CONFERENCE COVERAGE SERIES
Society for Neuroscience Annual Meeting 2010
San Diego, CA, U.S.A.
13 – 17 November 2010
CONFERENCE COVERAGE SERIES
San Diego, CA, U.S.A.
13 – 17 November 2010
The 40th annual meeting of the Society for Neuroscience came to a close on Wednesday, 17 November 2010 at the San Diego Convention Center in San Diego, California. Unusually for this conference, the buzz in the hallways and at the poster sessions was less about the latest hot research and more about the dismal funding scenario unfolding at the National Institute on Aging (NIA). “The meeting was dominated by this dreary feeling that the NIA is in freefall,” Stephen D. Ginsberg, New York University, told ARF. Every researcher ARF interviewed on the subject echoed this sentiment. Most Alzheimer’s disease researchers in the U.S. depend on the NIA to fund at least some of their research. While researchers expected a fall in the 2011 pay line—a measure of which is the percentage of requests funded—the extent of that drop is taking them by surprise. Talk of a 3 percent pay line drew fear and frustration from both junior and well-established investigators.
Researchers worry about funding for their own research and also about the impact on the field as a whole. The concern is that, just when aging research is becoming more important—baby boomers are reaching retirement age and older adults comprise a growing fraction of the population—researchers may be discouraged from starting or continuing to study both Alzheimer’s and aging. Several scientists said they expect colleagues young and old to leave the field in the next two years as their funding dries up. “You could not devise a more efficient way of killing basic aging research,” said Gary Landreth, Case Western Reserve University, Cleveland, Ohio. Landreth has decided not to apply for NIA funding next year. As reported recently in Nature, other researchers, including Karen Duff at Columbia University, New York, are requesting that their NIH grant applications not be funneled through the NIA, because the chances of getting funding are so low.
Program officers at the NIA are aware of the issue and share the concerns. “We are worried that people will turn off aging research exactly at a time when we need more funding for it,” said Marcelle Morrison-Bogorad, outgoing director of the NIA Neuroscience Division, in an interview with ARF. She said that the funding situation is at a point where it cannot be ignored, and that for the last nine months, at least, institute officers have been discussing ways to address the dilemma. On 25 October, NIA director Richard Hodes addressed the research community in an open letter explaining how the situation became so dire and outlining some austerity measures.
How did it come to this? The NIA claims it faces a triple whammy of sorts. Stimulus funds from the American Recovery and Reinvestment Act are running their course, more researchers are applying for more grants, and a bigger slice of the pie has been going to larger projects and non-modular grants, i.e., those over $250,000 per year. It is funding of larger grants that has some researchers particularly upset, as in their view, a majority of funds are finding their way to a minority of investigators. But it is not only large program project grants and clinical trials that are sucking up the dollars, as some researchers claim, but also non-modular grants of over $500,000 per year, explained Morrison-Bogorad. More of these grants have been awarded because the nature of the research has changed. “Because the field has matured and the science has gotten more complicated, the best science is a mixture of approaches, not all of which can be tackled by a single scientist,” she told ARF. “We have encouraged greater collaboration.” She also noted that some of the other NIH institutes have restrictions on funding mechanisms. The NIMH, for example, does not award program project grants, and the NINDS limits them.
Researchers are concerned that the funds for those projects come at the expense of regular rank-and-file researchers. “It is similar to what happens in the economy as a whole. The middle class gets squeezed,” said Ginsberg. Other researchers are not convinced that funding of non-modular grants is to blame for the funding crisis. It is unusual for R01 grants to be far above the modular cutoff of $250,000 per year, said Ilya Bezprozvanny, University of Texas Southwestern Medical Center, Dallas. Modular grants are now being squeezed off, he said, reducing the chances of novel ideas germinating in the AD field. “It is possible that ‘super large R01 grants’ with budgets over $500,000 per year are funding clinical studies and patient observational research, but quantitative information from the NIA is needed to better understand where the bulk of the money is going,” he told ARF.
Terrence Town, University of California, Los Angeles, noted that if these grants are funded over five years or more, then even a small number of them could drain the funding pool. He also lamented the fact that changes to the application process mean that it is harder for researchers to address questions that come up in review. “There is a continuity issue that needs to be addressed,” he told ARF. That’s because the NIH changed their policy and now only allow researchers to revise their submission once, rather than twice. If the study section panel reviewing the revision is different, then they often flag a whole new set of issues, said Town. “If you address an initial laundry list of concerns, and then you get another, you can’t address that and then the grant gets rejected,” he told ARF. This is particularly troubling for young investigators, he said.
Fixing this funding crisis will be neither quick nor painless. The NIA plans to limit funding for larger grants to try to improve the pay line for smaller modular grants (i.e., less than $250,000 per year), said Morrison-Bogorad. Since 2004, the institute has had a policy of an across-the-board cut of 18 percent, meaning every division paid out 18 percent less than study sections recommended. “That worked until people caught on and just asked for more money,” said Morrison-Bogorad. Going forward, the institute plans to take a closer look at requests for funding of higher than $500,000. Investigators have to seek permission from the NIA to apply for these grants. “We can say ‘yes’ or ‘no.’ From now on we’ll be saying ‘no’ more often,” said Morrison-Bogorad. That may limit expenditures on big projects, and it will also leave more of the decision-making process in the hands of NIA administrators rather than study sections.
The NIA will not know exactly what the pay line for big and small grants will be until mid- to late December, when they have a funding meeting. At that point they will model scenarios and decide how many large grants to cut or to skip over. “It is something we hate to do. Hopefully it will be temporary and we can come roaring back,” said Morrison-Bogorad. She stressed that a pay line of 3 percent means that grants scoring in the top three percentile get funded, not that only 3 percent of the grants will get funded. It all depends on how study sections score the grants, she said. A 3 percent pay line could mean that 6 percent of grants get funded.
How long a comeback might take is unclear. In the meantime, it seems unlikely that there will be sufficient funds from other sources to take up the slack. “If it does emerge that there is a 3 percent pay line, that is going to be extremely painful for the field,” said Bill Thies, Chief Medical and Scientific Officer at the Alzheimer’s Association, one of the large funders of Alzheimer’s research outside the NIH. “The likelihood that we could step in and make up for a shortfall is pretty small,” said Thies. Over the last few years, the Association has been struggling with its own funding issues since a major part of its donor base is particularly sensitive to swings in the real estate and the stock markets.
Researchers charge that more federal funding is required for aging research and for Alzheimer’s in particular, which is projected to affect 16 million Americans by 2050, according to some estimates. In Congress, the Alzheimer’s Breakthrough Act is languishing in Committee. “The most frustrating point of this is that the federal government is going to be stuck with the bill one way or the other,” said Thies. “They can pay that up front, but they are unable, or are deciding not to deal with the long-term threat. People will pay attention in 2048, but at that point it is going to be too late.” Ginsberg agreed. “Leadership has to come up with better answers,” he told ARF. He said that the NIH needs to make a greater commitment to AD, and if they split AD from the aging institute to do it, so be it. “It is beyond reason to expect progress in the face of the baby boom generation reaching the age of onset for dementia when funding AD research is pennies on the dollar.”—Tom Fagan.
Mirroring a trend in amyloid-β research, work on the tau protein seems to be moving from tangles toward that infamous “O” word—oligomers. At this year’s annual meeting of the Society for Neuroscience (SfN), held 13-17 November in San Diego, new studies in this area sprinkled life into hallway conversations otherwise laden with funding worries (see ARF related conference story). This story distills some of the science behind the latest tau talk.
Neurofibrillary tangles containing hyperphosphorylated tau have long stood alongside amyloid-β plaques as pathological hallmarks of AD. However, in fly, fish, and mouse tauopathy models, synaptic impairment and neuron loss precede tangle formation (Wittmann et al., 2001; Paquet et al., 2009; Yoshiyama et al., 2007), suggesting that NFTs themselves may not drive disease. Stereological studies in human AD have revealed neuron loss prior to appearance of NFTs as well (see Gómez-Isla et al., 1997). Meanwhile, a growing body of research makes the case that tau oligomers could be the most dangerous tau species of all (see Marx, 2007; Berger et al., 2007; Brunden et al., 2008; Meraz-Rios et al., 2009). “It’s no longer just about plaques and tangles,” said Rakez Kayed, University of Texas Medical Branch, Galveston, said in his SfN talk on characterizing tau oligomers. “It’s time to start thinking about different Aβ and tau aggregates in the AD brain. Pre-filament forms of tau may be the most toxic and pathologically significant.”
To enable more detailed studies of this particular tau species, Kayed’s lab generated antibodies specific for tau oligomers. The researchers made polyclonal (T22) and monoclonal (TOMA) antibodies to tau oligomers they prepared in vitro by seeding soluble tau with oligomeric Aβ (Lasagna-Reeves et al., 2010). The antibodies do not recognize tau monomers in Western analysis of brain samples from AD patients or P301L (JNPL3) tau transgenic mice, nor do they pick up AT8-positive mature tangles, as judged by AD brain immunohistochemistry. But the antibodies do co-localize with the Tau 5 antibody, which recognizes both non-phosphorylated and phosphorylated tau in NTFs—i.e., early stages of tau aggregation.
Using their new antibodies, Kayed’s lab found elevated levels of tau oligomers—by ELISA, Western, and immunostaining—in postmortem AD brains, compared to control specimens. About 10 to 20 percent of total tau in AD brains is oligomeric, and the few tau structures found in control brains all appeared to be “pre-tangles,” Kayed reported at the SfN meeting. Both monoclonal and polyclonal antibodies can also detect tau oligomers in human cerebrospinal fluid by direct ELISA.
Other studies bolster the idea that tau oligomers, much more so than monomeric or fibrillar forms, are neurotoxic. As described on a poster, Cristian Lasagna-Reeves, a graduate student in Kayed’s lab, injected each of the three forms of tau into the hippocampus of wild-type mice, six animals per group. One day later, Lasagna-Reeves and colleagues analyzed the mice for cognitive deficits and killed them to extract brain tissue for immunostaining and biochemistry. Compared to the groups injected with monomeric or fibrillar tau, tau oligomer-treated mice fared poorly on parts of the novel object recognition test involving memory encoding and consolidation. The oligomer-treated mice also had more severe mitochondrial alterations and synaptic dysfunction, as revealed by immunohistochemical and biochemical experiments.
In a slide talk, James Moe of Oligomerix, Inc., in New York, described a separate set of studies demonstrating the toxicity of tau oligomers made in vitro by incubating different tau isoforms overnight at 37 degrees Celsius. Collaborating with Ottavio Arancio, Taub Institute, Columbia University Medical Center, New York, Moe and colleagues introduced tau monomers and oligomers into wild-type mouse hippocampal slices, and showed dose-dependent reduction of long-term potentiation in the oligomer-treated specimens. Confirming these findings in vivo, the researchers found that bilateral hippocampal injections of tau oligomers, but not monomeric tau, disrupted associative fear memory in mice tested the day after treatment. The findings support tau oligomers as “a good target for immunotherapeutic approaches” in AD and related tauopathies, Moe said.
Immunotherapy Targeting Pathological Tau
Tau-based immunotherapy is fairly new, but interest in the approach is growing, in part because Aβ vaccination trials (e.g., AN1792) have sustained hopes while exposing the perils of this strategy for treating AD. At the SfN meeting, Diana Castillo-Carranza, a postdoctoral fellow working with Kayed, presented a poster with initial findings of her mouse immunotherapy studies using the lab’s tau oligomer antibodies. She injected anti-tau oligomer antibody or control antibody into the hippocampus of eight-month-old JNPL3 transgenic mice, which develop tau pathology and motor impairment due to expression of the human P301L mutant tau transgene.
Transgenic mice that got a single infusion of anti-tau oligomer antibody (1 microliter of TOMA at 1 mg/ml) did far better than their mock-immunized counterparts when tested four days post-injection on the rotarod test measuring balance and coordination. At this point, it remains unclear whether the passive immunotherapy improved motor performance back to normal levels, as wild-type mice were not included in these as controls in these experiments. These animals should be old enough to analyze in early 2011, Kayed told ARF (see ARF related news story).
TOMA immunostaining of hippocampal sections removed from the JNPL3 mice after behavioral testing showed that the antibody infusion did clear tau oligomers from CA1 and dentate gyrus neurons. Western blotting of brain homogenates from vaccinated and control JNPL3 mice using the lab’s polyclonal anti-tau oligomer antibody also demonstrated removal of tau oligomers. When the researchers analyzed the same brain homogenates with Tau 5 antibody, which recognizes both oligomeric and monomeric tau, they not only saw reduction of tau oligomers on the blots, but also a significant increase in tau monomers, in the vaccinated animals. This is surprising and significant, Kayed noted, because it suggests that tau toxicity is associated with its oligomerization, not simply modification of the monomer. The findings “advise us to be careful in describing tau as soluble or insoluble,” Kayed wrote in an e-mail to ARF. “We have to be more specific.”
Scientists who saw the poster seemed intrigued by the findings. “Their work on tau oligomers is exciting. Trying to use their new antibody for passive immunotherapy is a reasonable thing to do, and that they see improvement in motor behavior is encouraging,” said Gal Bitan, who develops molecular tools for studying Alzheimer’s, Parkinson’s, and other diseases of protein misfolding at the University of California, Los Angeles.
However, the current studies do not address a number of key issues—among them whether treatment with anti-tau oligomer antibodies could improve cognition. The JNPL3 model has a predominantly motor phenotype, with only minor cognitive defects. “It will be interesting to see if their approach can be applied to models that are more relevant to AD,” Bitan noted. Kayed said his lab plans to start TOMA injections in Tg2576 mice, a widely used AD transgenic strain, in a few months.
Einar Sigurdsson and colleagues at the New York School of Medicine have data suggesting that vaccination with an AD-specific phospho-tau peptide not only clears tau pathology but prevents memory decline in a new tauopathy model more suited for cognitive assessment. The hTau/PS1 mice in this study express all six human tau isoforms (Andorfer et al., 2003) as well as mutant presenilin-1. The M146L PS1 transgene accelerates disease progression in the hTau/PS1 strain, which, unlike JNPL3 mice, develops tau pathology predominantly in the hippocampus and cortex and much less so in motor areas. The work is in press in the Journal of Neuroscience, and has been reported at previous meetings (see ARF 2008 ICAD Chicago story), though not at SfN.
In a prior study (Asuni et al., 2007 and ARF related news story), Sigurdsson and colleagues improved motor deficits of JNPL3 mice by vaccinating them with the same phospho-tau peptide used in the hTau/PS1 work. And at this year’s International Conference on Alzheimer’s Disease held in July in Honolulu, Hawaii, the lab reported that passive immunotherapy using an antibody (PHF1) targeting the same phospho-epitope led to motor benefits in treated JNPL3 mice (see ARF related news story).
How Do Tau Vaccines Work?
Conceptually, it may seem puzzling that tau immunotherapy works as well as it does in mice, given that tau pathology develops inside the cell, where it is harder for antibodies to reach. Amyloid deposits, on the other hand, are extracellular and, not surprisingly, easily cleared from amyloid mouse models by antibodies injected directly into the brain (see Wilcock et al., 2003; Wilcock et al., 2004). “My perspective is that for treating Alzheimer’s, an initial infusion of antibodies intracranially to clear deposits, followed by low-dose systemic administration to prevent future accumulation, would be an ideal strategy,” commented David Morgan of the University of South Florida in Tampa. He said Kayed’s new data support this approach for tau antibodies, and suggest further investigation of immunotherapy against tau oligomers as a consideration for treating Alzheimer’s. Morgan himself has unpublished data showing that intracranial injection of anti-tau antibody reduces histological tau deposits in the Tg4510 mouse inducible tauopathy model.
As for how antibodies might clear intraneuronal protein aggregates, Kayed proposed in a recent review (Kayed, 2010) that “depletion of the extracellular pool of [tau] aggregates by the antibody will shift the equilibrium between the intra- and extracellular pools of aggregates, hence leading to removal of the intracellular aggregates.” (See also Asuni et al., 2007, and Sigurdsson, 2008, and Sigurdsson, 2009 reviews that propose similar and alternative hypotheses.) At the 2010 International Conference on Alzheimer’s Disease in Honolulu, Pavan Krishnamurthy of Sigurdsson’s group reported results from brain slice experiments suggesting that antibody-mediated clearance of tau aggregates involves the endosomal/lysosomal system.
AD biomarker studies suggest that tau aggregates are released into the extracellular space, perhaps by dying cells (see Trojanowski et al., 2010). How tau oligomers are released from cells is unclear. In an SfN meeting poster, Kaoru Yamada of David Holtzman’s lab at Washington University School of Medicine, St. Louis, Missouri, and colleagues showed that their in-vivo microdialysis method was able to detect interstitial tau even in young wild-type mice, suggesting tau is secreted in the absence of neurodegeneration, while both Moe and Kayed reported detection of oligomeric tau in the CSF of AD patients.
Along with these data, and prior work showing that antibodies to α-synuclein boost clearance of the protein in a PD mouse model (Masliah et al., 2005 and ARF related news story) and α-synuclein oligomers secreted from cells may seed aggregation (see Danzer et al., 2009; Emmanouilidou et al., 2010; and ARF related news story), Kayed’s findings “raise the possibility that secretion of intracellular aggregates is a general phenomenon in the brain,” suggested Ben Wolozin of Boston University. “One wonders whether a combined Aβ/tau vaccine might work better on humans than simply the Aβ vaccine.”—Esther Landhuis
TDP-43, a protein implicated in both frontotemporal lobar degeneration and amyotrophic lateral sclerosis, as well as Alzheimer’s disease (Brouwers et al., 2010), is an RNA-binding protein with an undefined role. If posters at the Society for Neuroscience (SfN) meeting and a recent publication are any indication, the field is poised to discover a wealth of clues to TDP-43 function (see also ARF related news story). Scientists are scanning the transcriptome for TDP-43 target RNAs, and early reports suggest there are quite a lot of them. A paper in the November 4 Journal of Biological Chemistry online describes more than 4,000 RNAs that interact with TDP-43 in cortical neurons, and at the SfN meeting, held November 13-17 in San Diego, California, several poster presentations described TDP-43 targets in cell lines. Scientists hope that they might link a small subset of TDP-43-binding RNAs to pathology. More specifically, altered gene expression might someday provide biomarkers for early diagnosis of ALS, suggested Shangxi Xiao of the University of Toronto, Canada, an author on one of the posters.
What RNAs does TDP-43 modify, how does it change them, and what proteins does it work with? “These are questions everybody is asking,” said Chantelle Sephton, first author on the JBC paper with senior author Gang Yu and colleagues at the University of Texas Southwestern Medical Center in Dallas. All lead up to the ultimate questions, she said: How do these processes cause pathology, and how might doctors target TDP-43 to treat disease? Therein lies the rub, since TDP-43 has so many binding partners.
“I do not believe every target plays a role in the disease process,” said Fen-Biao Gao of the University of Massachusetts Medical School in Worcester, who was not involved in the JBC paper or SfN posters. Indeed, transcriptome-scanning studies are merely hypothesis-generating tools, said Joachim Herz, another professor at Southwestern Medical Center and a coauthor on Sephton’s paper.
Foundational Studies
The JBC paper is the first published study to seek out TDP-43 targets without overexpressing the protein, the authors said. The authors isolated RNA from rat primary neuron cultures, then used TDP-43 antibodies to immunoprecipitate RNAs that interact with the protein. Collaborating with researchers in the group of Melissa Moore at the University of Massachusetts, the scientists sequenced the TDP-43 targets. “I was floored by the number of targets,” Sephton said. More than 4,000 genes appear to rely on TDP-43 to modulate their expression. “It’s really a foundation-type study,” Sephton said, noting that researchers can now pick and choose which targets they think are involved in disease for follow-up studies.
TDP-43 is bound to sequences of UG repeats in target RNAs, often with a UA in the middle, but Sephton said there may be other TDP-43 binding motifs, too. Using a standard gene ontology database, the researchers identified genetic categories that were enriched in their RNA sample. Genes involved in RNA processing, synapse function, and development were among the top hits.
Among the gene set was TDP-43 itself, suggesting it regulates its own expression via some sort of feedback loop. The authors suggest TDP-43 binds its own 3’-UTR, and alters either the stability or translational efficiency of the RNA. The exact mechanism for this feedback is unclear, Sephton said, but it dovetails nicely with animal studies. Animals heterozygous for TDP-43 often produce more than half the normal amount of the protein, suggesting the remaining allele compensates for the missing one (Sephton et al., 2010). Other TDP-43 targets readers may recognize include FUS, progranulin, tau, amyloid precursor protein, and α-synuclein, which are all related to neurodegeneration.
The researchers also identified proteins that interact with TDP-43. In collaboration with the laboratory of Junmin Peng at Emory University in Atlanta, Georgia, they used mass spectrometry to analyze proteins that co-immunoprecipitated with TDP-43 from rat brain nuclear extracts. They discovered 25 proteins that seem to buddy up with TDP-43 in the nucleus. Most were RNA-binding proteins such as splicing or translation factors, confirming previous results (Freibaum et al., 2010).
Cell Line Studies
Sephton and collaborators, as well as Clotilde Lagier-Tourenne and colleagues at the University of California in San Diego (see ARF related news story), are using brain tissue to investigate TDP-43 targets, but other scientists have started similar studies with cell lines. At the SfN meeting, Shangxi Xiao and Janice Robertson of the University of Toronto, Canada, presented work with TDP-43/RNA co-immunoprecipitation from the SH-SY5Y human neuroblastoma line. The scientists identified 102 TDP-43 targets, with UG repeat and pyrimidine-rich TDP-43 binding sequences. The majority of TDP-43 binding sites were within introns; 23 percent were outside introns. Using RT-PCR, Xiao showed that five out of 50 target genes were downregulated, or had altered splicing patterns, in the spinal cords of people who died of ALS compared to healthy control tissue.
Atsushi Shiga from Niigata University in Japan presented a poster on the gene expression and splicing changes in TDP-43-depleted cells. The researchers used short interfering RNA to dampen TDP-43 expression in HeLa human cervical cancer cells. They found that 123 genes, including several involved in inflammation, were up- or downregulated. Inflammation is a known factor in ALS as well as other neurodegenerative diseases (see ARF related news story). In addition, 892 genes were abnormally spliced in the absence of TDP-43. Many of the latter are involved in the Golgi and other endomembrane systems.
TDP-43, of course, is not the only RNA-binding protein implicated in ALS and FTLD. In a poster, Shinsuke Ishigaki and Gen Sobue reported on early studies knocking down FUS in the spinal cord neuroblastoma hybridoma NSC34. They have identified several FUS targets, Ishigaki told ARF, and hope to perform further studies with primary motor neuron cultures and knockout mice.
The multitude of TDP-43 targets indicates the complexity researchers face in untangling disease mechanisms Gao said. “All these toxic proteins affect so many pathways,” he told ARF. “It is a challenge for the field…to identify the key targets.”—Amber Dance
What causes Aβ to go rogue in Alzheimer’s disease? Some researchers believe that a modified, pyroglutamate form of Aβ is to blame. Pyroglutamate Aβ, first discovered almost 20 years ago, heated up as a research topic in recent years, with evidence suggesting that pyroGluAβ can seed the aggregation of Aβ peptides and initiate the amyloid cascade (for a recent review, see Gunn et al., 2010). The German biotech company Probiodrug AG, based in Halle/Saale, is developing therapeutics that target pyroGluAβ production. At the 2010 Society for Neuroscience annual meeting, held 13-17 November in San Diego, California, Probiodrug researchers were present in force and described the latest research in this field in several talks and posters. Though this company has driven much of this research, academic labs are also breaking ground in the field. Researchers discussed preclinical therapeutic work and presented data from animal models and cell cultures. Overall, the data strengthened the case for a causal role for pyroGluAβ in Alzheimer’s disease, and demonstrated that therapeutic efforts targeting this minor Aβ species are moving along, though it remains to be seen whether this strategy will pan out.
Scientists have known for some time that amyloid plaques alone correlate poorly with the severity of AD. However, the presence of pyroGluAβ, which lurks in plaques in both sporadic and familial AD, does correlate with disease severity, said Hans-Ulrich Demuth of Probiodrug. This modified Aβ species comes about when the first two N-terminal amino acids of Aβ42 are chopped off, and an enzyme called glutaminyl cyclase (QC) ties together the ends of the exposed glutamate residue, making pyroglutamate. This creates a highly stable, neurotoxic form of Aβ that aggregates faster than conventional full-length Aβ (see Piccini et al., 2005 and ARF related news stories: AD/PD 2007 Salzburg story; Keystone 2008 story; AD/PD 2009 Prague story; and Society for Neuroscience 2009 Chicago story). Recently, researchers led by Thomas Bayer at the University of Göttingen, Germany, developed an antibody that recognizes oligomeric forms of pyroglutamateAβ. They reported that this antibody binds to plaques in the brains of familial AD patients, and to a lesser extent to tissue from sporadic cases. Bayer and colleagues also reported that these oligomers are decreased in plasma taken from AD patients (see Wirths et al., 2010). Although there is broad agreement that pyroGluAβ is present in AD, whether it seeds aggregation remains somewhat controversial. PyroGluAβ production depends on QC, and this enzyme is increased in AD brains, making QC a potential target for therapeutic strategies.
Drug Companies Get Into the Pyro Act
Pharmaceutical and other biotech companies are now turning their attention to pyroGluAβ. Researchers from Merck & Co., based in West Point, Pennsylvania, developed a sandwich ELISA for pyroGluAβ, described in a poster by first author Guoxin Wu. This is a highly sensitive assay in which one antibody is used to capture the protein of interest and another is used to detect it. They tested it on APP/PS1 mice, as well as on human normal and AD brains, and verified the results using surface enhanced laser desorption/ionization time-of-flight mass spectrometry. The ELISA assay recovered nearly all the pyroGluAβ spiked into buffer, with a detection limit of 3 pM at the low end, the researchers said. With this method, the scientists determined that pyroGluAβ is elevated 8.5-fold in AD brains compared to controls. This contrasts with a 2.6-fold increase in all other forms of Aβ in Alzheimer’s brains.
Researchers at 21st Century Biochemicals in Marlborough, Massachusetts, described on a poster polyclonal antibodies they generated to be specific for two forms of pyroGluAβ, pE3Aβ and the smaller, truncated pE11Aβ. First author Eric Berg demonstrated specificity by Western blot and ELISA. The researchers then used these antibodies to stain AD and normal elderly brains. They found pyroGluAβ formed the center of plaques, with full-length Aβ at the periphery, suggesting that pyroglutamate forms of Aβ seed deposits.
Mouse Pyrotechnics? Modeling QC Role
For their part, the Probiodrug crowd added new data as well. Several researchers presented data from mouse models that appeared to strengthen the case for QC as a therapeutic target. Bayer, who consults for Probiodrug, crossed 5XFAD mice with mice that express human QC under the control of the neuron-specific Thy1 promoter. Normal 5XFAD mice already produce much more pyroGluAβ than do other AD models, but in the presence of human QC, the levels went up higher still, Bayer said. These mice developed worse motor and memory problems than the 5XFAD mice. Bayer and colleagues also generated 5XFAD mice with endogenous QC knocked out. The knockouts showed a significant rescue of wild-type behavior in the elevated plus maze, demonstrating a role for endogenous QC in the pathology of AD mice.
Similarly, Stephan Schilling of Probiodrug described a cross of an AD mouse that expresses human APP containing the Swedish and London mutations (APP-SL) with the mouse expressing neuron-specific human QC. By nine months of age, the amount of pyroGluAβ in the double transgenics was two- to fourfold higher than in APP-SL mice, Schilling said. At six months of age, double transgenics showed worse memory and behavior problems in Morris water maze and fear conditioning tests than did APP-SL mice, and a greater activation of microglia. When researchers gave a QC inhibitor starting at three months, however, they saw less pyroGluAβ at eight months.
PyroGluAβ Processing
Anca Alexandru, from the company’s Munich site, used mouse models to see whether different methods of inducing pyroGluAβ made a difference. She generated a transgenic mouse (TBA2.1) that expresses a secretory truncated Aβ missing two N-terminal amino acids. By two or three months old, these mice developed extracellular pyroGluAβ deposits, neuroinflammation, and severe neurodegeneration, Alexandru said, losing 35 percent of hippocampal neurons. These mice showed extreme motor problems as early as four to six weeks, making behavioral testing difficult. Alexandru and colleagues generated another mouse strain (APP-NLQ-10) that expresses full-length mutant human APP with the faster-cyclizing glutamine at position 3 of Aβ (normal Aβ has glutamate at that position). These mice modeled a later-onset, slower-progressing disease than TBA2.1, Alexandru said. At two to three months, they had significant intracellular Aβ and pyroGluAβ deposits, but no inflammation or degeneration in their brains. By 15 months, these mice carried a heavy load of pyroGluAβ and plaques and showed astrogliosis. The mice did not have motor problems, but demonstrated cognitive difficulties at a late age. Together, the results indicate that the subcellular localization of pyroGluAβ determines the type and severity of neuropathology, Alexandru said.
Vivian Hook of the University of California in San Diego also investigated pyroGluAβ secretion. She used primary cultures of neuronal-like chromaffin cells to show that various forms of Aβ, including pyroGluAβ, co-localize with secreted neuropeptides and neurotransmitters (such as galanin, somatostatin, dopamine, and epinephrine) in regulated secretory vesicles. These vesicles also contain full-length APP, β- and γ-secretases, and QC, indicating that the components necessary to process Aβ are present in these organelles.
Previous work has shown that QC not only injures neurons by producing pyroGluAβ, but also has a role in promoting inflammation. The enzyme acts on monocyte chemoattractant protein (MCP-1, also known as CCL2), making a pyroglutamate derivative that is stable and resists proteases. MCP-1 activates microglial migration and neuroinflammation, is upregulated in early AD, and has been found to contribute to degeneration in AD mouse models. Holger Cynis of Probiodrug described the discovery of an isoenzyme of QC that does not get secreted, but is confined to the Golgi apparatus inside cells. To dissect its role, Cynis and colleagues compared inflammation in QC knockout mice and isoQC knockout mice. In QC knockouts, they saw no difference in cyclized MCP-1 or monocyte counts, but in isoQC knockouts, both were sharply reduced. This suggests that isoQC controls the inflammatory response, while QC produces pyroGluAβ. This would appear to make QC a more attractive drug target.
Dissecting Molecular Mechanisms
One of the burning questions about pyroGluAβ is how it inflicts its harm. A talk by Justin Nussbaum, who works in the laboratory of George Bloom at the University of Virginia in Charlottesville, provided clues to the mechanisms of pyroGluAβ toxicity. The laboratory receives research support from Probiodrug. Nussbaum treated primary cortical neuron cultures with synthetic pyroGluAβ and conventional Aβ42, and found that pyroGluAβ poisoned cells at a fivefold lower concentration than did Aβ42. When Nussbaum mixed pyroGluAβ with a 19-fold excess of Aβ42 during oligomer formation, however, he achieved a mixture that was 10- to 50-fold more deadly to neurons than either peptide alone. Mixed oligomers of Aβ continued to promote the formation of toxic Aβ species even in the absence of pyroGluAβ, Nussbaum said, suggesting a prion-like propagation mechanism. Intriguingly, the toxicity of Aβ mixtures depended on the presence of tau, as neuronal cultures from tau knockout mice remained healthy at Aβ concentrations that killed normal neurons within 24 hours. This jibes with previous AD research showing that tau acts downstream of Aβ (see ARF related news story on Götz et al., 2001 and Lewis et al., 2000 and ARF related news story on Roberson et al., 2007).
Edging Toward Therapy
If pyroGluAβ does seed oligomers and Aβ protofibrils, then preventing pyroGluAβ production would be a logical therapeutic intervention. Probiodrug is pursuing a small molecule drug-discovery program for inhibitors of QC, Demuth said. They have synthesized and tested more than 1,300 compounds for their ability to specifically bind and inhibit QC and isoQC, and are identifying promising candidates. Demuth said he hopes for Phase 1 trials next year.
Researchers led by Cynthia Lemere at Brigham and Women’s Hospital are collaborating with researchers at Probiodrug to look at using the immune system to inhibit pyroGluAβ formation. First author Jeffrey Frost presented a poster describing a passive immunization study in two-year-old APP/PS1ΔE9 mice. Weekly injections of 200 micrograms of a monoclonal anti-pyroGluAβ antibody for seven weeks slashed total Aβ by 25 percent and fibrillar Aβ deposits by 50 percent in the hippocampus. Similar results were obtained in 3xTg AD mice, as described in a poster by first author Qiaoqiao Shi. The researchers are now testing prevention by means of passive immunization in younger AD mice, as well as examining the effects of active immunization in J20 mice. They plan to compare the effects of vaccination with pyroGluAβ versus Aβ40 and 42.—Madolyn Bowman Rogers.
No Available Comments
Move over, mice. There are now one, and possibly two, models for amyotrophic lateral sclerosis in man’s best friend. At the Society for Neuroscience annual meeting, held 13-17 November 2010 in San Diego, California, researchers learned about canines with idiopathic laryngeal paralysis (ILP), a disease that looks suspiciously similar to bulbar-onset ALS. If that is confirmed, these animals will join a group of dogs with degenerative myelopathy (DM), which share a genetic mutation with the human version of the disease.
“There is a need for large animal species models for ALS,” said Amelie Gubitz, Program Director for ALS research at the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland. “They fill an important gap,” she said, between the tiny brains and spinal cords of mice and the human-sized nervous system. Gubitz, who organized a satellite meeting to discuss ALS models on 12 November, noted that a golden retriever model has proved useful in several studies of Duchenne’s muscular dystrophy (Banks and Chamberlain, 2008).
At the satellite meeting, Bryden Stanley of Michigan State University in East Lansing presented a poster on dogs with ILP. These animals suffer problems in swallowing that mirror the bulbar onset in one-quarter of people who get ALS. There is no real animal model for bulbar-onset ALS, said Bob Brown of the University of Massachusetts in Worcester, although some mice may exhibit symptoms in the neck area. Thus far, Stanley has been unable to find a genetic cause for the ILP.
In another poster session during the SfN general meeting, Brandie Morgan of the University of Missouri in Columbia discussed her data on axon counts in dogs with DM. Morgan works with Missouri researcher Joan Coates, who presented her dog model two years ago at the SfN meeting in Washington, DC (see ARF related news story and ARF related news story on Awano et al., 2009; reviewed in Coates and Wininger, 2010). These animals have a missense mutation in superoxide dismutase 1 (SOD1); SOD1 mutations are responsible for some cases of inherited ALS.
The two models might turn out to complement each other, Gubitz said, with Coates’s model mimicking familial ALS and Stanley’s standing in for bulbar-onset, and possibly sporadic, forms.
Dog models offer many advantages. “The dog and human share an incredibly close environment,” Stanley told ARF in an interview. Thus, they may be exposed to the same toxins that some researchers suspect contribute to ALS. Because owners choose to euthanize their pets at different stages, dogs offer the opportunity to examine disease pathology before end-stage, which is impossible in people.
Dogs, given their inbred genomes, are also convenient for genomewide association studies (GWAS), said Dennis O’Brien, the director of the veterinary comparative neurology program at the University of Missouri. Dogs have less well-shuffled genomes than do people, he said, so their linkage disequilibrium groups are large, and scientists can find gene associations with a relatively small sample. “That is probably the biggest advantage that the dogs have,” O’Brien said. “The GWAS is so powerful.”
Bulbar Barks
Stanley originally set out to study not ALS but ILP, a common disorder in elderly dogs, particularly Labrador retrievers. Degeneration of the laryngeal nerves leads to paralysis of the laryngeal muscles, and symptoms include gagging, throat clearing, and a raspy or hoarse-sounding bark. Dogs tend to get sick around 10 years of age, and generally last for one to three years longer, Stanley told ARF.
Stanley and colleagues followed dogs with ILP for a year and performed neurological tests, looking at gait, muscle tone, and reflexes (see Stanley et al., 2010). Of 32 dogs with ILP, 10 had neurologic problems when they enrolled in the study. Half of the dogs had them by six months, and all had neurological signs within a year of enrollment. Eventually, the dogs with ILP became paralyzed. At autopsy, the dogs with ILP evinced muscle atrophy and loss of axons in the lumbar spinal cord’s ventral roots. In comparison, none of the 34 control dogs showed neurological symptoms.
Stanley found herself thinking that “There must be something in humans like this.” She started talking to neurologists, who immediately saw the similarities between ILP and bulbar ALS, which starts with problems in speaking and swallowing. “Their jaws dropped,” Stanley told ARF. “They were just taken away with the similarity.”
It is too early to consider ILP a definite ALS model, Stanley said, but it is promising enough to warrant further study. “It is going to be a good model for something,” O’Brien told ARF. “I do not know if we know enough yet to see if it is going to be a good model for ALS.”
Stanley is working to further characterize ILP, and recruited Michigan State neuropathologist Howard Chang to help analyze tissue samples. “Clearly, we have a problem of neuropathy,” he said. “The question is, where does the neuropathy come from?” He said he has only seen a few samples so far, and needs more control tissues from healthy animals before forming any conclusions. Stanley is searching for a genetic cause of ILP, and has tested several of the usual ALS suspects: SOD1, angiogenin, FUS, TDP-43, and FIG4. So far, she has had no hits, and plans to continue the search with a GWAS. Brown told ARF he hopes these studies will identify new ALS genes.
Dog Genes
The University of Missouri researchers do have a genetic link for degenerative myelopathy. This is an advantage, Gubitz said, because research colonies can be bred. Indeed, the scientists have already begun a proof-of-concept study of ALS treatment in bred-for-research animals, Coates told ARF, although she said it is too early to reveal any details.
Coates and colleagues first studied DM, which has its onset in the hind limbs, in Pembroke Welsh corgies and boxers. “We are continuing to find other breeds or dogs that are affected with this disease,” Coates told ARF in an interview. At least 17 breeds can get the disease, and more than two dozen breeds carry the missense SOD1 allele at a frequency greater than 20 percent. Most of the dogs have the same missense mutation—E40K—although the scientists may have found a second SOD1 mutation in one Bernese mountain dog, Coates said.
In people, a single faulty SOD1 gene amounts to a 100 percent certainty of getting ALS. That is not true in dogs, where few heterozygotes get sick and some homozygotes escape disease. This incomplete penetrance, Coates suggested, may be because some dogs die before the disease has time to develop. If that is true, she thinks there must be genetic factors that enhance or reduce risk. The researchers are conducting a GWAS to find those risk factors.
Coates and colleagues are also interested in finding biomarkers that would allow them to follow disease progression. At the ALS International Conference to be held in Orlando, Florida, later this month, Coates will present her work with a method she calls MUNE, for motor unit number estimation. Using electronic stimulation and recording, MUNE tells the scientists how many neuromuscular junctions a particular muscle has.
While much of the work in Coates’s group has focused on upper motor neuron degeneration, Morgan wanted to know whether axon fiber numbers drop in lower motor neurons. She counted axon fibers in cross-sections from T8 vertebra motor roots. Two healthy boxers had an average of 5,788 axons. But in seven boxers with DM, Morgan discovered an average of 4,603 axons. This reduction in fiber count is similar to ALS pathology.
Given the heterogeneity of ALS in people, it is unlikely that any single model will be the only one used in research, Gubitz said. Instead, she envisions a panel of models that researchers will use in parallel. Dogs just might be on the list.—Amber Dance.
Shoring up insulin responses is a tried-and-true strategy for helping people manage type 2 diabetes (T2D), but could it also prevent or delay Alzheimer’s disease? Strong epidemiological evidence suggests diabetes is a risk factor for AD, but capitalizing on that connection has not been plain sailing. While there is some early hint that intranasal insulin might offer some protection against dementia, sensitizing agents such as pio- and rosiglitazone failed to pass muster in larger clinical trials for dementia (ARF related news story). Could it be time for a new tack? At the 40th annual meeting of the Society for Neuroscience (SfN), held 13-17 November in San Diego, the promise of glucagon-like peptide agonists seemed to breathe new life into insulin signaling as a therapeutic target.
Because of new human and animal model data, it appears the field might see several clinical trials testing this drug class in the coming year, according to researchers. “These agonists have remarkable properties,” said Konrad Talbot, University of Pennsylvania, Philadelphia. “They do not affect cells that have normal insulin signaling, but they can boost synthesis of the insulin receptor, insulin receptor substrate-1 (IRS-1), and glucose transporter 4, and they enhance the response of cells to insulin stimulation. Boosting insulin responsiveness would help correct a major neuronal insulin signaling deficit we have found in the hippocampus of AD cases,” he told ARF. Whether that approach will be successful remains to be seen, but in an SfN nanosymposium organized by Talbot, speakers showed that glucagon-like protein 1 (GLP-1) agonists, which are approved for treating diabetes, protect against Aβ toxicity and rescue learning and memory deficits in model mice.
Just what are those insulin signaling deficits that crop up in AD? Insulin binds to cell surface receptors, kicking off a signaling cascade that relies on a number of kinases, commonly including phosphoinositol-3-kinase (PI3K) and Akt (see ARF related news story). At the SfN meeting, Talbot and Hoau-Yan Wang, City University of New York Medical School, reported on their collaborative work to examine the activation status of this pathway in the brain at baseline, and following insulin stimulation. The researchers used immunohistochemistry to look for phosphorylated epitopes in early postmortem tissue taken from normal controls, people with MCI, and people with AD. Tissue was obtained from the University of Pennsylvania and from the Religious Orders Study cohort at Rush University, Chicago, Illinois.
Talbot reported that in 24 age-matched samples from UPenn, AD cases displayed significant decreases in basal activation of the insulin receptor and increases in basal activation of downstream signaling molecules (Akt, mTOR, GSK-3β, and PKC λ/ζ). The most prominent AD feature was serine phosphorylation of insulin receptor substrate 1 (IRS-1), which helps propagate signals from the insulin receptor to downstream kinases. The phosphorylation, of serines 312, 636, and especially 616, was mostly extranuclear, which is unusual. Serine phosphorylation of these sites is inhibitory, Talbot explained, and in keeping with this, he saw reduced activation of IRS-1. The levels of S616 phosphorylation were also markedly elevated in MCI patients but not in tissue from normal controls or people with other forms of dementia. “This might be particularly interesting because it could be some sort of early marker of dysfunctional insulin signaling,” said Talbot. He reported similar findings using tissue samples taken from 30 normal, 29 MCI, and 31 AD cases from Rush.
Furthermore, the density of neurons containing extranuclear, serine-phosphorylated IRS-1 correlated with elevated oligomeric plaque load and deficits in global cognition, working memory, and especially episodic memory, that last having a tight correlation R value of 0.65, “which is phenomenal for postmortem tissue,” said Talbot, given the variability that can exist among samples. After the researchers adjusted for a variety of cofactors, including age, sex, years of education, density of neurofibrillary tangles, and oligomeric plaque load, the number of cells with inactivated IRS-1 still correlated with cognition, which suggests that dysfunctional insulin signaling is related to whatever is causing memory problems, said Talbot. In contrast, correlations between oligomeric plaque load and cognition disappeared when corrected for serine-phosphorylated IRS-1. “These results are consistent with evidence that oligomeric Aβ induces serine phosphorylation of IRS-1, and that this in turn disrupts insulin signaling at synapses, thereby resulting in cognitive deficits,” said Talbot.
Talbot collaborated with Wang, an expert in ex-vivo analysis, to study the ability of brain tissue to respond to insulin. They took early postmortem hippocampal tissue samples from six age-matched pairs—normal and AD—incubated them with low concentrations (1 and 10 nM) of insulin (to stimulate insulin but not insulin-like growth factor receptors), then looked for downstream effects. This ex-vivo analysis revealed a blunted insulin response in AD tissue. Tyrosine phosphorylation (activation) of IRS-1 was impaired. The AD tissue samples mounted a lackluster activation of a variety of downstream kinases, as judged by phospho-epitope analysis. Phosphorylation of Akt (serine 473), mTOR (serine 2448), and ERK2 (tyrosine 204) were all significantly lower than in normal tissue treated with insulin. Curiously though, baseline levels of these phospho-epitopes were higher in AD tissue, which could reflect an attempt to compensate for impaired insulin signaling. “This is the first direct evidence of insulin resistance in the brain of AD cases and suggests that basal levels of activated downstream insulin signaling molecules reflect inadequate compensatory responses to such upstream resistance,” said Talbot.
What causes dysfunctional insulin signaling in AD, and could correcting it help? A hint to the former came from Fernanda De Felice from the Federal University of Rio de Janeiro, Brazil. When working with William Klein at Northwestern University, De Felice found that Aβ oligomers reduce cell surface insulin receptors on neurons, and vice versa (see ARF related news story). Now, she has taken that one step further, looking at the downstream effects of Aβ on insulin signaling in hippocampal neuron cultures. At SfN, she reported that synthetic Aβ oligomers increase IRS-1 serine phosphorylation and decrease IRS-1 tyrosine phosphorylation, disabling the insulin signaling pathway. Aβ oligomers boosted phosphorylation of IRS-1 serines 636 and 639. These post-translational modifications are also found in muscle and adipose tissue in diabetes patients, said De Felice. In the periphery, IRS-1 inactivation is mediated by JNK and tumor necrosis factor α (TNFα); and De Felice presented evidence that those pathways might be involved in blunting insulin signaling in hippocampal neurons as well. Introducing a dominant-negative JNK, or an inhibitory TNFα antibody, prevented IRS-1 serine phosphorylation in neurons incubated with Aβ oligomers.
De Felice further reported that both insulin and exendin-4, an agonist of glucagon-like peptide (GLP-1), block the Aβ oligomer-induced increase in IRS-1 serine phosphorylation in cultured neurons. She also found that exendin-4 had the same effect when given to 13-month-old APP/PS mice, suggesting that the GLP-1 agonist might be a potential AD therapeutic.
Christian Holscher, University of Ulster, Coleraine, U.K., followed on that same theme in his talk. Holscher briefly reviewed some of the properties of GLP-1 and its mimics. These molecules increase insulin sensitivity by facilitating insulin release and re-sensitizing insulin receptors. They act in the brain, where neurons, particularly pyramidal neurons of the cortex and hippocampus, express GLP-1 and its receptors. The peptide has neurotrophic properties, boosting synaptic transmission and neuronal progenitors.
There are hints that GLP-1 analogues might protect against AD. Earlier this year, Holscher reported that a protease-resistant form (Valine[8]GLP-1) not only crosses the blood-brain barrier and increases long-term potentiation (LTP) in the hippocampus, but also reduces the number of dense core plaques in APP/PS transgenic mice (see Gengler et al., 2010). At the SfN meeting, Holscher reported that the GLP-1 analogue liraglutide, which is FDA approved for treatment of diabetes, has similar effects. An eight-week treatment restored novel object memory as well as spatial memory in nine-month-old APP/PS1 mice. Liraglutide also dramatically boosted LTP in anesthetized animals, said Holscher. Paired pulse facilitation, another correlate of learning, also improved. This is an indicator of altered GABAergic signaling, said Holscher, which would dovetail with expression of the GLP-1 receptor in pyramidal cells, as these are GABAergic. Interestingly, the agonist had little or no effect on LTP or PPF in wild-type controls, suggesting that the drug only works when normal LTP/PPF have gone awry.
Perhaps the most surprising effect of these agonists is on amyloid plaques. After the eight-week liraglutide treatment, plaque density was half that in untreated animals and the number of dense core plaques fell by two-thirds. Levels of soluble Aβ in the brain were down by about a third.
Previous results from Nigel Greig and colleagues at the National Institutes of Health, Baltimore, Maryland, suggest that GLP-1 has a similar effect (see Perry et al., 2003) and that exendin-4 reduced plaque load in 3xTG mice treated with streptozotocin to induce diabetes (see Li et al., 2010). During question time, Holscher said he did not know how these agonists influence Aβ load, but told ARF that exendin-4 reduces APP synthesis in cultured neurons (also see Perry et al., 2003). Whether this also happens in vivo needs to be tested, he said.
GLP-1 agonists are approved for diabetes, and trials testing exendin-4 in Parkinson’s disease patients are underway in London. Could trials in AD soon follow? “I find the prospect incredibly exciting,” said Talbot. “I have a vested interest,” he admitted, “but the effect on plaques that Christian Holscher reported was stunning, and it seems to me that [these drugs] are ready for clinical testing.”
Talbot told ARF that the NIH is planning an exendin-4 trial for AD. Across the pond, Holscher has been in discussions with colleagues at Hammersmith Hospital, London, to put forward a plan for a clinical trial of liraglutide. Novo Nordisk, which makes liraglutide under the trade name Victoza®, seems interested in the proposal, said Holscher, but whether it will sponsor such a trial is not clear at present. “With the agreement of my colleague Steven Arnold, who heads geriatric psychiatry and is associate director of the Alzheimer’s Disease Center here at the University of Pennsylvania, I am working with Christian to encourage Novo Nordisk to support Victoza trials in the U.S. and Europe,” Talbot told ARF. He noted that since exendin-4 and liraglutide have different pharmacological properties, trials of both are warranted.—Tom Fagan.
No Available Comments
Apolipoprotein E is the strongest genetic risk factor for late-onset Alzheimer’s disease, accounting for more than 95 percent of cases. But despite decades of research, it is still not clear how it ties in with AD etiology. At the Society for Neuroscience annual meeting held 13-17 November 2010 in San Diego, California, scientists presented their latest work using cutting-edge microscopy and new in-vitro methods to study ApoE-Aβ interactions, and their untoward effects, at human brain synapses. Postmortem studies of human brain tissue point to a scenario where ApoE, especially the E4 isoform, promotes Aβ oligomerization and targets Aβ oligomers to synapses. The study was “one of the most impressive presentations at SFN this year,” wrote Taisuke Tomita of the University of Tokyo in an e-mail to ARF. “The work is elegantly conducted, and suggests ApoE may have a ‘cargo receptor’-like function in Aβ-induced synaptic toxicity.” Moreover, other SfN data suggest that blocking the ApoE-Aβ interaction could help stem Aβ-induced synaptic toxicity, and efforts are underway to develop compounds to do just that. These agents have a way to go before entering the clinic, though, and scientists have much to learn about the mechanisms by which ApoE influences Aβ oligomerization and localization to synapses.
Conceptually, an ApoE-Aβ connection in AD has long seemed reasonable. ApoE is a lipid transport protein, and brain areas involved in learning and memory, which show heavy amyloid deposition in AD, also express abundant receptors for ApoE and other lipoproteins. ApoE has been shown to bind Aβ (e.g., Strittmatter et al., 1993 and others) and to promote its degradation (Jiang et al., 2008 and ARF related news story) and clearance (Deane et al., 2008) from the brain. Meanwhile, work by several groups solidified claims that Aβ oligomers are neurotoxic in vivo (see Shankar et al., 2008 and ARF related news story; McDonald et al., 2010 and ARF related news story). Robert Koffie, an M.D.-Ph.D. student in Tara Spires-Jones’s lab, in collaboration with Brad Hyman, at Massachusetts General Hospital, Charlestown, showed that Aβ oligomers shrink dendritic spines in the brains of APP/PS1 transgenic mice (Koffie et al., 2009 and ARF related news story). To achieve the high resolution needed to view individual synapses, the researchers used array tomography microscopy, which generates 3D images from stacks of 70 nanometer-thin tissue sections. The researchers stained these for pre-synaptic (synapsin 1) and post-synaptic (PSD95) markers, and with an antibody for oligomeric Aβ (NAB61) generated in the lab of coauthor Virginia Lee at the University of Pennsylvania, Philadelphia.
As shown on their SfN poster, Koffie and colleagues now extend this analysis to human AD brain tissue. In so doing, they focused on how ApoE might contribute to synapse loss induced by Aβ oligomers. They used array tomography to analyze postmortem tissue freshly isolated from 10 AD patients and five age-matched controls. All controls were ApoE3/3, and the AD group included three E4/4, four E3/4, and three E3/3 patients. As with the AD transgenic mice, oligomeric Aβ localized to synapses in both AD and control brains—even those without plaque pathology. Previous biochemical studies detected oligomeric Aβ in brain tissue from normal controls with plaque pathology (see McDonald et al., 2010), while using an antibody (A11) specific for prefibrillar oligomers, Charles Glabe and colleagues at the University of California, Irvine, detected prefibrillar oligomers in extracts from brains with little or no plaque pathology (see Tomic et al., 2009). “I am excited by the idea that oligomeric Aβ in the normal brain may play a role in LTD, but evidence is circumstantial so far,” Spires-Jones told ARF.
The researchers found that the oligomeric Aβ-positive synapses were abnormally small, and synapse loss around plaques correlated strongly with oligomeric Aβ burden. They showed that in the AD group, E4/4 brains had more synapse loss than did E3/3 brains, and that ApoE co-localized with synaptic Aβ oligomers more frequently in the E4/4 samples (see image below). ApoE was detected with the WU-E4 antibody developed in the lab of coauthor David Holtzman at Washington University School of Medicine, St. Louis, Missouri. Tomita did mention one concern about the findings. “Anti-Aβ oligomer antibodies sometimes show cross-reaction with several proteins, and synaptic terminals contain many proteins,” he wrote.

Double Trouble
Array tomography on human AD brain reveals that ApoE particles (red) and oligomeric amyloid-β (blue) co-localize at a subset of synapses (represented by synapsin 1 staining, green). Arrow points to one such example; scale bar is 5 μm. Image credit: Robert Koffie, MGH
In biochemical studies of the postmortem brain extracts, the team found that E4/4 AD patients had higher levels of Aβ oligomers, largely dimers and trimers, compared with E3/3 AD or control groups. To further address these isoform differences, the researchers cultured neurons with lipidated ApoE3 or ApoE4 particles, and with Aβ oligomers isolated from conditioned media of Tg2576 primary neuronal cultures. Immunostaining showed that, while both ApoE3 and ApoE4 co-localize to synapses, only the E4 associated with Aβ oligomers and enhanced their synaptic localization.
Taken together, the data suggest that ApoE4, more than other ApoE isoforms “causes oligomeric Aβ to form, and targets it to synapses, which then shrink and die,” Spires-Jones told ARF. That jibes with previous work of other groups showing that ApoE4 forms complexes with Aβ in vitro more quickly than ApoE3 or ApoE2 (e.g., Strittmatter et al., 1993; Sanan et al., 1994). Several studies (e.g., LaDu et al., 1994; Tokuda et al., 2000) have suggested the opposite, but that is probably due to differences in the lipidation status of ApoE used in the studies, Koffie said. Some studies are trying to address this very issue. On a poster, Philip Verghese and coworkers in the Holtzman lab presented in-vitro data suggesting that formation of ApoE/Aβ complexes depends critically on ApoE lipidation status, and on neuronal plasma membrane receptors.
In the meantime, array tomography looks to be a powerful tool for detailed postmortem studies of ApoE and Aβ oligomers at synapses. Stephen Smith and coworkers at Stanford University, Palo Alto, California, developed the technique (see ARF Enabling Technologies report; Micheva and Smith, 2007) and helped Spires-Jones’s team adapt it for their studies in AD transgenic mice (Koffie et al., 2009). Smith’s lab is now pushing the technological limits, increasing the numbers of markers that can be analyzed per specimen. As reported in the November 18 issue of Neuron (Micheva et al., 2010), first author Kristina Micheva and colleagues used the technique to examine single synapses in mouse neurons stained with 17 different antibodies. Their analysis allowed sub-classification of excitatory synapses into four groups. For their study, Spires-Jones and colleagues lumped all excitatory synapses into one group; however, in most neurodegenerative disorders, “you don’t lose all the synapses at once,” Smith said. “You lose some and gain others. The basis for that differential susceptibility is almost certainly a hallmark of AD and potentially of all other neurodegenerative disorders.” In future studies, Koffie said, array tomography can “explore additional synaptic markers and ask whether specific subsets of synapses are being affected in AD.”—Esther Landhuis
This is Part 1 of a three-part series. See also Part 2 and Part 3.
No Available Comments
Though it could well be the subcortical brain region hit hardest in Alzheimer’s and Parkinson’s, the blue spot, or locus ceruleus, has only recently begun to draw wider attention among neurodegeneration scientists. New work presented at this year’s annual meeting of the Society for Neuroscience, held 13-17 November 2010 in San Diego, could help place this lesser-known cluster of noradrenergic neurons more squarely on the AD research map. In a nanosymposium, Doug Feinstein of the University of Illinois, Chicago, reported that restoring dwindling supplies of brain noradrenaline reduced cognitive deficits in an AD mouse model that has robust pathology. Recent publications have also solidified connections between the locus ceruleus and AD.
Locus ceruleus (LC) neurons die and brain noradrenaline levels drop with normal aging (Marien et al., 2004), even more so in AD (German et al., 1992). AD mouse models also have these features (see German et al., 2005; O’Neil et al., 2007). Several recent studies have gone a step further by demonstrating functional consequences of LC degeneration in neurological disease models. Ahmad Salehi, Veterans Affairs Palo Alto Health Care System, and colleagues at Stanford University, restored contextual memory in APP-overexpressing Down’s syndrome transgenic mice (Ts65Dn) by giving subcutaneous infusions of L-threo-3,4-dihydroxyphenylserine (aka L-DOPS), a brain-penetrant noradrenaline precursor (Salehi et al., 2009 and ARF related news story). Though the mice already had significant LC degeneration, hippocampal neurons that were downstream targets of LC cells still responded to norepinephrine by firing when treated with the β1- and β2-adrenergic receptor agonist isoproterenol, the authors noted. More recently, Feinstein’s lab used a similar strategy to slow disease progression in experimental autoimmune encephalomyelitis (EAE) mice modeling multiple sclerosis (Simonini et al., 2010). At SfN, Feinstein presented new data suggesting the approach can help in AD mouse models.
This study used Bob Vassar’s 5XFAD strain, which has three mutations in its human APP transgene as well as two presenilin-1 mutations. Starting at six weeks of age, these mice develop robust amyloid pathology. They lose neurons and show massive glial inflammation leading to cognitive impairment by five to six months. Sergey Kalinin of Feinstein’s group established that the LC is compromised in these animals: LC neurons had rampant inflammation, as judged by immunostaining, which revealed a 50 percent increase in GFAP-positive cells compared to wild-type LC cells. Furthermore, 5XFAD LC neurons had unusually large cell bodies, likely an effect of stress, Feinstein said.
The researchers administered L-DOPS to 4.5-month-old 5XFAD mice, which are free of cognitive impairment but on the verge of decline. The treatment involved subcutaneous L-DOPS injections along with two other drugs given intraperitoneally. The first was carbidopa, which blocks conversion of L-DOPS to noradrenaline in the periphery. The second was atomoxetine, a noradrenaline reuptake inhibitor. The combination specifically boosts noradrenaline in the brain. The mice were treated three times a week for four weeks, and one month later, faced tests of spatial learning and memory before being sacrificed for biochemical and immunohistochemical analyses.
The postmortem analysis showed that the L-DOPS treatment increased CNS noradrenaline levels and reduced plaque burden and astrocyte activation in the hippocampus and frontal cortex. Behaviorally, the treated mice fared better than controls. In one set of experiments, control- and L-DOPS-treated 5XFAD mice were assessed on the Morris water maze four times daily for three days. “The control group basically didn’t learn,” Feinstein reported. After 12 trials, these mice were taking almost as long to find a hidden escape platform as they did in the first trial. By the tenth trial, however, L-DOPS-treated mice did start finding the platform faster than the control animals. But they did not perform as well as wild-type mice, which require fewer trials to learn the platform location, and once they learn, find the platform more quickly, Feinstein noted.
On a related water maze probe test, L-DOPS-treated transgenics showed better long-term memory than did the untreated 5XFAD group. This version of the test measures time spent in the quadrant that had contained the hidden platform seven days prior. During that one-week lag, untreated transgenic mice forgot where the platform was, whereas the L-DOPS-treated group showed some recall. At the meeting, Feinstein proposed that these cognitive benefits may involve increases in neurotrophin levels, as mRNA analysis showed higher expression of nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) in L-DOPS-treated mice relative to the control group. Expression of the Aβ-degrading factors neprilysin and insulin-degrading enzyme (IDE) also increased in treated mice, raising the possibility that upregulation of these enzymes may mediate L-DOPS’s amyloid-lowering effects, Feinstein said.
The use of L-DOPS in these studies is interesting, in part because it “could work even after many of the LC neurons degenerate,” commented David Weinshenker of Emory University in Atlanta, Georgia. This is because the enzyme that converts L-DOPS to noradrenaline (i.e., aromatic acid decarboxylase) is present in dopaminergic and serotonergic neurons, as well as noradrenergic neurons. Second, L-DOPS is already known to be safe. The compound (aka Droxidopa) has been used in Japan to treat “freezing gait” symptoms in PD, Weinshenker said. In the U.S., it is being tested in a Phase 3 trial of PD patients with neurogenic orthostatic hypotension, a condition where loss of peripheral adrenergic receptors in the sympathetic nervous system causes reduced blood pressure. “If L-DOPS has efficacy in the APP mice, it could quickly be translated to humans and open up a new avenue of treatment for AD,” Weinshenker noted.
A BioMed Central Medical Genetics paper published online on November 11 reinforces the link between noradrenergic neuron loss and AD. European researchers genotyped 1,757 AD patients and 6,294 elderly controls, and homed in on a single-nucleotide polymorphism that decreases activity of dopamine β-hydroxylase, which converts dopamine to noradrenaline. In their analysis, this SNP (-1021T) associated with increased AD risk (Combarros et al., 2010). The work was led by senior investigator Donald Lehmann of the Oxford Centre for Gene Function in the U.K. and first author Onofre Combarros of the University of Cantabria in Santander, Spain.
Furthermore, in a recent study, Steffen Rossner, University of Leipzig, Germany, and colleagues probed various regions of mouse and human AD brain for expression of glutaminyl cyclase (QC). This is the enzyme that catalyzes formation of N-terminally truncated Aβ peptides into pyroglutamate Aβ, a highly aggregation-prone species that has attracted increasing attention from AD researchers (see ARF Chicago story and ARF San Diego story). In the August issue of Acta Neuropathologica, first author Markus Morawski and colleagues report finding QC expression in locus ceruleus neurons, but not in adjacent brain structures, of mouse and human AD samples (Morawski et al., 2010). In line with this, AD brain samples had intraneuronal pyroglu-Aβ and extracellular pyroglu-Aβ deposits in the LC. The authors take these data as support for a “scenario in which human QC-expressing LC neurons are intoxicated by formation of pyroglutamate Aβ,” Rossner wrote in an e-mail to ARF. “Additionally, QC and/or pyroglu-Aβ may be released at hippocampal synapses of LC neurons, thus contributing to hippocampal pyroglu-Aβ deposition. This mechanism may contribute to the known degeneration of LC neurons in AD and in subsequent noradrenergic denervation of the hippocampus.” Alzforum will host a Webinar on the role of the LC in AD on 16 December 2010.—Esther Landhuis.
No Available Comments
Amyloid-β plaque load poorly associates with cognition in Alzheimer’s disease (AD) and in people with mild cognitive impairment. Synapse loss, on the other hand, is believed to be a major correlate of cognitive decline. At this year’s annual meeting of the Society for Neuroscience, held 13-17 November 2010 in San Diego, California, several presentations and posters focused on links between synapse health and apolipoprotein E (ApoE), a major risk factor for late-onset AD. Researchers presented evidence that in both animal models and the human brain, ApoE conspires with Aβ oligomers to destroy synapses. The findings expand the modi operandi of ApoE, which already include promoting Aβ aggregation and preventing clearance.
Work from Tara Spires-Jones’s lab at Massachusetts General Hospital, Charlestown, pinpointed ApoE and oligomeric Aβ together in synapses in postmortem tissue samples from AD patients. In people who carried an ApoE4 gene, which is the major genetic risk factor for late-onset AD, oligomeric Aβ-positive synapses were small, and synapse number was down (see Part 1 of this series). The results suggest that the two proteins conspired to damage synapses, a theory supported by work from Karen Gylys and colleagues at the University of California, Los Angeles. Gylys also presented data on synaptic localization of ApoE. By ELISA and flow cytometry, Gylys and colleagues detected increased levels of ApoE in synaptosomes isolated from postmortem brain tissue of AD patients and of 20-month-old APP/PS1 transgenic rats (Flood et al., 2009) compared with aged-matched controls.
The team also found isoform-dependent differences in synaptic ApoE levels. Analyzing 18-month-old ApoE targeted replacement (TR) mice developed by Patrick Sullivan of Duke University, Durham, North Carolina, the UCLA researchers found that synaptic ApoE levels were highest and Aβ levels lowest in E2/4- compared to E3/3- and E4/4-expressing TR mice. In addition, Gylys reported that synaptic cholesterol levels were “dramatically increased in the E2/4 animals, with E3/3 and E4/4 levels similar to each other but much lower,” Gylys noted in an e-mail to ARF (see also Part 3 of this story). The data hint that less cholesterol is available in E3- and E4-containing synapses than in E2-containing synapses. “This suggests that poorly lipidated E4 may impair Aβ clearance, and that insufficient cholesterol delivery may be a central problem in AD synapses.” In a more general sense, she wrote, “the modest ApoE deficit in AD synapses likely contributes over time to synaptic dysfunction and loss, and suggests the relevance of ApoE- and lipid-related therapeutic targets.”
Gylys’s results complement “our array tomography data showing that synaptic ApoE is found at large, presumably healthy, synapses,” Spires-Jones noted. “We propose that ApoE alone is trophic for synapses, and only when it targets Aβ to synapses, as in the case of E4, is it a bad thing.”
On a separate SfN poster, coauthor Tadafumi Hashimoto of the Hyman group presented additional biochemical data suggesting that ApoE promotes formation of Aβ42 oligomers in an isoform-dependent manner. Using a luciferase assay that allows specific and quantitative measurement of Aβ oligomerization in the presence of different ApoE isoforms, the researchers found the highest levels of Aβ oligomers in E4-transfected HEK293 cells, less in the E3 transfectants, and the lowest levels of oligomeric Aβ in E2-containing cells, as reported earlier this year (see ARF related conference story). Now, Hashimoto shows that cells transfected with an ApoE4 mutant (R61T) that mimics E3 structure had Aβ oligomer levels comparable to E3-transfected cells and lower than in the E4 transfectants, offering further support that ApoE enhances Aβ oligomerization in an isoform-dependent fashion. Studies in HEK293 cells transfected with various ApoE fragments showed that the C-terminal lipid-binding region of ApoE is needed for facilitation of Aβ oligomer formation, Hashimoto and colleagues reported.
What could all of this mean for therapeutic approaches? At SfN, Martin Sadowski, New York University School of Medicine, and colleagues reported on a poster that blocking ApoE-Aβ oligomer interactions can stem Aβ-induced synaptic toxicity in vitro. Since the structure of ApoE’s Aβ docking site is unclear, the researchers targeted ApoE-Aβ interplay using a peptide homologous to the fragment of Aβ that binds ApoE, i.e., Aβ12-28. This peptide (Aβ12-28P) crosses the blood-brain barrier and reduces amyloid plaques and vascular Aβ burden in AD transgenic mice (Sadowski et al., 2006 and ARF related news story).
The new data presented at SfN show how the peptide works at the level of neurons and synapses. The NYU scientists added synthetic Aβ1-40 and Aβ1-42 monomers to co-cultures of astrocytes and hippocampal neurons from wild-type mice. In this in-vitro system, lipidated ApoE secreted by astrocytes promoted buildup of intraneuronal Aβ oligomers, as detected by Aβ oligomer-specific ELISA and by dot-blot densitometry using oligomer-specific antibody (A11). ApoE also impaired Aβ degradation, as determined by pulse-chase experiments, and led to downregulation of post-synaptic proteins involved in synaptic plasticity and memory formation—namely, the NR1 subunit of NMDA receptor, PSD-95, and synaptophysin. Addition of Aβ12-28P peptide curbed these effects.
The researchers repeated the experiments using co-cultures of neurons and astrocytes from targeted replacement mice producing specific human ApoE isoforms. The E4 isoform had the strongest effect on promoting intraneuronal Aβ accumulation, whereas E3 showed a modest effect, and E2, no effect. Importantly, Aβ12-28P abolished the E4-induced effects, suggesting the approach may hold promise in E4 carriers, which constitute nearly half of AD patients, Sadowski noted.
Ongoing studies in the Sadowski lab are focused on developing an Aβ12-28P derivative that could be used in human studies. In the best of worlds, the researchers hope to tease out an active structure within Aβ12-28P and convert it into a brain-penetrant drug that could be taken by mouth. They will use peptidomimetic technology to replace particular amino acids with non-natural mimics that resist degradation, and test potential compounds in the astrocytic/neuronal cell culture model used in their SfN research, Sadowski told ARF.—Esther Landhuis.
This is Part 2 of a three-part series. See also Part 1 and Part 3.
No Available Comments
Cholesterol not only clogs up arteries and leads to heart disease, but it can be bad for your brain as well. Evidence for cholesterol’s role in Alzheimer’s disease has been growing in recent years, and many cardiovascular risk factors have popped up also (for a recent review of cholesterol genes in AD, see Wollmer, 2010). Apolipoprotein E, a cholesterol transporter, is the primary genetic variant associated with sporadic AD risk, supporting a central role for the sterol in the disease. Yet despite a large body of research since 1993 (see, e.g., ARF Live Discussion from 2002 and ARF related news story and ARF news story), this role remains mysterious. The 2010 Society for Neuroscience annual meeting, held 13-17 November in San Diego, California, showcased the cholesterol-AD link in a nanosymposium, where researchers explored multiple aspects of the relationship between ApoE, cholesterol, and AD. Yet again, in 2010, the talks failed to add up to a unified picture of disease mechanisms; rather, they presented diverse ideas. Some researchers discussed roles for ApoE in AD pathogenesis that have nothing to do with cholesterol transport, while others focused directly on the effects and location of cholesterol in the brain. Overall, the symposium provided more questions than answers, but pointed to some intriguing new directions for research.
Is It the Coziness Between Cholesterol and Aβ?
Where does cholesterol go in the brain? First author Santiago Solé Domènech and Björn Johansson at the Karolinska Institute in Stockholm, together with Peter Sjövall at the SP Technical Research Institute of Sweden, Boras, described a new method for visualizing cholesterol and Aβ deposits in AD brains of both humans and 3xTgAD mouse model. Solé Domènech and colleagues first identified Abeta deposits using an amyloid-specific fluorescent probe (p-FTAA) and confocal laser scanning microscopy. Then they performed time-of-flight secondary ion mass spectrometry (ToF-SIMS) on adjacent brain sections to determine cholesterol content. The sections analyzed by ToF-SIMS were then incubated with p-FTAA to detect amyloid. By overlaying ToF-SIMS and fluorescent images from the same section, Solé Domènech and colleagues could examine the relationship between Abeta and cholesterol. They found that cholesterol accumulations often surround Abeta plaques at a distance of 0 to 50 microns from the plaque center. At least some of this cholesterol is extracellular, Solé Domènech said. In addition, p-FTAA fluorescence imaging revealed structural differences between human and mouse amyloid deposits. Solé Domènech hypothesized that the cholesterol deposits around plaques might be released primarily by astrocytes, the main providers of ApoE and cholesterol in the central nervous system, an idea he is testing in ongoing experiments. The findings dovetail with recent work led by Christian Duyckaerts at the Pitié-Salpêtrière Hospital in Paris, France, that shows increased cholesterol concentrations in human AD plaques by mass spectroscopy (see Panchal et al., 2010). At the SfN meeting, researchers led by Tara Spires-Jones at Massachusetts General Hospital, Charlestown, also reported that ApoE also co-localized with oligomeric Aβ (see Part 1 of this series).
Is It Inflammation?
Why is the ApoE4 allele a risk factor for AD? Robert Bell, working with Berislav Zlokovic at the University of Rochester, New York, presented data that suggested ApoE4 fails to repress a proinflammatory cytokine, cyclophilin A, that damages blood vessels. Previously, the lab had shown that the ApoE4 allele slows the clearance of Aβ across the blood-brain barrier (see Deane et al., 2008). In the current work, Bell focused instead on ApoE’s direct effect on the vasculature. Bell showed how ApoE knockout mice, as well as mice with the ApoE4 allele, lose blood-brain barrier integrity and have poorer cerebral blood flow. Overexpression of ApoE2 or ApoE3 alleles rescued this phenotype, but the ApoE4 allele did not.
Looking for the mechanism behind this effect, Bell turned to cyclophilin A. This cytokine has been shown to promote atherosclerosis and vascular damage in mouse models (see Jin et al., 2004 and Satoh et al., 2010). Bell and colleagues found higher cyclophilin A levels in the microvasculature of ApoE knockouts and ApoE4-expressing mice compared to wild-type animals. In primary brain endothelial cell cultures from ApoE knockout mice, ApoE2 and ApoE3 repressed cyclophilin A expression, but ApoE4 did not. Inhibition or knockout of cyclophilin A restored blood-brain barrier function, and improved cerebral blood flow and synaptic connections in ApoE knockout mice. The authors also showed that cyclophilin A directly injures mouse brain endothelial cells in culture, but not cortical neurons, implying that the cytokine exerts its effects by damaging endothelial cells. Bell suggested that since breakdown of the blood-brain barrier and reductions in microvasculature could lead to neurodegeneration, inhibition of cyclophilin A could be a promising therapeutic strategy in people who carry the ApoE4 allele, because it could help prevent vascular damage and neurodegeneration. Prior studies have linked microhemorrhages to dementia, while cerebral amyloid angiopathy is a major form of the disease (see, e.g., ARF related news story and ARF news story).
Is It Estrogen?
Qun Zhao, in the lab of Carol Colton at Duke University in Durham, North Carolina, took a different approach to studying ApoE effects. Zhao wondered whether ApoE genotype could help explain the contradictory data on estrogen replacement therapy, which has shown a cognitive benefit in some studies but not in others (see, e.g., a review by Gleason et al., 2005, and a report from the Women’s Health Initiative Memory Study by Shumaker et al., 2003). Zhao suggested that several factors may influence estrogen’s effects. One of these is the timing of hormone replacement. For example, a recent study found that hormone replacement therapy in midlife protects against dementia, while in late life it increases dementia risk (see Whitmer et al., 2010). Other factors, Zhao said, include the hormone type and a woman’s ApoE genotype. An earlier study on atherosclerosis found that women with ApoE2 or ApoE3 alleles benefited from estrogen therapy, while women with an ApoE4 allele did not (see Lehtimäki et al., 2002). Zhao and colleagues wondered if ApoE status may also impact estrogen’s effect on cognition. “In general, this has been a neglected area of research that may have a great deal of public health impact,” Colton wrote in an e-mail to ARF.
Previous work in the Colton lab demonstrated that 17β-estradiol has weaker anti-inflammatory effects on microglia from mice that have their ApoE gene replaced by a human ApoE4 gene, compared to microglia from ApoE3 targeted replacement mice (see Brown et al., 2008). In vitro studies showed that ApoE binds to the estrogen receptor (ER), suggesting it may act as an estradiol coactivator. Zhao tested this idea in cell cultures using the mouse neuroblastoma cell line N2a, which expresses only ERα, and a mouse catecholaminergic neuronal cell line, CAD, which expresses only ERβ. Co-immunoprecipitation studies showed that ApoE bound to ERβ but not ERα. Zhao is now studying the effect of the ApoE allele in cell cultures containing both estrogen receptors using reporter assays, and will look for any difference in estrogen response in the presence of ApoE3 or ApoE4 alleles. She speculated that ApoE may modulate the estrogen response by directly interacting with ERβ and indirectly affecting downstream ERα pathways involved in neuroprotection. (For more on estrogen, see ARF Live Discussion from 2006.)
Is It the Diet?
Other talks focused directly on the effects of cholesterol. Lifestyle factors such as cholesterol intake and exercise are known to alter AD risk. Amanda Kiliaan of Radboud University Nijmegen Medical Center in The Netherlands described published work that examined the effects of diet on cerebral blood volume, Aβ deposition, and learning and behavior (see Hooijmans et al., 2009), as well as recent unpublished experiments. Kiliaan and colleagues fed APP/PS1ΔE9 and wild-type mice either a standard diet of rodent chow, a cholesterol-rich diet resembling a typical Western diet, or a diet rich in docosahexaenoic acid (DHA), a omega-3 fatty acid found in fish oil and believed to be good for brain health (see ARF related news story and ARF news story). The first differences between mice on alternate diets appeared at 15 months, with AD mice eating high-cholesterol chow showing greater Aβ deposition than AD mice on standard diets. AD mice receiving DHA, on the other hand, demonstrated improved spatial memory, greater cerebral blood volume, and fewer Aβ deposits than those on standard chow.
Kiliaan also presented new work, in which AD mice were fed enhanced DHA diets that included various B vitamins, antioxidants, and other essential precursors and cofactors, in a formulation called Fortasyn ConnectTM (see ARF related news story on human trials of this mixture). On Fortasyn, six- to eight-month-old AD mice showed behavioral improvement compared to mice on standard diets or given DHA alone, demonstrating better memory and less anxiety. Kiliaan speculated that Fortasyn may target more mechanisms than simple DHA, and may improve the health of cell membranes. Kiliaan and colleagues are also analyzing data from ApoE knockout mice and ApoE4 targeted replacement mice fed cholesterol-rich or Fortasyn-supplemented diets.
Kiliaan’s results agree with a recent paper from researchers led by Christian Humpel at Innsbruck Medical University in Austria, in which rats fed a high-cholesterol diet had poorer spatial memory than rats on normal chow (see Ullrich et al., 2010). Rats eating cholesterol-rich food had higher levels of Aβ42 and phosphorylated tau in the cortex, as well as increased inflammation and more cortical microbleeds, Humpel found. These rats also lost cholinergic neurons. Overall, the effects of high cholesterol on the brain resemble those of AD, Humpel and colleagues concluded.—Madolyn Bowman Rogers.
This concludes a three-part series. See also Part 1 and Part 2.
No Available Comments
Spinal cord injury creates a rift that has proven extremely difficult to bridge—once the spine is severed or crushed, the neurons on either side remain forever divided. But perhaps there are ways to close the gap, researchers reported at the Society for Neuroscience annual meeting, held in San Diego, California, 13-17 November 2010. They agreed it will likely take a combination of approaches to truly repair spinal injuries.
Several researchers reported that they boosted axon regeneration in rodents by tapping a neural fountain of youth. In a nanosymposium, Kai Liu of Children’s Hospital in Boston, Massachusetts, presented one way to rejuvenate neurons. Liu and colleagues targeted mTOR, which promotes growth in developing rodents. Enhancing mTOR activity in injured adults led to axon regeneration that impressed several scientists in attendance. “The magnitude of the effect is quite large” in terms of how many axons sprouted beyond the lesion, commented Stephen Strittmatter of the Yale University Medical School in New Haven, Connecticut. Other researchers adopted different approaches (see Part 2 of this series). Ryan Williams and Mary Bunge, of the University of Miami Miller School of Medicine in Florida, described how they rejuvenated adult neurons by promoting other genes that are active early in development. In a poster presentation, Paul Lu and Mark Tuszynski, both of the University of California in San Diego, discussed their use of embryonic neuron grafts to repair spinal cord lesions. Finally, Martin Oudega of the University of Pittsburgh presented work performed mainly in the laboratory of his collaborator Barbara Grimpe at Heinrich Heine University in Düsseldorf, Germany. Grimpe and colleagues used an RNA knockdown technique to prevent the formation of scar tissue that normally inhibits regeneration.
Past research indicates that axon growth depends on the balance of growth-promoting and growth-inhibiting factors in the central nervous system (reviewed in Deumens et al., 2005; see ARF related news story on Alto et al., 2009 and Peng et al., 2009). Researchers have enhanced axonal regeneration with growth factors (Hiebert et al., 2002) and drugs that promote cell plasticity (García-Alías et al., 2009). Others have taken the complementary approach, using genetic alterations (Cafferty and Strittmatter, 2006) or antibodies (Thallmair et al., 1998) to block the proteins that would normally inhibit growth.
The new approaches at SfN are a step forward, but progress in spinal cord repair continues to be slow. As Bunge put it in an interview with ARF: “The rats were not climbing out of the cage.”
No PTEN, No Problem
Liu worked with joint first author Yi Lu and senior author Zhigang He, also at Children’s Hospital. The trick, Liu said, is to knock out phosphatase and tensin homolog (PTEN), a negative regulator of mTOR. With mTOR’s cell growth-promoting activity enhanced in mice, Liu showed, neurons grew right up to the edge of injury and several leapt the fissure to the other side. Once there, they appeared to form pre-synaptic boutons, although the researchers do not know if it is possible to regenerate fully functional synapses. Liu’s presentation covered work published in Nature Neuroscience in September (see Liu et al., 2010).
Collaborator Oswald Steward of the University of California in Irvine has been studying regeneration since 1974. “The corticospinal tract has always been thought of as the most resistant to treatments,” he told ARF in an interview. “Nothing like this has ever been seen,” he said of the findings.
In an e-mail to ARF, Liu added: “Reproducible regeneration of ascending sensory tract, bulbospinal, and propriospinal tracts after spinal cord injury have been shown previously, but not in the corticospinal tract…. The kind of regeneration seen before was still quite limited and often required a combination of strategies.”
Two years ago, the He lab published a paper on PTEN deletion and regeneration of optical nerves (Park et al., 2008). Optical nerves are a convenient system for study, Liu told ARF, but what scientists really want is to repair damaged spinal nerves.
Expression of mTOR decreases gradually with age, Liu said, and with it drops the body’s ability to regenerate nerves. “The drive is gone, the pathway is silent,” he said. But PTEN deletion could offer a way to artificially amplify the regeneration pathway in adults.
The researchers used mice with floxed PTEN and AAV-Cre injections in the motor cortex to obliterate the PTEN gene in neonatal mice. Then, in six-week-old animals, they tried two injury models: dorsal hemisection and complete crush. In the hemisection, the researchers sliced apart the dorsal part of the spinal column but left the ventral portion intact, creating a cleft. For crush injury, they compressed one part of the spinal column. The severe crush injury is more clinically relevant, Steward said, because the spinal cord bruising that it mimics is more common than a hemisection. The researchers injected the tracer biotinylated dextran amine (BDA) into the sensorimotor cortex to stain uninjured axons upstream of the injury. Any BDA-labeled fibers downstream of the injury would indicate the axons crossed the damaged site.
Normally, axons retract from an injury, and growing axons halt before they come right up to a lesion. In nine control animals, which had an intact PTEN gene, the researchers observed no axons crossing the lesion eight weeks after injury. In two other control mice, only a couple of axons reached across the divide. But in all 11 PTEN-knockout mice, Liu and colleagues saw many axons on the far side of the lesion. “This is a dramatic proximal sprouting,” Liu said during his presentation. Some axons traversed the lesion directly, while others crossed via the intact ventral spinal cord. This is exciting, Bunge told ARF, because most axon regeneration protocols only lead to axons crossing the intact tissue, not the gap. Axons grew an average of one millimeter beyond the lesion.
The researchers sought confirmation, recruiting Jae Lee in the laboratory of Binhai Zheng at the University of California in San Diego to repeat the hemisection experiments. Lee also saw axons traveling across and beyond the injury.
Back in Boston, Liu also tested his theory in the crush injury model. In eight control animals, not a single axon bridged the divide, 12 weeks after injury. “I have never seen any single corticospinal tract axon ever grow across the lesion” in control animals, Liu told the audience. But in eight PTEN knockouts, he saw plenty of lesion-crossing (see image below).
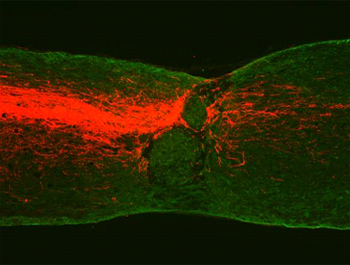
Crossing the Great Divide
Axons (red) grow into and beyond a spinal cord crush lesion in animals lacking PTEN. Image credit: Kai Liu, Children's Hospital Boston
Since mTOR expression declines with age, the researchers checked if regeneration was possible in older animals. In mice that lost PTEN after birth, but did not suffer injury until five months of age, axons still regenerated. In another experiment, the researchers waited until the mice were four weeks old to inject the AAV-Cre. Although the PTEN deletion was less efficient, they nevertheless observed some regeneration.
“The discovery of the PTEN pathway as a powerful controller of CNS axon regeneration is a landmark advance,” wrote Ben Barres of Stanford University in Palo Alto, California, in an e-mail to ARF. “Now, the big question is whether the regenerated axons will reform their specific synaptic connections and restore neurological function.”
The He lab researchers collaborated with Steward to examine their animals for any indication of synapse formation. When they stained sections for the pre-synaptic marker vGlut1, they saw it along axons and in the bouton-like axon tips, a promising sign. Further, in electron micrographs, the scientists observed that the axons had contact zones holding pre-synaptic vesicles as well as nearby post-synaptic densities. However, the researchers are still working to discover if these synapse-like structures can actually transmit action potentials. Strittmatter said he would really like to see some evidence of recovery in the injured animals. Liu told ARF those experiments are ongoing, but it is too early to form any conclusions.
Another possible explanation is that the neurons, having grown up without PTEN, were more likely to regenerate, Tuszynski suggested in an e-mail to ARF. He wondered, “Could this be resistance to injury, instead of enhanced regeneration?”
Having shown regeneration in both optic and corticospinal nerves, the authors suspect it will work in other systems. “It is highly likely that [Liu and Lu] have found a core mechanism of axonal regeneration,” Steward said.
There are many future challenges in converting these results into a therapy. For one, the researchers noticed that growing axons avoided areas occupied by macrophages and fibroblasts. They appeared to prefer growing alongside an unidentified cell population expressing glial fibrillary acidic protein (GFAP), a common marker for astrocytes. Macrophages seem to cause axons to retract in control animals, Liu said: “I believe that if you had a way to eliminate the macrophages, likely you may increase axonal growth.” In addition to Liu’s evidence that axons grow along GFAP-positive cells, Bunge told ARF she has observed a role for astrocytes in regeneration as well.
Another challenge, Steward noted, is to find a therapy that would work several months post-injury, which would be more applicable in a clinic. Doctors would also need a way to activate mTOR without genetic engineering. The researchers have tested a few PTEN inhibitors in vitro, Liu told the audience, but thus far found none that support regeneration. RNA interference is also a possibility, Steward said.
Liu told ARF he hopes the research in He’s lab will entice more researchers to study the topic. “It has been a sort of dark ages in the regeneration field,” he said, but he thinks the new results herald a renaissance.
What does any of this mean for neurodegeneration? Steward thinks it is likely the PTEN pathway could help there, too. “What you’re doing is essentially rejuvenating the neuron,” he said. “I cannot imagine that would not contribute to maintenance of a sick cell.” Already, researchers have found that siRNA for PTEN boosted motor neuron numbers in a mouse model of spinal muscular atrophy (Ning et al., 2010).—Amber Dance
This is Part 1 of a two-part series. See also Part 2.
No Available Comments
Paralyzing spinal cord injuries are often no longer than an inch. They might as well be a mile. Repairing severed or severely damaged spinal cords is an intractable medical problem, and progress in solving it has been slow. Using rodent models, researchers are trying various means of coaxing neurons to extend their axons across spinal cord lesions, and several new methodologies were explored at this year’s annual meeting of the Society for Neuroscience, held 13-17 November in San Diego, California. Strategies ranged from rejuvenating neurons by reinitiating developmental pathways to breaking down physical barriers to axon growth in the damaged spine.
Researchers led by Zhigang He at Children’s Hospital, Boston, Massachusetts, achieved some success in rodents by knocking out the phosphatase and tensin homolog (PTEN), which is a negative regulator of mammalian target of rapamycin (mTOR) and a major driving force in neurodevelopment (see Part 1 of this series). At SfN, other researchers reported similar rejuvenation attempts. Paul Lu and Mark Tuszynski, both of the University of California in San Diego, simply grafted young nerves. They transplanted spinal cord neurons from embryonic day 14 (E14) rats into adult cords that are severed halfway through at the neck. Transplantation is a well-known method to enhance nerve repair, Lu told ARF in an interview, but until now, researchers were unsure how far axons from the transplanted neurons spread. “It is very hard to track these cells,” he said. To get an accurate picture of the transplants, Lu used E14 rat neurons that express green fluorescent protein. In addition, Lu improved upon previous graft techniques by including a fibrin matrix to support the transplanted cells at the injury site.
Lu observed glowing green axons spreading out from the graft site. He estimated that 1,000 axons were growing in a 30-micron section of spinal cord. These grew as far as 30 millimeters when he examined the tissues at three months post-treatment. Moreover, he observed synapse formation, and the treated rats were able to move their hind limbs.
Ryan Williams and Mary Bunge of the University of Miami Miller School of Medicine in Florida used cell transplants of a different sort, combined with a transcription factor, to make adult neurons act like young’uns again. Bunge’s laboratory has long been interested in using Schwann cell transplants to provide fertile ground for axon regeneration (see ARF related news story; Xu et al., 1997; Takami et al., 2002). The Schwann cells prevent secondary damage to remaining neurons and myelinate any remaining axons as well as new, regenerated ones. However, they do not allow for complete recovery, and Bunge believes a multifactorial approach combining Schwann cell transplants plus other treatments will be most effective (reviewed in Bunge, 2008).
In their study, the researchers combined the Schwann cell treatment with a transcription factor they hoped would nudge the adult neurons toward a regeneration-capable phenotype. Mammalian achaete-scute homolog-1 (Mash-1) is expressed during development, when brain stem neurons are sending processes toward the spinal cord. “What we’re trying to do, basically, is make the nerve cell bodies younger,” Bunge said.
Williams injected AAV vectors carrying the Mash-1 transgene into rats’ brainstems up to six weeks before completely transecting the thoracic spinal cord. He examined the animals six weeks after injury, when he stained tissues for dopamine-beta hydroxylase (DBH), a marker for neurons that reside in the brainstem, far from the lesion site, and send axons down the spinal cord. Those axons from these neurons were cut at the time of injury, but some then grew across the lesion. In the Mash-1-treated animals, there were 2.5-fold more DBH-positive axons, which grew at least 0.25 millimeters into the Schwann cell bridge, than in animals that received Schwann cell transplants alone. A handful of axons grew as far as 2.5 millimeters into the bridge. Moreover, Mash-1-treated animals moved their hind limbs better than the transplant-only rats.
Bunge was cautious about overinterpreting the results. “It is not a really strong improvement,” she told ARF. “We probably need to change a few other genes, as well.”
Another challenge in regrowing axons is to knock down the barriers in their way. Scar tissue, composed of proteoglycans, provides both a physical and biochemical barrier to regeneration, Bunge said, because the tissue produces growth inhibitors. One method to dissolve the scar is to use chondroitinase, an enzyme that breaks down proteoglycans. Barbara Grimpe and colleagues at Heinrich Heine University in Düsseldorf, Germany, took a different approach to attack scar tissue. As described by coauthor Martin Oudega of the University of Pittsburgh, the researchers blocked scar tissue formation by targeting the enzyme xylosyltransferase-1 (XT-1), which adds glycosamino glycan side chains to proteoaminoglycans.
Grimpe and colleagues used a deoxyribozyme to knock down XT-1 transcription. These so-called “DNA enzymes,” widely used in cancer and virus research, are single-stranded DNA molecules that bind and digest a specific target mRNA. Compared to other RNA interference technology, the authors say, DNA enzymes are easier to administer, and cells, including neurons, naturally import the single-stranded DNA by endocytosis. This treatment blocked proteoglycan formation and, in combination with peripheral nerve grafts, boosted descending axon growth nearly 10-fold (Hurtado et al., 2008). Moreover, the scientists reported at SfN, the treatment was safe, and injured rats on the XT-1 treatment were better able to traverse a horizontal ladder than were untreated animals.
“It is important that many laboratories investigate many different repair approaches,” Grimpe and Oudega wrote in an e-mail to ARF. Bunge agreed: “There is great promise in the combination of cell transplantation, administration of growth factors, manipulation of genes, and the application of the enzyme chondroitinase, which will attack the scar,” she said.—Amber Dance.
This is Part 2 of a two-part series. See also Part 1.
No Available Comments
Like squirmy toddlers dodging Grandma’s repeated attempts to photograph them, Aβ oligomers have proven evasive to the doting biophysicist. Indeed, progress has been slow for researchers toiling to discern the physical makeup of these neurotoxic clumps, widely seen as the form of amyloid-β most dangerous in Alzheimer’s disease. At the Society for Neuroscience (SfN) annual meeting held 13-17 November 2010 in San Diego, several labs presented new efforts to tease out the structure of Aβ oligomers. Surprisingly, a computational approach fingers the N-terminus as especially important for mediating Aβ42 neurotoxicity. Compared to the identical region in Aβ40, a less pathogenic Aβ peptide, the N-terminal end of Aβ42 flops around and is more exposed to solvent—making it potentially more susceptible to enzymatic cleavage and other modifications. The differences are captured in brief video clips (see below). Some scientists wonder how the results fit with prior work suggesting that the N-terminus is not involved in Aβ aggregation. Others seem captivated by the new data’s synchrony with growing evidence that N-terminally truncated forms may be the deadliest of Aβ oligomers. Further clarity should come with future experiments addressing whether amino acid substitutions that change solvent exposure correspondingly influence toxicity of the mutant Aβ peptides.
Having just two extra amino acids at its C-terminal end, Aβ42 seems to give neurons far more trouble than does Aβ40, but the reasons for this remain unclear. It would help if scientists could see the structures of these proteins. However, Aβ oligomers are constantly on the move, coalescing into larger assemblies and falling back apart—making them terribly unwieldy for high-resolution methods such as x-ray crystallography and solution-state nuclear magnetic resonance (NMR). While some scientists have used molecular tricks such as photochemical crosslinking (see Bitan et al., 2003) and disulfide bridges (see Sandberg et al., 2010; O’Nuallain et al., 2010) to tame Aβ oligomers for structural analysis, others are trying to manhandle the misfits from the comfort of their own laptops.
But even in the realm of computational modeling, Aβ oligomers have put up a challenge. The proteins are too complex for all-atom molecular dynamics—the computational models giving the most detailed structural information. “There are too many atoms, and the analysis would take way too long,” Brigita Urbanc, Drexel University, Philadelphia, Pennsylvania, told ARF after presenting her work at an SfN nanosymposium. To tackle the problem, Urbanc and colleagues went with a simplified approach called discrete molecular dynamics (DMD). This technique models atoms in motion as if they were colliding billiard balls. Proteins are reduced into strings of amino acids, each represented by a maximum of four beads, where one bead denotes, for example, a chemical group.
Of course, peptides do not exist in a vacuum, but are surrounded by solvent under normal physiological conditions, which makes molecular modeling even more complex. The analysis involved “no explicit solvent,” but instead used “a force field describing hydrophobic and hydrophilic effects,” Urbanc said. “With these simplifications, we can capture the full process of oligomerization, starting from non-interacting Aβ peptides and ending in a steady state where monomers and oligomers of various sizes are present.” As oligomers “form” on the computer screen, the scientists can separate them by size and do more detailed structural analysis on specific species such as dimers or trimers.
In earlier simulations of Aβ40 and Aβ42 oligomer formation using the four-bead protein method (Urbanc et al., 2004), “…the intriguing thing is that Aβ42 seemed to have more exposed N-termini than Aβ40 even though the structural differences are at the C-terminus,” Urbanc told attendees. Folding and assembly of even short proteins like Aβ are complex processes, she noted, and a sequence change at one position can cause structural changes anywhere in the sequence. This was also exemplified in her recent study (Urbanc et al., 2010), which used the same four-bead simplified approach to characterize oligomers of Aβ40 and Aβ42 and their Arctic mutants. With this mutation, “the amino acid substitution (E22G) is at the center of the peptide, and yet the [structural] differences are in the N-terminus,” Urbanc said. Specifically, her team found that the N-terminal end of Aβ40 has a short β strand that does not appear in Aβ42, or in the Arctic mutants of either full-length Aβ40 or Aβ42. These findings hinted that the N-terminus “presents the free energy barrier…that may be the reason Aβ40 doesn’t so readily form larger oligomers,” Urbanc said.
To test this prediction, Bogdan Barz, a postdoctoral fellow in Urbanc’s lab, selected 10 Aβ40 and 10 Aβ42 dimers formed in silico with the four-bead protein modeling, and examined their stability in a shorter (50-nanosecond) simulation using the more detailed all-atom approach. Aβ oligomers are typically too dynamic and complex for this comprehensive method, but in this case the researchers chose limited numbers of a specific Aβ species (i.e., dimers), making the analysis more manageable. Consistent with their structural data from the simplified model, the N-terminus “is doing some kind of dance, moving around and returning to its original position. This is typical for Aβ42, but not Aβ40,” Urbanc said as she showed video clips of each dimer “immersed” in water (see movies below).
Click on the images below to view each movie. (Please be patient; the movies may take several seconds to load.)


Aβ42 N-Termini—Movin’ and Groovin'
In computational models of Aβ oligomer formation, N-terminal region (with position 1 aspartic acid shown as red spheres) of Aβ42 dimer (right panel) is more flexible and less structured than the same region of Aβ40 dimers (left panel). Aβ peptides within each dimer appear in blue and green. Image credit: Bogdan Barz and Brigita Urbanc, Drexel University
In another round of analyses, researchers applied the four-bead protein method to study Aβ42 oligomer formation in the presence of three Aβ-derived C-terminal fragments (Aβ30-40, Aβ31-42, and Aβ39-42) previously shown to block Aβ42 neurotoxicity in cell culture studies done in the lab of coauthor Gal Bitan at the University of California, Los Angeles (see ARF related conference story and Fradinger et al., 2008).
In these simulations, the C-terminal toxicity inhibitors stuck themselves in between Aβ42 molecules, relaxing the β-strand structure of the full-length peptides and hindering their aggregation. Each toxicity inhibitor “acted like a glue that clamped the N-termini of Aβ42 and prevented them from moving around,” Urbanc told ARF. “Aβ40 is less toxic and has smaller solvent exposure at the N-terminus than Aβ42. But toxicity inhibitors seem to make Aβ42 have less solvent exposure and behave more like Aβ40.” The scientists also tested the effect of a control peptide, Aβ21-30, which did not affect Aβ42 toxicity in cell cultures, and showed that Aβ21-30 increased the solvent exposure of Aβ42 N-termini. “We conclude that perhaps it’s the exposure of the N-terminus to solvent that is in some way mediating the toxicity of Aβ42 oligomers.”
The new data are “extraordinarily compelling,” Thomas Bayer of the University of Goettingen, Germany, wrote in an e-mail to ARF. Urbanc and colleagues “demonstrated that the N-terminus of full-length Aβ oligomers is sticking out, and therefore exposed to potential peptidases for N-terminal truncation.” Given increasing evidence from Bayer’s and other labs that N-terminally truncated AβX-42 oligomers are highly toxic, aggregating more quickly than full-length Aβ (see ARF related conference story on pyroglutamate Aβ), Bayer noted that in Urbanc’s studies, “structural form and potential pathological function nicely fit together.”
Michael Nichols of the University of Missouri, St. Louis, was also intrigued by the new data, but more in a head-scratching sort of way. Whereas Urbanc’s results suggest that the N-terminus may affect Aβ oligomerization, “many studies, including our own (Touchette et al., 2010), indicate that the N-terminus is not involved in Aβ aggregation and remains available for modification after fibrils are formed (see Petkova et al., 2002 and ARF related news story; Whittemore et al., 2005; Kheterpal et al., 2001),” he wrote in an e-mail to ARF. In support of the SfN studies, researchers led by coauthor David Teplow at UCLA reported that two familial AD-linked mutations at the peptide’s N-terminus (English and Tottori) hastened Aβ assembly and increased toxicity (Ono et al., 2010).
As follow-up to the current work, Urbanc said her team plans to use both computational and experimental methods to test the predictions implicating the solvent exposure of Aβ42’s N-terminus in mediating toxicity. “Computationally, my group will explore single or double amino acid substitutions to predict how these changes affect solvent exposure of both Aβ40 and Aβ42, and then correlate our findings with available toxicity data on these mutated peptides,” Urbanc told ARF. “My group will also collaborate with the Teplow and Bitan labs to experimentally characterize structural changes induced by selected substitutions.”
At SfN, Leonid Breydo of Charles Glabe’s lab at the University of California, Irvine, described his latest efforts to characterize Aβ oligomers in “wet lab” structural studies. The Glabe lab has developed a number of conformation-specific Aβ antibodies (see Kayed et al., 2003 and Kayed et al., 2007), which they use to classify oligomeric Aβ as prefibrillar or fibrillar. The current study used site-specific denaturation and spectroscopy methods to compare the stability of Aβ40 fibrils, fibrillar oligomers, and prefibrillar oligomers. (All experiments were done with Aβ40 instead of Aβ42 because the latter aggregates so quickly that it is hard to work with, Breydo said during question time.) For the denaturation experiments, the researchers made 13 Aβ40 mutants, each with a different acrylodan-labeled cysteine substitution, and generated fibrils, fibrillar oligomers, and prefibrillar oligomers from each mutant peptide. Acrylodan is a polarity-sensitive fluorescent dye used here to measure local hydrophobicity of different regions in the tested proteins. The researchers measured hydrophobicity by using fluorescence spectroscopy to monitor the various Aβ species under various denaturing conditions. These data support the idea that fibrillar oligomers are structurally similar to Aβ fibrils, whereas prefibrillar oligomers look different. Their N-termini are more exposed, since “the acrylodan environment there is very polar—as polar as it is in water,” Breydo noted in an e-mail to ARF. “Prefibrillar Aβ oligomers may have a micelle-like structure,” Breydo said. “We think this because hydrophilic N-termini of prefibrillar oligomers are fully exposed, while hydrophobic regions of the structure are buried.”
Bitan mentioned a potential technical issue with this study—the possibility that the results could be influenced by experimental manipulations that compromise biological relevance. “The substitution by cysteine and introduction of the acrylodan fluorophore, which is quite bulky compared to most amino acid side chains, may alter the structure of the oligomers,” he wrote in an e-mail to ARF. He acknowledged that such concerns plague nearly every structural study of Aβ that uses molecular trickery to coax the peptide into forms amenable for analysis. And, perhaps more importantly, since the study only looked at Aβ40, he suggested, it is unclear whether the data “advance our understanding of the Aβ oligomer structures that are relevant to AD.” Breydo told ARF his team may examine Aβ42 oligomerization in future studies.—Esther Landhuis.
In Alzheimer’s disease and other neurodegenerative disorders, misfolded proteins are seen banding together en masse, wreaking havoc in neurons. These findings have prompted research on potential treatments that rev up intracellular pathways to degrade and clear the rogue proteins. At the Society for Neuroscience annual meeting (SfN) held 13-17 November 2010 in San Diego, scientists talked about boosting autophagy in mouse models. One pharmacological approach improved pathology and behavior in tauopathy mice, while a genetic strategy showed similar benefits for a strongly amyloidogenic AD model.
Led by Wai Haung (Ho) Yu and Karen Duff, researchers at Columbia University Medical Center, New York, induced autophagy in the JNPL3 and rTg4510 tau transgenic models using trehalose, a disaccharide made by plants, fungi, and invertebrates. This compound has been shown to promote clearance of mutant huntingtin and α-synuclein in cell culture experiments (Sarkar et al., 2007), and to relieve motor dysfunction when administered orally in a Huntington’s mouse model (see Tanaka et al., 2004 and ARF related news story). The Columbia researchers wondered if the sugar could also relieve tauopathy in P301L transgenic mice with predominantly motor (JNPL3) or cognitive (rTg4510) phenotypes. Yu reported the results of these studies at SfN.
As a proof of concept, the scientists treated JNPL3 brain slice cultures with trehalose and other compounds that inhibit or induce autophagy. Trehalose did, in fact, promote autophagy (as judged by upregulation of LC3-II, a marker of autophagic vacuoles) and clear tau aggregates, as measured by a reduction in sarkosyl-insoluble tau detected using the human-tau specific antibody CP27 (Duff et al., 2000).
For the in vivo studies, the Columbia team then tested trehalose in preventive and therapeutic paradigms. They did the prevention study in JNPL3 mice, which develop neurofibrillary tangles, and behavioral and motor deficits that mimic human tauopathies (Lewis et al., 2000). Yu and colleagues gave JNPL3 mice 2 percent trehalose (or sucrose as a control) in their drinking water starting at 3.5 to four months of age, before tau pathology develops. After eight weeks, the treated group had lower levels of hyperphosphorylated tau (PHF1) and sarkosyl-insoluble tau (CP27) in the cortex than did the sucrose-fed controls. The trehalose group had better motor skills, as measured by rotarod and by how the animals fared on a hanging wire and when held by the tail. The researchers saw the same benefits in a therapeutic study in which they gave a four-week trehalose treatment to five-month-old mice with early-stage tau pathology.
Yu and colleagues also studied treatment in rTg4510 mice, which model AD and frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) by showing tangle pathology, neuron loss, and cognitive impairment. They dripped trehalose in the drinking water starting at four to five months of age, when the mice already have aggregated tau. After eight weeks, treated rTg4510 mice had reduced levels of insoluble tau and aggregated p62, which is thought to bind tau and ferry it into the autophagic system for eventual degradation. Importantly, compared with sucrose-fed rTg4510 mice, the trehalose group also did better in the Morris water maze test of spatial learning and memory, though not quite as well as non-transgenic mice. Yu said his lab has a Tg4510 prevention study underway.
Company researchers seemed intrigued by the data. Yu told ARF he “fielded many questions from pharma,” including representatives from Pfizer, Bristol-Myers Squibb, Amgen, and Eli Lilly. Steve Jacobsen, who moved to Proteostasis in Cambridge, Massachusetts, after Pfizer bought his former company, Wyeth, said of the SfN data, “Overall I had a very favorable impression.” On a broader level, he noted that if a therapeutic strategy can be worked out for clearance of one misfolded, aggregated protein, there is a “good possibility it may clear other misfolded, aggregated proteins as well.”
In an SfN symposium entitled “AD-360°: Nonamyloid Mechanisms in Alzheimer’s Disease Pathogenesis” (see also Pimplikar et al., 2010), Ralph Nixon, New York University School of Medicine, spoke about a genetic approach for stimulating autophagy in TgCRND8 mice. This model has amyloid pathology starting at 8-10 weeks, with spatial memory loss developing soon after. Dun-Sheng Yang, of Nixon’s lab, established that TgCRND8 mice have impaired autophagy, as shown by grossly enlarged lysosomes, overabundance of ubiquitinated proteins, and defective proteolytic clearance of neuronal autophagic substrates including Aβ.
To reinvigorate the autophagy system in the TgCRND8 model, Yang and colleagues crossed them to mice genetically lacking cystatin B. Not to be confused with cystatin C, an AD genetic risk factor, cystatin B (CstB) inhibits the activity of cathepsins, enzymes that help break down proteins within lysosomes. Prior work in people (Rinne et al., 2002) and mice (Kopitar-Jerala and Turk, 2007) showed that reduced CstB activity correlates with increased cathepsin activity. Breeding TgCRND8 transgenic mice with CstB-knockout mice lifted the brakes on cathepsin activity, leading to enhanced autophagy as judged by high protein turnover rates in the crossed animals. The cross “represents what we consider a targeted intervention at the level of the lysosome,” Nixon said.
The NYU researchers analyzed the progeny at six months of age, when amyloidosis is well underway in the TgCRND8 strain. The autophagy defects were much reduced in the CstB-deleted transgenic mice, as shown by less intraneuronal Aβ, ubiquitinated proteins, and other autophagic substrates—including Aβ (see Yu et al., 2005)—clogging the system. CstB-deficient TgCRND8 mice also had less extracellular amyloid deposits and lower brain Aβ40 and Aβ42 levels. And on two cognitive/memory measures—fear conditioning and olfactory habituation (a newer test developed at NYU to measure memory for odors)—CstB-deleted TgCRND8 mice showed “significant improvement,” and were barely distinguishable from wild-type and CstB knockout controls, Nixon said. The findings will be published in the December 15 issue of Brain.
Nixon mentioned one caveat with the data. “Cystatin B deletion from birth creates its own pathology,” he told ARF. CstB-deleted mice model a form of childhood epilepsy, though they seem cognitively intact and their seizures are clinically invisible. Still, Nixon and colleagues were pleased to find that, despite the potential for “superimposed problems” from using CstB knockout mice, “our intervention was very effective in reversing the pathology [in TgCRND8 mice],” Nixon said. “One would not want to eliminate cystatin B from birth in a human to treat AD, but it's quite possible that once you get over the developmental period, a drug that mimicked the effect of eliminating cystatin B would be therapeutic.” To model a therapeutic paradigm, Yang and colleagues are now using viral vectors to induce RNA-mediated silencing of cystatin B in the brains of TgCRND8 mice.—Esther Landhuis.
No Available Comments
Since the approval of cholinesterase inhibitors and memantine, no new therapy for Alzheimer’s disease has passed the clinical trial litmus test. But that does not stop ideas from coming. At the 40th annual meeting of the Society for Neuroscience (SfN), held 13-17 November 2010 in San Diego, California, researchers laid several new strategies on the table, from glucagon-like peptide agonists that tweak insulin signaling pathways (see ARF related news story), to molecular tweezers and other compounds that prevent amyloid-β oligomerization and toxicity. Numerous compounds have been identified that seem to block Aβ aggregation, but the mechanism is often unclear and nothing has made it into the clinic. “The important thing is that we invest in trying to understand how these compounds work,” said Gal Bitan, University of California, Los Angeles, who chaired an SfN nanosymposium dedicated to therapies targeting Aβ and Aβ oligomers. “It is crucial to find out what those molecules bind, where they bind, and how they inhibit. This will help us find compounds that will be a success,” Bitan told ARF.
Case in point are small fragments from the C-terminus of Aβ42 (Aβ28-42 and smaller) that block Aβ aggregation (see Fradinger et al., 2008). Bitan co-founded Clear Therapeutics, and holds patents on lead compounds for AD therapy. Working in Bitan’s lab, Huiyuan Li and colleagues have used rational drug design to generate second-generation versions of these small peptides, systematically modifying the amino acids and looking at structure-function relationships. In her talk, Li noted that Aβ39-42 potently inhibits Aβ neurotoxicity, but is likely easily degraded and would make a poor drug candidate. Since those amino acids have mostly aliphatic side chains, one would not expect them to do anything other than act as a hydrophobic glue, said Bitan. However, a D-amino acid analogue of the peptide had little anti-Aβ activity, said Li, which suggests that specific peptide-peptide interactions are crucial for inhibiting Aβ neurotoxicity. Li showed that the side chain of isoleucine 41 is necessary, for example, and she has a variety of new derivatives that are being tested. So far, data from light-scattering experiments suggest that these compounds work by facilitating formation of non-toxic “hetero-oligomers” composed of a mixture of Aβ and the short peptide molecules. At SfN, collaborator Birgita Urbanc at Drexel University, Philadelphia, Pennsylvania, reported that the modified C-terminal peptides relax the β-strand structure of the full-length Aβ and hinder its aggregation.
Another strategy the Bitan lab pursues is to use molecular “tweezers” to block Aβ from forming multimers. In her talk, Aida Attar described the tweezers as C-shaped molecules that wrap around lysine side chains. Originally introduced by collaborators Thomas Schrader and Frank-Gerrit Klärner, University of Duisburg-Essen, Germany, the tweezers have hydrophobic arms that interact with lysine’s butylene side chain. A negatively charged cavity that separated the arms sits over lysine’s positively charged amino group. With this structure, the tweezers bind to lysine with high affinity, said Attar. Bitan, Schrader, and colleagues have been modifying these compounds to find molecules that can reduce protein aggregation. Attar focused on CLR01, a compound showing some efficacy in transgenic AD mice and in-vitro assays, she said.
In thioflavin T fluorescence tests of protein aggregation carried out by coauthor Sharmishta Sinha, CLR01 blocked the assembly of Aβ into β-sheet structures. Electron microscope pictures showed that the compound prevented Aβ from forming fibrillar structures in vitro. Similarly, while Aβ incubation normally yields oligomers that react with the A11 antibody on dot blots (A11 is specific for protein oligomers), Attar and Sinha found no A11 cross-reactivity when CLR01 was in the mix (the 6E10 antibody that reacts with most forms of Aβ tested positive). CLR01 also blocked tau aggregation as judged by thioflavin assays and electron microscopy, Attar said.
At concentrations up to 400 μM, CLR01 had little effect on the viability of PC12 neuroblastoma cells, but at substantially lower concentrations it completely rescued cell loss driven by 10 μM Aβ. The compound almost completely prevented Aβ-induced dendritic spine loss in primary hippocampal neurons, as well. And in single-cell recordings in primary hippocampal neurons, CLR01 rescued loss of mini-excitatory post-synaptic current (mEPSC) frequency that occurs in the presence of Aβ.
Early indications are that the compound can prevent protein aggregation in vivo, too. Attar and colleagues used subcutaneous administration of CLR01 into 14- to 16-month-old triple transgenic mice (see Oddo et al., 2003). On autopsy 28 days later, plaques and tangles were reduced compared to placebo-treated controls. The reductions occurred in most affected regions of the brain, including the CA1 of the hippocampus. The researchers have no behavioral data because they did not find any memory deficits in the 3xTG animals at the recommended age for study, said Attar. (Other researchers have also reported loss of the memory phenotype in these mice.)
Whether these compounds will make it into human testing remains to be seen. Their ability to bind generically to any lysine might raise safety flags, requiring preclinical controls to ensure that they do not interfere with normal protein function. Attar reported that at 30 times the effective dose, CLR01 had no obvious toxic effects on mice.
Andreas Müller-Schiffmann presented a different strategy for blocking Aβ oligomerization. Müller-Schiffmann works with Carsten Korth at the Heinrich Heine University of Düsseldorf, Germany. The researchers are taking a synergistic approach, combining two different classes of molecule that independently prevent Aβ aggregation in the hope of finding a much more potent compound. One class, aminopyrazoles (APs), are small molecules that bind the cross-β-sheets stabilizing β amyloid and other amyloid structures. APs, which like the molecular tweezers were developed by Schrader’s group, recognize the KLVFF amino acid motif of Aβ, said Müller-Schiffmann, but while they decrease Aβ aggregation in vitro, they work less well in vivo.
The researchers combined APs with a dodecapeptide called D3 that was found in a mirror phage display screen by researchers at Dieter Willbold’s group, also at Heinrich Heine University (see ARF related news story). Molecular simulation suggested to the German scientists that adding APs to D3 would yield a compound that bound very stably to Aβ and could disrupt aggregation. A hybrid molecule, JM169, then prevented oligomerization of Aβ secreted from 7PA2 cells, which are used as a natural source of Aβ (see Walsh et al., 2002). Neither D3 nor aminopyrazoles by themselves had strong effects. The researchers reported some of these results in the November Angewandte Chemie (see Müller-Schiffmann et al., 2010).
In addition, JM169 rescued long-term potentiation deficits in hippocampal slices treated with Aβ. The hybrid also partially restored mESPC in these slices. Bitan complimented this rational approach to drug design and the synergy in the hybrid, but wondered whether the size of the molecule might limit its use in vivo.
In her talk, Susan Catalano from Cognition Therapeutics, Inc., Pittsburgh, Pennsylvania, took a different tack. Instead of addressing Aβ oligomerization/aggregation, Catalano focused on preventing downstream effects. Aβ oligomers affect the rate of membrane trafficking, for example, which is essential for proper synaptic transmission and plasticity. “Depending on the Aβ oligomer, they can accelerate exocytosis, they can inhibit endocytosis, they have a variety of different actions on membrane trafficking,” she told ARF. Catalano’s team used a membrane trafficking screen (dyed cycling vesicles) to search for compounds that prevent Aβ oligomers from interfering with these processes. The researchers tested a variety of Aβ oligomer preparations, including those derived postmortem from human AD patient brain. “We use every Aβ oligomer preparation we can get our hands on, because though there are certain hypotheses about what the active oligomer species is, I don’t think it is yet established which ones play what role at what point in the development of AD. So we take a comparative approach,” Catalano told ARF.
One of the compounds, CT0093, restored trafficking in primary hippocampal neurons that were treated with 130 pM Aβ oligomers derived from human tissue but had no effect in the absence of Aβ. Catalano said that the CT0093 seems to work by blocking Aβ from binding the cell surface. It also prevented synapse loss in Aβ-treated neurons.
These compounds seem to be effective in vivo. Catalano reported that they prevent learning and memory deficits in an acute toxicity model involving injection of Aβ into wild-type mouse hippocampus. “We find that pharma is increasingly interested in acute models,” Catalano told ARF. “What is going on with transgenic animals is not exactly clear, and the wild-type background offers advantages, including the ability to look at off-target effects. Clearly, both transgenic and wild-type animals are important.”
When the compounds were injected one hour prior to the Aβ oligomers in this model, the mice performed as well as controls in a fear-conditioning test of learning and memory, whereas mice treated with oligomers only did poorly. The company is currently testing the compounds in transgenic models.
Catalano does not know which cell surface receptors these compounds bind. They represent a first in class and readily cross the blood-brain barrier. The company is continuing preclinical studies, she said, but it will be a while before the required safety studies in advance of any clinical trial are done.—Tom Fagan.
No Available Comments
Progranulin mutations cause some forms of frontotemporal dementia, but exactly what the protein does and how it fails in disease are open questions. The dying neurons possess one good progranulin gene, but do not make enough of the protein. New research presented at the annual Society for Neuroscience meeting, held 13-17 November 2010 in San Diego, California, suggests that the deficiency stems from a vicious feedback loop that keeps progranulin expression in neurons low, even as neighboring microglia produce plenty of the protein.
Ezra Rosen, a student in the laboratory of Dan Geschwind, and independent researcher Eric Wexler, all at the University of California in Los Angeles, presented a pair of posters detailing the PGRN-Wnt connection. Premature stop codons in the granulin gene (GRN) that encodes progranulin are a common cause of frontotemporal dementia (FTD). In a chromosomal coincidence, GRN sits just a megabase away from tau, another FTD-related gene on chromosome 17 (see ARF related news story on Cruts et al., 2006 and Baker et al., 2006). Progranulin’s role in neurons is unknown. “Our very simplistic hypothesis is that progranulin is some sort of trophic factor,” Wexler told ARF in an interview. Unique among the dementias, Wexler said, PGRN-associated FTD appears to be caused by a haploinsufficiency, with one valid GRN gene still present. Yet in the brains of people who had FTD, PGRN expression was actually higher than normal, Wexler said, mostly due to PGRN in microglia (see also Philips et al., 2010). “It does not correlate,” he said. “If the microglia can make it from the gene that is still good, why can’t the neurons?”
A clue turned up through Wexler’s separate interest in Wnt signaling, which is essential throughout the body, with roles in neurogenesis (see ARF related news story on Lie et al., 2005) as well as in dementia (see ARF related news story on De Ferrari et al., 2007). As outlined in one poster, Wexler and Rosen started with a study of Wnt1 signaling in fetal human neural progenitors (hNPs) that they differentiated into neurons. After stimulating the cells with Wnt1, the researchers used an unbiased expression screen to examine changes to the transcriptome over periods ranging from two hours to three days. They found widespread alterations in RNA levels of genes related to cell death processes and to neurodegeneration, including both GRN and presenilin-1, which rose and fell at different time points.
The researchers were particularly interested in the Wnt-PGRN connection. They confirmed, via Western blotting, that Wnt1 reduced PGRN protein levels in the hNP-derived neurons. They discovered that knocking down PGRN by half, via RNA interference, increased Wnt1 expression by at least twofold in the hNP-made neurons. The result is a feedback loop in which ever-increasing Wnt1 levels repress PGRN expression more and more, leading to even greater induction of Wnt1. This, the authors suggest, is one possible reason why progranulin might be down in the neurons of people with FTD, even when they have one good GRN gene. Microglia, which make abundant progranulin, may not rely on this pathway to regulate GRN expression. The researchers confirmed their results in postmortem tissue from people who had GRN-mediated FTD; in this tissue they found upregulation of Wnt signaling-related genes compared to control tissues.
In their second poster, Rosen and colleagues described hunting for downstream consequences of progranulin deficiency that might help explain its role in dementia. They infected the hNP-derived neurons with a lentiviral construct carrying a doxycycline-inducible GRN RNAi. Cells treated with the inducer had less than 10 percent of the normal levels of progranulin and altered expression of 153 other genes, compared to untreated cells. Using a standard gene ontology database, the researchers determined related gene groups, or modules, that were most affected by progranulin knockdown. “The only disease category that is represented is dementia, and the only signaling pathway that is represented is Wnt signaling,” Wexler said. Among the dementia-linked genes with altered expression were glycogen synthase kinase 3β (GSK-3β) protein phosphatase 2 A (PP2A), and APC, which are scaffolds mediating presenilin-1/β-catenin interactions. Other affected genes were Wnt1, Frizzled-2, and other signaling and pro-apoptotic genes.
Thus far, the researchers knew that Wnt1, Frizzled-2, and PGRN were all involved together in lab-grown neurons. Next, they sought to confirm their finding in human and animal studies. They compared their GRN-influenced gene set to data from a previously published study on gene expression in brain samples from people who had FTD (Chen-Plotkin et al., 2008). Between the two datasets, a handful of genes overlapped. Frizzled-2 was one of the most highly expressed in both the human and cell studies, so the researchers analyzed this gene further.
For an animal model, coauthor Robert Farese of the University of California in San Francisco provided an as-yet unpublished GRN knockout mouse. These animals show microglial activation by six months and neural loss by 18 months of age, but they had elevated Frizzled-2 in the neocortex at six weeks, suggesting Frizzled-2 upregulation is an early response to GRN deficiency. Frizzled-2 upregulation might be a contributor to neurodegeneration, or might be an insufficient attempt to compensate for GRN loss, the scientists posited. They found that Frizzled-2 knockdown in GRN-deficient hNP-derived neurons led to an increase in cell death, suggesting it could be a neural protector of some kind.
Frizzled-2 upregulation might be a useful early marker for FTD, Wexler speculated. The researchers also looked at other dementia model mice, he told ARF, but only saw upregulation of Frizzled-2 in GRN knockout animals. “It seems to be highly related to this disease,” he said. Indeed, gene association studies have not implicated Frizzled-2 in any other major neurodegenerative disease.
Interestingly, the Frizzled-2 gene is one of a handful of genes that sit between the tau and GRN genes on chromosome 17. “This does seem like a real coincidence,” Wexler said. He hypothesizes that non-coding RNAs in the same region might somehow affect these neighboring genes in a manner that leads to disease, although he admits that is “hand waving” at this point. To address this hypothesis, Wexler is collecting tissue samples from people with GRN-mediated FTD and their unaffected siblings. He plans to make pluripotent stem cells, then neurons, from these samples and prepare RNA libraries. Then, he intends to sequence the RNA to look for chromosome 17 patterns that are common among FTD cells.
“The identification of the interaction between progranulin and Wnt signaling is important and requires further study,” wrote Philip Van Damme of VIB Leuven, Belgium, who also studies GRN-mediated neurodegeneration, in an e-mail to ARF. “An important issue will be to what extent Wnt signaling mediates the functional consequences of progranulin deficiency. Is it a secondary effect or integral part of the detrimental effects of progranulin deficiency?”
In sum, the research points to a role for Wnt1 in silencing GRN in FTD neurons, potentially aided and abetted by Frizzled-2. The Wnt-GRN pathway is thus a potential therapeutic target, but the widely interconnected signaling networks could complicate the search for a therapy, Wexler suggested. “This is as much a disease of dysregulation as of anything else,” he said. “It may not be as easy as giving people more progranulin.”—Amber Dance.
No Available Comments
Mutations in superoxide dismutase-1 (SOD1), which lead to amyotrophic lateral sclerosis, cause SOD1 to buddy up with mitochondrial proteins and interfere with normal mitochondrial conductance. Researchers expanded their understanding of these interactions as described in posters at the Society for Neuroscience annual meeting, held 13-17 November 2010 in San Diego, California. Two groups presented results that laid out how mSOD1 interacts with the normally protective Bcl-2 and the mitochondrial voltage-dependent anion channel (VDAC) to alter ion traffic across the outer mitochondrial membrane. Using different techniques, scientists in the laboratories of Piera Pasinelli at Thomas Jefferson University in Philadelphia, Pennsylvania, and Don Cleveland at the University of California in San Diego acquired somewhat contradictory results with respect to SOD1/Bcl-2/VDAC interactions. They agree that somehow or other, mSOD1 interferes with mitochondria, and that can’t be good.
Some 1 to 2 percent of SOD1 localizes to mitochondria (Pasinelli and Brown, 2006), where it binds to the outer mitochondrial membrane in general (Vande Velde et al., 2008) and to Bcl-2, a pro-survival pore protein, in particular (see ARF related news story on Pasinelli et al., 2004 and Liu et al., 2004). Researchers in the Pasinelli lab recently showed that mSOD1 twists Bcl-2 into a new conformation, exposing a toxic BH-3 domain (see ARF related news story on Pedrini et al., 2010). Nicole Naniche, working with Pasinelli, followed up on that research, adding VDAC/Bcl-2 binding and altered mitochondrial conductance to the story.
Naniche mainly used HEK293T human embryonic kidney cells as a model. These cells do not normally express Bcl-2, so Naniche was able to compare Bcl-2-negative cells to those that she stably transfected with the gene for the pore protein. She treated the cells for half an hour with recombinant SOD1-G93A and looked for mitochondrial cytochrome c release, an indicator of mitochondrial dysfunction. Mutant SOD1 had little effect on the Bcl-2-negative cells, but in the Bcl-2-expressing cells, it doubled the cytochrome c output. In addition, she found that co-transfecting both SOD1-G93A and Bcl-2 deformed mitochondria, disrupting membranes and causing vacuolization, which did not occur in cells expressing mSOD1 only.
Bcl-2 was recently shown to bind the outer mitochondrial membrane anion channel VDAC (Arbel et al., 2010), so Naniche wondered if mutant SOD1 would alter the interaction between the two. She confirmed Bcl-2/VDAC binding in N2A mouse neuroblastoma cells by co-immunoprecipitation. When she transfected the cells with wild-type or mutant human SOD1, she discovered that SOD1-G93A boosted not only the Bcl-2/mSOD1 interaction, but also the binding between Bcl-2 and VDAC. The researchers were unable to co-immunoprecipitate mSOD1 and VDAC, so they suspect that mSOD1 boosts the Bcl-2/VDAC interaction without touching VDAC itself.
What about in vivo? To examine the interaction in animals, the researchers traced the Bcl-2/VDAC partnership in ALS model mice carrying the human SOD1-G93A gene. Naniche isolated spinal cord mitochondria from animals at pre-symptomatic stage, disease onset, mid-disease, and end stage. As the disease progressed, the amount of VDAC pulled down in Bcl-2 immunoprecipitates steadily increased, suggesting this interaction is somehow associated with disease progression.
Naniche and colleagues suspected that the mSOD1-Bcl-2 interaction would have functional consequences for mitochondria, so they isolated the organelles from HEK293T cells for patch-clamp studies of ion conductance. In the control, Bcl-2-negative cells, the addition of SOD1-G93A had no effect on outer mitochondrial membrane (OMM) conductance. But in Bcl-2-positive mitochondria, mSOD1 increased conductance. This difference in ion exchange could be from any channel, not necessarily VDAC, the authors noted. They suspect this altered conductance could be detrimental to mitochondrial activities.
Just around the corner from Naniche’s poster, Adrian Israelson from the Cleveland lab presented his own data on the mSOD1/VDAC partnership (see ARF related news story on Israelson et al., 2010). At first glance, the two posters seemed to clash. Israelson’s data say that mSOD1 binds VDAC directly and blocks the channel, reducing its conductance. Israelson told ARF that, unlike the Pasinelli group, he has been unable to see a Bcl-2/SOD1 interaction by co-immunoprecipitation. This dovetails with previous research (Gould et al., 2006). Israelson and colleagues also recently found that mSOD1 does not require VDAC to bind the outer mitochondrial membrane; mSOD1 also interacts with mitochondrial protein import machinery (Li et al., 2010).
Despite the discrepancies, both sets of authors agree that it is inappropriate to compare the research side-by-side. “The experimental conditions used in our work are very different,” Pasinelli wrote in an e-mail to ARF. She suggested the groups should compare technical notes before drawing conclusions.
Immunoprecipitation conditions—including the specific antibodies used—could explain the different co-IP results, Pasinelli wrote. The mitochondrial voltage methods were also different. Naniche used purified mitochondria, while Israelson put VDAC alone into reconstituted, artificial lipid bilayers. “This means that the experiment is very clean,” he wrote in an e-mail to ARF. “On the other hand, this means that many other factors that might influence the OMM conductance are not tested.”
Either way, both groups agree that mSOD1 alters mitochondrial current. “The bottom line is that, if there is a change in the current, this can certainly affect mitochondria,” said Giovanni Manfredi of the Weill Medical College of Cornell University in New York, New York, who was not involved in either study. “It will affect mitochondrial metabolism.” In addition, Manfredi noted that mSOD1 not only causes problems on the outer mitochondrial membrane, it also gets into the matrix where it likely leads to other damage.
How to rein in damaging misfolded SOD1? Researchers at Brandeis University in Waltham, Massachusetts, suggest a possible treatment in a paper published online by PNAS November 22 (Auclair et al., 2010). The team, led by first author Jared Auclair and senior author Jeffrey Agar, sought to chemically stabilize the wayward protein. Researchers have theorized that, once settled in its healthy dimer formation, mSOD1 would be less able to partner up with other proteins and cause damage (see ARF related news story). Using both the sulphydryl compound, maleimide, and thiol-disulfide exchange, Auclair and colleagues cemented the monomers’ cysteines together, stabilizing the dimer. The authors suggest this might be a potential route for treating mSOD1-based ALS.—Amber Dance.
No Available Comments
In an ideal future, physicians would diagnose people with incipient Alzheimer’s disease long before memory problems appear, and prescribe treatments that would slow or halt the progression of the disease. That future still appears a long way off, but at a November 16 press conference held at the 2010 Society for Neuroscience annual meeting in San Diego, California, four researchers presented work that could bring us closer to that vision. Two talks described the use of MRI imaging techniques to identify brain changes that may presage Alzheimer’s, potentially enabling earlier diagnosis of AD. Another group discussed efforts to develop a vaccine against Aβ while avoiding the inflammatory side effects that hobbled earlier attempts (see Part 2 of this series). The final talk focused on tau, showing that tau oligomers can disrupt memory processes and may be a promising therapeutic target as well.
Reiterating human AD research by other groups, session chair Sangram Sisodia of the University of Chicago, Illinois, pointed out that AD may begin more than 10 years before the onset of symptoms. Positron emission tomography (PET) with Pittsburgh Compound B, which decorates amyloid plaques, shows that amyloid deposition can start years before memory loss (e.g., see ARF related news story). And scientists have found other imaging markers that may help flag people at risk. PET imaging with the glucose analog FDG reveals that brain metabolism drops before the onset of dementia, while functional MRI makes apparent an early loss of connectivity (see ARF related news story and Drzezga et al., 2010). Another telltale warning sign is a shrinkage of the hippocampus, as seen by MRI (e.g., see ARF related news story). Some scientists believe synaptic damage may occur even earlier in some forms of dementia (see ARF related news story). All these data highlight the importance of intervening early, the speakers emphasized. Identifying people in this prodromal phase could be important in two ways. In the present day, it would boost the power of clinical trials by recruiting the people most likely to benefit from therapy. In the future, once reliable treatments for AD are available, it could enable treatment before the disease causes irreversible brain damage.
The hippocampus may not be the only part of the brain that gets smaller. Sarah Madsen, a graduate student in the lab of Paul Thompson at the University of California in Los Angeles, showed data indicating that shrinkage of the caudate nucleus, a C-shaped structure that curls around the lateral ventricles deep in each hemisphere of the brain, can predict disease progression as well (see Madsen et al., 2010). The caudate nucleus is part of the basal ganglia, and nestles next to the thalamus and amygdala. It participates in motor control, attention, and other cognitive processes. It is known to deteriorate in Parkinson’s disease, contributing to motor problems. Madsen was interested in it because the caudate nucleus develops amyloid plaques and neurofibrillary tangles in AD brains. The caudate is also believed to play a role in some forms of learning and memory.
Madsen used data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) to analyze structural brain MRIs of 100 people with AD, 200 people with mild cognitive impairment (MCI), and 100 healthy elderly. She found that the caudate nucleus was 4 percent smaller in people with MCI than in healthy elderly, and 7 percent smaller in people with AD. The right caudate consistently showed greater volume loss than the left, for reasons that are not clear, Madsen said. Volume loss in this structure also correlated with increased age, high body weight, and poor scores on the Mini-Mental Status Exam. In most of these conditions, the head of the caudate nucleus showed the greatest volume loss. Intriguingly, however, Madsen found that in people with mild cognitive impairment who went on to develop AD within the next year, the middle of the caudate nucleus displayed the greatest loss. Madsen suggested that combining MRI measurements of hippocampal and caudate nucleus volume might increase the predictive power of these scans and lead to more accurate early diagnoses. Another implication of the findings, Madsen said, is that maintaining a healthy body weight might help to protect against AD. Madsen is now looking at genetic data from ADNI participants to try to dissect the role that genes play in structural brain changes.
Another brain region of interest is the substantia innominata (SI), a small structure deep in the brain that contains cholinergic neurons and degenerates in AD. The SI sends projections to most regions of the cortex and the hippocampus, delivering the neurotransmitter acetylcholine and modulating attention, motivation, and cognition. Sarah George, working with Leyla Detoledo-Morrell at Rush University in Chicago, Illinois, had previously found that the SI is smaller in people with AD, but not in those with mild cognitive impairment (see George et al., 2009). George and colleagues wondered whether they might see a volume difference in the SI in people with MCI who went on to develop AD, as compared to those whose impairment did not progress. George followed 52 people with amnestic MCI (i.e., MCI with memory impairment) for six years. Within this time frame, 23 of them developed AD. SI volume was no different among people who converted to AD than those who did not, George found. However, several regions of the cortex that receive projections from the SI were significantly thinner in people who later developed the disease. Because this thinning is apparent before AD develops, it could serve as an early warning sign, George suggested. Cortical thinning also implies that AD pathology travels retrogradely down processes back to the SI, which atrophies later in the disease, George said.
Elliott Mufson of Rush University Medical Center in Chicago, Illinois, a coauthor with George, said the new findings will help improve the accuracy of early AD diagnosis, and should be combined with previous imaging biomarkers such as changes in hippocampal size. With this combination, “You can begin to see a pattern of changes, an MRI signature of early AD,” he told ARF. “If you add that information to PIB scanning and CSF tau, you’re beginning to build a series of biomarkers that can give you a clinical diagnostic. You need to have multiple surrogate markers for the disease to do that.”—Madolyn Bowman Rogers
This is Part 1 of a two-part series. See also Part 2.
No Available Comments
Scientists are developing better ways to predict who will develop Alzheimer’s disease (see Part 1 of this series), but earlier diagnosis of the disease will be of limited benefit without viable treatments. In a November 16 press conference held at the 2010 Society for Neuroscience annual meeting in San Diego, California, two researchers described possible ways to tackle the Aβ and tau pathology believed to underlie disease progression. In the first talk, Charlie Glabe, of the University of California in Irvine, and also a consultant to biotechnology company Kinexis, Inc., discussed efforts to develop a safe vaccine against Aβ. In theory, vaccination could harness the immune system to clear Aβ deposits and slow the course of the disease. In mice, vaccination has worked to reduce memory loss and learning difficulties (see ARF related news story). In 2002, however, clinical development of an Aβ vaccine by Elan Corporation was halted because some patients developed brain inflammation due to autoimmune effects (see ARF related news story).
Since then, many newer immunotherapy approaches have entered the clinic. For his part, Glabe and colleagues took a different tack to immunization. They focused on Aβ oligomers, rather than fibrillar or monomeric Aβ, as an antigen. Oligomeric Aβ, believed by many to be the most toxic form of the peptide, assembles itself into β-sheet structures, Glabe said. “When they form β-sheet oligomers, the peptides stick together precisely, just like Lego blocks, and the amino acid sequence is in exact alignment with the peptide above and below in the stack,” Glabe said in the press conference. The side chains of each amino acid stick out perpendicularly to the sheet, giving rise to vertical bars on the surface of the sheet that resembles a product bar code. Since the amino acids are lined up, each bar is unique to a specific amino acid. The immune system recognizes the vertical bars, Glabe said, but not their sequence. “This means that when you vaccinate an animal with a β-sheet oligomer of one sequence, you get an immune response that recognizes all β-sheet oligomers, as long as it has the same bar code present somewhere on the surface of the sheet.” In other words, the immune response is not specific to the peptide sequence.
Glabe and colleagues generated several random, 20-amino-acid peptide sequences that form β-sheet oligomers but are not found in the human genome. They screened for the oligomer (3A) that was best recognized by oligomeric Aβ antibodies. To test the effectiveness of this peptide as an antigen, first author Suhail Rasool immunized 3xTg AD mice with 3A oligomers, Aβ oligomers, islet amyloid polypeptide oligomers, or Aβ fibrils, at ages ranging from three to 14 months. Vaccination with 3A oligomer worked as well as the Aβ forms in preventing plaque formation, lowering total tau and hyperphosphorylated tau, and improving cognitive function. Rasool and colleagues found high levels of oligomer-specific antibodies in 3A-vaccinated mice, and these antibodies recognized Aβ oligomers, but not monomers or fibrils. This demonstrated that a random peptide sequence can generate antibodies against amyloid oligomers.
Because the 3A sequence is not in the human genome, it should not create an autoimmune response. Mice vaccinated with 3A oligomers showed less activated microglia and astrocytes than mice receiving Aβ peptides, Rasool said. Glabe suggested an additional safety feature: Because antibodies to 3A oligomers do not bind to fibrillar plaques, they are less likely to lead to microhemorrhages.
Glabe said he wants to move the 3A peptide toward a human trial once he can secure funding. Since oligomer-specific antibodies cannot distinguish between oligomeric forms of different peptides, these antibodies might be equally effective at clearing other kinds of pathogenic proteins, such as those present in Parkinson’s disease and frontotemporal dementias. Glabe said his group is starting to study this vaccine in Parkinson’s animal models.
Several other types of Aβ vaccine are currently in clinical trials. Elan and Wyeth (now part of Pfizer) are conducting a Phase 2 trial of a second-generation vaccine (see ARF related news story). An anti-Aβ antibody known as bapineuzumab has been shown to reduce amyloid plaques in AD patients (see ARF related news story), and Johnson & Johnson is testing bapineuzumab in numerous clinical trials. Eli Lilly has an anti-Aβ antibody in Phase 3 trials (see ARF related news story). Additionally, Novartis is running a Phase 2 trial, and United Biomedical has a vaccine in Phase 1 trials. Swedish pharmaceutical company BioArctic Neuroscience AB, headquartered in Stockholm, and Eisai Co., Ltd. in Tokyo, Japan, announced in September they will begin a Phase 1 clinical trial of an antibody that recognizes soluble Aβ aggregates (see press release).
With the exception of the BioArctic approach, none of the vaccination strategies currently in clinical trials use oligomeric Aβ as an antigen; rather, they use part of the native structure of APP as an epitope, Glabe said in a telephone interview. This means that the antibodies they raise “don’t necessarily distinguish between the pathologically misfolded form and the native form” of Aβ. This could lead to off-target effects, especially if the native form of the peptide is more abundant. Vaccinating with oligomeric forms targets only the pathological species, Glabe said.
Thomas Wisniewski of New York University in New York City is pursuing a similar strategy using a different antigen. Wisniewski and colleagues immunized APP/PS1 AD mice with oligomeric British amyloidosis related peptide, which has no sequence similarity to normal Aβ and therefore should not produce autoimmune effects. Treated mice showed reduced amyloid load and improved cognition (see Goñi et al., 2010).
In the final talk of the press conference, Ottavio Arancio of Columbia University in New York City showed that oligomers of tau cause memory problems and disrupt synaptic plasticity in mice. This suggests that vaccines targeting tau might also be effective against AD (see ARF related SfN story).—Madolyn Bowman Rogers.
This is Part 2 of a two-part series. See also Part 1.
Comments
UTMB
Boutajangout et al. demonstrate that targeting phosphorylated tau by active immunization prevents cognitive decline in their new htau/PS1 mouse model. This is the third study demonstrating the efficacy of active vaccination using phosphorylated tau fragments in different animal models and confirms their (Asuni et al., 2007) and other previous findings (Boimel et al., 2010). The authors did an outstanding job in sensorimotor and memory testing of all groups; still, the biochemical analysis of the vaccinated and unvaccinated animals is incomplete.
The authors vaccinated the new model with the same immunogen use in the their earlier study (Asuni et al. 2007), the tau fragment (379-408) phosphorylated at Ser396 and Ser404. What is surprising and worrying is the antibody response (Figure 1A). Although the initial response at T1(one week) showed high antibody response toward the phosphorylated sequence (tau 379-408), after several boosts, the antibody response was stronger toward the unphosphorylated sequence (tau 379-408) than the phosphorylated sequence at the end of the study (Tf). That may lead to the depletion of functional tau and may interfere with its functions and cause other complications.
References:
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007 Aug 22;27(34):9115-29. PubMed.
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010 Aug;224(2):472-85. PubMed.
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007 Aug 22;27(34):9115-29. PubMed.
View all comments by Rakez KayedNew York University School of Medicine
We thank Rakez for his comment.
Antibody response towards non-phosphorylated regions of the immunogen can be expected considering its length and immunogenicity. This should not cause major concerns, as we address in the fourth paragraph of the Discussion (pp. 16564-5). However, we are certainly looking more closely into this interesting issue.
View all comments by Einar SigurdssonMake a Comment
To make a comment you must login or register.