healthy volunteers took placebo, 1 mg, 3 mg, or 7 mg LY3372689 daily for 14 days. Again, the drug appeared safe and well-tolerated at all doses tested, with only mild adverse events occurring with no relationship to dose. Leveraging this PET data with plasma pharmacokinetics, the investigators settled on 0.75mg LY3372689 as a low dose for Phase 2.
Called Prospect-ALZ, this trial began in 2021 to evaluate two doses of the inhibitor for 76-124 weeks in people with early AD. The trial uses a two-step screening process—plasma p-tau217 followed by tau-PET—to identify participants with tau pathology while minimizing the number of tau-PET scans. The first pass with p-tau217 paid off. Of the 2,177 people recruited for the trial, 1,850 failed screening, most because they had normal levels of plasma p-tau217. Mergott said that more than 1,000 tau-PET scans were avoided by adding this plasma screening step. Ultimately, 327 participants with moderate to high cortical tau pathology based on PET scans were randomized, and the fully enrolled trial expects to read out in late 2024. Change in the integrated Alzheimer’s Disease rating scale among people with moderate cortical tau accumulation serves as the primary endpoint, although secondary endpoints will assess disease progression in the full cohort, which also includes people with high tau tangle burden.
At least one other OGA inhibitor, Asceneuron’s ASN51, is moving toward Phase 2. The company reported favorable safety and target occupancy findings at the CTAD meeting in October 2023 and plans to start a Phase 2 trial this year.
Tau Immunotherapies Branch Out
Small molecule drugs like OGA inhibitors may be relatively cheap and easier to smuggle into the brain than antibodies, but what the latter lack in maneuverability, they gain in specificity. At AD/PD, scientists presented incremental findings on several. These approaches have moved beyond infusing full-size antibodies into the blood. Instead, researchers presented strategies to boost the chances of reaching the target in the brain, such as provoking an enduring immune response with an active vaccine, using small antibody fragments, or encasing full-size antibodies within slippery micelles to help them pass into cells.
AC Immune’s active vaccine, ACI-035.030, is the only one of these approaches in clinical trials. ACI-035.030 comprises liposomes with an antigenic phospho-tau peptide anchored to their lipid bilayer. Two adjuvants, as well as an antigen to rally T-helper cells, are also embedded. The package provokes a robust antibody response against phosphorylated tau (Dec 2022 conference news). The vaccine cleared safety hurdles and demonstrated immunogenicity against phospho-tau in a recently completed a Phase 1b/2a study. At AD/PD, AC Immune’s Marija Vukicevic said that a Phase 2b trial, led by partner Johnson & Johnson, is underway.
She presented no trial data, but detailed the seed-stopping capacity of antibodies raised by the vaccine in nonhuman primates. Using a cell culture model in which filamentous tau extracted from the human brain instigates the aggregation of endogenous tau in primary rat neurons, Vukicevic reported that sera from monkeys injected intramuscularly with ACI-035.030 effectively stopped tau propagation. The more times animals had been treated, the more antibodies they produced. Through a process called affinity maturation, whereby B cells produce antibodies with greater and greater affinity as an immune response matures, these antibodies also became more specific for p-tau antigens, and more adept at stopping seeds in their tracks with each vaccination, suggesting the specificity and functionality of the anti-p-tau antibodies had improved. ACI-035.030 was designed to target extracellular, seed competent tau.
Targeting Intracellular Tau
Until recently, Rakez Kayed of the University of Texas in Galveston had focused his efforts on going after extracellular tau as well. Specifically, he sought to dispatch soluble tau oligomers, which he sees as the primary agents of tau propagation and toxicity. As such, his group developed a suite of tau-oligomer-specific antibodies, aka, TOMAs. Previously, Kayed reported that while TOMAs block tau seeding and propagation in cell culture and animal models, the antibodies were not so good at removing established, intracellular tau pathology (Bittar et al., 2022; Castillo-Carranza et al., 2014). Notably, like most full-size antibodies, TOMAs do not efficiently get into cells.
In Lisbon, Kayed explained what happened when he shifted efforts toward targeting oligomers and other forms of tau inside cells. To do this would require both a different antibody and a new method of delivery. First, he went back to his antibody library and selected a likely candidate that they had previously dismissed. Called tau toxic conformation specific monoclonal-2 (TTCM2), this antibody was not 100 percent oligomer-specific like TOMAs. Instead, it latched onto oligomers, misfolded monomers, and small fibrils of tau. To get TTCM2 into cells, postdoc Sagar Gaikwad worked with scientists at InnoSense, Torrance, California, to package the antibodies within micelles. Made of a mix of polymers, these 100-nanometer-wide particles can slip into cells because their polymer coat melds with the cell membrane. Finally, to sneak past the blood-brain barrier, Gaikwad gave fluorescently labeled “TTCM2-ms” to mice intranasally. Three hours later, the fluorescent micelles had spread widely throughout the brain, inhabiting the olfactory bulb, hippocampus, cortex, cerebellum, and thalamus.
In 15-month-old hTau mice, a single sniff of these TTCM2-ms dramatically lowered existing levels of tau pathology, including aggregates of insoluble, hyperphosphorylated tau as measured by several different antibodies. The treatment also boosted flagging levels of synaptic proteins PSD95 and synaptophysin, and even revitalized memory. Relative to mice that sniffed micelles loaded with control antibodies, those that received TTCM-ms were able to better recognize novel objects, and remember which arms of a maze they’d explored before. Looking closer at synapses in the mouse brain with immunofluorescence, the researchers found that TTCM2-ms treatment reduced the amount of tau aggregates crowding synpases by a third, while doubling total synapse numbers. This is critical, Kayed said, because recent studies have implicated synaptic tau oligomers as a culprit in the spread of tau pathology (May 2023 news; Oct 2023 news).
Targeting Tau for Destruction. TTCM2-ms slip into cells, where the antibody latches onto various forms of pathological tau. TRIM21 binds the Fc fragment of the antibody, and ubiquitinates it, targeting the entire complex to the proteasome. [ Courtesy of Rakez Kayed, UTMB.]
How did TTCM2-ms find and destroy its intracellular targets? Gaikwad found that this depended on TRIM21, a cytosolic Fc receptor that also serves as an E3 ubiquitin ligase. It whisks antibodies that end up in the cell, and their cargo, to the proteasome for destruction. Broadly expressed in neurons and other cell types, this atypical Fc receptor may have evolved to deal with viruses that bust into the cytoplasm with antibodies clinging to their capsids (McEwan, 2016). Gaikwad found that in seeding assays in tau biosensor cell lines, knocking down TRIM21 thwarted the seed-stopping effects of TTCM-ms. In hTau mice treated intranasally with TTCM-ms, Gaikwad found TTCM2, TRIM21, and tau aggregates comingling within neurons. The findings jibe with a recent report that TRIM21 is required for the effectiveness of tau immunotherapies (Mukadam et al., 2023).
Kayed told Alzforum that his lab continues to investigate the mechanisms involved in the coordinated takedown of intracellular tau by TTCM2 and TRIM21. With an eye toward clinical development, TTCM2 has been fully humanized.
Other scientists with their sights on intracellular tau are taking a leaf out of the TRIM21 book. In Lisbon, Bengt Winblad of the Karolinska Institute, Stockholm, described how he developed proteolysis targeting chimeras, aka, PROTACs, for tau. First described more than 20 years ago, these engineered molecules comprise three connected parts: a ligand that binds to a target of interest, e.g., tau; a linker; and a ligand that binds an E3 ubiquitin ligase (Sakamoto et al., 2001). Once the PROTAC binds the target and the ligase, the latter adds ubiquitin, diverting the target to the proteasome for disposal. Winblad has generated a library of these molecules, pairing small molecules that latch onto paired helical filaments of tau, with others that ensnare an E3. He is currently testing out the top contenders in neuronal cell culture studies and mouse models of tauopathy.
Attack of the PROTACs. A PROTAC links a protein-binding domain with a ligand for E3 ubiquitin ligase. Once both the protein target and E3 are bound, E3 adds ubiquitin residues to the target protein, relegating the whole complex to proteasomal degradation (right). [Courtesy of Bengt Winblad, Karolinska Institute.]
Others are using the PROTAC method to promote the proteasomal degradation of tau-targeted single-domain antibodies, aka nanobodies. Produced naturally by camelids such as camels and llamas, these pared-down antibodies contain only a single variable heavy domain (VHH). Like full-size antibodies, they are exquisitely specific for their targets, however, they are small enough to easily slip across the blood-brain barrier and even enter cells via bulk endocytosis. Buée has generated a library of such nanobodies against tau. Previously, he reported that one of them, Z70, recognizes the filament-driving, microtubule-binding region of tau, and that it thwarted tau aggregation intracellularly and vanquished tauopathy in a mouse model (May 2023 conference news). In Lisbon, Buée said that preliminary findings from his lab suggest that the efficiency of nanobodies can be bolstered significantly by rigging them up with PROTACs, in which the anti-tau VHH serves as the tau-nabbing portion of the PROTAC. Unlike the full-size antibodies that Kayed smuggles into cells with micelles, nanobodies lack the Fc domain that binds TRIM21, making the PROTAC approach critical to rev proteasomal degradation of the nanobody and its cargo.
Although he maintains that intracellular targeting is critical to stop the progression of tau pathology, Buée has also explored whether nanobodies might squelch tau inside of cells by preventing its uptake from the outside. While he found that Z70 blocked uptake, another nanobody, H3-2, did so even more efficiently. Upon binding to tau’s C-terminus, H3-2 forms a dimer, effectively preventing tau from being taken up into cells. He did not explain how the single-chain nanobody dimerizes, but he said other nanobodies do not do this. The findings raise the possibility of using combinations of nanobodies to interfere with different stages of tau propagation.
Einar Sigurdsson of New York University also uses the single-domain antibody approach to target both tau and α-synuclein pathologies (Congdon et al., 2022). At previous meetings, and more recently in preprint articles, he reported that PROTACs significantly enhanced the clearance of pathogenic targets in mouse models of tauopathy and synucleinopathy (May 2023 conference news; Jiang et al., 2024; Sigurdsson et al., 2024). In Lisbon, he reiterated that, and also reported on yet another approach, expressing nanobodies from viral vectors. In the A53T mouse model of synucleinopathy, intravenous injection of an adeno-associated virus vector carrying a gene for an α-synuclein-specific sdAb not only prevented synucleinopathy, but reversed it in older mice that had substantial Lewy body pathology.—Jessica Shugart
Mukadam AS, Miller LV, Smith AE, Vaysburd M, Sakya SA, Sanford S, Keeling S, Tuck BJ, Katsinelos T, Green C, Skov L, Kaalund SS, Foss S, Mayes K, O'Connell K, Wing M, Knox C, Banbury J, Avezov E, Rowe JB, Goedert M, Andersen JT, James LC, McEwan WA.
Cytosolic antibody receptor TRIM21 is required for effective tau immunotherapy in mouse models.
Science. 2023 Mar 31;379(6639):1336-1341. Epub 2023 Mar 30
PubMed.
Mouse Models and Markers for Cerebral Amyloid Angiopathy, ARIA
With people now being treated with amyloid immunotherapy in the clinic, reducing the risk of ARIA has taken on a new sense of urgency. This inflammatory side effect occurs in people who have amyloid in small to medium-sized blood vessels of the brain, aka cerebral amyloid angiopathy. However, CAA cannot be easily detected, making it hard to screen patients for the condition. To better manage amyloid immunotherapy, scientists are seeking fluid biomarkers for vascular amyloid, as well as ways to mitigate the risk of microhemorrhages that underlie ARIA. They would also like better mouse models for both CAA and ARIA. At the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 5 to 9 in Lisbon, Portugal, progress in all these areas impressed attendees.
Shinobu Kitazume of Fukushima Medical University, Japan, debuted a mouse model with extensive vascular amyloid deposits that could be used to study CAA, while Thierry Bussiere of Biogen described mice that develop ARIA-like vascular lesions. On the biomarker front, Marcel Verbeek of Radboud University Medical Center, Nijmegen, The Netherlands, suggested the protein TIMP4, aka tissue inhibitor of metalloprotease-4, as a possible CAA biomarker. Regarding mitigation, Anna Bonaterra-Pastra of the Vall d'Hebron Research Institute, Spain, reported that intravenous injections of Apolipoprotein J, aka clusterin, in aged amyloidosis mouse models cut the number of microhemorrhages in half, perhaps by suppressing matrix metalloproteases that damage blood vessels.
The talks attracted a lot of interest. Costantino Iadecola of Weill Cornell Medical College, New York, believes the two mouse models will advance research. “Both may tell us more about the dynamics of Aβ vascular accumulation and clearance, and help us better understand the impact that Aβ antibodies may have on vascular amyloid,” he told Alzforum.
Mice With CAA? APP knock-in mice (bottom) have parenchymal amyloid (green) and none in blood vessels (red), but when crossed with mice expressing endothelial APP (top), amyloid preferentially deposits in vessels (overlay appears yellow, right). [Courtesy of Shinobu Kitazume, Fukushima Medical University.]
Are Endothelial Cells the Key to Vascular Amyloid?
Recent data suggest that before anti-amyloid antibodies begin removing Aβ from plaques, they first interact with amyloid in the vasculature, triggering inflammation that leads to damaged blood vessels, edema, and microhemorrhages (Aug 2023 conference news). Autopsies of people who died with ARIA jibe with this, showing inflamed blood vessels in people with severe CAA (Jan 2024 news). It has been difficult to study CAA and ARIA in mice, however, since they do not develop much vascular amyloid.
Endothelial APP. Neurons make a short, 695 amino acid version of APP (top), while endothelial cells (bottom) make a long 770 amino acid version containing two extra domains, Kunitz protease inhibitor (tan) and OX-2 antigen (purple). [Courtesy of Shinobu Kitazume, Fukushima Medical University.]
In Lisbon, Kitazume suggested that this species difference comes down to endothelial cells. In people, endothelial cells make a version of amyloid precursor protein, APP770, that is longer than the one neurons make—APP695. Kitazume and colleagues generated an antibody specific for this longer version, which contains two additional domains near the N-terminal end totaling 75 amino acids (see image above at right). Immunostaining revealed APP770 coating blood vessels in the human cortex (Kitazume et al., 2010; Kitazume et al., 2012; Miura et al., 2020). This implies that endothelial cells might be a primary source of vascular amyloid, Kitazume noted. The secreted α/β cleavage product sAPP770 is abundant in human blood, with sAPP770α predominating, but nearly absent from cerebrospinal fluid, in keeping with an endothelial origin. By contrast, rodents have almost no sAPP in their blood, hinting that their endothelial cells make little APP.
Could boosting endothelial APP production in mice promote vascular amyloidosis? To test this, Kitazume and colleagues generated mice that expressed human APP770 specifically in their endothelial cells. These EC-APP770+ mice pumped out high levels of serum sAPP770. At two years of age, they had about six times as much Aβ40 in their blood as did wild-type mice. However, they did not deposit cerebrovascular amyloid.
To spur amyloidosis, the researchers crossed the mice with APPNL-F knock-ins. That did the trick. By 15 months of age, the offspring had developed extensive vascular deposits in the cortex, particularly in meningeal blood vessels (see CAA image above; Tachida et al., 2022). When the researchers crossed EC-APP770+ mice with APPNL-G-F knock-ins, which have more aggressive amyloidosis, vascular amyloid formed by 7 months of age.
“I think these might be great models to study ARIA,” noted Cynthia Lemere at Brigham and Women’s Hospital, Boston, one of the ADPD organizers. For example, in the EC-APP770+/ APPNL-F crosses, endogenous IgG antibodies coated vascular deposits, suggesting the potential for an inflammatory response. EC-APP770+ mice are available from RIKEN.
Mouse ARIA Resembles Human
Bussiere took a different approach to model ARIA. Biogen researchers treated 5xFAD mice with weekly injections of aducanumab, gantenerumab, or 3D6, the mouse version of bapineuzumab, for up to 12 weeks. The researchers assessed ARIA using 7 or 9.4 T MRI scans.
ARIA incidence varied with dose and antibody. With all mice on 5 or 10 mg/kg developing edema, 3D6 produced the most. On 50 mg/kg aducanumab, half the mice got ARIA, and on 50 mg/kg gantenerumab, about a third did. On MRIs, the researchers saw bright spots indicating vascular lesions, as well as more diffuse patches of edema. As in people, edema either resolved with time, or worsened and caused blood vessels to spring leaks. As with human ARIA, large hemorrhages were rare.
Histochemical analysis revealed damage to meningeal arterioles, including the presence of enlarged macrophages containing amyloid, and patches of the plasma protein fibrin in degenerating vessel walls. These morphological changes correlated with MRI signals. Overall, vascular changes in 3D6-treated mice resembled those in people with severe ARIA, Bussiere noted (Solopova et al., 2023; Castellani et al., 2023). The model reproduces many of the characteristics of human ARIA and could be useful for research, he concluded.
Iadecola agreed. “I find the data of interest because they used MRI to assess brain injury and edema, which is relevant to the way ARIA is diagnosed in humans,” he told Alzforum. Lemere noted the importance of being able to detect both ARIA-E and ARIA-H in mouse models. Her group has seen ARIA-H on MRI, but so far not ARIA-E. “It was nice to see it can be done,” she told Alzforum.
Fluid Biomarkers for CAA
In the clinical setting, detecting CAA would help identify people who are not good candidates for amyloid immunotherapy (Aug 2023 conference news). In a meta-analysis of 170 studies covering 73,000 people, Verbeek found that almost a quarter of people over the age of 55 have moderate to severe CAA on postmortem analysis, and most of those cases are missed on MRI. The majority are people with cognitive impairment, of whom many would be eligible for anti-amyloid immunotherapy (Jäkel et al., 2021).
What fluid markers correlate with CAA? Numerous studies have flagged matrix metalloproteases and their inhibitors. This large family of proteins chews up extracellular matrix, aiding tissue repair. In CAA, expression and activity of several MMPs, particularly 2 and 9, rises, correlates with degradation of the extracellular matrix and damage to blood vessels (Jung et al., 2003; Zhao et al., 2015). MMP inhibitor expression also goes up, perhaps in compensation. One MMP inhibitor, TIMP4, caught Verbeek’s eye as a potential CAA marker because it is specifically expressed in brain and heart, and rises in the plasma in people with vascular dementia.
Microhemorrhage Marker? Metalloprotease inhibitor TIMP4 (brown) is absent from healthy blood vessels (left), elevated in cerebral amyloid angiopathy (middle), and higher still in CAA vessels with microhemorrhages (right). [Courtesy of Lieke Jäkel, Radboud University.]
First, Verbeek investigated whether TIMP4 was expressed in vessels with CAA. His group examined human postmortem occipital lobe sections from 39 people with CAA, 18 with CAA and intracerebral hemorrhages (ICH), and 42 unaffected controls. Their average age was 78. As he had hypothesized, people with CAA had more TIMP4 in cerebral blood vessels than did controls. Those with CAA-ICH had even more (image above). TIMP4 associated with neither plaques nor tangles, nor with superficial siderosis, i.e., the leakage of blood on the surface of the brain.
To find out if TIMP4 would make a good fluid biomarker, the researchers measured it in CSF and in serum from 38 people with CAA and from 37 controls. The CAA cohort had more TIMP4 in the serum, and less in the CSF. The worse the CAA, the lower CSF TIMP4 was. This pattern resembles that of Aβ, which drops in CSF as the protein deposits in plaques. In fact, TIMP4 correlated with Aβ40 in CSF, albeit weakly, with an r value of 0.3.
The ratio of CSF/serum TIMP4 distinguished CAA and control groups better than did CSF or plasma TIMP4 alone, but the overlap between the groups was large. TIMP4 could be combined with other markers to create a more specific measure, Verbeek suggested. He previously reported that several Aβ peptides, including Aβ38, Aβ40, Aβ42, and Aβ43, are lower in CAA CSF than in CSF from AD patients or controls (De Kort et al., 2023; van den Berg et al., 2024). “Combined with TIMP4, these might constitute a specific panel to identify CAA and predict ARIA,” he suggested in Lisbon.
Protection Against Blood Vessel Damage?
Vascular breakdown and larger hemorrhages are the most dangerous aspect of ARIA. Could these be prevented? Bonaterra-Pastra and Mar Hernández-Guillamon, also at the Universitat Autònoma, have proposed ApoJ as a candidate therapy because this chaperone co-deposits with vascular amyloid, and is elevated in the plasma of CAA patients (Camacho et al., 2019; Bonaterra-Pastra et al., 2023). Hernández-Guillamon and colleagues previously found that treating 14-month-old APP23 mice with recombinant human ApoJ prevented the accumulation of plaques and vascular amyloid (Fernández de Retana et al., 2019).
To find out if ApoJ could ameliorate established CAA, Bonaterra-Pastra moved to 21-month-old mice, which develop extensive vascular amyloid and spontaneous microhemorrhages. She injected 1 mg/kg recombinant human ApoJ twice weekly into mouse veins for three months. This cut the number of microhemorrhages from an average of 12 per mouse to six, as seen by MRI. For larger bleeds, those greater than 300 μm in diameter, there was an even bigger drop, to about a sixth as many as in untreated mice.
In addition, ApoJ-treated mice had half as many damaged, fibrinogen-positive blood vessels in their cortex, and more healthy smooth muscle actin cells, compared to untreated controls, indicating less damage to vessel walls. There was no difference in the amount of vascular amyloid, however, indicating the treatment was not clearing it.
How, then, did ApoJ protect mice? Bonaterra-Pastra found less MMP12 in treated animals. The amount of MMP12 correlated with the number and volume of microhemorrhages in both treated and untreated mice, at r=0.6 to 0.8. Other studies have linked MMP12 to blood-brain barrier breakdown as well (Power et al., 2003; Wells et al., 2005; Chelluboina et al., 2015). In Lisbon, Lemere likewise reported that MMP12, along with the complement protein C3, were the two most upregulated proteins in mice that developed microhemorrhages after treatment with 3D6.
To find out if the same might be happening in people, Bonaterra-Pastra analyzed MMP12 levels in plasma from 22 volunteers with lobar ICH, 18 with hemorrhages in deeper brain regions, and 17 healthy controls. Lobar ICH associates with CAA, while deeper bleeds do not. She found no consistent difference in MMP12 levels between groups. However, in the lobar ICH group, the amount of MMP12 correlated with larger, irregularly shaped hemorrhages. That, in turn, correlated with a worse prognosis. Data presented by Ines Hristovska of Lund University, Malmö, Sweden, supported this idea. She showed that in the BioFinder study MMP12 is elevated in people who have microbleeds or white-matter lesions.
Bonaterra-Pastra thinks ApoJ could protect against microhemorrhages by lowering MMP12, and that it would be worth testing if giving patients ApoJ before amyloid immunotherapy could lessen the risk of ARIA.
“Her data look exciting, both in terms of a potential mitigation strategy for ARIA and for blood-based biomarkers for CAA, both of which are sorely needed,” Lemere told Alzforum.—Madolyn Bowman Rogers
van den Berg E, Kersten I, Brinkmalm G, Johansson K, de Kort AM, Klijn CJ, Schreuder FH, Gobom J, Stoops E, Portelius E, Gkanatsiou E, Zetterberg H, Blennow K, Kuiperij HB, Verbeek MM.
Profiling amyloid-β peptides as biomarkers for cerebral amyloid angiopathy.
J Neurochem. 2024 Jul;168(7):1254-1264. Epub 2024 Feb 16
PubMed.
At ADPD, Scientists Dissect the Ins and Outs of Tau Propagation
The revelation that tau aggregates can pass between cells, corrupting their intracellular counterparts, changed how the Alzheimer’s field studies tau pathology. At this year’s AD/PD meeting, held in March 5-9 in Lisbon, Portugal, researchers focused less on how the microtubule-binding protein travels from place to place, and more on what happens after it arrives. For example, they presented evidence that an ATPase—valosin containing protein—plays decisive roles in dismantling, and amplifying, incoming tau seeds in the cytoplasm. Others described how intracellular tau seeding takes place within astrocytes, and that blocking the release of extracellular vesicles, including those carrying tau, curbs tau pathology in the mouse brain. Further evidence implicated tau oligomers, rather than tangles, in neuronal demise. The focus on the intracellular mechanisms underlying tau propagation comes at a time when preclinical and clinical drugs’ discovery efforts increasingly focus on intracellular tau (see Part 3 of this series).
Seed Fate Hinges on VCP
A decade has passed since Marc Diamond of UT Southwestern in Dallas generated the now widely used tau biosensor cell lines, which detect intracellular tau seeding via FRET signals emitted when two molecules of tau snap together (Oct 2014 news). Since then, Diamond and other researchers have used these HEK293 cells, along with mouse models, to investigate the mechanisms involved in the seeding and propagation of tau pathology. Most recently, his lab reported that tau aggregates spread retrogradely—from post- to presynapses —between neurons (Jun 2023 news).
In Lisbon, Diamond focused on the intracellular events that go on after tau passes from one cell to another. He noted that most internalized tau aggregates are destined for degradation in the endolysosomal system. However, according to recent work from the lab, a small proportion of tau escapes the confines of these digestive vesicles and crosses into the cytosol, where they corrupt endogenous tau (Dodd et al., 2022). Once set in motion, the seeds replicate in the cytoplasm with incredible efficiency, he said. “The question of the day is, what is the cellular machinery that allows this to happen?”
To answer this, postdoc Sushobhna Batra and colleagues ran a screen for proteins that greet tau fibrils upon their arrival in the cytoplasm. They used the so-called split APEX2 system developed by Alice Ting at Stanford University. Essentially, instead of using FRET to detect tau aggregation, this system brings together two parts of a peroxidase enzyme when two units of tau join. Then, by adding biotin-phenol along with peroxide, all proteins in proximity to the tau pair become biotinylated, allowing the researchers to isolate and identify them. This way, Batra hunted for proteins associated with the earliest stage of tau seeding, at five hours after fibril exposure. The top hit, by several orders of magnitude, was valosin containing protein (VCP). The AAA+ ATPase supports a multitude of cellular processes. Chief among them is the plucking of individual proteins from membranes or larger complexes for the purpose of refolding, recycling, or disposal. The protein has also been tied to neurodegenerative tauopathies, and just last year, Diamond, in collaboration with Ulrich Hartl of the Max Planck Institute of Biochemistry, nabbed VCP as a protein that disaggregates tau fibrils (Saha et al., 2023; Apr 2023 conference news).
How might this disaggregase influence tau seeding? In biosensor cell lines, Batra found that lowering VCP expression cut tau seeding efficiency by half. Treatment with the VCP inhibitor NMS-873 had a similar effect. Surprisingly, ML240, a different VCP inhibitor, did the opposite. It skyrocketed seeding from 2 to 3 percent of cells, to 95 percent of cells. Neither of these inhibitors had any effect on tau seeding if given eight or more hours after seed exposure, implying that VCP plays a role in the earliest stages of seeding. The inhibitors had similar effects in iPSC-derived neurons expressing the tau biosensor constructs.
What explains the opposing effects of the VCP inhibitors? A potential explanation emerged when Batra methodically knocked down each of the 30 known VCP cofactors in HEK293 biosensor cells. While most had no effect on seeding, one, called FAF2, increased seeding by 30 to 40 percent when knocked down, while six others did the opposite. Diamond proposed a model whereby some cofactors help VCP yank tau monomers from the end of a growing fibril, relegating them to the proteasome for degradation and slowing seeding. In contrast, other VCP cofactors might help the enzyme pluck tau monomers from the middle of a fibril, thus splitting the fibril in half and doubling the number of seeds, effectively accelerating seeding (image below). Diamond suggested that perhaps NMS-873 inhibits the seed-promoting VCP cofactors, while ML240 hinders the enzyme’s seed-stopping partners.
Seed Model. When tau seeds enter the cytoplasm, they encounter VCP. In this model, depending on which cofactors VCP is associated with, the ATPase removes monomers of tau from the end of the fibrils (bottom half), leading to fewer seeds, or from the middle of fibrils (top half), leading to more seeds. [Courtesy of Marc Diamond, UT Southwestern.]
Together, the inhibitor and knockdown findings paint VCP as a nexus that determines the fate of tau seeding, Diamond said. While the processes that govern expression of different VCP cofactors remain a mystery, Diamond told Alzforum that cofactor availability could contribute to selective vulnerability of some neurons to tau pathology. As such, Diamond believes VCP cofactors could make good druggable targets. What’s more, ongoing work in his lab suggests this VCP-related mechanism might be afoot not only for propagation of tau, but also for other proteopathic proteins, such as α-synuclein and TDP-43. He noted that James Shorter of the University of Pennsylvania in Philadelphia previously uncovered a similar relationship between Hsp104—a yeast AAA+ATPase—and the seeding of prion and prion-like proteins, and between nuclear transportins and seeding of RNA-binding proteins such as FUS in animals (Aug 2014 news; May 2017 news).
Astrocytes as Tau Seed Incubators
Neurons aren’t the only cells that deal with the menace of tau aggregates. Astrocytes have been found to shoulder the lion’s share of tau pathology in several neurodegenerative diseases, particularly in 4R tauopathies such as progressive supranuclear palsy, aging related astrogliopathy (ARTAG), and even in AD, noted Aurélien Lathuilière of the University of Geneva (Kovacs et al., 2016; Oct 2023 news; Nov 2020 news). The glial cells also express known tau aggregate receptors, such as LRP1 and heparin sulfate proteoglycans, at higher levels than their neuronal counterparts. For these reasons and more, Lathuilière said, studying tau uptake, seeding, and aggregation within astrocytes is critical. To do this, he created an astrocyte version of Diamond’s biosensor cell lines. In the astrocytoma line CCF-STTG1, Lathuilière used a lentiviral vector to stably express P301L tau fragments fused with complementary FRET probes, which, as a postdoc in Bradley Hyman’s lab at Massachusetts General Hospital in Charlestown, he had previously modified to enhance sensitivity to seeds (Lathuilière et al., 2023).
In Lisbon, Lathuilière reported that these astrocyte biosensor cells readily internalized tau seeds from AD brain lysates. Using immunostaining to detect the internalized phospho-tau, he found the internalized material in a dotted pattern, suggesting it inhabited vesicles of some kind. Staining with LAMP1 indicated that while some of these tau-carrying compartments were lysosomes, many others were not. Lathuilière suggested they could be endosomes destined for lysosomal fusion. Next, Lathuilière measured seeding efficiency of the internalized seeds via FRET. He found that inhibiting lysosomal function with hydroxychloroquine dramatically enhanced seeding within astrocytes. Revving up metabolic stress by feeding the astrocytes fatty acids also enhanced tau seeding. Blocking the LRP1 receptor only dampened tau aggregation by 30 percent, suggesting that other receptors, in addition to LRP1, help internalize tau seeds.
Nuclear Tau? Tau aggregates associated with the nuclear membrane in a small proportion of astrocyte biosensor cells. [Courtesy of Aurélien Lathuilière, University of Geneva.]
Lathuilière was in for a surprise when he looked closer at the tau aggregates that formed within the biosensor astrocytes. While most of the visible inclusions resided in the cytoplasm, in about 10 percent of the cells, aggregates formed within the nucleus, where they associated with the nuclear membrane (image above). Tau has been spotted loitering in and around the nucleus before, including within nuclear speckles, which are hubs for the cell’s splicing machinery (Sep 2018 news; Jan 2019 news; Apr 2021 news). However, Lathuilière said that the pattern of tau’s association with the nucleus in the astrocyte biosensor cells appeared far more extensive compared to previous reports. Ongoing studies in his lab are focused on understanding this odd form of tau aggregation.
Finally, Lathuilière treated the astrocytes with high-molecular-weight tau oligomers extracted from human AD brain lysates, which he believes are the most potent form of tau seeds. He then looked for changes in their transcriptomes. When faced with tau seeds, the cells adopted a gene-expression profile much like astrocytes in the AD brain. This suggests that these astrocytoma cells approximate physiological responses to tau pathology, Lathuilière said.
Given the proposed endolysosomal localization of tau aggregates in the astrocytes, Doo Yeon Kim of Massachusetts General Hospital asked Lathuilière if he had investigated whether astrocytes might secrete tau seeds via exosomes, which derive from lysosomal compartments. Lathuilière said that his lab is studying if and how astrocytes secrete internalized tau seeds.
Previously, Roberto Piacentini of Università Cattolica del Sacro Cuore in Rome found that, relative to neurons, astrocytes have a voracious appetite for tau oligomers, which the cells internalize via glypican-4 (GPC4), a heparin sulfate proteoglycan. Once internalized, the oligomers bungle the release of gliotransmitters, which causes synaptic dysfunction in associated neurons (Dec 2018 conference news). In Lisbon, Piacentini reported that none other than the C-terminal domain of amyloid precursor protein, AICD, induces the expression of GPC4. Once cleaved from APP, AICD binds directly to the GPC4 promotor, Piacentini said. When astrocytes were deprived of APP, they downregulated GPC4 and lost their appetite for tau oligomers, curbing synaptotoxicity nearby. Piacentini reported some of this last year (Puliatti et al., 2023). His work suggests that when astrocytes take up tau oligomers directly, the cells sour neuronal function indirectly. No spewing of tau seeds required.
Stopping Tau in its Tracks
For better or worse, there is plenty of evidence that tau aggregates do travel, and that they ride in extracellular vesicles. Seiko Ikezu and Tsuneya Ikezu of the Mayo Clinic in Jacksonville, Florida, previously reported that tau trafficked to neurons via exosomes released from microglia (Oct 2015 news). The researchers later pegged the ATP-gated cation channel, P2X purinoceptor 7 (P2RX7), with triggering exosomal secretion in microglia, and reported that a P2RX7 inhibitor reduced tau accumulation and rescued memory loss in PS19 mice (Ruan et al., 2020). Later, Seiko Ikezu reported similar benefits could be had by deleting from microglia TSG101, a protein that forms extracellular vesicles (Apr 2023 conference news).
At AD/PD, she focused on P2RX7. Scientists in her lab have generated mice lacking the receptor, and have crossed them to PS19 tauopathy mice. At 9 months of age, the hippocampal region of PS19 controls is inundated with intracellular, hyperphosphorylated aggregates of tau. Deleting the cation channel substantially squashed this pathology, with one exception: the mossy fiber region, where P2RX7 knockouts had a higher burden of tau aggregates (image below). Ikezu does not have an explanation for this, but noted that mossy fibers typically have high expression of P2RX7. In a barrage of behavioral tests, deletion of P2RX7 restored the flagging learning and memory in PS19 mice to wild-type levels, suggesting an overall salubrious effect. It also prevented shrinkage of the cortex and hippocampus.
No P2RX7, No Problem. PS19 mice have profound brain shrinkage (left) and accumulation of hyperphosphorylated tau (AT8, pink) runs amok in several regions of the hippocampus (top right). Both problems are substantially reduced in PS19 mice lacking the P2RX7 cation channel (P2rx7-/-). [Courtesy of Seiko and Tsuneya Ikezu, Mayo Clinic.]
Did P2XR7 knockout influence the quality and quantity of extracellular vesicles released by cells in the brain? Ikezu has some preliminary findings on this. For one, cryo-EM analysis of EVs isolated from the mouse brain revealed that, compared to wild-type animals, PS19 mice had larger EVs, and more of them. Knocking out P2RX7 restored EV size and quantity back to wild-type levels. A proteomic analysis uncovered nearly 200 proteins that were more abundant in PS19 EVs relative to wild-type, but these were normalized by P2RX7 knockout. Tantalizingly, MAPT and ApoE were among these proteins, as were several involved in mitochondrial function. Finally, Ikezu expressed a fluorescently tagged EV marker—CD9—under control of either microglial or neuronal promoters, to assess how P2RX7 influenced EV release from both cell types. She reported a profound reduction in EV release from both, with the strongest effect in microglia. In ongoing work she is focused on deciphering which cell types and subtypes are responsible for P2RX7-mediated spread of tau pathology in EVs.
“How EVs secreted from each neuronal cell type affect each other is still a mystery, and many investigators, including in our own lab, are trying to shed light on intercellular communication by EVs,” Ikezu told Alzforum. She and colleagues are also developing P2RX7 inhibitors with an eye toward clinical development. In addition to carting tau aggregates, EVs also transport inflammatory molecules, such as cytokines, between cells, so it’s possible that inhibiting EV release with P2RX7 inhibitors might stem both tau propagation and inflammation, she said.
One attendee asked Ikezu whether blockage of EV secretion altered the overall state of microglia. Ikezu said that they are planning single-cell RNA-Sequencing studies to answer this question.
Invisible Tau and Shunned Neurons
Which forms of tau promote propagation of pathology across the brain, and which are most harmful to neurons? These questions were recurrent throughout this year’s AD/PD meeting, with several scientists taking the view that oligomers, as opposed to fibrils, play a leading role. Case in point, Hyman recently reported that both fibrillar and oligomeric tau are capable of seeding tau aggregation when injected into the PS19 mouse brain. However, he found that of the two, high-molecular-weight oligomers were far more adept at triggering widely disseminated tau aggregates (Mate de Gerando et al., 2023). In Lisbon, Hyman shifted gears away from seeding, and focused instead on another fundamental question: Which species of tau aggregates are neurotoxic? He noted that while correlations between tau tangles and neurodegeneration are rock-solid at the brain region level, some findings suggest that this correlation might not hold up at the level of individual cells. Hyman suspects that oligomeric tau, which he calls “the tau we cannot see,” could be more neurotoxic than bona fide tangles.
Dying Alone. Neighboring neurons appear to scoot away from a neuron on the precipice of death (center). [Courtesy of Bradley Hyman, Massachusetts General Hospital, Boston.]
To investigate, the Hyman lab used in vivo multiphoton imaging to track the growth of tangles, as well as the loss of individual neurons, over time. In both rTg4510 and Thy-tau22 mouse models, the researchers used a fixed camera to check in on the same neurons week after week. When a neuron turned up missing, it was presumed dead, and researchers could then refer back to images from earlier sessions to look for signs of that neuron’s impending demise. Using this technique, they found that, as expected, the number of tangle-bearing neurons increased week after week. Neuronal death was a rare event, but the researchers were able to document 64 incidents in rTg4510 mice over four weeks. Strikingly, they found that few doomed neurons had tangles prior to their passing. Rather, the rate of neuronal loss was threefold higher among neurons without tangles than it was for neurons with them. Hyman said the findings suggest that tangles are not a death sentence, and that their formation might even be a marker of resilience.
If not tau tangles, were there any other telltale signs of impending neuronal death? Indeed, Hyman and colleagues found that their closest neighbors appeared to ditch them at their darkest hour. Using three-dimensional longitudinal imaging, the researchers found that one to two weeks before a neuron disappeared, the space between the dying neuron and its nearest neighbors increased by up to 60 percent, creating a “hole” surrounding the ill-fated neuron. Hyman still doesn’t know how this happens, but he wondered if this morphological feature could help identify and study dying neurons in postmortem human brain samples. In support of that idea, in thick, cleared sections from AD brain samples, the researchers spotted these holes surrounding about 2 percent of neurons, roughly matching the calculated rate of neuronal loss in the AD brain. These loners rarely had tau tangles. In control brains, few if any of these deserted neurons were spotted. Hyman hopes to use this marker to study how tau tangles, and other characteristics, relate to neuronal death in the human brain.—Jessica Shugart
Multiple Strategies Seek to Banish α-Synuclein Aggregates
For synucleinopathies, which include Parkinson’s disease, multiple system atrophy, and dementia with Lewy bodies, scientists currently have no way to slow the underlying disease progression. Many groups are on the hunt for ways to prevent or clear α-synuclein aggregates. At this year’s AD/PD meeting, held March 5 to 9 in Lisbon, Portugal, speakers presented a variety of approaches that are in early stage trials or heading that way. Some use small molecules to break up aggregates. These include an oligomer-busting drug entering Phase 2, and a new candidate that disassembles fibrils by targeting a co-aggregated synaptic protein. Other approaches use antibodies, but with a twist. One immunotherapy strategy, rather than directly targeting intracellular α-synuclein, blocks its spread by binding a cellular receptor. Another antibody binds only the nitrated form of α-synuclein, which is linked to toxicity. While it remains to be seen which approaches will pan out, the talks demonstrated that synucleinopathy researchers have disease modification squarely in their sights.
Stopping Spread by Blocking Uptake
Immunotherapy approaches against α-synuclein face a challenge—unlike in Alzheimer’s disease, where plaques are extracellular, α-synuclein deposits form inside cells. Antibodies are large molecules that do not readily cross the plasma membrane and thus may not reach aggregates. One solution is to target α-synuclein spread rather than the protein itself.
One Receptor, Multiple Actions. α-Synuclein aggregates from neurons (blue) bind Toll-like receptor 2 (red) on other cells, leading to internalization of the aggregates and causing death in other neurons, demyelination in oligodendrocytes, and neuroinflammation in microglia. [Courtesy of Seung-Jae Lee, Neuramedy.]
Toll-like receptor 2 (TLR2) has been blamed for bringing α-synuclein aggregates into neurons, as well as for sparking harmful inflammation in microglia (Apr 2020 conference news; Oct 2020 news; Kim et al., 2021). The antibody tomaralimab, developed by Opsona Therapeutics, Ireland, and acquired by the South Korean company Neuramedy, binds TLR2 and blocks its activity. It lowered α-synuclein deposits and gliosis while improving motor abilities in a mouse model of neuronal synucleinopathy (Apr 2022 conference news). In Lisbon, Seung-Jae Lee of Seoul National University, South Korea, extended the findings to multiple system atrophy (MSA).
In this fast-progressing synucleinopathy, aggregates known as glial cytoplasmic inclusions (GCIs) form in oligodendrocytes. As in Parkinson’s disease, dopaminergic neurons die, and motor abilities deteriorate. Lee, who works for Neuramedy, noted that the origin of GCIs has always been mysterious, because oligodendrocytes make very little α-synuclein. The leading hypothesis is that these cells take up α-synuclein aggregates released by neurons.
Supporting this idea, Lee found that cultured human oligodendrocytes internalized α-synuclein oligomers added to their media, triggering GCI formation (Yoon et al., 2020). Similarly, in mice that expressed human mutant A53T α-synuclein only in neurons, phosphorylated α-synuclein showed up in oligodendrocytes, too, indicating it had passed between cells.
Lee theorized that TLR2 was the gateway for α-synuclein entry into oligodendrocytes. In keeping with this, pretreating cultured oligodendrocytes with tomaralimab before adding α-synuclein prevented GCIs, he reported. Likewise, injecting A53T mice with 10 mg/kg tomaralimab weekly from 6 to 10 months of age cut phosphorylated α-synuclein in white matter by half. Tomaralimab also prevented gliosis. Treated mice maintained their grip strength and balance, and they survived as long as did wild-type mice.
Lee next wanted to know how α-synuclein damages oligodendrocytes. RNA-Seq analysis of cultured human oligodendrocytes, as well as postmortem samples from MSA patients, found that glia containing GCIs were likelier than healthy glia to have expression profiles characteristic of immature cells. In particular, they expressed fewer myelination genes. This suggested that α-synuclein triggers oligodendrocytes to de-differentiate, causing demyelination. Treating cultured oligodendrocytes with tomaralimab restored their mature phenotype, Lee found. In mice, treatment maintained healthy myelin.
Targeting TLR2 could have multiple benefits in synucleinopathy, keeping neurons healthy, myelin robust, and gliosis down, Lee concluded. Clinicaltrials.gov lists a planned Phase 1 trial of tomaralimab in healthy volunteers in the U.K. to test safety and pharmacokinetics, but Lee told Alzforum that study has been shelved. Instead, Neuramedy will run the Phase 1 trial in South Korea. Lee said the company will focus first on MSA trials, rather than PD, as the former is faster-progressing and may soon have a PET tracer available (Mar 2022 conference news; Smith et al., 2023).
Disease Marker? In CSF from Parkinson’s patients (red, left) total α-synuclein is only a little lower than in healthy controls (blue). In contrast, nitrated α-synuclein (right) in patient CSF dwarfs that in control CSF. [Courtesy of Sheerin Shahidi-Latham, Nitrase Therapeutics.]
Could Nitrated α-Synuclein Hold the Key to Toxicity?
In contrast to this indirect TLR2 immunotherapy strategy, Sheerin Shahidi-Latham of the biotech Nitrase Therapeutics, Brisbane, California, proposed directly binding α-synuclein—but only its nitrated form. Nitrated α-synuclein was first implicated in neuron damage in PD more than two decades ago, but few have pursued this target (Giasson et al., 2000; Yu et al., 2010; Apr 2011 conference news).
In Lisbon, Shahidi-Latham made a case for nitration being tightly linked to disease. She reported that nitrated α-synuclein (nSyn) was present in cerebrospinal fluid from 50 PD patients, but virtually absent from the CSF of 50 healthy controls (see image above). Moreover, having more than 15 pg/mL nSyn in CSF correlated with having a positive α-synuclein seed amplification assay (Apr 2023 conference news; Aug 2023 conference news). Notably, three PD patients who were below this nSyn threshold were SAA-negative as well, strengthening the link between nSyn and aggregates (see image below). “This points to nSyn having a pathogenic role in PD,” Shahidi-Latham suggested.
Nitrated α-Synuclein and Aggregation. People with more than 15 pg/mL nitrated α-synuclein in their CSF (dotted line) also had positive α-synuclein seed amplification assay (SAA) tests (black), while those with less did not, even if they had been diagnosed with PD. [Courtesy of Sheerin Shahidi-Latham, Nitrase Therapeutics.]
The company generated several antibodies against mouse nSyn, picking the most selective and potent candidate to test. Researchers injected preformed α-synuclein fibrils into the striatum of A53T M83 mice to stimulate α-synulcein aggregation, and waited one week. Then they injected 30 or 100 mg/kg antibody weekly for 12 weeks. The lead antibody candidate dose-dependently suppressed α-synuclein phosphorylated at Ser129, a marker for aggregated protein, and reached statistical significance at the higher dose. By contrast, Roche and Prothena’s α-synuclein antibody prasinezumab had no effect in these mice. Prasinezumab had been negative in a Phase 2 Parkinson’s trial, but had modest benefits on secondary outcomes and in subgroups (Apr 2020 conference news; Apr 2021 conference news).
Shahidi-Latham noted that in Parkinson’s patients, only 3 percent of α-synuclein in the CSF is nitrated. Why, then, would targeting this form work better than binding all forms of the protein? Shahidi-Latham drew a parallel with donanemab, which targets a pyroglutamate, aggregating form of Aβ and showed high efficacy in removing plaque in a Phase 3 trial (May 2023 news; Jul 2023 conference news). By selectively targeting a toxic form, she believes her antibody prevents secreted nSyn from seeding aggregation and promoting the spread of pathology.
The company has now humanized the antibody, and optimized it to make it more potent. That version, NDC-0524, has a half-life in human plasma of 18 days, compatible with monthly dosing. Modeling suggests that a dose of less than 30 mg/kg will reach effective concentrations in people, Shahidi-Latham said. The company plans to take NDC-0524 into Phase 1 next year, using change in CSF nSyn and SAAs as readouts for efficacy.
Breaking Up Toxic Couple. Small-molecule syntacasyn (red dots) binds synaptic protein synapsin III (gray) and detaches it from α-synuclein fibrils (purple), causing fibrils to fall apart. Syntacasyn then encourages binding of synapsin III to physiological α-synuclein, restoring dopamine homeostasis. [Courtesy of Arianna Bellucci.]
Busting Aggregates by Hitting a Synaptic Protein
Small molecules have an advantage for tackling synucleinopathies, since they can enter cells to interact directly with deposits. Arianna Bellucci of the University of Brescia, Italy, debuted one such molecule that takes a unique approach to busting up fibrils—by first making them split up with a co-aggregating synaptic partner. Bellucci noted that α-synuclein loiters in pre-synapses, putting it in the right place to bind synaptic proteins, and that its presence there correlates with neurodegeneration (Schulz-Schaeffer, 2010). She previously reported that α-synuclein interacts with synapsin III in nerve terminals to regulate dopamine release (Zaltieri et al., 2015). Moreover, she detected synapsin III in Lewy bodies, suggesting it plays a role in pathology (Longhena et al., 2018).
To investigate synapsin III’s role, Bellucci overexpressed α-synuclein in the nigrostriatum of wild-type and synapsin III knockout mice. While the wild-types developed α-synuclein aggregates and lost neurons, the synapsin III knockouts were protected (Faustini et al., 2018). Lowering synapsin III also helped mice that already had synuclein pathology. In transgenic mice expressing human truncated α-synuclein, silencing synapsin III in the nigrostriatum broke up existing aggregates. Dopamine release recovered, and neuron number stabilized (Faustini et al., 2022).
This led Bellucci and colleagues to search for small molecules that could interfere with synapsin III binding to α-synuclein fibrils. The monoamine reuptake inhibitor methylphenidate, which binds synapsin III, emerged as the best candidate. MPH is approved to treat attention deficit hyperactivity disorder, a disease associated with synapsin III polymorphisms, Bellucci noted. In synucleinopathy mice, MPH lowered fibril load and improved movement. This effect did not depend on its monoamine transporter activity (Faustini et al., 2020).
The researchers fiddled with MPH’s structure to develop derivatives that bound synapsin III more strongly, while not affecting monoamine transport. They named their lead candidate syntacasyn. Syntacasyn detaches synapsin III from α-synuclein fibrils, causing them to unravel. In computer simulations of protein interactions, it nudges α-synuclein back toward a physiological, α-helical shape, allowing it to interact with soluble synapsin III, Bellucci said. This restores normal dopamine release. In addition, syntacasyn enters the brain well and has a favorable pharmacokinetic profile, Bellucci said. In mice, the compound was not toxic, even at 10 times the effective dose.
The researchers tested syntacasyn in 50-day-old midbrain organoids made from patients with an α-synuclein gene triplication. After 30 days, 200 nM syntacasyn had lowered α-synuclein aggregates by about 80 percent, while maintaining dopaminergic neurons at the same level as in control organoids. Similarly, mice expressing human C-terminally truncated α-synuclein that were treated for four weeks had about one-quarter as much aggregated α-synuclein as untreated mice, and did not lose dopaminergic neurons.
“We can destabilize α-synuclein aggregates by controlling the interplay between α-synuclein and synapsin III,” Bellucci concluded. She told Alzforum that she is planning to test syntacasyn in clinical trials.
Aggregation Inhibitor Heads to Phase 2
Further along the development pipeline is a molecule familiar to Alzforum readers. In Lisbon, Johannes Levin of Ludwig Maximilian University, Munich, presented the latest on the oligomer-busting drug anle138b, now rechristened emrusolmin. In earlier studies in Parkinson’s disease models, this small molecule entered the brain well, bound α-synuclein, and broke up aggregates by changing the protein’s shape (Apr 2011 conference news; Mar 2015 conference news).
Armin Giese at Ludwig Maximilian, who first identified emrusolmin, co-founded the pharma company MODAG GmbH to take the compound into trials (Aug 2014 conference news). The company has completed Phase 1 studies in healthy volunteers and PD patients. Emrusolmin had a half-life of 12 hours, compatible with once-daily dosing, and achieved plasma levels above those shown to be effective in mouse models. Doses up to 300 mg were well-tolerated (Levin et al., 2022).
Levin said the company will next test the drug in MSA. PLP-haSyn mice model MSA by driving α-synuclein expression in oligodendrocytes (Kahle et al., 2002). When fed emrusolmin from two to six months of age, these mice maintained their dopaminergic neurons, mounted little microgliosis, and could balance on a narrowing beam as well as wild-types (Heras-Garvin et al., 2019).
A Phase 2 trial dubbed Topas-MSA will enroll 160 people at early disease stages, when they are still able to walk at least 30 feet unassisted. Participants will take 300 mg emrusolmin or placebo once daily for 48 weeks. The primary outcome will be the modified unified MSA rating scale part 1. The trial will take place in the U.S., France, Italy, Germany, Spain, Israel, and Japan, and will start at the end of this year, Levin said.—Madolyn Bowman Rogers
Scientists are expanding strategies for targeting misfolded tau. At this year’s AD/PD conference, held March 5 to 9 in Lisbon, Portugal, speakers highlighted a protein modification approach—sumoylation. This post-translational modification adds a small ubiquitin-like modifier (SUMO) to lysine residues. This modifier comes in two main flavors, SUMO1 or SUMO2, which have different effects. Sumoylation has been studied in Huntington’s and Parkinson’s diseases, but so far has received scant attention in Alzheimer’s and other tauopathies.
According to talks in Lisbon, that may be about to change. Ottavio Arancio of Columbia University Medical Center, New York, linked sumoylation to tau pathology, with SUMO1 worsening it, and SUMO2 protecting against it. He noted that tauopathy mice have an abundance of SUMO1-conjugated tau, but a dearth of SUMO2-tau. Luana Fioriti of the Mario Negri Institute of Pharmacological Research in Milan reported that adding SUMO2 to tau prevented its aggregation. This rescued synaptic function and memory in a tauopathy mouse model. Meanwhile, Paul Fraser of the University of Toronto extended the findings to Alzheimer’s, a secondary tauopathy. In a mouse model of amyloidosis, boosting SUMO2 protected synapses. Fioriti’s lab has developed a biologic analog, SBT02, that Fraser said restored synaptic health and memory in mice. The three labs collaborated on these studies.
“SUMO1 and SUMO2 impact tauopathies and amyloid pathology in quite different fashions,” Fraser told Alzforum. “SUMO2 maintains tau in a more soluble state and mitigates the synaptic damage associated with AD.”
Amy Pooler of Sangamo Therapeutics, San Francisco, noted that earlier reports of SUMO1 sumoylation excited the field. “Perhaps surprisingly, these data suggest that SUMO2 appears to have an opposing effect … However, more work will need to be done to understand the underlying mechanism and whether SUMO2 represents an important novel target for treating tauopathies,” she wrote to Alzforum (comment below).
Two Types of SUMO. SUMO1 (left) is shaped differently from SUMO2 (right), with distinct electrostatic potential and hydrophobicity. [Courtesy of Luana Fioriti.]
Sumoylation can change a protein’s shape and stability, as well as influence its interactions with other proteins. It was first implicated in AD about two decades ago, but it was unclear what substrates were altered (Jan 2003 news). Fraser later showed that tau was one, becoming sumoylated at lysine 340, but not other lysines (Dorval and Fraser, 2006). Another group reported that SUMO1 promoted tau phosphorylation and lowered its solubility, causing tau to aggregate (Luo et al., 2014). Fraser generated transgenic mice that overexpress SUMO1, and found they lost synapses and developed memory problems with age (image below; Matsuzaki et al., 2015).
SUMO1 Snuffs Synapses. Transgenic mice overexpressing SUMO1 (top) have degenerating dendritic spines (red arrows) and fewer mushroom spines (yellow arrows) than do wild-type mice (bottom). [Courtesy of Shinsuke Matsuzaki and Paul Fraser.]
Even less is known about SUMO2 and tau. Unlike SUMO1 sumoylation, which occurs constitutively, SUMO2 conjugation needs a trigger, such as cellular stress. Arancio noted that 8-month-old PS19 mice, which express mutant P301S tau, have less SUMO2-conjugated tau, and more SUMO1-conjugated tau, than do wild-types. The same held true in neurons generated from a patient with a R406W tauopathy; the cell work was done in the lab of Kenneth Kosik at the University of California, Santa Barbara.
Could increasing SUMO2 conjugation ameliorate tauopathy? Yes, according to Fioriti. When Franca Orsini in her lab expressed SUMO2 in neurons that carried mutant tau, phosphorylated tau and aggregated tau were cut in half. Mutating lysine 340 of tau abolished this protection, demonstrating that tau sumoylation was the key factor. The strategy worked in vivo, too. When Orsini crossed PS19 mice with transgenic mice generated by Fraser that overexpress SUMO2, the offspring had half as much p-tau and insoluble tau as did typical PS19 mice (image below).
Preventing Tau Pathology. P-tau (brown) accumulates in the hippocampi of PS19 tauopathy mice (left), but when SUMO2 is overexpressed (right), less forms. [Courtesy of Rosaria Pascente and Luana Fioriti.]
How does SUMO2 protect against tauopathy? In part, by keeping tau in the right place, Fioriti claimed. In tauopathies such as Alzheimer’s, tau strays into synapses, leading to loss of dendritic spines (Sep 2010 news; Jan 2011 news). In the PS19-SUMO2 crosses, however, tau stayed put on microtubules and the mice maintained as sharp a memory as wild-types, Fioriti said (image below). Many of these findings are summarized in a preprint on bioRxiv (Orsini et al., 2022).
Spines Off-Limits. In tauopathy mice (left), mutant tau (purple) accumulates in dendritic spines, but when SUMO2 is overexpressed (right), tau stays out. [Courtesy of Franca Orsini and Luana Fioriti.]
SUMO2 also rescues flagging mitochondrial metabolism due to tau toxicity, according to Fioriti. Previous studies reported that wild-type tau cozies up to proteins in these cellular powerhouses. Mutant tau does not, leading to sluggish energy production (Dec 2017 conference news; Jan 2022 news; Apr 2023 conference news). Collaborating with Catarina Quinzii at Columbia, Fioriti found that SUMO2 overexpression restored mitochondrial energy production to normal levels in the brains of PS19 mice.
Scientists in Lisbon wondered if the primary effect of SUMO2 is to stop phosphorylation of tau, preventing its misfolding and negative downstream effects. Fioriti said she believes this is the case, but still needs to demonstrate it experimentally.
“It would be interesting to know whether the protective role of SUMO2 is only due to tau sumoylation, or if sumoylation of other proteins contributes to the effects observed,” Tara Tracy at the Buck Institute for Research on Aging, Novato, California, told Alzforum. In Lisbon, Fioriti noted that there are other candidates, such as synaptic proteins, that might play a role as well.
PS19 mice model primary tauopathies such as frontotemporal dementia. Would the same principles hold in models of Alzheimer’s disease, where aggregated Aβ abounds? Fraser addressed this, crossing his SUMO2 mice with APP mice. As in primary tauopathy models, SUMO2 rescued synaptic plasticity and memory. Notably, it had no effect on amyloid. APP-SUMO2 mice had as many plaques as APP controls. “SUMO2 makes synapses resistant to amyloid toxicity,” Fraser concluded.
Fioriti has developed a synthetic protein analog of SUMO2, SBT02, that becomes conjugated to tau in vitro. It readily enters the brain in animal models. When the researchers injected APP mice with 20 mg/kg SBT02 under the skin three times per week from the age of 3 to 6 months, it prevented synaptic damage and memory problems. Could it do as well in mice that had already lost synapses? Fioriti and colleagues injected the drug starting at 6 months, when the animals had established plaques. Three months later, synaptic plasticity, as measured by long-term potentiation in brain slices, was restored to wild-type levels, and memory nearly to that level. “Treatment can rescue damaged and dysfunctional synapses,” Fraser said in Lisbon.
Fraser told Alzforum that the researchers have finished preclinical testing and initial toxicology studies on SBT02, and are preparing to do comprehensive toxicology testing. If all goes well, they plan to advance the biologic to clinical trials.—Madolyn Bowman Rogers
Over the Span of AD, Roles of Astrocytes and Microglia Change
As neuronal supporters and immune surveyors, astrocytes and microglia are no mere bystanders to the neuronal mayhem that unfolds in Alzheimer’s disease. At the AD/PD meeting, held March 5-9 in Lisbon, scientists presented new twists on the relative contributions of the two cell types to various aspects of AD, including plaque and tangle accumulation, synaptic deterioration, brain atrophy, and cognitive decline. The upshot? Reactive astrocytes spell trouble for neurons and their synapses, and this holds true even among people who have no Aβ plaques. Microglia, on the other hand, transform from neuroprotectors to slayers only once plaques inundate the brain. In mouse models, where researchers could deplete and restore the cells, microglia polarize into several transcriptional states over the course of amyloidosis. These evolving states are paralleled by functional transitions, in that the cells initially seed Aβ plaques, and then compact them later on.
Several presentations that attempted to disentangle the role of astrocytes versus microglia came from collaborators and scientists in the lab of Tharick Pascoal at the University of Pittsburgh. The researchers looked for connections between fluid and imaging biomarkers of Aβ and tau pathology, glial activity, and synaptic loss among several human AD cohorts.
Bruna Bellaver, an assistant professor in Pascoal’s lab, studies how astrocyte reactivity contributes to AD pathophysiology. Previously, she reported that among cognitively healthy people who have amyloid plaques, tangle pathology only showed up on PET scans among those whose astrocytes were fired up, as gauged by elevated plasma GFAP (Jun 2023 news). As she described in Lisbon, Bellaver has extended this line of work to decipher how astrocyte reactivity mediates correlations among plaques, plasma p-tau217, ApoE, and cognitive impairment across the AD spectrum
Bellaver’s newest analysis relied on data from more than 2,000 participants in four cohorts in the U.S., Canada, South Korea, and Chile. About half were cognitively healthy, and half had been diagnosed with mild cognitive impairment or AD. First, Bellaver investigated how astrocyte reactivity influenced the relationship between amyloid status, as gauged by PET or plasma Aβ42/40 levels, and plasma p-tau217. She found that across the AD continuum, plasma p-tau217 increased as a function of plaque burden, but only if astrocytes were reactive, as gauged by elevated plasma GFAP. In those with calm astrocytes, plaques did not rouse plasma p-tau217. This mediating effect was strongest among people who were cognitively impaired. These glia also mediated ApoE effects, such that ApoE4 potentiated the plaque-driven plasma p-tau217 only in people with reactive astrocytes.
Finally, Bellaver reported that astrocyte reactivity determined the heft of the cognitive blow dealt by amyloid and tau pathologies. While MMSE scores were unaffected by amyloid or p-tau217 alone, they dropped if reactive astrocytes accompanied either marker (image below). People with all three—Aβ plaques, high p-tau217, and reactive astrocytes—fared worst.
The Astrocyte Factor. Astrocyte reactivity (Ast+) potentiates the effect of amyloid (A+) and/or p-tau217 (Ptau+) on cognition (MMSE). [Courtesy of Bruna Bellaver, University of Pittsburgh.]
Bellaver speculates that astrocytes change when they sense amyloid buildup in the brain. “They get reactive and progressively lose neuroprotective functions and/or gain novel neurotoxic properties, disrupting brain homeostasis,” she wrote to Alzforum. This reactivity might preclude their ability to contain tau pathology, she speculated.
In Pascoal’s lab, Francieli Rohden used biomarker data to explore a related question: How do astrocytes and microglia contribute to synaptic loss that worsens over the course of AD? Rohden examined relationships among CSF biomarkers measured in 105 participants in the TRIAD cohort, and 373 from ADNI. Volunteers in both cohorts were classified based on cognitive and amyloid status. Rohden used CSF GFAP to gauge astrocyte reactivity, soluble TREM2 as a proxy for microglial activation, and pre- and post-synaptic proteins GAP43 and neurogranin released into the CSF as markers of damaged synapses. At AD/PD, Rohden reported that astrocyte reactivity correlated with both markers of synaptic destruction, regardless of amyloid or cognitive status.
The story was different for microglial activation, which only associated with GAP43 and neurogranin among people who had amyloid. In these participants, sTREM2 rose in step with GAP43, regardless of cognitive status, and with neurogranin among those who were cognitively impaired. These associations hinted that microglial activation might spur deterioration of pre-synapses earlier in disease, eventually compromising post-synapses later.
Showdown at the Synapse. Reactive astrocytes (top) and activated microglia (bottom) influence synaptic deterioration, as measured by the release of GAP43 from pre-synapses (left) and neurogranin from post-synapses (right). [Courtesy of Francieli Rohden, University of Pittsburgh.]
What explains ties between glial activity and synaptic damage? Rohden found that levels of CSF p-tau181 could fully predict the association between astrocyte reactivity and synaptic markers, regardless of Aβ or cognitive status. CSF p-tau181 also linked microglial activation with synaptic markers, but only among those with amyloid. Given that CSF p-tau181 is strongly linked to amyloid, how might it connect astrocytes to synaptic damage among those without a substantial plaque burden? The scientists speculated that Aβ oligomers might rile astrocytes, which then instigate the phosphorylation of tau. When p-tau congregates in pre-synapses, it gloms onto synaptic vesicles and might erode synaptic integrity.
One interpretation of Rohden’s findings is that once plaques are established in the brain, microglial activation becomes a liability. Shifty microglia were the focus of several presentations in Lisbon. Guilherme Povala, also of the Pascoal lab, used data from the TRIAD cohort to assess how activated microglia—this time, measured by TSPO-PET—related to brain atrophy throughout the AD continuum. TSPO is an outer mitochondrial membrane protein that rises in activated microglia, and can be measured with the tracer [11C]PBR28. For several regions of interest across the brain, Povala looked for associations between TSPO-PET and brain atrophy, which was measured with serial MRI scans over two years in 80 normal and 54 cognitively impaired volunteers.
What did he discover? Among those with normal cognition, the relationship between microglial activation and brain atrophy depended on amyloid status. In those without plaques, TSPO-PET correlated with higher regional volume, suggesting microglial activation was protective. In people with plaques, the relationship flipped, such that regions with higher TSPO-PET had more atrophy.
Among those with impaired cognition, all of whom had Aβ plaques, Povala uncovered a similar relationship, this time relative to tangle pathology. In people whose tangles were limited to early Braak stage regions, microglial activation correlated with more brain volume. The opposite was true when tau tangles had spread into later Braak regions, beyond the temporal lobe. For those people, riled microglia associated with brain shrinkage. Finally, Povala reported that regardless of clinical disease stage, microglial activation in the presence of amyloid and tangles predicted future cognitive decline.
Delphine Boche of the University of Southampton, U.K., noted that the findings seem consistent with a cascade in which Aβ triggers microglial activation, which then promotes tangle pathology and neurodegeneration. Povala agreed, adding that it appears microglia mount two distinct reactions, first to amyloid and then to tau. Interestingly, although they used two different markers for microglial activation, Povala and Rohden’s findings fit together, as both cast microglia in the amyloid-ravaged brain as a neurodegenerative force. At last year’s AD/PD meeting, researchers had suggested that perhaps TSPO reflects a harmful form of microglial activation (May 2023 conference news).
To better understand the effects of elevated TSPO, Boche used immunohistochemistry to characterize cells in postmortem brain samples from 60 people who had died with varying degrees of plaque and tangle pathology. In Lisbon, she reported that in microglia, TSPO congregated around nuclei and sometimes appeared in cytoplasmic processes, consistent with its mitochondrial localization. Curiously, TSPO was also occasionally expressed by smooth muscle endothelial cells lining blood vessels, but was not found in astrocytes or perivascular macrophages. TSPO increased in the temporal lobe with increasing Braak stages, and was not found in the cerebellum, where amyloid, but not p-tau, accumulated. Co-staining with antibodies for other microglial markers revealed that TSPO+ microglia tended to express CD68 or macrophage scavenging receptor-A (MSR-A), markers of phagocytic and scavenging microglia, respectively. Boche believes that TSPO+ microglia may be responding to p-tau and/or to neurodegeneration in AD.
Mariko Taga of Columbia University in New York also scrutinized postmortem brain sections to hunt for disease-related microglial phenotypes. Specifically, she wanted to know how microglia with ramped-up expression of CD74—a cell surface receptor that stokes antigen presentation—were related to different AD traits. Previous transcriptomic studies identified CD74 among elevated genes in disease-associated microglial subsets (Dec 2020 news). Because microglia are a diverse lot and make up only a small fraction of cells in the brain, Taga and colleagues developed an automated technique to scan an entire brain section at high resolution, pick out all of the microglia based on their expression of Iba1, and then study other features of the cells (De Jager et al., 2024). In Lisbon, Taga detailed her findings, applying the technique to dorsolateral prefrontal cortex sections from 64 donors in the ROSMAP cohort, and 91 from Columbia University’s brain bank. The analysis illuminated nearly 500,000 microglia within samples from both cohorts. In addition to Iba1, the sections were immunostained for GFAP, p-tau, and CD74. S
First and foremost, Taga found no major differences in the density of microglia based on pathological diagnosis. Across all Iba1+ microglia, expression of CD74 and Iba1 were strongly correlated, suggesting CD74 rises in step with microglial activation. Taga zeroed in the microglia with extraordinarily high CD74, i.e., more than two standard deviations above the mean. These cells were found in all of the samples, and did not increase in proportion across Braak stages. This suggested that tau pathology did not spur expansion of the cells. Oddly, the proportion of CD74-hi microglia did correlate with clinical diagnosis of AD, and with the rate of cognitive decline. The findings hint that CD74-hi microglia somehow potentiate the cognitive blow inflicted by AD pathology.
Tracking Human Microglia … in Mice
Microglia are known to have an intimate, and tumultuous, relationship with Aβ plaques. Researchers have extensively probed this interaction in mouse models by depleting microglia. As Nóra Baligács of KU Leuven in Belgium pointed out in her talk in Lisbon, conclusions have conflicted, with some casting microglia as plaque builders, and others as plaque destroyers (for example, Zhao et al., 2017; Jul 2019 conference news; Sep 2019 news). The differing results have been chalked up to differences in mouse models and microglia depletion protocols. However, Baligács, a graduate student in Bart De Strooper’s lab, hypothesized that answer might lie in timing—microglial involvement in plaque deposition may change throughout the disease process.
To test this idea, she treated APP-NL-G-F knock-in mice, which start to develop Aβ plaques just prior to 2 months of age, with the CSF1R inhibitor PLX3397 at different ages. Microglia rely upon this receptor for their survival, and inhibiting it docked their numbers by 83 percent. When this microglial culling was done in 1-month-old mice, before plaques had formed, Baligács found fewer plaques, and an overall lower plaque burden, in 4-month-old mice than in untreated NL-G-F mice. This early depletion of microglia also substantially reduced the amount of insoluble Aβ peptides detected in brain extracts, and curbed the development of dystrophic neurites. In contrast, depleting microglia when the mice were 3 months old—and plaque-ridden—resulted in a higher overall plaque burden. This boost was driven by an increase in plaque size, rather than plaque numbers, Baligács reported. Depriving mice of microglia at this later stage also exacerbated the numbers of dystrophic neurites.
Baligács thinks the findings reflect microglia’s shifting role from plaque seeders to plaque compactors. In support of this idea, depriving mice of microglia from 1 to 7 months of age resulted in a combination of effects: Mice had fewer plaques, but they were larger than in their microglia-replete counterparts.
To validate their inhibitor findings in a genetic model of microglial depletion, the researchers turned to FIRE mice. These animals have a deletion in the enhancer for the CSF1R receptor gene, resulting in a lack of receptor expression and completely snuffing out microglial survival, while macrophages remain unscathed. These animals were bred on an APP-NL-G-F background, and were also rendered immunodeficient, to allow for transplantation of human microglia. At both 6 weeks and 3 months of age, these microglia-deficient FIRE mice had fewer plaques than NL-G-F mice, again casting microglia as plaque seeders. When Baligács transplanted human microglia into the brains of FIRE mice a few days after they were born, the cells repopulated the brain. What’s more, by 3 months of age, plaque numbers increased relative to mice without the xenotransplants, though not to levels in age-matched NL-G-F mice. The findings further support the idea that microglia, and in this case human ones, build plaques.
Baligács plans to use these chimeric models to study the contributions of microglia throughout amyloidosis, and to investigate how different AD risk genes in human microglia influence this interaction.
Other ongoing work in the De Strooper lab focuses on how transplanted human microglia respond to amyloidosis. In Lisbon, collaborator Renzo Mancuso of the University of Antwerp described results from these studies, which he conducted with De Strooper lab postdoc Anna Martinez-Muriana and graduate student Nicola Fattorelli, who now works as a postdoc in Mancuso’s lab. In Lisbon, Mancuso described their newest findings, which were published in Nature Neuroscience on March 27 (Mancuso et al., 2024). Alzforum previously covered the bulk of the findings as reported on bioRxiv (Oct 2022 news). Essentially, Mancuso reported that upon exposure to amyloidosis in APP-NL-GF mice, transplanted human microglia gradually ditch their homeostatic signature in favor of a number of transcriptional states. These included two related cytokine response microglia (CRM) states, which expressed a bevy of inflammatory chemokine and cytokine genes; a disease-associated microglia (DAM) state; and an antigen-presenting profile, which involved the upregulation of HLA genes. Mancuso reported that transitions into these states proceeded on two separate transcriptional trajectories, one leading to CRMs, and the other to DAM and then to HLA. Based on these and other findings, he speculated that perhaps CRM arise in response to Aβ oligomers, while they embark on the DAM/HLA trajectory in response to plaques. In support of this idea, the CRM state appears to arise earlier and hold steady as mice age, while the DAM/HLA state emerges later.
Collectively, these AD/PD presentations make it clear that microglia are a moving target, shifting their roles as disease progresses. Despite this, scientists are still developing therapeutics to coax the cells into neuroprotective states. Read Part 9 of this series (to come) to learn about microglia-targeted contenders presented in Lisbon.—Jessica Shugart
Rubbing Microglia the Right Way? At ADPD, Scientists Showcase New Strategies
Microglia seem to play a hand in every aspect of Alzheimer’s pathogenesis, from the seeding and clearing of plaques to the traveling of tau to the dying of neurons. Scientists are pursuing therapeutics that both promote their neuroprotective functions and put the kibosh on their neurotoxic behavior. At AD/PD, held March 5-9 in Lisbon, Portugal, researchers detailed preclinical findings on a handful of potentials coming down the pike. One was an antibody, discovered in the blood of cognitively sharp centenarians, that blocks the inhibitory receptor CD33, thus freeing microglia to carry out their protective duties with gusto. Other approaches, including an antibody and small molecule, activated TREM2, promoting microglial phagocytosis and plaque clearance. Finally, a new antibody reportedly mops up ASC specks, which are inflammatory, amyloid-seeding protein complexes unleashed upon activation of inflammasomes in microglia.
Ralph Minter of London-based Alchemab Therapeutics presented preclinical findings on ATLX-1088, an antibody that blocks CD33 signaling. As an inhibitory receptor CD33 quashes phagocytosis and other microglial functions, so blocking it seems a promising strategy. However, Minter and colleagues did not set out to do just that. Rather, they started by hunting for autoantibodies tied to resilience to AD. That might sound a bit strange, because as Minter noted autoantibodies generally get a bad rap for causing a plethora of diseases, including multiple sclerosis, Type 1 diabetes, and rheumatoid arthritis, among others. However, in some cases, autoantibodies can protect against disease. For example, anti-HER2 antibodies protect against breast cancer. Likewise, the anti-amyloid antibody aducanumab was discovered in healthy older people who had little or no amyloid in their brains (Apr 2013 conference news).
To find more of these resilience antibodies, Minter and colleagues sequenced B cell receptors—the transmembrane precursors of antibodies—found in the blood of participants resilient to amyloid plaques in the European Prevention of AD consortium. Who was considered resilient? Any older person with blood biomarker evidence of Aβ accumulation but high scores on the MMSE and normal plasma p-tau. Minter said the goal was to find people who appeared stalled in the preclinical stage of AD, perhaps protected from progression. Antibodies to CD33 emerged as some of the few that were common among resilient people, but were not found among those with AD, Minter said. Next, Minter collaborated with Henne Holstege of Amsterdam University Medical Center to look for potentially protective antibodies in healthy centenarians in the 100-plus observational study (Oct 2022 conference news). Again, CD33 antibodies stood out. From this pool, the researchers selected their lead ATLX-1088.
In Lisbon, Minter reported that the antibody boosted phagocytosis of Aβ by human iPSC-derived microglia. In tri-cultures with neurons, astrocytes, and microglia, the antibody dampened the rise in multiple inflammatory cytokines in response to adding lipopolysaccharide and interferon-γ, suggesting it evokes a general anti-inflammatory effect, Minter said.
By mutating residues in CD33, the researchers narrowed down the epitope to CD33’s ligand-binding site, suggesting ATLX-1088 competes with the natural CD33 activator, sialoglycan. Other CD33 antibodies, including gemtuzumab, approved to treat acute myeloid leukemia, did not engage the ligand-binding site. Rather, several triggered internalization of CD33 by myeloid cells, effectively stripping the receptor from the cell surface and bringing the antibodies along for the ride. ATLX-1088, on the other hand, did not change levels of CD33 on the cell surface, functioning instead as a classic stoichiometric inhibitor of ligand binding. Minter said this bodes well for pharmacokinetics. Since CD33 is expressed on myeloid cells in the blood, any internalization would whittle down the amount that makes it to the brain. Ongoing experiments in mice expressing humanized CD33 in place of the mouse version support that idea, Minter reported.
How might naturally occurring CD33 antibodies confer resilience to amyloid? That is not known. Minter also does not know if these antibodies get into the brain in natural carriers, but for therapeutic development, Alchemab plans to target microglia.
What about other autoantibodies that might be floating around in healthy centenarian plasma? Michael Heneka at Luxembourg Centre for Systems Biomedicine wanted to know if TREM2 antibodies were among them. Minter said they have not found any, yet. He also noted that blocking a receptor, such as CD33, is generally more straightforward than activating one, as scientists are trying to do with TREM2.
Turning On TREM2
Indeed, scientists continue to move TREM2 agonist candidates toward the clinic, with Alector’s antibody, AL-002, furthest along, being evaluated in an ongoing Phase 2 trial in people with AD. Denali halted its DNL919 program last year after reports that the TREM2 antibody caused anemia; Novartis has a new candidate to fill the void. In Lisbon, Dominik Feuerbach from the company’s site in Basel, Switzerland, presented preclinical findings on VHB937. A fully human antibody, it latches onto TREM2’s ligand-binding, IgSF domain. Feuerbach reported that in iPSC-derived microglia and in cultured human M2A macrophages, VHB937 stabilized TREM2 expression on the cell surface, which boosted signaling through the receptor. This activation bolstered TREM2-related function, including phagocytosis of microglial “prey,” such as bacteria and apoptotic neurons, Feuerback reported.
In hTREM2-APP23-PS45 mice, which develop aggressive amyloidosis and express humanized TREM2, Feuerbach reported that administering weekly abdominal injections of VHB937 to 3-month-old mice resulted in fewer dystrophic neurites around Aβ plaques two months later. When the researchers injected these mice with a dye to label the plaques and then isolated microglia 24 hours later, they found more plaque material in the microglial innards if the mice had been treated with VHB937. This suggested that the antibody upped microglial appetite for plaques.
Boosting TREM2 signaling in this way also protected neurons in other disease models. In a PD model, the antibody spared neurons in the substantia nigra from the neurotoxin MPTP. It also accelerated remyelination in the wake of cuprizone treatment, a model for multiple sclerosis, Feuerbach reported. Collectively, the findings suggest that TREM2 agonism by VHB937 protects neurons from a range of insults, he said.
Vigil Neurosciences, based in Watertown, Massachusetts, has been developing small molecules to turn on TREM2 in people with AD. At last year’s AD/PD meeting, Vigil’s Christian Mirescu reported that the company’s top TREM2 agonist compounds appeared to work like “molecular glue,” by huddling TREM2 receptors together on the cell surface and boosting their signaling. In humanized TREM2 mice, and in nonhuman primates, Mirescu reported that the agonists crossed efficiently into the brain. Levels of the extracellular domain of TREM2, aka soluble TREM2, fell in the CSF, an indication that the agonist slowed processing and internalization of the receptor (Apr 2023 conference news).
The company has since selected a lead compound—VG-3927—as a candidate for AD trials. In Lisbon, Mirescu reported that when 4.5-month-old, plaque-ridden 5xFAD mice were fed with the agonist daily for six weeks, plaque area, as well as levels of insoluble Aβ42, fell by around 40 percent. The microglia adopted a neuroprotective, disease-associated microglia (DAM)-like signature, Mirescu said. Vigil has begun Phase 1 dosing studies with VG-3927 in healthy volunteers, with data due later this year.
One attendee asked Mirescu if he thought that chronic treatment with a TREM2 agonist might provoke tau pathology. This possibility was raised by findings from some mouse studies. For example, one study reported that activation of TREM2 with an antibody made tau pathology worse (Oct 2022 news). However, the relationship between TREM2 and tau is far from settled, as other mouse studies came to conflicting conclusions, or found that the relationship between TREM2 and tau may change as disease progresses (Oct 2017 news). Mirescu acknowledged these equivocal findings from mouse studies. To his mind, genetics from the tauopathy field speak for themselves, in that mutations that hobble TREM2 function raise disease risk (Guerreiro et al., 2013).
Shooting at Specks
In addition to targeting the dueling TREM2 and CD33 surface receptors, scientists are attempting to modulate microglia by way of inflammasomes. These multiprotein complexes contain nod-like receptors such as NLRP3, which serve as intracellular sentinels that detect danger signals, including Aβ and signs of cellular damage. Once triggered, inflammasomes set off a cascade that unleashes a storm of inflammatory cytokines that wreak havoc on neurons. Previously, studies led by Heneka implicated the NLRP3 inflammasome in AD, and various inhibitors are being developed to curb activation of the pathway (Dec 2012 news; Nov 2019 news; Li et al., 2023). In Lisbon, Heneka focused on apoptosis speck-like protein complexes, aka ASC specks, which are protein conglomerates that help form the inflammasome. They are released once microglia die a pyroptotic death. Heneka previously reported that ASC specks fuel Aβ aggregation in mouse models of amyloidosis, and that ASC proteins reside within the cores of Aβ plaques in the human brain (Dec 2017 news).
Heneka showed this aggregation frenzy happening in real time. Scientists in his lab generated transgenic APP/PS1 mice in which microglial CX3CR1 and ASC specks are fused to different fluorophores. Then, they injected methoxy-XO4 dye to label Aβ plaques, and monitored what unfolded in the brain with in vivo, live, two-photon imaging through cranial windows. Within the brain, they spotted microglia with ASC specks inside, as well as extracellular ASC specks that had presumably been released upon microglial death. Importantly, at the heart of every single blue, methoxy-labeled Aβ plaque, lay a ruby-red ASC speck.
To investigate whether the ASC specks were truly seeding plaques, the researchers checked the brain at different times after injecting the methoxy dye. They found that over the course of nine weeks, the amount of amyloid around each ASC speck grew, suggesting that ASCs had helped seed the plaques, which then proceeded to expand around the ASC core (image below).
Seeded by a Speck. In APP/PS1 mice injected with methoxy-XO4, ASC specks (pink) formed the core of plaques (blue) at baseline (left). Three (middle) and nine (right) weeks later, the ratio of plaque surface area to ASC speck volume increased. [Courtesy of Michael Heneka, Luxembourg Centre for Systems Biomedicine.]
Finally, Heneka reported that this toxic interaction could be thwarted with an antibody directed against the pyrin domain of ASC, which binds Aβ42. In an in vitro assay, the antibody blocked ASC-induced Aβ42 aggregation. Heneka would not disclose any information about the antibody or its development as a therapeutic.
Heneka does believe that ASC makes a good therapeutic target for AD, and that severing its ties with Aβ could thwart the seeding of Aβ plaques. While the inflammasome plays an important physiological role in fending off infections in the rest of the body, Heneka said that in the context of neurodegenerative brain, they do more harm than good. “I believe that you don’t need ASC specks in your brain,” he said.
Along those lines, Davide Basco from AC Immune, Switzerland, described preclinical findings from the company’s anti-ASC antibody, ACI-6635. Scientists at AC Immune used their SupraAntigen vaccine platform, which consists of liposomes embedded with antigens of choice, to evoke anti-ASC antibodies in mice. One, ACI-6635, bound to both mouse and human ASC with high affinity.
In Lisbon, Basco explained that ASC specks released from dying microglia not only go on to seed Aβ plaques, they can also be taken up by neighboring cells, where they propagate the inflammatory cascade. Nipping both processes in the bud is the goal of developing an anti-ASC antibody, he said. To see if the ACI-6635 antibody could break the inflammatory cascade of released specks, Basco treated cultured macrophages with human recombinant ASC aggregates to simulate release of specks. In response, the cells activated the inflammasome, and released IL-1β. Treatment with ACI-6635 potently blocked this response. It appears to work by promoting phagocytosis of the antibody-ASC complex via the antibody’s effector function.
Next, Basco tried the antibody in the ARTE10 transgenic mouse model of amyloidosis. It carries mutant human APP and PS1 genes. Starting at 3 months of age, when plaque growth was starting to ramp up, the mice were given 14 weekly injections of 60 mg/kg ACI-6635 or an isotype control antibody. In the controls, gliosis exploded, as gauged by levels of both Iba1 and GFAP detected in brain extracts. Immunostaining revealed diffuse ASC staining around Aβ plaques, surrounded by activated microglia and astrocytes. Basco emphasized that these diffuse deposits differ from the well-defined structures of specks. ACI-6635 reduced the amount of ASC protein in the vicinity of plaques, and reduced plaque size. The treatment also blocked the gliosis response almost entirely, as it dramatically stemmed the recruitment of microglia and astrocytes (image below).
Calling Off Glia.In control ARTE10 mice (top), plaques cores (red) are surrounded by ASC (green), microglia (pink), and astrocytes (yellow). Treatment with ACI-6635 (bottom) lowers ASC, shrinks the size of plaques, and interrupts recruitment of glia. [Courtesy of Davide Basco, AC Immune.]
After the talk, Minter asked if the antibody’s Fc receptor, which was necessary for it to promote phagocytosis, might inadvertently provoke a damaging pro-inflammatory cascade, as has been seen with other active and passive immunotherapies. Basco said that had yet to be tested. Delphine Boche of the University of Southampton, U.K., wondered if Basco had tested for markers other than Iba1 and GFAP, to ensure that the antibody had calmed, but not killed, microglia and astrocytes. Basco said no, but he thinks the antibody works by changing the way microglia and astrocytes respond to plaques, and by keeping the cells in a healthier, less reactive state.—Jessica Shugart
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group.
TREM2 variants in Alzheimer's disease.
N Engl J Med. 2013 Jan 10;368(2):117-27. Epub 2012 Nov 14
PubMed.
















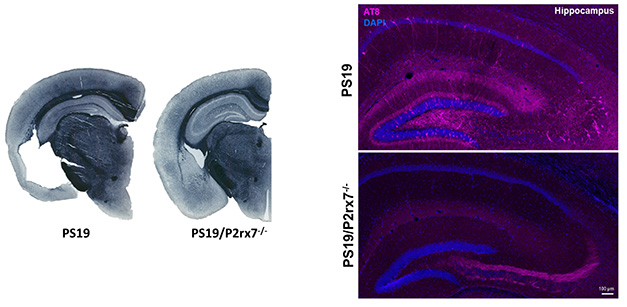












Comments
No Available Comments
Make a Comment
To make a comment you must login or register.