Held July 22 to 28 in Toronto, the Alzheimer’s Association International Conference showcased a field in transformation. At the clinical level, groups from Europe, North America, and Japan are attempting to coalesce around new ways to recruit preclinical populations for large observational and trial platforms for late-onset AD, while the smaller but more established DIAN initiative is growing into a worldwide movement. At the biological level, research is set to expand thanks to funding increasing in response to national plans. Health economics research is pressing in. Topically, tau ruled the roost, though genetics, vascular contributions to dementia, and efforts to define ever-earlier stages of the decades-long disease continuum advanced, as well. On the clinical trials front, the only Phase 3 study appears to have been largely a bust, while some Phase 1 presentations of new antibodies, BACE inhibitors, and a small-molecule tau modifier drew quiet praise.
In First Phase 3 Trial, the Tau Drug LMTM Did Not Work. Period.
Results of the eagerly awaited Phase 3 clinical trial of LMTM, a derivative of the dye methylene blue, generated consternation at the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto. Speaking to a packed audience on behalf of the study’s sponsor, TauRx Pharmaceuticals based in Aberdeen, Scotland, Serge Gauthier reported that LMTM failed to slow cognitive or functional decline in people with mild to moderate Alzheimer’s disease. On the main primary results slide, disease progression curves for both doses of drug and the placebo were practically identical. Scientists’ disappointment at this finding soon turned into disbelief when Gauthier went on to present a subgroup analysis that held no statistical credence yet purported to show a strong benefit on cognition and brain atrophy. LMTM is also known as TauRx0237 or LMT-X; Gauthier is at McGill University in Montreal, and chairs the scientific advisory board of TauRx.
Many researchers in the room, and later in the hallways, were dismayed not only at the way the data were being parsed, but also at media stories about the drug’s ostensible “success.” Outlets such as The Times, New Scientist, and the Huffington Post variously announced the arrival of the first drug to halt Alzheimer’s, or that the drug slowed disease by 80 percent, even while leading Alzheimer’s researchers at AAIC were challenging the company’s leader, Claude Wischik, for overstating the trial’s result.
Some media stories ran before the scientific session had even taken place and leaders in biostatistics had a chance to see and discuss the subgroup analysis. That is because press briefings at AAIC are held at 7 a.m. the day of the formal scientific presentation, which in this case happened at 4:30 p.m. Reporters who had attended the press briefing on the trial data and wrote more accurately were left scratching their heads at how their colleagues could have gotten the story so wrong.
Here is the bottom line on this trial: The drug missed its co-primary endpoints of slowing cognitive and functional decline in mild to moderate Alzheimer’s disease, as measured by the ADAS-cog and ADCS-ADL batteries. “The only important finding is that treatment was ineffective,” Paul Aisen, University of Southern California, San Diego, told Alzforum. “The results are disappointing,” said David Knopman, Mayo Clinic, Rochester, Minnesota, who chaired the press briefing but was not involved in the trial. “In my opinion, the only thing that counts is the primary outcome,” Knopman added. “Secondary results are interesting, but our experience in this field, and in trials in general, tells us that secondary analyses are fraught with hidden bias.” John Hardy of University College London was blunt with Alzforum, writing, “This was a misleading presentation of data which failed to show signs of achieving its primary endpoint.”
Suspect Subgroup Analysis
What was wrong with the subgroup analysis? Gauthier told the audience that about 15 percent of the 891 patients in the trial were not taking standard therapy, namely memantine or one of three acetylcholinesterase inhibitors. A pre-specified subgroup analysis suggested that among this group of 136 people, those taking 75 or 125 mg LMTM twice a day had a slower cognitive and functional decline over the 15 months of the trial. He said their brains atrophied less, as well, which he took as an indication that neurodegeneration was slowed. While this analysis prompted the sensational media reports, experts said that it was flawed in two important ways.
First, the researchers lacked any statistical basis for it. They had, in trial parlance, “spent their alpha on the primary analysis,” said Aisen. Wischik conceded that this was the case to biostatisticians and trialists who asked him directly after Gauthier’s talk, and he confirmed it with Alzforum.
What does this mean? Alpha refers to type 1 errors, i.e., false positive results that occur by chance. Researchers must take these potential errors into account, otherwise clinical trial results could be meaningless. Biostatisticians typically power trials to limit the chance of getting a false positive result to 5 percent or less. If they intend to run several different analyses with the trial data, the appropriate way is to pre-specify that, and then ensure the total “alpha” for all those analyses adds up to that 5 percent. There is no free shot at a second analysis. As Aisen explained, one could run analyses ad infinitum until a positive outcome came up by chance. “If you ran 20 analyses with an alpha of 5 percent for each, then one of those would, by chance, be positive," said Aisen, adding, “That would not be a true positive.” The correct approach splits the alpha among the 20 analyses, setting it to 0.25 percent for each. Because the TauRx researchers set their alpha for the primary analyses only, they left the secondary analyses open to false positives and as such, should have deemed the data “exploratory,” as other AD trial sponsors have done in the past.
That aside, the secondary analysis used an inappropriate placebo group, said clinicians at the conference. It compared people on LMTM therapy alone with the placebo group for the whole trial, which includes people taking standard AD drugs. “The correct analysis is to compare the LMTM monotherapy group against people on placebo who are also not on standard AD drugs,” said Aisen. Experts Alzforum spoke with at AAIC agreed that the comparison with the placebo group was inappropriate. “Comparing all control subjects to a subgroup of active subjects is like comparing apples to oysters,” wrote Suzanne Hendrix, Pentara Corporation, to Alzforum (see full comment below).
Gauthier explained that there were too few people in the placebo group who were not on AD drugs to run that analysis. His data slide showed that of 54 people who started in this group, 34 completed the trial. By comparison, 26 and 25 people on the low and high doses of LMTM monotherapy, respectively, completed the trial. Others commented to Alzforum that TauRx could have done the analysis, but chose not to because the results would have missed statistical significance. “If you were to publish this data, you’d have to do it with the correct placebo group,” said Aisen.
Researchers had further reasons to question the secondary analysis. Aisen noted that typically in AD trials, participants with a diagnosis of AD who are not on standard AD drugs may have very slowly progressing disease, or they may not have AD at all. “The need for treatment is an indicator of more rapid decline; for this reason the secondary analysis may simply reflect slow decliners,” he said (see Schneider et al., 2011). If that is what is happening here, then the group on LMTM monotherapy would be expected to show slower decline in any marker of disease, including atrophy, he added. Other leading trialists agreed that the monotherapy group may represent the slow progressors.
Aisen further pointed out that the fraction of people in the trial who carried an ApoE4 allele—48, 42, and 53 percent among the placebo, low-, and high-dose LMTM, respectively—seemed low. “In ADCS trials, ApoE4 prevalence among people with mild to moderate AD is typically 60-70 percent,” he said. “Since there was no biomarker to support the diagnosis of Alzheimer’s disease, the low ApoE4 numbers make one wonder about the accuracy of the diagnosis,” Aisen said. “If you rely solely on clinical diagnosis, then you had better have highly experienced clinicians,” he added. In ADNI, the bapineuzumab and first set of solanezumab Phase 3 trials, about a quarter of participants, especially the ApoE4 non-carriers, were later found to be amyloid-negative.
Reisa Sperling, Brigham and Women’s Hospital, Boston, asked if there was something unique about the people who were not on standard AD treatments. On this point, there seemed to be some confusion. During his conference presentation, Gauthier said nothing distinguished this group, but earlier during the press briefing he had said that many of them were from Eastern Europe and Malaysia. “I think it is very possible that people who entered the study who were not on standard care may have been from areas not receiving good health care, and when they entered the trial they experienced a placebo effect,” suggested Knopman. “There have been previous examples of that,” he added. Dimebon was reported to have had a treatment benefit in a Phase 2 trial in Russia before failing in Phase 3 trials conducted in the Americas, Europe, Australia, and New Zealand (see Mar 2010 news).
Others wondered if, as part of regular checkups in the context of a trial, those patients had begun to receive treatment for other conditions that may affect mental status, such as hypertension or diabetes. “That is certainly a valid consideration,” said Ron Petersen, who is also from the Mayo Clinic.
Gauthier reported that about 30 percent of participants had dropped out. Side effects were primarily gastrointestinal and urinary, and Gauthier called the drug’s safety profile acceptable. Among all treated patients, 25 percent reported diarrhea and 10 percent dysuria, a burning sensation related to the drug.
In an interview with Alzforum, Wischik acknowledged that the results were disappointing. He also accepted that because the alpha of 0.05 was expended in the primary analysis, the secondary analyses were therefore “formally hypothesis-generating and provide only nominal p-values (albeit corrected for multiple comparisons).” A TauRx press release stated “clinically meaningful and statistically significant reductions in the rate of disease progression were observed across three key measures in patients who were treated with LMTX® as their only Alzheimer’s disease medication.” The closing slide of Gauthier’s presentation to the field stated “LMTM as monotherapy is a safe and effective treatment for mild to moderate AD with larger effect size than currently available treatments.”
Where does this leave LMTM? Wischik told Alzforum that going forward, the most important step was to modify the primary analysis of a second, completed AD trial to make it a monotherapy analysis. He said that two-thirds of the patients in the second trial are from North America and one-third are from Europe, and that 20 percent of them are not taking cholinesterase inhibitors or memantine. Combining data from both trials should yield a large enough monotherapy placebo group for statistical comparison with LMTM monotherapy, he said. He said the second trial had the same subgroup result, namely that LMTM as monotherapy slowed decline and brain atrophy. “The first study generated that hypothesis, and the primary analysis of the second study was modified accordingly in the SAP [statistical analysis plan],” noted Wischik. This modified primary analyses would cut the number of patients available.
That data will be presented at the upcoming CTAD meeting in December. Data on the third trial with this drug—in frontotemporal dementia—was originally scheduled to be presented at AAIC, as well, but Wischik withdrew this second talk. The FTD results will instead be presented at the ICFTD meeting starting August 31 in Munich.
Staging of Alzheimer’s, the Second: Neurodegeneration Does Not Equal Tauopathy
The use of biomarkers has transformed scientists’ view of Alzheimer’s disease, revealing that pathology begins to accumulate more than 20 years before clinical symptoms. This knowledge paved the way for secondary prevention trials. Now, tau PET is further changing conceptions of the disease. The new data also challenge the view that tau accumulation and neurodegeneration are synonymous. In fact, in many older people, the brain atrophies in the absence of neurofibrillary tangles. At the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto, Cliff Jack of the Mayo Clinic, Rochester, Minnesota, argued that AD staging criteria need to change to account for this new knowledge.
Jack proposed a new classification system that considers tau pathology separately from markers of neurodegeneration, such as brain atrophy and hypometabolism. In this scheme, all older adults could be classified based on the presence or absence of three key markers: amyloid-β (A), neurofibrillary tangles (T), and neurodegeneration (N). This ATN system would be a descriptive classification scheme rather than a diagnostic one, Jack said. It does not include cognitive status, although he suggested that could be added later as a fourth variable. Such a system might better describe people with suspected non-AD pathology (SNAP), who fall out of the current classification method, he noted. Jack described the proposed system in the Aug 2 Neurology, in a paper co-authored by many other experts in the imaging and biomarker fields.
Overall, researchers at AAIC expressed enthusiasm for the proposal and said they think the field needs a system like this. Some questioned whether continuous variables can truly be dichotomized as either present or absent. Several talks at AAIC supported this idea of a cutoff to define negative and positive imaging scans. Others stressed the importance of using multiple biomarkers to pin down a diagnosis. Victor Villemagne of the University of Melbourne, Australia, appreciated the ATN scheme, telling Alzforum, “Combining biomarkers will increase [diagnostic] specificity.”
Refining AD Classification. The proposed ATN system divides people with dementia according to their biomarker profile, allowing for more nuance than the NIA/AA system.
Evolving Disease Concepts
The current staging criteria for Alzheimer’s have been around for only about five years. In 2011, the National Institute on Aging and the Alzheimer’s Association debuted new diagnostic guidelines for AD that recognized preclinical and prodromal phases and used biomarkers to stage patients for research purposes (see Aug 2010 conference news; Apr 2011 news). In the preclinical period, stage 1 was marked by amyloid accumulation alone, stage 2 by amyloid plus a marker of neurodegeneration or neuronal injury, including tau accumulation, and stage 3 by those markers plus subtle cognitive impairments. Meanwhile, a separate effort by the International Working Group for New Research Criteria for the Diagnosis of Alzheimer’s Disease, spearheaded by Bruno Dubois at Pierre and Marie Curie University, Paris, defined a distinct but similar system (see Jun 2011 webinar). In several longitudinal studies, the NIA/AA criteria proved useful for predicting who among a cognitively healthy group was likely to progress to cognitive impairment and dementia (see Aug 2013 conference news; Dec 2014 conference news; Apr 2016 news).
However, real-world data also made clear that many people did not fit into this classification scheme, because they had markers of neurodegeneration in the absence of amyloid. Called SNAP, this group had a distinct prognosis. Most people with SNAP maintain relatively stable cognition over time, progressing slowly if at all, although some studies have reported exceptions (see Aug 2013 conference news; Sep 2015 conference news). Now, tau imaging has found that most people in this group lack neurofibrillary tangles, as well as amyloid, Jack noted. Instead, they seem to suffer from a diverse array of neurodegenerative conditions or brain injuries.
The ATN system would better reflect this reality, Jack suggested. In this scheme, the presence of amyloid would be determined by amyloid PET or analysis of CSF Aβ42, and neurofibrillary tangles by tau PET or CSF phospho-tau. More generic neurodegeneration would be marked by hippocampal atrophy, hypometabolism in the brain, or CSF total tau. Some scientists have objected to the separation of CSF ptau and t-tau, Jack noted. However, studies by Kaj Blennow and colleagues at the University of Gothenburg, Sweden, as well as others, indicate that total tau reflects neuronal injury, as it spikes after head trauma or stroke. Phosphorylated tau, on the other hand, signals the presence of neurofibrillary tangles, and is more specific for AD than total tau or even CSFAβ42, Henrik Zetterberg at Gothenburg noted in a talk at a separate session. CSF Aβ42 levels drop in neuroinflammatory conditions and normal-pressure hydrocephalus, while ptau does not rise in non-AD tauopathies, Zetterberg said.
Most researchers who have worked on Alzheimer’s classification schemes now support separating tau pathology from neurodegeneration, Jack claimed. Evidence for this separation has been building for a while (e.g., Sep 2015 conference news on total tau). However, some point out that markers of degeneration, such as structural MRI and FDG PET, can disagree with each other and may be measuring different things. Jack argued that these markers reflect a similar underlying process, which is the loss of synapses and neurons. Other researchers at AAIC suggested that “neurodegeneration” is a misnomer, and the third category in the ATN system should perhaps be called brain injury or damage instead, reflecting the fact that it typically does not progress.
Jack did not address how the new ATN system affects the models of AD biomarker progression that have become widely used in the field (see Jan 2010 webinar). There is also no data yet on how existing cohorts such as ADNI would break out under this categorization system. Jack suggested, however, that because the ATN model does not assume anything about the order in which biomarkers appear, or what pathology causes which symptoms, it could better capture the varied diseases that can afflict the brain. “More goes wrong with the aging brain than just Alzheimer’s disease,” he noted.
In an August 2 Neurology editorial on Jack’s paper, Alison Murray of the University of Aberdeen, U.K., praised the system’s reliance on objective biomarkers rather than clinical judgments of cognitive impairment for staging AD. “[This] can only be helpful,” she wrote, noting that factors such as education and early life experience can confound staging by cognitive assessments. She suggested that future iterations of the system should include biomarkers for vascular disease and Lewy body accumulation as well, to more fully encompass all pathologies of aging.
Making Sense of SNAP
One of the main inspirations for the new system was to decipher the puzzle posed by the SNAP group, Jack told Alzforum. The ATN system divides the heterogeneous SNAP group into three categories, according to whether a person has neurodegeneration only, tau pathology only, or both. This may help researchers pin down underlying conditions. Because the ATN system is untested, Jack had no data on what proportion of the SNAP group might fit into each category. He suggested that some people in this group could have primary age-related tauopathy (PART) and be “T” positive. Because PART accumulates the same type of tau aggregate that AD does, the deposits would be detectable by tau scans. Others might be “N” positive due to any of several conditions such as vascular disease, TDP-43 or Lewy body accumulation, hippocampal sclerosis, or argyrophillic grain disease, Jack said. Some of these diseases would be expected to progress quite slowly, as SNAP does.
The question of what makes up SNAP matters because the group is large. In the Mayo Clinic Study of Aging, which comprises roughly 450 participants, one-quarter are classified as SNAP, Jack said at AAIC. Other studies report similar numbers. A recent paper from the Australian Imaging, Biomarker, and Lifestyle (AIBL) study bolsters the view of SNAP as a collection of slowly progressing disorders. Almost a quarter of the 573 cognitively healthy AIBL participants are classified as SNAP based on low hippocampal volume and absence of amyloid. Although this group had slightly lower cognitive scores at baseline compared with people without any pathology, they did not decline any more than controls over the six years of the study. Only 9 percent of them developed amnestic mild cognitive impairment or AD, about half the progression rate of the amyloid-positive groups (see Burnham et al., 2016).
A MeTeR for Tau. Tau tracer binding in three composite regions may determine whether a person is positive or negative for tangles. [Courtesy of Victor Villemagne.]
Can Yes/No Calls Capture Complex, Mixed Disease?
The objections to defining biomarkers as positive or negative center around the idea that any threshold is arbitrary, hence questionable. Jack counters that medicine uses arbitrary thresholds for many other continuous biomarkers—blood pressure, cholesterol, blood glucose. William Jagust at the University of California, Berkeley, agreed. “A lot of researchers don’t like the idea of classifying people as positive and negative on all these markers. But I think we have to do it,” he told Alzforum.
Other talks discussed specific methods for setting thresholds for positivity. Shruti Mishra of Washington University School of Medicine, St. Louis, described a data-driven method to define a pattern of tauopathy that distinguishes disease stages. She took AV1451 scans from 84 cognitively normal participants in WashU aging studies and applied a clustering algorithm to find the brain regions that best separated people into high and low tau signal groups. Four regions stood out: the entorhinal cortex, lateral occipital cortex, inferior temporal lobe cortex, and precuneus. From these she calculated a summary measure. Higher values correlated with lower scores on tests of episodic, visuospatial, and attentional memory. In these regions, an AV-1451 SUVR cutoff of 1.28 cleanly separated amyloid-positive and -negative participants, Mishra reported. Recent studies report that tau becomes widespread only in the presence of plaques, so the two pathologies often go hand-in-hand (see Jul 2016 news).
In the AIBL study, researchers use a similar method for determining the tau imaging cutoff. Chris Rowe of the University of Melbourne explained the “MeTeR” scale, based on tau pathology in three defined regions: a mesial temporal composite (M), a temporoparietal composite (T), and the rest of neocortex composite (R). Tau levels in two of these three regions, as seen with either AV1451, THK5317, or THK5351, have to be in the top 25 percentile of the cognitively normal group to be considered “positive.” This measure distinguishes cognitively normal participants from cognitively impaired and AD patients, Rowe reported. According to the MeTeR scale, high regional and global tau levels associated with worse performance on all cognitive tests. In addition, everyone who had high cortical tau (the T and R regions) also had high amyloid, as in Mishra’s study. In the SNAP group, the percentage of tau-positive people depended on their clinical classification. About 10 percent of those who were cognitively healthy had high tau, but among those with mild cognitive impairment this rose to 40 percent.—Madolyn Bowman Rogers
Coming to a Center Near You: GAP and EPAD to Revamp Alzheimer’s Trials
Part 1 of a three-part story.
The biggest story at the Alzheimer’s Association International Conference, held July 22-28 in Toronto, unfolded rather quietly. It took place in off-site or pre-meetings, and in a sparsely attended session on the last morning, when a majority of conventioneers had left. It is the story of how multiple powerful interests on either side of the Atlantic and Pacific oceans have coalesced to try to fundamentally reorganize the way clinical trials will be done on Alzheimer’s disease starting in the near future.
Here is the vision: Rather than neurologists waiting for symptomatic patients to come through their doors, and perhaps suggesting a trial after delivering a diagnosis, the new modus operandi for recruitment will be one of massive efforts to tap into aging but outwardly healthy segments of societies. In other words, the new initiatives are about locating people who do not know they are heading toward Alzheimer’s dementia, and who had no plans to show up at a memory clinic anytime soon. “This really changes how the sites do business. It becomes a game of outreach. It is very exciting,” said Stephen Salloway of Butler Hospital at Brown University, Providence, Rhode Island.
Drug companies and academic leaders, egged on by advocates and funders, are setting in motion this reorganization of Alzheimer’s clinical research. “We are at an inflection point in terms of how people are coming together. It really is true that academics, pharma, and even individual governments are realizing they cannot do it alone. The barrier toward collaborating and sharing and adopting standardization, and enabling the larger initiatives, is coming down in pharma,” said Luc Truyen of Janssen Research & Development, a company of Johnson & Johnson.
George Vradenburg, who leads the Global Alzheimer’s Platform (GAP), views it like this: “The Alzheimer’s field has been thinking too small. We need to link up patient registries, health plans, large physician groups, and trial sites to solve the fundamental challenge of getting participants into prevention trials.” Academic leaders agree. “This is about creating and executing a whole new paradigm for how to do trials for AD,” said José Luis Molinuevo of Barcelonaβeta Brain Research Center in Barcelona, Spain, who co-leads the European Prevention of Alzheimer’s Dementia (EPAD). Industry is actively on board. “We have to change how we evaluate our compounds,” said Truyen. He previously co-led the development of galantamine and bapineuzumab; his job nowadays is to oversee Janssen’s role in EPAD, GAP, and related efforts in the United Kingdom. In essence, the pharma industry’s support for these joint initiatives reflects an acknowledgement, expressed in 2014 by Sanofi executive and former National Institutes of Health director Elias Zerhouni that “pharma has to stop playing solo.”
[Courtesy of GAP.]
What are GAP and EPAD? Centered in the United States and Europe, respectively, they are the two largest and most advanced AD trial reform initiatives, though similar efforts are coming along in Canada, Australia, and Japan, as well. GAP grew out of USAgainstAD and the Global CEO Initiative, patient advocacy and industry groups, respectively, that were founded by Vradenburg and have since come to exist under the umbrella of the GAP Foundation, a 501c3 organization. The foundation aims to revamp the trials system so it can evaluate more candidate drugs faster, less expensively, and with a greater chance for success. It has secured commitments for $23.2 million from nine companies and several foundations thus far, and on July 1 resubmitted a large National Institute on Aging grant. For its part, EPAD is a project of the Innovative Medicines Initiative (IMI), the public-private partnership funded jointly by the European Commission and the continent’s pharmaceutical industry association, EFPIA. In January 2015, EPAD received a five-year grant of €64 million.
Both projects engage leaders from all sectors of AD research in Europe and North America. GAP has attracted academic trial specialists such as Jeffrey Cummings, Reisa Sperling, Paul Aisen, Salloway, and Rachelle Doody, industry scientists including Andy Satlin, Steve Paul, Richard Mohs, Russ Barton, and Truyen, as well as business leaders; it partners with some 40 companies, academic sites, and philanthropy groups. EPAD is organized into eight topical teams called “work packages” that engage leaders throughout the AD research community in Europe. EPAD’s academic leaders include Simon Lovestone, Craig Ritchie, and Molinuevo; its industry leads include Truyen and Satlin, and it partners with an equally large, and overlapping, set of academic and pharma groups as GAP.
Why attack the problem of AD therapy research at the systems level? Essentially, GAP and EPAD leaders cite the law of holes, that is, “If you find yourself in a hole, stop digging.” In 2013, companies and academics alike were reeling from a string of late-stage trial failures, and when bapineuzumab went under in Phase 3 as well, they did decide to stop digging. They stepped back to acknowledge that AD pathology was present in the brain earlier than they had thought. They asked themselves how they were going to find these early stage patients to test their disease-modifying therapies. They decided to act because their drug portfolios were broadening, and they needed a better way to study those medicines.
“It used to be ‘fire and forget.’ Industry would start huge Phase 3 programs without taking the time to learn if was going the right way, and we’d end up four years later with a negative study,” Truyen told Alzforum. Richard Mohs agreed. “When a trial ends, its infrastructure goes away, and staff leave the sites. There is no system,” said Mohs, who since retiring from Eli Lilly splits his time between the biotech startup AgeneBio and GAP. The field was ready to put something into place that allowed researchers to learn faster. Conversations in 2013 led to a consensus that to gain access to the right patients, and test more drugs in them efficiently, a standing, integrated, continuously active trials platform would be built, and new methodology and trial designs implemented. The result in Europe was the IMI EPAD grant in December 2014, in the United States it was GAP’s launch in 2014.
EPAD and GAP have three main components in common, though they use different terms.
1. They are exploring ways to access large numbers of participants who are well-characterized and enter a longitudinal cohort study. Both EPAD and GAP are intending to funnel at-risk people into deeply phenotyped biomarker cohorts, and from there into therapy trials.
2. They are certifying networks of sites that use streamlined procedures with a standing staff. EPAD is bringing online 30 so-called trial delivery centers; GAP is expanding a pilot “GAP Net” of 11 sites by adding 20 this year and another 30 in 2017.
3. They will innovate by testing adaptive trial designs. On this goal they differ in more than terminology. EPAD intends to run Bayesian adaptive, Phase 2 trials in pre-dementia populations. The trials will feature interim analyses every three months to evaluate whether the intervention at hand slows cognitive decline. Different EPAD trials will adapt on different intermediate phenotypes specific to the intervention, for example lowering Aβ for an anti-amyloid drug or lowering tau for a tau-based drug. EPAD considers the first Bayesian Phase 2 proof-of-concept trial of BAN2401 a potential model (Satlin et al., 2016). A drug will “graduate” from an EPAD trial if it meets a predefined cognitive outcome, and the company would then take it through confirmatory Phase 3 trials and regulatory approval on its own. In contrast, GAP intends to run both Phase 2 and Phase 3 trials of preclinical, prodromal, and dementia stages of AD, using drugs aimed at the disease’s cognitive and behavioral symptoms. GAP trials will start off with parallel group designs. Both EPAD and GAP hope to start their first trials in late 2017.
Flow chart of the standing trials platform being built by the European Prevention of Alzheimer’s Dementia project. [Courtesy of EPAD.]
At AAIC this summer, discussions focused on progress on points 1 and 2, as does this news series. For more on how EPAD and GAP are trying to find trial participants, see Part 2 of this story. For the latest on how site networks are forming, see Part 3.—Gabrielle Strobel
Not to be confused with the TV series about chasing, preparing, and cooking wild food, researchers at the Alzheimer’s Association International Conference, held July 22-28 in Toronto, jokingly invoked this title phrase to express just how differently they will have to go about enrolling participants into future AD trials. Facetiousness aside, how will the Global Alzheimer’s Platform (GAP) and the European Prevention of Alzheimer’s Dementia (EPAD) find the people who may be on the trajectory toward Alzheimer’s dementia but do not know it, and prepare those people for intervention trials?
[Courtesy of EPAD.]
EPAD has built a registry as a tool to prescreen potential participants. It starts by gathering so-called parent cohorts maintained locally throughout Europe—population-based cohorts, aging studies, research cohorts enriched for family history, etc.—and exporting and harmonizing their de-identified data set into a virtual platform. On the register’s central search portal, a software program called PREPAD can query these federated data sets to find members of parent cohorts who are at different risk stages of Alzheimer’s dementia. This is being done with customized search algorithms that use the particular data points being collected in each contributing parent cohort. For some parent cohorts, the algorithm uses little more than age, a memory score, and family history, but for others it can incorporate more detailed data. For example, the UK Biobank comprises verbal recall scores, and of its 490,000 members, 17,000 are known to be more than one standard deviation below the norm and older than 55, said LucTruyen of Janssen Research & Development, a company of Johnson & Johnson. Those people would be candidates for the EPAD longitudinal cohort.
When such candidates pop up, EPAD staff sends its encrypted IDs back to the owners of the respective parent cohorts. The local owner re-identifies the participants and invites them to join the EPAD longitudinal study for standardized clinical/cognitive and biomarker tracking of their Alzheimer’s risk. Once they have been followed in this study for one year, and their amyloid and tau biomarkers indicate they are at high risk of cognitive decline, they can join a PoC trial.
At AAIC, Lisa Vermunt of VU University Medical Center, Amsterdam, showed off the current status of this register. Vermunt works for Pieter Jelle Visser at VU Amsterdam and Maastricht, who co-leads this EPAD work package. As of this summer, the EPAD register contains 17,460 of its target number of 24,000 people age 50 and up drawn from parent cohorts in Spain, the United Kingdom, the Netherlands, Italy, and Sweden. Of the 17,460 people, 15,570 are cognitively normal, 940 have subjective cognitive impairment, and a few hundred have MCI or unknown status. The goal is to ramp up that number with additional cohorts in France, Switzerland, and other countries, Vermunt told Alzforum.
Will the parent cohort owner cooperate and pass its study subjects on to EPAD? And will these subjects come? Yes and yes, EPAD leader José Luis Molinuevo of Barcelonaβeta Brain Research Center in Barcelona, Spain, told Alzforum. The owners have signed an agreement, the first patients in Edinburgh and Barcelona have entered the EPAD longitudinal cohort study via a parent cohort (see May 2016 EPAD press release; July 2016 EPAD release), and eight more have joined in the last few weeks. One of the original parent cohorts is the Barcelona ALFA cohort of 3,000 people aged 45 and older who are cognitively normal but have a family history, and its nested ALFA+ cohort of more deeply phenotyped people (Molinuevo et al., 2016, Alzheimer’s & Dementia, in press).
Until now, EPAD’s goal was to create the register, the algorithms, and the longitudinal cohort infrastructure, and to show that the funnel system works, Molinuevo said. To do this, EPAD started with centers and parent cohorts related to its leaders. Now it is scaling up to include additional sites and to ramp up participant numbers. Geneva and Toulouse are being activated this month, Amsterdam and Stockholm in September, and sites in additional cities will follow this winter and next year. Ultimately, the cohort is to include 6,000 people with well-characterized preclinical dementia. Importantly, EPAD sites continually recruit new people into their local registers and parent cohorts. “We need a system to replenish local cohorts, so they can keep feeding people into the EPAD cohort and the trials,” Molinuevo told Alzforum.
For its part, GAP is exploring a range of recruitment processes. Like EPAD, it intends to tap multiple parent cohorts—“feeders” in GAP parlance. They include the Brain Health Registry at the University of California San Francisco, the Alzheimer’s Prevention Registry of the Banner Alzheimer’s Institute in Phoenix, the Healthy Brains registry of the Cleveland Clinic, and local ones such as the Alzheimer’s Prevention Registry at Butler Hospital in Providence, Rhode Island. The idea is to “white label” registries so they can combine a local and a GAP brand identity. This is necessary because registries have been found to recruit most strongly in their local catchment area.
Besides registries, GAP also welcomes data from large clinical studies such as IDEAS, which is conducting PET scans on 18,000 people in hopes of persuading the Centers for Medicare & Medicaid Services to reimburse clinical use of amyloid PET (Apr 2015 news). Also wanted are the many people who fail the elaborate screening procedure for large, early stage clinical trials such as A4 and the new programs. These include GENERATION, which evaluates two Novartis drugs, and EARLY, which tests a Janssen drug. “We want to put into the GAP registry all those people who have shown so much interest already. We want to engage them and find another trial that they do qualify for,” George Vradenburg, who leads the Global Alzheimer’s Platform (GAP), told Alzforum.
The idea is that from among a target number of 200,000 “feeders,” a quarter who are interested in clinical trials will consent to a GAP registry. This is to be a new virtual registry managed by staff at the GAP foundation. It will assign every member a Global Universal ID, capture a set of Web-based baseline data, genotype participants for ApoE, and attempt to collect cognitive follow-up data quarterly or biannually, depending on the test. An adaptive algorithm will be run periodically against this data to hone the algorithm’s ability to pick out those people who are most likely to have brain amyloid deposition. This serves to reduce the number of negative PET scans or lumbar punctures. This process will select a “trial-ready cohort” initially comprising 1,000 people with prodromal and 1,000 people with preclinical AD who are invited to be seen in person at GAP Net clinical sites. After biomarker testing, eligible people can move into trials; all others will revert to the GAP Registry and may be able to join a future trial.
The virtual registry and its public-facing website are currently being built, said Vradenburg, and the grant for the cohort is under review at the National Institute on Aging. GAP calls this cohort the Trial-Ready Cohort for Preclinical/Prodromal Alzheimer’s Disease, aka TRC-PAD; Reisa Sperling at Boston’s Brigham and Women’s Hospital, Paul Aisen at the University of Southern California, San Diego, and Jeffrey Cummings at the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas are its main drivers. “Despite the differences in details, everyone’s theory is recruit broadly, identify through risk algorithms those people who should get deeply phenotyped at higher cost, and move those who are suitable into trials,” Vradenburg said.
[Courtesy of GAP.]
Most online Alzheimer’s registries are but a few years old. They are beginning to deliver registrants to clinical trials, but have had little success thus far, said Jessica Langbaum of Banner. GAP calculates that in order to recruit the 4,500 people into trials who are being sought by current AD therapeutic studies, nearly 7 million would need to sign up for a registry. That’s because only 10 percent of registry members tend to get referred to sites, 4 percent of those get prescreened, a fifth of those drop out, and 80 percent of the remaining 22,500 people hit a snag during screening. Successful virtual registries invest extensive effort into building trust so their members will contact a study site and join a trial; successful local registries cultivate personal relationships to do so, said GAP’s president John Dwyer, an entrepreneur and business executive who joined GAP because his extended family has suffered from late-onset AD.
In one of several pilot studies to boost recruitment, GAP has invested $1 million in marketing for the Brain Health Registry. It broadcast public-service announcements with President Ronald Reagan’s son Ron and celebrities Paula Abdul, Leeza Gibbons, and Linda Gray, and it pursued various types of digital marketing and local media sponsorships. This netted 5,781 registrants, of whom nearly 1,000 have some data that hint they may be prodromal. Of those, 500 people agreed to be referred to three clinical trial sites, three have been enrolled, and more are in screening, Dwyer said. This experience suggests, though, that many people are reluctant to take cognitive tests online. Of those who do and later contact a site, some get disaffected because the site’s staff and institutional review board make them wait. “The sites can be a bottleneck. We have to engage fast, otherwise we lose people. When participants do this upfront work, their phone has to ring,” said Dwyer.
Beyond registries, GAP is experimenting with other “feeder” systems for recruiting people. Partnerships with health insurers could identify at-risk people based on their electronic health records, yielding referrals. Engaging minority communities could boost their participation rates. State governors could issue Medicaid directives, and the federal government could provide access to records by people insured via Medicare and the Veterans Affairs system.
In one such pilot, GAP engaged Jeffrey Burns at Kansas University Medical Center in Kansas City, well as Kansas City Mayor Sly James, and the CEO of its BlueCross/BlueShield insurance provider. Starting this September, GAP will target a marketing campaign to Kansas City BC/BS members, to city employees, and to large administrative services only (ASO) companies, whose employees are self-insured. The goal is to send interested people to Burns’ center or to their primary care providers for pre-screening and referral into the GAP registry, if appropriate. To help with processing the hoped-for spike in inquiries, GAP leaders may interpose a call center. How to bulk up capacity so sites are equipped to deal with successful outreach efforts has become a general concern as EPAD and GAP gear up.
To pre-screen large numbers of people quickly, GAP is currently using the AD8, a validated five-minute screening test (Galvin et al., 2007). They will also explore easy-to-use, inexpensive digital pen technology that captures subtle decrements in how people with preclinical AD draw; it could become a sensitive tool to log longitudinal change electronically while people see their primary care provider. For more on how site networks are forming, see Part 3 of this story.—Gabrielle Strobel
Playing Where the Puck Is Going to Be: Trial Sites Skate Toward GAP Net, EPAD TDCs
Part 3 of a three-part series.
“A good hockey player plays where the puck is. A great hockey player plays where the puck is going to be.”
At the Alzheimer’s Association’s International Conference, held July 22-28 in Toronto, this quote by Ontario’s native son Wayne Gretzky echoed in the hallways as clinicians discussed upgrading their infrastructure and operations in anticipation of a clinical trials platform being set up by the European Prevention of Alzheimer’s Disease (EPAD) and Global Alzheimer’s Platform (GAP) Net (see Parts 1 and 2 of this series). A crucial component of these initiatives that is currently moving into place is the clinical site networks on either side of the Atlantic Ocean. EPAD is certifying a network of 30 sites throughout Europe; in the United States, GAP is expanding a pilot of 11 sites to add 20 sites this year, and 30 more in 2017. “Eighty sites, both academic and commercial ones, have expressed interest to join,” GAP founder George Vradenburg told Alzforum. GAP Net is a not-for-profit site management organization that is independent of trial sponsors or specific drugs. Both GAP and EPAD are public-private partnerships.
There is much room for improvement in how clinical trials in Alzheimer’s disease are being run today. A sponsor finds, contracts, and activates hundreds of sites separately every time a new drug program starts. When a program fails, teams disband, people are let go, and the whole process starts anew when a sponsor comes along with a new drug. Sites, and sponsors, use different contract language, ethics boards, and training methods. Besides slowing things down, these inefficiencies make it difficult for sites to maintain a steady work flow and budget personnel and infrastructure, and they drive up the cost for sponsors.
Some sites take three months to process contracts and institutional review board (IRB) assessment, others up to a year, and their performance varies greatly on other measures, as well. Variability leads to noisy data, requiring larger trials—it’s a familiar problem to Alzheimerologists. “Current trials are slow, redundant, inefficient, expensive. If a biotech has to make a decision between a cancer drug and an AD drug, they will choose the former because it is cheaper to test. We need change,” said Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas, Nevada.
EPAD dealt with this problem by establishing its network of certified sites, which it calls trial delivery centers, or TDCs. A TDC must be located near a hotspot of the EPAD Register, and its ability to serve as an EPAD TDC is tested in feasibility runs as the register begins to recruit locally. They need to have PET scanners nearby, and must be able to recruit 200 people into the EPAD cohort and 50 of those into the first EPAD proof-of-concept trial. The sites must have experience running Phase 2 trials and sign best practice agreements. The sites will open in three waves, as EPAD works out the kinks of the system, said José Luis Molinuevo of Barcelonaβeta Brain Research Center in Barcelona, Spain. This summer, EPAD is bringing on the second wave.
GAP is starting up GAP Net. In a pilot study with 11 U.S. sites in the past year, GAP leaders asked the sites to name their worst “pain points.” They heard about lack of recruitment personnel, having exhausted limited site staff in pre-screening large numbers of trial candidates, four out of five of whom are rejected. They also heard about too much paperwork for contracts and ethics approval. In turn, each site received $100,000 from GAP to spend as it saw fit and to report back. At a GAP Net site expansion meeting at AAIC, Richard Mohs of GAP told site leaders from across North America that just by hiring an outreach or recruitment coordinator, administrative support, and taking out advertisements, the pilot sites had doubled the number of people they were able to screen and enroll. Some sites started trials up faster, had fewer discontinuations, or were able to accommodate additional trials.
Going forward, however, GAP will not simply fund sites locally. Its leaders also want to bring sites into line by mandating common “best practice” procedures across the network. GAP leaders intend to institute templates to streamline budgeting, agreed-upon common contract language, and administrative tasks. GAP Net sites will have to train raters annually through a central company, though this will eliminate the current burden of having to train them anew, and differently, for each drug program. Sites will use the CDISC2 data standards to support pooling, sharing, and, importantly, regulatory submission, Vradenburg said. They will also have to provide some concierge services to patients, Cummings said. This includes not just helping with parking but also with finding one’s way around the study site and general personal assistance to make study visits less stressful, perhaps even pleasant. There is a business feel to GAP Net, with an emphasis on performance expectations and monitoring. Quantitative performance data as well as qualitative considerations will guide the selection of the next batch of 20 GAP Net sites from the 80 applications that have come in, Cummings told the audience at the GAP Net meeting in Toronto.
GAP Net will require its sites to switch from using their own IRBs to a central, nationwide IRB. While some institutions may initially resist giving up local control of ethics oversight, others may welcome the simplification. A survey of 45 academic sites commissioned by GAP indicates that the majority have already used a central IRB for other trials. On June 21, the National Institutes of Health mandated use of a single IRB for multi-center research funded by the federal government starting in May 2017. “The goal of this policy is to enhance and streamline the IRB review process in the context of multi-site research so that research can proceed as effectively and expeditiously as possible. Eliminating duplicative IRB review is expected to reduce unnecessary administrative burdens and systemic inefficiencies without diminishing human subject protections,” the policy declares.
Large IRB organizations exist to provide this service. One of them was commissioned for the White House’s Cancer Moonshot 2020 initiative to run Phase 2 cancer immunotherapy trials in 20,000 patients within the next three years, which was championed by Vice President Joe Biden. In a recent series, The New York Times chronicled a history of cancer immunotherapy replete with early setbacks and side effects, but with tremendous potential going forward. At AAIC, immunotherapy presentations evoked a field at an earlier stage, but perhaps with similar potential.
“We have to do this. The central IRB, and funding for a permanent recruiting staff alone can really speed things up,” said Pierre Tariot of the Banner Alzheimer’s Institute in Phoenix.
Whether all participating sites, in both the United States and Europe, fall fully in line with the new plan remains to be seen. “To make this successful, centers have to be completely engaged. This is not business as usual. This is creating and executing a new paradigm for how to do AD trials. It takes extra energy, and everyone has to be fully committed,” Molinuevo told Alzforum.
Similar initiatives to broaden recruitment and reform clinical trials are forming around the world. [Courtesy of GAP.]
Different Countries, Same Idea
Other countries are developing similar initiatives, and they are communicating with GAP and EPAD to harmonize the overall approach, if not the details. For example, the Canadian Consortium on Neurodegeneration and Aging, led by Howard Chertkow at McGill University in Montreal, is that country’s national dementia initiative. With five years’ worth of government funding, CCNA is charged with engaging 350 Canadian researchers and clinicians to advance research, treatment, and care. CCNA provides resources to establish registries and a cohort of 1,600 people with various neurodegenerative diseases called COMPASS-ND. To be fully enrolled by 2018, this cohort will deliver deeply phenotyped people for prevention or prodromal trials, Chertkow said at an AAIC pre-meeting. To add the trials and the sites that would make COMPASS-ND a full equivalent of EPAD— aka Canadian Pipeline for AD Therapeutics, or cPAD—Howard Feldman earlier this year submitted a grant to the Canadian government. Feldman moved to University of California, San Diego, this past April (see Jan 2016 news), but stayed affiliated with the University of British Columbia in Vancouver. He proposes a master protocol to evaluate multiple compounds through Phase 2 adaptive proof-of-concept trials; this would be done with a network of up to 30 C5R sites. As does GAP, Feldman’s proposal emphasizes broad-based recruitment efforts through family practice and provincial health networks, as well as patient engagement to keep presymptomatic or only mildly impaired people interested in participating in research for long periods of time.
Across the Pacific, Australian researchers have applied for government funding to transition the Australian Imaging, Biomarkers, and Lifestyle Study of Aging into becoming a trial-readiness cohort. AIBL will change its name to Australian Prevention of Alzheimer’s Disease Partnership, APAD for short. The AIBL longitudinal cohort started in 2006 with 1,822 participants, 1,201 of them cognitively normal at the time, and is currently conducting 90-month (7.5-year) follow-up assessments. To date, 642 people have died or withdrawn, but 503 are left, 419 of them still cognitively normal. With these deeply phenotyped people, AIBL has not only produced highly regarded progression data, but is also now recruiting some of them into early stage trials, including A4 and EARLY.
More broadly, for APAD, Australia currently has 14,000 people in a cognitive-disorders registry, 2,000 people screened for a longitudinal readiness cohort, and 500 in a deeply phenotyped trial-ready cohort. The capital cities of all Australian territories participate in APAD, Colin Masters of Melbourne University said at AAIC.
Japan, which expects 7 million dementia cases by 2025, in 2015 established a nationwide clinical register. Its name is a mouthful: Organized Registration for the Assessment of dementia on Nationwide General consortium toward Effective treatment. ORANGE, for short, unites existing registries, such as the multi-site Japan Gerontological Evaluation Study cohort, which is expected to soon double its current size of 3,000 people with preclinical AD. It is funded by the Japan Agency for Medical Research and Development, AMED. In 2015, AMED announced funding until 2020 for ORANGE2, a beefed-up, more comprehensive platform for prevention and drug discovery research similar to EPAD and GAP, whereby registrants are followed longitudinally, funneled into a trial-ready cohort, and enrolled in prevention drug trials. The organization to support this process, for those readers who can take on board yet another acronym, is called J-DCS, or Japanese Dementia Clinical study Support. In J-DCS, leading Japanese researchers Atsushi Iwata and Takeshi Iwatsubo consider advice from international colleagues at ADNI, DIAN-TU, and GAP.
“Similar processes are beginning to be built in all these countries, and they are all aiming to prevent Alzheimer’s disease at greater speed, lesser cost, and better-quality data,” said Vradenburg.
Can these international initiatives come together? They won’t all merge into one global effort. For their part, GAP and EPAD hold joint meetings to make sure the two initiatives are developing compatible components so they will be able to run global trials, said Luc Truyen of Janssen Research & Development. Both initiatives maintain formal contacts. For example, Truyen is on EPAD’s executive committee and in this role answers to EPAD’s funder, Innovative Medicines Initiative, while also heading GAP’s management advisory council. Likewise, Simon Lovestone at the University of Oxford is EPAD’s academic co-lead and also serves as EPAD liaison for GAP. Eisai’s Andy Satlin is on the EPAD executive committee as well as in a GAP trials development group, and has worked with Cummings and others to develop ADCOMS, a clinical outcome measure derived from the ADAS-cog (Wang et al., 2016).
With Canada, Australia, and Japan, the idea is for each initiative to obtain funding regionally and set up their own governing structure, but to consult with the other initiatives and build similar capacities where possible. In a field known for strong personalities, as well as a history of debates and at times even truculence, the growing sense of consensus and a shared commitment marks a notable change. Molinuevo summed it up this way: “Some people say this is an ‘interesting project.’ It is not just an interesting project. I believe very strongly that this is the future. Around the world, we have to be ‘all in.’ As we would say in Barcelona, ‘You have to put all the meat in the pan.’”—Gabrielle Strobel
Results of the six-year Prevention of Dementia by Intensive Vascular Care (PreDIVA) trial, presented at the Alzheimer’s Association International Conference, held July 22-28 in Toronto, indicated that the multi-domain intervention aimed at reducing cardiovascular risk factors had no overall benefit. The incidence of all-cause dementia recorded in the intervention arm matched that in control group. “The main conclusion is that this type of intervention for this primary outcome in this study group does not show an effect,” said Edo Richard, University of Amsterdam, who presented the data.
However, he told Alzforum that this interpretation lacks nuance. “We also saw in per-protocol subgroup analysis that the intervention seemed more effective in people who had untreated hypertension at baseline,” he said. People with high blood pressure who stuck to the trial regimen over the six to eight years of follow-up reduced their risk of dementia by 46 percent.
Researchers at AAIC urged caution in interpreting the subgroup analysis. “It would have been nice if this trial had clearly shown a positive response, but we need to keep the many methodological issues in mind,” said Miia Kivipelto, Karolinska Institute, Stockholm. “We need to learn more about prevention trials, including what is the right type of intervention, how much should be given to make a difference and when, and who stands get the most benefit,” she told Alzforum. Some took the results to mean that broadly targeting populations with modest interventions may not the best way to prevent dementia.
PreDIVA is one of several trials testing multi-domain interventions and the first to suggest any effect in reducing dementia incidence. Participants in the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), run by Kivipelto, and in the Multidomain Alzheimer’s Prevention Trial (MAPT) being run in France gained modest improvements in cognition, but it remains unclear if those will translate to protection against Alzheimer’s disease or other dementias (see Nov 2015 conference news).
Together with principal investigator Willem van Gool, Richard, and co-investigator and Eric Moll van Charante designed PreDIVA to test interventions that could be easily adopted by routine clinical practices. All interventions were carried out at general health-care facilities in the Netherlands. The study recruited 3,526 volunteers, age 70-78, to one of 116 clinics that were cluster randomized to standard care or vascular care. At 63 of them, nurses assessed 1,853 volunteers for cardiovascular risk factors during routine visits (three per year), gave tailored advice on healthy lifestyle, and optimized treatments for hypertension, dyslipidemia, and type 2 diabetes, if necessary. At another 53 clinics, 1,601 people received standard care over the length of the trial. For primary outcomes the researchers measured dementia incidence and disability according to the Academic Medical Center Linear Disability Score (ALDS). The trial design and outcome were published online in The Lancet on July 26.
Overall, the intervention failed to reduce dementia incidence, with 121 cases in the intervention group (6.5 percent) and 112 in the control (7 percent). Likewise, no difference in disability emerged, with mean ALDS scores in both groups being 85.7 at the end of the trial. The hypertension effect surfaced in a per-protocol analysis, in which investigators limited the treatment group to people who had attended at least two-thirds of their planned visits, and restricted the control group to those who had attended less than three unplanned doctor visits for cardiovascular risk management. Richard explained that extra visits would in essence move people from the placebo to the treatment arm. “We wanted to examine why we might not see a treatment effect, so we left those people out,” he said.
The per-protocol analysis turned up an effect in people who had untreated hypertension at baseline. If they were randomized to treatment, their dementia incidence was 4.8 percent versus 6.9 percent in the control arm. It dropped to 4.3 percent in patients who strictly adhered to the intervention for the duration of the trial. Compared with the control group, that drop in incidence was statistically significant. “When you think that the control group is already getting good care, seeing a difference in the treatment group is actually a very positive signal,” suggested Kivipelto. She said the data bodes well for countries where hypertension is not routinely treated.
It is unclear what led to the reduction in dementia incidence. While the intervention very slightly reduced blood pressure in the overall treatment group by 2.1 mmHg compared to the control group, data on blood pressure for the per-protocol analysis has not been released yet. Richard told Alzforum that this data is being analyzed.
Why did the intervention not work overall? Richard attributes this, at least partly, to high-quality health care in the Netherlands. Other researchers agreed that trying to reduce dementia by improving care in a country that already has top-quality medicine presents a challenge. This is especially true against the backdrop of falling incidence in many developed nations, which may be due to an overall improvement in lifestyle and health, including cardiovascular health (see Apr 2016 news; Feb 2016 news). In fact, Richard said that the new incidence data that has emerged since PreDIVA was designed suggest the trial was underpowered. “We would actually need a much larger sample size to see a difference,” he said.
How does this data inform population-based intervention trials going forward? In an accompanying Lancet commentary, Lon Schneider, University of Southern California, Los Angeles, questioned the value of the population-based approach. “Providing modestly enhanced care to non-selected or non-targeted patients already connected to a medical practice, and identifying and trying to mitigate risk, does not seem to be effective or to reduce overall dementia,” he wrote. He suggested focusing intervention on people at high risk, not the general population.
Richard partially agrees with this. He said biomarkers such as amyloid PET or CSF analytes are a way to select for at-risk people, but are unsuitable for this type of intervention. “Selection should be based on readily obtainable clinical data, such as hypertension or family history,” he said. Schneider agreed, but cautioned that just surveying for undiagnosed high blood pressure and then ensuring it was treated might not necessarily get the same result as in the multi-domain PreDIVA because the context of the therapy is very different. He noted that in a multi-domain trial subjects may also be quitting smoking, or getting diabetes treatment. “If you just treat for hypertension, you might not get similar results,” he said. “We would need to do that kind of trial.” In the Systolic Blood Pressure Intervention Trial (aka SPRINT), treating older adults who are about the same age as those in PreDIVA reduced cardiovascular events such as stroke and heart attack but did not measure any cognitive outcomes (see Williamson et al., 2016).
Richard has teamed up with Kivipelto and MAPT investigators Bruno Vellas and Sandrine Andrieu at the University of Toulouse to pool PreVIVA, FINGER, and MAPT data. “The goal is to see what we can learn from these trials and to use that going forward,” said Richard. Kivipelto coordinates MIND AD, a multimodal intervention trial based on the FINGER design but targeting people with prodromal AD, i.e., more cognitively impaired than participants in FINGER. It includes sites in Finland, France, Germany, and Sweden. MIND AD begins in September with a six-month pilot study to optimize protocols. The full study will run two years.
Richard, Kivipelto, and the MAPT investigators also collaborate on the Healthy Ageing through Internet Counselling in the Elderly (HATICE) trial, which aims to improve cardiovascular health through interactive Internet counseling. HATICE recruited 2,700 people in Finland, France, and the Netherlands, and will run for 18 months. “This is a very large proof-of-concept trial,” said Richard. “If we can improve cardiovascular risk profiles, which should reduce the risk for cardiovascular diseases and for cognitive decline and dementia, then we will have to test in a much larger study to see if it can lead to prevention,” he said. He believes it will be feasible to do this on a very large scale because volunteers will not need to come to a research clinic so long as they have an Internet connection. “It’s the next step in the pragmatic design of implementable interventions,” he said.—Tom Fagan
At Age 8, DIAN Is Churning Out Data and Growing into a Movement
Part 1 of a five-part report.
At age 8, DIAN has grown up. In 2008, when the Dominantly Inherited Alzheimer’s Network formally began to study autosomal-dominant AD, the notion of assembling globally dispersed families afflicted with this rare form of AD into a standing research platform seemed radical to observers. In theory, the idea of characterizing the preclinical course of their disease, and using that knowledge to conduct prevention trials, was enticing. But could it really be done? In 2016, at the Alzheimer’s Disease International Conference July 22-28 in Toronto, the answer to that was plainly on display.
DIAN’s first therapeutic trial, testing two investigational antibodies, is fully enrolled and expecting a preliminary biomarker readout this winter. The second stage of the trial is slated to start enrolling next spring, and plans for a primary prevention trial—in people in their 20s—are taking shape. Meanwhile, the observational DIAN study has logged some 1,500 participant visits to date. It is beginning to churn out the longitudinal data across all observed markers of disease that will put the progression rates researchers had estimated based on cross-sectional data on a more definitive footing. From this in-person serial data, a disease progression model is being built to undergird intervention trials.
A New Generation’s Fighting Spirit.
On February 25, 2015, Lori DeMoe McIntyre died of Alzheimer’s disease at age 56, after participating in DIAN research for nearly eight years. Her daughter Jessica McIntyre memorialized her mom with a large tattoo of her amyloid PiB PET scan. The younger McIntyre and one of her sisters participate in DIAN and DIAN-TU. [Courtesy of Jessica McIntyre, art by Joey Borger, Certified Customs, Denver, Colorado.]
DIAN scientists have begun an ambitious effort to compare their data to comparable data sets in late-onset AD (LOAD). It is their attempt to settle—for regulators and the field at large—the long-standing debate about how similar, or different, early onset and late-onset Alzheimer’s disease truly are. The DIAN network itself is expanding not only the number of its member sites and countries, but also the sample, resource, and data collections that it offers to outside scientists for independent analyses. “DIAN is gathering tremendous momentum,” said Nick Fox of University College London, an original DIAN site, adding, “This is exciting to see, after years of small studies that had too little power to answer questions and deliver subjects to trial.”
The network’s families are meeting in gatherings that increasingly feel like reunions. Relatives are gaining confidence in sharing their experiences. They inject hope, humor, even spunk into serious conversations about intensely painful topics. Increasingly, their young adult members are joining, and getting to know each other. Updates on all of these fronts came in the form of a daylong meeting that drew 118 family members and as many scientists and other stakeholders, a meeting of DIAN’s steering committee with family representatives present, as well as separate meetings for international sites wishing to join, existing sites prepping for the next therapy trial, a featured research session, and a slew of individual talks and posters throughout the AAIC conference itself. Below are selected highlights.
First, the treatment trial. It is being run by the DIAN Trials Unit, a spin-off of the original observational study, DIAN-TU and DIAN-Obs for short. This four-year trial is the first intervention study designed solely for autosomal-dominant AD. Last December, it met its enrollment target of 210 people who range between 15 years before and 10 years after their particular mutation’s expected age of clinical onset. The trial compares the immunotherapies solanezumab and gantenerumab to a pooled placebo group, and it does so without requiring that participants find out their mutation status.
Unlike early stage trials in LOAD, which grapple with screen failure rates of up to 80 percent, only 19 percent of people who tried to join the DIAN trial ran afoul of an inclusion or exclusion criterion, Randy Bateman of Washington University, St. Louis, said at AAIC. About half the trial participants previously had been in the DIAN observational study, 15 percent entered directly via clinical sites, and 38 percent found the trial by way of the DIAN Expanded Registry.
The ideal scenario is for participants to join the observational study for a while so researchers can collect some data on their biomarker trajectories, and then to enter an intervention trial that is suitable for them. At the end of the trial, participants can return to the observational cohort or join another trial until a drug is found that works, said Bateman. The whole public-private enterprise of DIAN is a continually operating research and trials machine that does not waste data by disbanding at the end of a given drug study. To make this possible, DIAN and DIAN-TU take harmonized assessments on many markers, such that run-in data from the observational study, and placebo data from a trial, can feed into a disease progression model the scientists have built.
The DIAN participants understand the network’s mission well. In a field where dropout rates of 30 percent are routine, 2 percent per year have dropped out of the current DIAN-TU treatment trial, Bateman said. Another handful had to leave per the trial’s protocol, because they decided midway through to find out their genetic status and discovered they had not inherited the family’s Alzheimer’s disease mutation. Their departures will not reduce the trial’s power, because non-carriers are included to help keep the overall group blinded to everyone’s mutation status, not to contribute to the actual drug evaluation data. Importantly, the trial’s completion rate for its intensive battery of clinical, cognitive, fluid and imaging measures is nearly 100 percent. “This is unheard of in AD clinical trials, and it speaks to the commitment of family members and centers,” Bateman said. For a clinical trial as small as this, a near-perfect completion rate greatly helps its statistical power.
The second stage of the DIAN-TU trial, dubbed NexGen, is widely expected to add a third drug in early 2017. Despite ample speculation about which one it will be, DIAN investigators at AAIC kept mum on the subject. They readily talked about the new features of the NexGen stage of the trial, though. It will also treat people for four years, with a biomarker readout at year one and potential interim cognitive readouts at years two and three. NexGen can adjust the administered dose up to achieve maximal effect. The point of doing this is to avoid a scenario that has happened before in AD, whereby a trial proceeds to its end only for scientists to discover the dose had been too low all along. These trials are long, and the families keep saying their situation is too urgent for conventional fixed-dose designs. NexGen will pool placebo data from the first two drug arms in order to further increase power and keep the fraction of participants who don’t get drug low, at 25 percent. That is being done because the prospect of being on placebo for four years is perhaps the families’ top complaint. The new trial will enable cognitive testing at home in an effort to improve cognitive estimates and reduce the burden on its middle-aged participants, who are working and raising children.
Most importantly, perhaps, NexGen uses an ADAD-specific disease progression model WashU scientists have been building with longitudinal data from the observational study. To date, DIAN-Obs has tracked in-person change on cognition and biomarkers for a period of up to six years. For their research, DIAN scientists anchor where along the trajectory to AD symptoms each DIAN participant is, based not on their biological age but on how far away they are from their specific mutation’s mean age at symptom onset. This expected year of onset (EYO) is calculated from the mean age at onset of all known carriers of that particular mutation, not only the carrier’s own family. In essence, plotting a given participant’s cognitive, clinical, and biomarker trajectories relative to their EYO tightens up the variance of the raw data.
Built from this data, the disease progression model essentially lays out a quantitative path for the disease. This then allows scientists to predict what a drug effect of, say 30, 50, or 70 percent should look like in terms of cognition, the primary outcome measure for these trials. “Using data from DIAN-Obs in this way gives our trials a substantial increase in power,” Bateman said, adding that regulators have been supportive of evaluating both the EYO concept and the use of the disease progression model for the trial. This so-called NexGen trial received funding from the Alzheimer’s Association and has a grant submission pending at the NIA.
With one trial halfway done and one about to start, Eric McDade at WashU has set his sights on the horizon. That horizon is primary prevention. Such a trial would treat people in hopes that they never develop amyloid deposition, or the subsequent pathophysiology of Alzheimer’s disease, in the first place. Essentially, it would mean giving investigational Alzheimer’s medications for many years to some college-age people and others in their 20s to early 30s. Why is that not an outlandish prospect anymore? For several reasons, McDade said.
Number one, the targets are in hand. Longitudinal data analyses from the DIAN observational cohort indicate that the cognitive tests used in DIAN pick up a subtle decline in cognition starting as early as 15 years before a person’s EYO. That is a big change from what was known before. It opens up an earlier window for therapeutic intervention, McDade said. Scientists are also gaining a more robust handle on the trajectories by which preclinical biomarkers diverge between mutation carriers and non-carriers prior to 15 years EYO, and on the order by which biomarkers change. These trajectories essentially become targets for intervention, McDade believes.
Number two, the participants may be there, too. Even as 89 people left DIAN-Obs to enter the solanezumab/gantenerumab trial, 289 are currently in DIAN-Obs and are hoping for a trial. In a survey Joshua Grill of the University of California, Irvine, conducted among DIAN participants, 90 percent said they would stay in trials that last longer than five years, and a majority said people younger than 15 years before their EYO should be offered a trial. “The families want trials for their young. It is a very important point for them,” McDade said.
Anecdotally, the second DIAN family meeting held in conjunction with AAIC featured a substantial number of young adults who are starting to get to know each other. One young man spoke about traveling from his home in Canada to Boston, and using his love of U.S. history as a balm to overcome the apprehensions of a first DIAN study visit. Another young man articulated a widely felt sentiment when he described how hearing his mom utter certain stock phrases long after her cognition had failed made him question the veracity of his childhood and adolescent memories of her saying those phrases in moments he had held dear. A 20-year-old woman, whose symptomatic father spoke of writing everything down at work so he could continue to provide for his family, was there the second year in a row. An 18-year-old man, who also has been watching his father decline, told this reporter he would “absolutely” enter a primary prevention trial if there was one for him. McDade received a grant from the GHR Foundation to prepare for such a trial.
For an AAIC news update on DIAN-Obs, and other data garnered on dominantly inherited Alzheimer’s disease, see Part 2 through Part 5 of this series as each is posted this week.—Gabrielle Strobel
DIAN Longitudinal Data Say Cognition Goes Earlier Than Previously Thought
Part 2 of a five-part report.
With therapy trials of the dominantly inherited Alzheimer’s network gearing up (DIAN-TU, see Part 1 of this story), what is the status of the network’s observational study (DIAN-Obs)? Its size fluctuates somewhat. From a high point of 465 participants in 2013, 89 transferred into the treatment trial but 111 have discontinued. Some people died, others had to leave because their disease had advanced too far for them to travel, take tests, and put up with the needles and scanner. In fact, some participants declined so rapidly that, on the current DIAN-Obs protocol of visits every other year in the symptomatic phase, they were able to return only twice after their onset, leaving the DIAN progression model with a dearth of data for rates of change in this late phase of the disease. To fill this gap, DIAN will from now on invite symptomatic patients back for annual visits. Yet other participants, however, missed visits because scheduling was tricky, or the tests were burdensome, or for unknown reasons. At an internal DIAN meeting, John Morris of Washington University, St. Louis, asked all site leaders to redouble their efforts to engage and support participants in their concerns, so that they will choose to stick with the study and contribute longitudinal data.
DIAN is replenishing its ranks as additional family members come forward. Moreover, sites in Germany, Argentina, and Japan have begun enrolling, with 55 new participants between them thus far. A site in Seoul, South Korea, is about to begin enrollment, and sites in Mexico, Israel, and Taiwan have expressed interest in joining. The University of Antioquia in Medellin, where Francisco Lopera’s team already conducts an Alzheimer Prevention Initiative therapy trial in the world’s largest ADAD cohort of families carrying the E280A “Paisa” presenilin-1 mutation also is considering becoming a DIAN site. After all, Colombia is home to families with other autosomal-dominant AD mutations, as well, Lopera told Alzforum. Morris said that going forward, he aims to offset attrition or departure into trials recruitment in order to maintain a DIAN-Obs cohort of about 300 people.
In the observational study, three-quarters of participants are asymptomatic at entry; of those, three-quarters carry the family mutation. They are in their 20s, 30s, and 40s, with an average 14.7 years of education. Roughly 30 percent are ApoE4 positive. This risk gene exerts a far smaller influence on age of onset in ADAD than in LOAD, possibly because it is a surrogate for amyloid deposition, which is already driven far more strongly by the APP or presenilin mutation, said Randy Bateman of WashU.
DIAN participants handle the study’s intense regimen of tests well, though some sites are more successful than others at obtaining CSF and PET scans, Morris said. Lumbar puncture headaches have annoyed quite a few among these young-adult and middle-aged people. At AAIC, Morris showed results from a recent survey suggesting that once participants had decided to undergo spinal fluid sampling, they were more likely to do it on follow-up visits, regardless of whether they had suffered a headache the previous time. Recent research on lumbar punctures suggests that using particular atraumatic needles and letting the CSF drip rather than pulling it into a syringe reduces the risk of this unpleasant consequence of donating CSF for research. “This data has busted some myths about CSF collection,” Morris said. (Moulder et al., submitted; Duits et al., 2016.)
Throughout AAIC, scientists presented data from the observational cohort study. Jason Hassenstab of WashU described how cognitive performance in the long preclinical period changes over time. Traditionally in AD research, cognitive decline has been measured with less-sensitive tests against group norms. In staging diagrams, cognition has been pegged to start changing only a few years before clinical diagnosis, certainly after CSF tau goes up. In many instances, cognitive decline was conceptualized as mild cognitive impairment and applied to a heterogeneous mix of people who had different underlying diseases affecting their cognition. In contrast, when measuring cognition repeatedly in people who are known with certainty to harbor the pathogenic process of AD, it turns out that changes can be well-characterized early in the disease process, suggesting that cognition changes early, at least in the purer form of AD seen in DIAN participants, Hassenstab showed.
Hassenstab used results of 16 cognitive tests that tap episodic memory, attention control, working memory, processing speed, and semantic memory, plus a DIAN cognitive composite of four tests. So far, 432 participants have taken these tests at least once, 235 at least twice, 76 at least three times, and 35 people sat them at least four times. Some of these people became clinically symptomatic while in DIAN. Hassenstab compared rates of cognitive decline between asymptomatic and symptomatic mutation carriers, and non-carriers, and he modeled the rates in carriers relative to their expected year of onset (EYO). Overall, cognitive decline in otherwise asymptomatic carriers was detectable as far back as 15 years prior to EYO, Hassenstab said at AAIC. The first test to flag change was on ability to remember word lists; other tests of episodic memory and attention control revealed a signal nearly as early. Rates of cognitive decline accelerated as people edged closer to their EYO.
Scientists are now drilling down on whether different mutations in PS-1, PS-2, or APP affect progression differently. This research is one way of trying to explain the variance seen within the DIAN data set; however, thus far rates of cognitive decline look similar for the three genes in question, Hassenstab reported. Overall, the longitudinal data mirror the published cross-sectional data, he said.
CSF analysis is generating longitudinal data, as well. At AAIC, Anne Fagan updated her surprising discovery that the known increase in CSF tau prior to EYO does not continue, but rather levels off and may even reverse once people have been symptomatic for some years. This was published based on 75 longitudinal samples from 37 DIAN participants (Fagan et al., 2014). “I hoped and prayed it would hold up in a larger number of people,” Fagan quipped to the DIAN steering committee. It did. As of now, 329 samples from 134 DIAN participants confirm the same finding. CSF Aβ42 goes down over time and stays down after EYO; but both phospho-tau and, two years later, total tau increase as people approach EYO, then both markers plateau and decrease again.
Modeled as rates of change, Fagan said, the data reveal a linear decrease for Aβ42. For phospho-tau, they reveal a kink—a change point in biostatistics lingo—at six years prior to EYO. This finding is puzzling because tau PET indicates increased tracer uptake well after that; tau PET data thus far is largely cross-sectional. Many researchers at AAIC emphasized that hypotheses of how a given marker might change over time that are drawn from cross-sectional comparisons must be tested with in-person, serial measurements. Cross-sectional data compare different people to each other, with the individual variability this brings.
Emerging CSF biomarkers are starting to round out the picture. Nelly Joseph-Mathurin of WashU showed new data on the neuronal calcium sensor protein VILIP-1. “This calcium sensor protein could be a preclinical marker of neuronal injury,” Joseph-Maturin said. VILIP-1 is thought to change similarly to tau, but much less is known about it.
In the DIAN cohort, most VILIP-1 samples analyzed thus far, from 202 participants, are cross-sectional. Levels are highest in preclinical mutation carriers who already have brain amyloid deposition, Joseph-Mathurin showed. Intriguingly, in the first batch of repeat measurements analyzed thus far, from 41 participants, VILIP-1 appears indeed to behave like tau. It rises while a person approaches his or her EYO, but then drops again during the symptomatic phase of disease. It is almost as though the most acute neuronal death occurred before overt clinical illness, followed by a relative slowing, said Fagan.
CSF VILIP-1 appears correlated to CSF total and phospho-tau in LOAD, as well. In the first few participants who received tau PET scans within DIAN, VILIP-1 again was highest in asymptomatic carriers who were positive for both amyloid and tau tracer uptake, but much more data is needed on these emerging studies.
In LOAD, additional markers correlate with VILIP-1, Fagan said. They include neurogranin, YKL-40, and the synaptic protein SNAP-25. “There are many interesting comparisons to be made in the DIAN dataset, but this was underfunded,” Fagan noted. Doing those types of analyses on the DIAN samples could help scientists slot a rapidly growing panel of injury and inflammation markers downstream from Aβ42 into the disease’s evolving staging diagram. This would flesh out the field’s understanding of what happens during the crucial years before and around clinical onset of AD.
For example, at AAIC, Christian Haass, Ludwig-Maximilian University in Munich, used DIAN samples to advance the story of sTREM2. This is the shedded, soluble stub of the microglial transmembrane receptor that has been genetically linked to a range of neurodegenerative diseases. Haass reported in Toronto that CSF sTREM2 levels in AD appear to be most dynamic during the five years before and five years after clinical symptoms begin. sTREM2 creeps up somewhat as healthy people get older, reflecting a mild immune stimulation with age. In LOAD, a much steeper increase is seen in preclinical stage 2 of the NIA-AA preclinical research diagnostic criteria, i.e., in people who have both amyloid and tau pathology. sTREM2 is not particularly elevated in the amyloid-only preclinical stage 1, Haass reported. In this study, sTREM2 is also up in SNAP, i.e., people with an as-yet vaguely defined brain atrophy that is not triggered by amyloid.
Haass’ lab obtained aliquots of DIAN cross-sectional CSF samples, and in Toronto, he showed results. Mutation non-carriers had the same steady, slight elevation with age seen previously in other people. Carriers have a much steeper increase during the decade of minus five to plus five years from their EYO. “That is the window of time for TREM2,” Haass said. CSF sTREM2 correlates with tau, but starts its rise a few years after tau does.
But is sTREM2 truly a marker of microglial activation? The answer might be yes, Haass believes, at least according to an unpublished mouse triple PET study visualizing the new microglial tracer TSPO together with amyloid and tau pathology in presenilin/APP transgenic lines. Based on these data, it would appear that microglial activation, and sTREM2 as an indicator of it, fits into the amyloid cascade after tau. “You must have cellular debris from neurodegeneration to fully activate microglial phagocytosis,” Haass told the AAIC audience on the last morning of the conference.
Like CSF research, brain imaging is also grappling with the confluence of neurodegeneration and inflammation in early AD. In one presentation at AAIC, Juan Domingo Gispert of Barcelonaβeta Brain Research Center in Barcelona, Spain, reported that CSF sTREM2 levels were up at the same time that MRI indicated brain swelling in AD areas as judged by increased gray-matter volume and decreased water diffusivity. Gispert’s new data came from people at the amnestic MCI stage of LOAD, but a similar finding was reported last year at AAIC by Philip Weston and Natalie Ryan at University College London in presymptomatic people with autosomal-dominant AD, as well (see Aug 2015 conference news). For more data on imaging in dominantly inherited AD, see Part 3.—Gabrielle Strobel
Duits FH, Martinez-Lage P, Paquet C, Engelborghs S, Lleó A, Hausner L, Molinuevo JL, Stomrud E, Farotti L, Ramakers IH, Tsolaki M, Skarsgård C, Åstrand R, Wallin A, Vyhnalek M, Holmber-Clausen M, Forlenza OV, Ghezzi L, Ingelsson M, Hoff EI, Roks G, de Mendonça A, Papma JM, Izagirre A, Taga M, Struyfs H, Alcolea DA, Frölich L, Balasa M, Minthon L, Twisk JW, Persson S, Zetterberg H, van der Flier WM, Teunissen CE, Scheltens P, Blennow K.
Performance and complications of lumbar puncture in memory clinics: Results of the multicenter lumbar puncture feasibility study.
Alzheimers Dement. 2016 Feb;12(2):154-63. Epub 2015 Sep 11
PubMed.
Brain Imaging in DIAN: Atrophy Rates—Check. Tau PET—Not Yet.
Part 3 of a five-part report.
Overall in the Dominantly Inherited Alzheimer’s Network (DIAN), the brain imaging news was twofold. On the larger goal of putting cross-sectional data on a longitudinal footing, Nick Fox of University College, London, showed that for brain atrophy, longitudinal data on the DIAN cohort are confirming the hypotheses suggested by cross-sectional and previous, much smaller serial studies. The new data is mostly adding precision for rates of change of this “final common pathway” of neurodegeneration, Fox said during the Alzheimer’s Association International Conference held July 22-28 in Toronto. In contrast, tau PET data is emerging and less consistent across mutations thus far.
Broadly, the annual gray-matter atrophy among mutation non-carriers in DIAN matches the 0.3 percent per year that has been found in huge population studies of aging. In contrast, in symptomatic mutation carriers, the brain shrinks a whopping six times faster than that, Fox said. In asymptomatic mutation carriers, atrophy rates differ by region and by disease stage. For example, the precuneus, which crops up frequently in imaging studies of preclinical AD, thins noticeably starting a decade before a person’s estimated year of onset (EYO). Atrophy there, and in other areas, accelerates as people approach their EYO.
For modeling rates of shrinkage in the whole brain and hippocampus, as well as modeling the corresponding expansion of the brain’s ventricles, change-point analysis is proving useful, Fox said. This kind of statistical approach also works well for the modeling of cognitive and CSF trajectories in DIAN (see Part 2 of this story). In particular, for MRI, change-point analysis indicates an inflection point around 7.5 years prior to EYO, when many markers appear to begin changing more rapidly.
The other imaging news of DIAN concerns tau PET. For the past three years, DIAN leaders tried to get tau PET up and running across DIAN sites and in the DIAN-TU trial in order to get baselines for longitudinal data collection toutde suite. Alas, DIAN got bogged down by similar difficulties that many groups throughout the AD field have experienced in getting access to the first widely studied tracer—T807, aka AV1451, now named flortaucipir in analogy to Avid/Lilly’s florbetapir. A new arrangement negotiated since then does offer this tracer, though only to U.S. and Australian sites, and in the form of an ancillary study that has to be taken under contract directly between Avid and each site. The sites can then donate their tau PET data back to the DIAN database. At AAIC, DIAN investigators decided to pursue this deal to get the science started, but they will also explore other tracers and possibly switch once a better or more easily available one comes to the fore. The German sites and some others have already decided to use the Tohoku/GE tracer THK5351, according to Johannes Levin of Ludwig-Maximilian-University in Munich.
Thus far, only Tammie Benzinger at Washington University, St. Louis, has had access to flortaucipir for DIAN participants for long enough to be able to present cross-sectional data on the first 19 participants. And the small data set available thus far is not straightforward. At AAIC, Benzinger explained that she is testing whether the regional spread of tau pathology in ADAD follows the Braak and Braak stages—after all, stages I and II of this classification scheme are often attributed to age, and the asymptomatic DIAN mutation carriers are in their 20s and 30s. She also asks whether the spread of tau pathology follows the proposed stages of the NIA-AA criteria of preclinical AD, or is better described by the more recent A/T/N scheme proposed by Cliff Jack at the Mayo Clinic in Rochester, Minnesota, and colleagues (Sperling et al., 2011; Jack et al., 2016). Benzinger’s ability to interpret the data is somewhat limited because, to stay blinded to who carries the AD mutation, she looks at all asymptomatic participants in one group. But that is not the main reason the initial findings don’t quite fit expectations, she told Alzforum.
Some of the asymptomatic people are in early stages of amyloid deposition; they retain no tau tracer. Some are positive on both amyloid and tau scans, but have much less tau than the symptomatic participants. “This confirms that amyloid is first and tau comes later,” Benzinger said. Beyond that, it gets complicated.
One person has most of his or her tau uptake in the brain’s occipital area, not in the four areas where tau is typically thought to spread first once it leaves the entorhinal cortex. (They are the entorhinal, lateral occipital, inferior temporal lobe cortex, and precuneus). Confusing matters further, this person has a normal FDG scan but is mildly symptomatic. Another symptomatic person with high amyloid load retained a low level of tau tracer on one side of his or her occipital lobe, but was hypometabolic in both temporal lobes. A third amyloid-positive person fit conceived wisdom better in that he or she had high tau and hypometabolism in the same area, but the location of that coupling was again in more posterior regions than Benzinger expected to see. “We see little tau tracer uptake in the medial temporal lobe, and more in the precuneus and posterior cortex,” she said. More data in more people, serial scans, and also analyses by gene or mutation may solve the questions raised by these early data.
Curiously, tau PET imaging of the PS-1 E280A “Paisa” mutation afflicting families in Colombia shows less variability. Like Benzinger, Yakeel Quiroz of Massachusetts General Hospital had presented a first, small data set at the last Human Amyloid Imaging conference in Miami Beach, Florida (Feb 2016 conference news), and at AAIC Quiroz followed up with results from a larger group. In her ongoing project, a handful of relatives fly from Medellin to Boston every few months for PiB and AV1451 scans. They are not enrolled in the Alzheimer Prevention Initiative trial of Genentech’s antibody crenezumab because they failed screening, or for other reasons; others are already symptomatic and hence are excluded from the trial. Yet they still participate in Quiroz’s research.
Of the 21 Colombians Quiroz has scanned so far, eight are asymptomatic carriers aged 28 to 44, two carriers have amnestic MCI. In contrast to the variability emerging in DIAN, Quiroz continues to see a consistent pattern. Tau in non-carriers matches that in age-matched controls, but in mutation carriers it starts in the medial temporal lobe, especially the entorhinal cortex. PiB uptake starts around age 28. Until age 35 people have very little tau tracer uptake in the entorhinal cortex, but starting at age 36, tau pathology increases and spreads rapidly to the inferior temporal and lateral temporal lobe, and from there to other cortical regions—all while people remain asymptomatic. The two people with MCI have much higher levels and a broader cortical distribution of tau uptake.
While her scans thus far broadly resemble what is seen in LOAD, Quiroz said she needs many more to fill in current gaps in the age range before she can obtain at least a cross-sectional picture of exactly how tau pathology appears to develop before clinical onset. “My goal is to have 50 subjects in this study,” she said. With that number, she can analyze the relationship between tau, other biomarkers, and cognitive decline, and compare the E280A data set to the LOAD data currently being gathered within the ongoing Harvard Aging Brain Study.—Gabrielle Strobel
Monogenetic and Sporadic Alzheimer’s Disease: Same or Different?
Part 4 of a five-part report.
Asking whether autosomal-dominant Alzheimer’s disease is essentially the same disease as sporadic late-onset AD is a sure-fire way to spark debate among researchers. This debate has raged since the 1970s, when Bob Katzman just about equated Auguste Deter’s middle-age affliction to the “senility” that ailed millions of older Americans. “Lumpers” among the scientific community emphasize the abundant similarities in biomarker trajectories and the clinical/cognitive course between the rare and the common forms of AD; “splitters” cite differences ranging from a different starting region of amyloid PET to clinical variations such as spastic paraparesis and epilepsy. And then there is age. Does aging mean LOAD is a heterogeneous syndrome, essentially a collection of many diseases? Or does the aging process contribute separate, age-related comorbidities to the central pathway of Alzheimer’s disease?
Many scientists maintain that LOAD is heterogeneous, but others disagree. “There is a mythology out there that AD is heterogeneous. I think it is homogeneous, with added variables of aging,” Colin Masters, University of Melbourne, Australia, told an audience at the Alzheimer’s Association International Conference, held July 22-28 in Toronto.
The question is far from academic. As soon as the first drug shows evidence of working in ADAD, regulators will confront the quandary of whether to approve it only for this small subgroup or for all AD patients. “If we find a drug that works in ADAD, will it work in LOAD?” asked Virginia Buckles of Washington University, St Louis.
The literature on the peculiarities of autosomal-dominant AD, or dominantly inherited AD (DIAD) as DIAN researchers call it, comprises small patient series or individual case reports, and it is marked by reporting bias. One ADAD family suffers spastic paraparesis—that makes a paper. The next four do not—that’s unremarkable, hence no paper. However, eight years into the DIAN project, observational data on 465 participants followed in a standardized way make for a comprehensive data set to analyze in search of a more definitive answer. For LOAD, large data sets exist by now, as well, such as those from the Alzheimer's Disease Neuroimaging Initiative (ADNI) or the National Alzheimer’s Coordinating Center (NACC). This DIAD-LOAD comparison aims to build a knowledge base that regulators can consult to support an informed decision, said Randy Bateman of Washington University, St. Louis, who leads the DIAN initiative. It is a multipronged project that will go on for some time, but at the AAIC conference the first glimpses were visible.
Consider postmortem pathology. How do the brains of people who died from either disease compare to each other? Nigel Cairns, who heads the neuropathology core for both DIAN and ADNI, showed the data available so far. Twenty-four DIAN participants or relatives who carried the family mutation have come to autopsy. Unsurprisingly, each of them had a neuropathologic diagnosis of AD; 13 of them also met diagnostic criteria for dementia with Lewy bodies (DLB). Importantly, they had no other discernable pathologies, Cairns said.
From ADNI, 50 cases have come to autopsy. Of those, two never had Alzheimer’s at all. They had argyrophilic grain disease (AGD) plus another age-related tauopathy called PART. Eighteen had pure Alzheimer’s disease with amyloid plaque and neurofibrillary tangle pathology, 22 had AD with DLB. The remaining 22 cases all had AD plus another comorbidity—either TDP-43, AGD, an age-related tau astrogliopathy called ARTAG, hippocampal sclerosis, or infarcts. At death, the ADNI participants were 30 years older than DIAN participants on average, 81 versus 51. What might be going on? Cairns suspects that with advancing age, other lifestyle or genetic factors come to bear on AD pathogenesis.
Another emerging research trend is that white-matter hyperintensities appear to start early in both ADAD and LOAD. Cairns is now trying to relate the last MRI FLAIR scans taken before death in DIAN and ADNI cases against postmortem signs of white-matter degeneration.
What about during life? Does cognition decline similarly or differently? At AAIC, Buckles showed a comparison of progression rates before and after symptom onset between 129 people in DIAN on the one hand and 853 people in the NACC data set on the other. She was able to compare cognition directly, because DIAN uses some tests from the Unified Data Set (UDS) that were instituted to standardize assessments across the nationwide network of some 30 federally funded Alzheimer’s Disease Centers (ADC), where people throughout the United States seek Alzheimer’s care.
The NACC’s UDS database contains cognitive decline data on more than 33,000 people, but only a subset met the criteria Buckles had set for this comparison. For one, people had to have had neuropathologically confirmed AD in order to be equivalent to the DIAN data set. For another, they had to have been cognitively normal at the time they started being followed at an ADC, so that their whole progression from asymptomatic through dementia could be calculated.
By aligning each person with ADAD or LOAD at their respective point of symptom onset, and then calculating rates of longitudinal change prior to and after that point in time, the researchers were able to sidestep the 30-year age difference of the two groups. “ADAD represents AD without the comorbidities of aging, hence we compared groups on disease course instead of age,” Buckles said.
Of the 13 cognitive tests in this comparison, progression on 10 was the same between ADAD and LOAD, Buckles said. Three were different: on the Boston naming test and both the Trials A and B tests, LOAD progression rates in the NACC data were steeper. A composite of these three also showed faster decline in LOAD. Then Buckles realized that these are speeded components, and aging is marked by a slowing in thinking and processing information. When Buckles subtracted the three speeded tests, ADAD and LOAD progression slopes both before and after onset became almost identical. This was true not just for progression but also for people’s performance scores at symptom onset: Except for the three speeded components, the scores were highly similar for ADAD and LOAD. “We found the expected age difference on speeded tasks, but without those tasks, ADAD and LOAD declined at the same rate,” Buckles said.
Other researchers praised this talk as “compelling,” and AAIC attendees later mentioned it as standing out in their minds; however, Buckles cautioned that further analyses, particularly of the role of vascular disease and other comorbidities, still need to be made.
Finally, consider genetic modifiers. For the most part, the field of pinning down variant genes that influence the onset or course of Alzheimer’s disease remains in its infancy; however, one genetic commonality between LOAD and ADAD reportedly comes in the form of the Val66Met variant of the gene for brain-derived neurotrophic factor (BDNF). In the Australian AIBL cohort of LOAD, this common variant has been implicated in accelerating cognitive decline once a person has brain amyloid deposition (Lim et al., 2013; Lim et al., 2014). At AAIC, Yen Ying Lim of the University of Melbourne said that she wanted to see if this was true in other study cohorts, as well, or rather was due to a national quirk she cheerfully called “beer-derived neurotrophic factor.”
Lim was eager to see whether the BDNF Val66Met variant made a difference in DIAD, because these families lack the comorbidities of aging. She analyzed a sample of 95 presymptomatic DIAN participants who carried the Val66Val variant and 48 with the Val66Met variant. The BDNF Val/Met carriers had the same amyloid load as Val66Val carriers, but their episodic memory performance, glucose metabolism, and tau pathology were more abnormal. “BDNF does not act on amyloid deposition; rather, it acts on the brain’s ability to withstand the downstream effects of amyloid. It takes homozygous Val/Val carriers longer before the full clinical syndrome of disease presents itself. This is the same in LOAD and ADAD,” Lim said. In both forms of AD, BDNF may exert its effect via tau.—Gabrielle Strobel
DIAN’s Tightrope Act: Sharing Data While Protecting Your Wish Not to Know
Part 5 of a five-part report.
Besides generating data, DIAN is charged by its funders to make its information available to scientists around the world. Now that resources have been built and data are pouring in, DIAN leaders are focusing on how to open its doors more widely. They are working to simplify and accelerate sharing, and voted in specific measures at the Alzheimer’s Association International Conference, held July 22 to 28 in Toronto. At the same time, knowledge about even earlier stages of Alzheimer’s disease is growing and becoming ever more widely known. This may gradually squeeze the space for many people’s wish not to know if Alzheimer’s is in their future.
To broaden access to its data, DIAN will retire a three-tiered access system. Until now, DIAN’s core leaders, who acquire and process the data, have enjoyed exclusive access to test hypotheses for a predetermined set of core publications before the networks’ larger group of site leaders could run and publish their own analyses. In turn, sites had one year of exclusivity before outside investigators could obtain access. At AAIC, DIAN leaders decided to no longer distinguish between cores and participating sites, though they stopped short of voting to lift the one-year access exclusivity vis-à-vis outside investigators just yet.
“As a core leader, I am not funded to do all the mutation-specific analyses that we need in order to understand the variance and find modifying genes. I want others to use this data so more science gets done, though I would offer expertise on how to conduct these analyses,” said Alison Goate of Icahn School of Medicine at Mt. Sinai University in New York City, who leads genetics within DIAN.
Outside scientists will still have to submit their projects for review, in part to ensure that data and samples remain blinded. In the past, DIAN has fielded criticism for its slow review of resource and data requests. The process has already sped up this year, with 10 requests granted since April, but opening the DIAN database by eliminating exclusivity for core leaders should make sharing faster still. “We want DIAN resources to be widely shared,” Bateman said.
With regard to tissue samples, the genetics core has banked 79 fibroblast lines representing 26 mutations in APP, PS1, and PS2 from DIAN participants who underwent a punch biopsy to create this resource for scientists around the world. Also available are eight iPSC lines, one each from a carrier and a non-carrier of three different PS-1s and one PS-2 mutation. Researchers can obtain these biospecimens with a brief description of the research rationale here. DIAN’s CSF biorepository currently contains 2,276 fluid samples collected from 15 sites, 268 longitudinal CSF, 393 longitudinal plasma, and 340 longitudinal serum samples, said Washington University’s Anne Fagan, who leads the DIAN biomarker core. Thus far, the core has filled 650 CSF and 120 plasma sample requests.
Another step to broaden access is an anonymized data set Virginia Buckles of Washington University, St. Louis, and colleagues are preparing. It took some years to assemble this, because each field must contain data from at least five people to render re-identification impossible. The data set is not quite ready yet, but once it is, it will be freely downloadable without application. Called the Anonymized DIAN Data Set, it will be so thoroughly scrubbed of identifying information that not even Buckles will be able to trace data back to the individuals from whom it came. It will contain dummy ID numbers, EYO, carrier status, and other variables, but not age, ethnicity, family relationship, etc. This data set can support exploratory analyses. To continue privacy protection of participants, about half of whom do not know their mutation status, deeper work with restricted DIAN data will still require a traditional data request, Buckles explained.
Preventing anyone from figuring out who is behind a given result in DIAN, either accidentally or intentionally, has been a central concern for DIAN from the outset. This commitment has animated everything DIAN scientists do, from its internal procedures to restricted outside access and even to limitations on how research results are graphed and published. Even so, as more data comes flooding into the project’s databases, it is becoming apparent that keeping a lid on unblinding will become trickier over time. After all, DIAN is a massive project to study particularly the preclinical decades of ADAD, and as it succeeds, the disease will eventually become well known in all its early features to scientists and participants themselves. In general, cohort studies of other autosomal-dominant diseases do not guarantee indefinite blinding.
A long-term forgetting test reveals differences in carriers and non-carriers of dominantly inherited AD mutations seven years before their expected year of clinical onset. [Courtesy of Philip Weston, UCL.]
Already, preclinical cognitive hints of Alzheimer’s disease are under intense scrutiny in many research groups. They could become highly useful to science but also lead people to realize early on whether they are on the road to AD. To cite but one example, Philip Weston of University College, London, showed at AAIC that among his center’s local cohort of 35 at-risk relatives of families with autosomal-dominant AD, mutation carriers could be picked out as early as seven years before EYO by way of a long-term memory loss that unmasks a failure of memory consolidation. Carriers and non-carriers were in their 30s and performed equally well on standard cognitive testing. The two groups also did equally well learning a word list and remembering it 30 minutes later. Alas, seven days later, the list tended to be wiped from the minds of the mutation carriers while their relatives still remembered it. Carriers were twice as likely to have forgotten, and their group’s 95 percent confidence interval did not overlap with that of the non-carrier group. The result recapitulated a recent mouse study (Jun 2016 news on Beglopoulos et al., 2016.)
“We can distinguish preclinical AD from normal aging by assessing long-term forgetting,” Weston told the audience. He cited three phases of memory: encoding (tested right after the word list), early retention (tested after 30 minutes), and long-term retention. On the long road from healthy cognition to AD dementia, long-term retention fails first, early retention fails at the CDR 0.5/aMCI stage, and encoding fails at the dementia stage. This long-term forgetting may be part of what troubles people when they voice subjective memory complaints, an early stage of dementia that is drawing increasing research attention across the field at large. At AAIC, several dozen presentations attempted to define the cognitive, imaging, and other features of this state, called, variously, subjective cognitive decline or subjective cognitive impairment.
Weston’s AAIC presentation was news to researchers because they do not generally assess word-list memory days after the study participant has left their clinic. But word-list recall—the 30-minute, short-term kind—is the memory skill that tends to crumble first in DIAN longitudinal observation, as well. In fact, Weston’s finding is an extension of studies reporting as early as 1998 that verbal memory such as remembering lists preceded other cognitive problems in ADAD mutation carriers years before they became symptomatic (Fox et al., 1998). If long-term forgetting was replicated as a specific alarm bell of future Alzheimer’s dementia in late-onset disease, it could become useful to help the large asymptomatic LOAD therapy trials that are ramping up internationally, and which are mightily challenged to recruit participants. Trial leaders at AAIC said that with a simple follow-up phone call after a trial participant’s screening visit, this feature could possibly pick out people who are likely to have AD pathology in their brain, and reduce the screen failure rate for these trials.
Even so, the flip side of Weston’s finding was not lost on researchers at AAIC. Could a DIAN participant guess their mutation status by way of this long-term forgetting? Nick Fox of University College, London, does not think so, because one would first need to know what the age-related normal range is. But more broadly, as scientists are learning more about the nature of the preclinical phase of AD, families have become more observant as well. “In my experience, these families continually monitor themselves for memory problems, and both carriers and non-carriers can be convinced they are going to get the disease because of memory lapses,” Fox wrote to Alzforum.
The field’s collective push toward ever-earlier trials in the Alzheimer’s prodome brings with it a general realization that even without direct genetic disclosure or a data-handling mistake, research results such as this and future ones will increasingly reveal features of preclinical AD that may tip off carriers to their mutation status earlier than was previously the case.—Gabrielle Strobel
As data from tau PET studies come rolling in, they paint a remarkably uniform picture of how tau accumulation fits into the pathophysiology of Alzheimer’s disease. Speakers at the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto, presented the latest findings from longitudinal and cross-sectional studies of aging and AD. Without exception, they reported that widespread tau pathology depends upon the presence of Aβ, and that the tau signal correlates closely with brain atrophy and declining cognition, even at preclinical disease stages. Researchers lauded the robustness of the findings, which held true across diverse study populations and for different tracers and methodologies. Together, the data strengthen the idea that tau PET could serve as a marker of AD progression and perhaps an outcome measure in trials, although this remains to be tested.
In all, the imaging data generated some of the biggest excitement at the conference. “I was really impressed by how the amount of tau imaging data has exploded in the last two years,” Christian Sorg of the Technical University of Munich told Alzforum. Not only do the data match previous postmortem findings about the patterns of neurofibrillary tangle spread, but tracers now allow researchers to track progression of tau pathology in people at preclinical disease stages and link it with other biomarkers, Sorg noted. Clifford Jack of the Mayo Clinic in Rochester, Minnesota, believes such data will open new frontiers for research. “Tau imaging is a transformational technology,” he said.
New View of Alzheimer’s.
Tau imaging reveals patterns of tangle accumulation in AD brains. [Courtesy of Clifford Jack.]
Amyloid Sets Off Tangles
Although tau is not the only cause of neurodegeneration (see Aug 2016 conference news), widespread tangles in the brain generally spell trouble. Initial tau PET studies had already suggested a close association between regional tau pathology and brain atrophy, in agreement with past autopsy studies (see Feb 2015 conference news). The extent of tangles, unlike amyloid plaques, also correlates with cognitive decline (see Nov 2013 conference news; Aug 2014 conference news; May 2016 news). Plaques play a key role as well, however, as numerous groups have now reported that tangles spread through the brain only when amyloid has accumulated (see Mar 2016 news; Jul 2016 news; Aug 2016 news).
At AAIC, researchers continued to build the case that amyloid plaques act as a trigger for tau pathology. Mark Mintun of Avid Radiopharmaceuticals, Philadelphia, a subsidiary of Eli Lilly, described a study of 57 cognitively normal and 96 cognitively impaired people, and 46 with possible or probable AD. Averaging around 70 years old, they all underwent PET scans with AV1451 (aka T807, aka flortaucipir) at baseline and again at nine and 18 months later. Those without evidence of amyloid on florbetapir PET had an average AV1451 standard uptake value ratio (SUVR) of 1.01 in the cortex, and this remained stable over the course of the study. Those with positive florbetapir scans bound more cortical AV1451 already at baseline (average SUVR of 1.32), and the higher their tau signal was, the likelier it was to rise further on follow-up visits (correlation of r = 0.74). Over time, tau deposits typically spread to new regions, rather than intensifying in existing ones, Mintun noted.
Similar data emerged from 474 participants in the Mayo Clinic Study of Aging, which studies 50- to 90-year-olds who are cognitively normal or have been diagnosed with mild cognitive impairment or dementia. People free of brain amyloid, regardless of cognitive status, had similarly low AV1451 uptake, said David Knopman of the Rochester Mayo Clinic. However, in cognitively normal people with amyloid, the tau tracer lit up the medial temporal and lateral temporal lobe. In cognitively impaired people with amyloid, signal intensity was even higher and extended to parietal regions as well. The researchers saw an even bigger tau signal in amyloid-positive people with dementia. Because tau PET correlates with disease stage, it might be useful for selecting patients for trials and as a potential outcome measure, Knopman suggested.
Many talks noted that the spread of tangles depends on disease stage and follows specific spatial patterns. Agneta Nordberg of the Karolinska Institute, Stockholm, presented data from a small cohort of 38 people who ran the gamut from cognitively normal to AD. They underwent scans with tau tracer THK5317, developed by Tohoku University, Sendai, Japan. About half the group came back for follow-up THK5317 and FDG PET scans one to two years later. At the group level, brain metabolism waned, but THK5317 retention did not change. On the individual level, however, participants displayed heterogeneous regional patterns of change in THK5317 retention. In people in the prodromal stage, tau tangles built up mostly in the medial temporal lobe, while in those with AD, the pathology spread throughout the brain, Nordberg noted (see image below). In addition, the more brain amyloid a person had at baseline, the faster tau accumulated, she reported (see Chiotis et al., 2016).
Tracking Tau Accumulation. Tau PET ligands allow researchers to see regions (red) where tangles build up in an individual as prodromal Alzheimer’s progresses. [Courtesy of Agneta Nordberg.]
Hanna Cho of Yonsei University, Seoul, South Korea, and colleagues wanted to test whether the spread of amyloid and tau followed the patterns described by Braak staging, as previous PET studies suggest. At AAIC, Cho described a cross-sectional study that compared amyloid PET with florbetaben (trade name Neuraceq™) and AV1451 tau scans in 195 people who ranged from cognitively normal to AD. The researchers assumed that the brain regions that most commonly contain plaques and tangles were the sites of earliest accumulation of those proteins, while regions where pathology occurs less often would be those that succumb later in the disease. Using this logic to evaluate the data from her cohort, she determined that amyloid spread rapidly through the cortex, later reaching the medial temporal lobe, while tau accumulation followed a stepwise pattern through different regions, starting in the entorhinal cortex and spreading to temporal and parietal cortices before reaching the neocortex. Atrophy followed the same stepwise pattern as tau (see Cho et al., 2016). The pattern of tau accumulation mirrored Braak staging and disease severity and may serve as a useful biomarker, Cho suggested.
Tau Tangles Track with Atrophy and Cognition
Speakers at AAIC consistently reported that tau pathology correlated with declining cognition. Mintun noted that higher AV1451 SUVRs associated with cognitive decline in his cohort. In the Swedish cohort, high tau signal correlated with worse performance on tests of episodic memory, as well as poorer global cognition, Nordberg said.
Shannon Risacher of Indiana University School of Medicine, Indianapolis, reported similar findings from the Alzheimer's Disease Neuroimaging Initiative (ADNI). This long-running initiative added tau imaging only recently, scanning participants two to five years after they entered the study. Risacher compared baseline and longitudinal measures of brain Aβ accumulation, hypometabolism, atrophy, and cognition to these later scans with AV1451. She found that baseline Aβ best predicted a high tau signal. The next-best predictors of tau pathology were the rates of change in three measures: memory scores, the CDR-SB, and hippocampal volume. This agrees with prior PET findings tying tangles to cognitive decline and degeneration.
In several studies, tangles in specific brain regions correlated with atrophy and hypometabolism in that same area. In a cohort from the Australian AIBL study, comprising 89 controls, 24 people with mild cognitive impairment, and seven with dementia, those who had high tau signal in regions such as the hippocampus, entorhinal cortex, prefrontal cortex, and anterior cingulate also had smaller volumes in those same regions. This was true even in non-demented participants, said Vincent Doré of CSIRO, Canberra, Australia. In one case where a participant had more tau signal in the right hemisphere, atrophy was also localized to the right side, he noted. The researchers saw similar results with both AV1451 and THK5351, a successor to THK5317. “The association between regional tau and regional atrophy is already evident at the early stages of tau deposition,” Doré concluded.
Can tau accumulation flag the earliest, subtle memory impairments? Rachel Buckley of Harvard Medical School, Boston, addressed this in her talk. She scanned 106 cognitively normal AIBL participants using AV1451, assigning them to high and low MTL tau groups, and asked all if they had concerns about their memory. People in the high tau group tended to answer yes, and the tendency was even stronger in those with positive amyloid scans. High MTL tau correlated with poor episodic memory and weak executive function, and people scored even worse on cognitive tests if they also reported memory concerns. The results suggested that tau pathology may underlie subjective memory complaints. This might prove useful for staging preclinical disease, Buckley said, though she acknowledged the study was small, and a simple yes/no question might not capture the complexity of early cognitive problems. In amyloid studies, subjective cognitive concerns have been found to correlate with amyloid burden (see Amariglio et al., 2012; Sep 2014 news).—Madolyn Bowman Rogers
Even as tau imaging forever changes the way researchers study Alzheimer’s disease and other tauopathies, the first batch of PET tracers are plagued by problems such as noisy data and off-target binding. At the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto, researchers delved into technical aspects of tracer uptake and how to deal with these vexing problems. Mark Mintun of Eli Lilly’s subsidiary Avid Radiopharmaceuticals, Philadelphia, described a new method for subtracting non-specific background signal by using a different anatomical reference region. This, he said, produced more reliable results. Melissa Murray of the Mayo Clinic in Jacksonville, Florida, tackled non-specific binding by Lilly’s AV1451, still the most commonly used tau tracer; she argued that these signals are spatially distinct from tau tangles and do not cut into AV1451’s usefulness for AD. Other tau tracers are entering human trials; Genentech researchers described preliminary data from their tracer GTP1 that indicated it could detect small changes in tau load. Meanwhile, researchers from Merck presented two posters suggesting their tracer MK-6240 is more specific for neurofibrillary tangles than is AV1451. In addition, MK-6240 may recognize the tau aggregates found in Pick’s disease, which are structurally different from those found in AD. Researchers still do not know exactly what forms of tau aggregate current tracers bind in vivo, noted Christian Sorg of the Technical University of Munich.
“This is a rapidly moving field with a lot of momentum,” Gil Rabinovici at the University of California San Francisco Memory and Aging Center wrote to Alzforum. “There are a number of new tracers just entering human studies, and over the next year or two we will learn a lot about their specific strengths and weaknesses compared to each other.” (See full comment below.) Other companies including Roche and Piramal are developing tau tracers, as well. This spate of tau tracer development was sparked in part because Lilly/Avid is proving difficult in making flortaucipir available for other groups to use.
Noise Control
Data noise creates problems in any study, but particularly in longitudinal PET scans, it can obscure the subtle changes in tracer uptake over time that are the point of the whole exercise. Researchers try to reduce this by picking an anatomical reference region with a low and stable signal for comparison. For both amyloid and tau PET, early studies have typically used the cerebellum, which accumulates less AD pathology than other brain areas. Recently, however, several groups have reported that using white matter as a reference region for amyloid PET lowers scan-to-scan variability and allows researchers to detect small changes over time (see Jul 2014 conference news; Landau et al., 2015). Some researchers speculate that white matter works better because it lies in the same plane as the regions harboring pathology and any “wobble” in the scan data, such as from a slight change in head position, will affect both equally. The cerebellum lies lower in the brain, and may be subject to a different level of noise.
Finding True Background.
AV1451 pixels in white matter separate into high-intensity (blue curve) and low-intensity (red curve) populations; the latter is thought to represent non-specific binding of tracer to brain. [Courtesy of Sudeepti Southekal and Mark Mintun.]
At AAIC, Mintun described how Avid has adapted white matter as a reference for use with tau PET. The challenge was that, unlike amyloid tracers, tau ligands light up white matter. This is probably not because tau aggregates there, but because the signal from tracer binding to nearby gray matter bleeds into the white matter, Mintun told Alzforum. The resolution of PET scans is too low to discriminate between close signals. To deal with this, the researchers analyzed the intensity of the signal in all the pixels in the white-matter regions. They separated pixels into two populations with high and low intensity, respectively (see image at left). The researchers decided the higher-intensity pixels reflected noise from the gray matter, and the lower-intensity pixels represented true non-specific background. The researchers used these latter pixels as their reference region. “The method identifies a subset of the white-matter pixels that appear to best estimate the reference uptake,” Mintun wrote to Alzforum. Use of this reference region lowered variability in test-retest data. It also heightened the difference in tau signal between cognitively normal and impaired people, resulting in more robust, reliable data, he said. “We believe improved methods for estimating reference region uptake should make PET tau imaging a more powerful biomarker in Alzheimer’s disease research,” Mintun wrote.
Besides their problem with random noise, existing tau tracers tend to bind to things other than tau tangles. Many speakers noted that AV1451 produces non-specific signals in the striatum and brainstem (see Feb 2016 conference news). Murray investigated this off-target binding by comparing autoradiography and immunohistochemistry in postmortem brain slices from 38 people who had a variety of neurodegenerative disorders, including Alzheimer’s, frontotemporal dementia, primary age-related tauopathy (PART), parkinsonism, Lewy body disease, and multiple-system atrophy. AV1451 bound neurofibrillary tangles, which comprise tau with both three- and four-repeat domains, in AD and PART brains, but it poorly labeled pure 3R and 4R tau aggregates present in other tauopathies. In addition, the tracer did not bind less-mature neurofibrillary tangles recognized by the CP13 antibody, she reported. AV1451 binding more closely matched the pattern of staining seen with the PHF-1 antibody, which recognizes mature neurofibrillary tangles. Several researchers told Alzforum that this is a potentially important finding that needs to be independently reproduced.
However, AV1451 lit up many areas that the tau antibodies PHF-1 and CP13 did not. She saw evidence of off-target binding to neuromelanin-containing cells in the substantia nigra, as well as lipofuscin in the lateral geniculate nucleus, hemoglobin in red blood cells, and melanin in the subpial membrane. Other researchers have previously speculated the tracer binds neuromelanin, a pigment found in dopaminergic and noradrenergic neurons. The tracer also bound to regions of calcification, such as in the blood vessels of the choroid plexus, which may explain why signal is often seen there, she suggested. These non-specific signals do not abrogate the tracer’s usefulness, Murray emphasized. Radiologists have to learn what to ignore on scans, she said. In good news for AD research, AV1451 did not label the four-repeat tau deposits found in aging-related tau astrogliopathy (see Lowe et al., 2016). Called ARTAG, this pathology can be widespread in the aging brain, and would have been a significant confounder, Murray said (see Kovacs et al., 2016).
Some researchers are hoping that other tau tracers in development may be more specific for tangles. In two posters, Merck researchers made the case that their tracer MK-6240 selectively binds the neurofibrillary tangles found in AD. In postmortem studies of AD brains, the tracer bound tangles tightly, while producing no signal in control brains, reported Idriss Bennacef. In non-human primates, the ligand entered the brain rapidly and washed out quickly, and did not light up white matter (see Walji et al., 2016; Hostetler et al., 2016). Zhizhen Zeng directly compared MK-6240 binding with AV1451 in slices from frozen AD and control brains. Zeng described similar binding to tangles by both tracers, but less off-target binding with MK-6240. In control brain tissue, clorgyline, a monoamine oxidase inhibitor, competed with AV1451 for binding to control brain tissue, but it did not compete with MK-6240, suggesting the latter has better specificity for tau aggregates.
“The Merck compound looks very interesting because of its unique chemical structure, which is quite different from existing compounds,” Rabinovici wrote to Alzforum. “The very early data presented in normal controls suggests it may have low ‘off-target’ signal. That said, we still need to see more human data, including positive signal in AD patients, to judge whether this will be a useful tracer.”
Bennacef noted the possibility that this tracer may detect forms of tau other than the paired helical filaments present in neurofibrillary tangles. In vitro, MK-6240 also reacts with pure 3R tau, which forms straight filaments, he reported. This form accumulates in Pick’s disease. The researchers are currently evaluating binding in healthy elderly and AD patients in a Phase 1 trial. Biogen recently announced that it will partner with Merck to test this tracer in drug studies.
Other tracers are also entering human studies. Sandra Sanabria Bohorquez of Genentech, South Francisco, presented data on the company’s tau tracer GTP1. In a study of six cognitively normal people, six with prodromal AD, five with mild AD, and five with moderate, tracer binding intensity increased at each disease stage and correlated with the amyloid PET signal, she reported. In preliminary longitudinal data, tau signal did not increase over six to nine months in a healthy control, but rose about 9 percent in a person with mild AD, and 5 percent in one with moderate AD. Accumulation was most notable in the temporal lobe and hippocampus. The results suggest the tracer is sensitive to small changes in tau load, Sanabria Bohorquez noted.
Brian Gordon of Washington University, St. Louis, appreciated seeing the data on the new tracers, but noted more histological and autoradiography work will be needed to understand exactly what each one is binding. “There were similarities between the tau patterns across the different tracers, but they were not exactly the same. While I am optimistic about their ultimate potential, we need to be cautious with these new tracers,” he wrote to Alzforum.—Madolyn Bowman Rogers
New Ways to Target Aβ and BACE Show Promising Phase 1 Data
Amyloid β, in its various incarnations, remains a favorite target of big pharma. This year’s AAIC meeting, held July 22-28 in Toronto, saw the arrival of both new BACE inhibitors and the first therapeutic antibody against pyroglutamate forms of Aβ found in plaques.
The latter seems to clear aggregated Aβ from the brain, but researchers expressed concern that this passive immunotherapy turns into active antigen, eliciting a peripheral immune response from nearly every volunteer who receives it. Researchers were unsure what this means for the future of the therapy; some pronounced it dead on arrival but others said this sort of problem can be overcome.
In the increasingly crowded BACE inhibitor arena, researchers from both Novartis and Eli Lilly and Company reported first-in-human studies of their newest compounds, while Janssen reported the first trials of its hopeful in Alzheimer’s patients. These three BACE inhibitors join several others that are well on their way through the trials pipeline. “It is a good thing to have so many BACE inhibitors in clinical trials,” Robert Vassar, Northwestern University, Chicago, told Alzforum. “It is an indication of the high level of confidence that the pharmaceutical industry has in the target. The field is moving forward, and over the course of the next several years, we will know one way or the other whether BACE inhibitors will be safe and effective for Alzheimer’s disease.”
Specificity, Potency, Early Intervention Key for BACE Inhibitors Johannes Streffer from Janssen Research and Development in Beerse, Belgium, described the first trial of the company’s BACE inhibitor JNJ-54861911 in people with early AD. Streffer previously had introduced the compound at the AD/PD meeting in Nice, where he described the first safety and tolerability study in healthy elderly. Janssen researchers had used state-of-the-art diagnostics to monitor the effects of the inhibitor on APP processing (see Apr 2015 conference news). At AAIC, Streffer reported first results in people who either have prodromal AD or are at risk for the disease.
For the trial, Streffer and colleagues selected 37 people with prodromal AD based on their having a clinical dementia rating (CDR) score of 0.5, and 19 cognitively healthy people who were at risk for AD based on their biomarker profile. The volunteers were randomized (1:1:1) to receive placebo, 10, or 50 mg of JNJ-54861911 once daily for four weeks. Plasma Aβ fell by 83 and 92 percent, respectively, for the low and high dose, and by 67 and 90 percent in the CSF. Similar reductions occurred across the board for all Aβ peptides tested, including Aβ1-37, Aβ1-38, Aβ1-40, and Aβ1-42. Streffer said levels of sAPPβ, the fragment of APP left over by BACE cleavage, fell by similar amounts while there was a dose-dependent uptick in sAPPα, which is shed by α-secretase. The volunteers on the drug performed no better than those on placebo in cognitive testing, but Streffer said he would not expect to see a difference in such a short trial.
All the volunteers completed the trial. Streffer said there were eight adverse events in the high dose, three in the low, and four in the placebo group. He said the pattern did not suggest any safety concern and that the drug was well tolerated.
Streffer said the data matches the lowering seen in Janssen’s previous study on healthy elderly. In a poster, Alberto Russo described how pharmacokinetic/pharmacodynamic modeling of data from that trial accurately predicted lowering of CSF Aβ in the prodromal AD and at-risk population. Streffer told Alzforum that the modeling allows researchers to predict the range of reductions of CSF Aβ in treated populations to ensure that an adequate dose is given to achieve a desired effect. This helps the researchers set dosing levels in upcoming trials. DIAN is pursuing a similar approach of building a disease-progression model from observational data, and using it to predict responses in the DIAN-TU trials (see Part 7 of this series).
Janssen has already begun recruiting for the Phase 2/3 EARLY trial, a safety and efficacy study of people who are asymptomatic but at risk for AD. They plan to recruit more than 2,000 volunteers to receive placebo, 5, or 25 mg of JNJ-54861911 over 4.5 years. Reisa Sperling, Brigham and Women’s Hospital, Boston, told Alzforum she is very excited about this trial, dubbed A5 because it broadens the horizon of the current A4 trial by treating younger, more cognitively intact people. EARLY will recruit volunteers as young as 60. There is no exclusion of “supernormals,” i.e., people who score particularly high on cognitive tests, as there was in A4. Because the trial focuses on younger people and runs longer than A4, those restrictions could be relaxed, noted Sperling. Recruits will have to test positive for amyloid, either by PET scan or through CSF analysis for low Aβ. “CSF is more important in countries that place less emphasis on PET,” said Sperling. “Evidence also suggests Aβ in CSF drops before PET scans test positive, potentially moving us even earlier in disease course,” she added.
EARLY has begun recruiting in Europe and Australia and starts up in Japan today under the leadership of Takeshi Iwatsubo at the University of Tokyo; it will start in the United States and Canada this fall. Its leaders chose the Preclinical Alzheimer Cognitive Composite, or PACC, as the primary outcome. This test battery was designed to detect subtle changes in cognitively normal people and is being used in the A4 trial as well (see Jun 2014 news). EARLY will measure a wide range of secondary outcomes, including safety parameters, PET and MRI imaging for amyloid and neurodegeneration, and a number of different cognitive and functional tests.
Ulf Neumann from Novartis, Basel, Switzerland, presented the first-in-human data for the company’s BACE inhibitor CNP520. Novartis lags behind some of the other pharmaceutical companies that have BACE inhibitors already in Phase 2 and 3 trials, including Merck, Lilly, and AstraZeneca. “Because we are a bit late to the field, we can learn from what others have discovered along the way to help us better tailor our compounds,” Neumann told Alzforum. He believes since all the BACE inhibitors in development are very potent, safety may be the distinguishing characteristic. “We want to test the compound in the early phase of AD when the chances of slowing the disease are best, but where we need long, large trials. Therefore the drug has to be very safe,” Neumann added.
CNP520 was chosen for the Generation prevention trial being conducted by Novartis, the Banner Alzheimer’s Institute in Phoenix, and Amgen. The trial, which began enrolling this month, will test the BACE inhibitor and CAD106, Novartis’ anti-Aβ active immunotherapy in asymptomatic people who carry two copies of the ApoE4 gene (see July 2014 conference news). “CNP520 was selected because of its relatively preferential selectivity for BACE-1, preliminary indications of its safety, tolerability, and potent Aβ-reducing effects,” wrote Eric Reiman from Banner, who is the principal investigator of the study. Generation is incorporating genetic screening and counseling, and will run for five years. Primary outcome measures are time to MCI or AD and change in the Alzheimer's Prevention Initiative Composite Cognitive test score.
In his AAIC talk, Neumann reviewed preclinical safety data for CNP520. He claimed that CNP520 has about threefold selectivity for BACE1 over BACE2. In contrast to NB-360, an earlier Novartis compound, CNP520 caused no coat discoloration in mice. Hypopigmentation can occur if BACE2 is blocked because it helps with processing the melanocyte protein PMEL that is essential for pigmentation (see Dec 2013 conference news). Vassar said that as far as he knows, all other BACE inhibitors cause hypopigmentation to a degree, but whether this will be a significant problem in people remains to be seen. Neumann thinks CNP520 spares melanocytes partly because of its selectivity for BACE1 and partly because it distributes poorly to the skin. Other scientists said the compound’s high brain penetrance might be as important as its selectivity in reducing the risk of peripheral side effects.
CNP520 had even better selectivity over other proteases, including pepsin, renin, and cathepsin D, a close relative of BACE1, Neumann reported. Both are aspartic proteases and have similar pH optima. CNP520 bound to BACE1 with a 1,000-fold specificity over the cathepsin. Vassar noted that most BACE inhibitors now in development show similar selectivity over this protease. Poor selectivity over cathepsin D likely scuppered some early BACE inhibitors, because the protease is essential for the rapid turnover of photoreceptors in the eye and blocking it can cause lipofuscinosis of the retina (see Mar 2011 conference news).
What about the human data? Novartis tested CNP520 in a Phase 1 safety and tolerability trial consisting of three parts. In one, 38 healthy adults received a single ascending dose up to 1,125 mg; 14 received placebo. The second part repeated the single ascending dose up to 750 mg in 50 healthy older adults, while 17 got placebo. In part three, multiple ascending doses up to 300 mg were given to 55 older adults (20 received placebo) over two weeks. The volunteers were monitored for safety, tolerability of the drug, and pharmacodynamics based on CSF sampling. In Toronto, Neumann said adverse events were generally mild. In the single-ascending-dose arm, one volunteer who received 30 mg of CNP520 became anxious and delusional, but on review these symptoms showed up in the person’s medical history and should have excluded him or her from the study, claimed Neumann.
As for efficacy, the compound dose-dependently lowered Aβ40 in the CSF by up to 95 percent after multiple doses. The drug appears to be cleared relatively slowly, with half of it still remaining in the blood of adults after 70 hours. “That can be both a positive and a negative,” Neumann told Alzforum. On the plus side, he said, it’s a forgiving drug—once it reaches a steady state, its concentration will not change very much, so even if a person forgets to take his or her medication one day it will not matter that much. The downside is that if there is an adverse reaction and the patient needs to stop taking the medication, it will take days for it to clear the system. Novartis has yet to run a trial to test the effect of the drug on cognition, but has run other safety and tolerability trials, including a three-month Phase 2a trial in healthy elderly and an ethnic sensitivity study in Japan.
Lilly has taken a new tack in BACE-inhibitor development, debuting a highly potent compound at AAIC. As Lilly’s Brian Willis reported, 1 mg/day of LY3202626 reduced CSF Aβ1-40 by half after two weeks. “This should be an advantage, because lower dosing means reduced potential for off-target side effects,” Vassar told Alzforum. Neumann agreed, noting that the potency Lilly achieved in vitro translated into improved potency in people. Others claimed this was the first time they had seen this work out for a BACE compound. Neumann noted it has no selectivity over BACE2.
Nevertheless, the inhibitor seems to have a good safety profile. Willis reported results of a Phase 1 trial conducted on healthy controls and people with AD. The trial comprised four sequential parts. Parts A to C enrolled 92 healthy people, while part D enrolled two people with AD. In part A, subjects received placebo or 0.1 to 45 mg of the drug as a single dose and were monitored for safety. For part B, volunteers were given 1.6 to 26 mg or placebo as a single dose, and serial plasma and CSF samples were taken for pharmacokinetic and pharmacodynamic analysis. Part C entailed multiple dosing of 1 to 26 mg or placebo for two weeks. Lastly, the two AD patients received 6 mg/day for two weeks. Willis and colleagues took CSF at baseline from volunteers in parts C and D, and again 24 hours after their final dose.
Willis reported that volunteers tolerated LY3202626 well at all doses, without adverse events up to 42 days after the last dose. The compound’s half-life in plasma was 21 hours, and it readily crossed the blood-brain barrier. The compound dose-dependently reduced Aβ40 and Aβ42 in plasma and CSF. At 1, 6, and 26 mg/day over two weeks, it reduced Aβ40 in CSF by 50, 75, and 90 percent, respectively.
Passive Immunotherapy Turns Active Antigen Mike Irizarry from Eli Lilly and Company, Indianapolis, presented results of a Phase 1 safety and tolerability trial of LY3002813, an antibody against pyroglutamate-modified forms of Aβ. LY3002813 is a humanized version of a mouse monoclonal that purportedly recognizes only plaques (see Dec 2012 news). For the Phase 1, Irizarry and colleagues administered LY3002813 or placebo intravenously to 49 patients, average age 74, who had prodromal to moderate Alzheimer’s disease. All had memory deficits and a positive florbetapir scan for brain deposits of Aβ. Thirty-seven volunteers in five cohorts received a single initial dose of between 0.1 and 10 mg/Kg of the antibody, while 12 received placebo. During this single-ascending-dose phase, clinicians monitored them for three months for any adverse signs, then in the next multiple-dose phase gave them up to four additional monthly injections. For this phase, each person was given the same dose they got originally, except for people in the 0.1mg/Kg cohort—they received 0.3mg/Kg in subsequent injections.
The good news was that at the highest dose the antibody seemed to reduce plaque load in the brain. Irizarry stressed that at six, the number of people in this group was small. Overall, the mean standard uptake value ratio for florbetapir in the high-dose group was 1.65 at baseline, and this fell by 0.26 over seven months. Irizarry said this translates into a mean 40 percent reduction on the centiloid scale of amyloid positivity (see Nov 2014 news). Researchers at the meeting were impressed by the data, which seemed stark on florbetapir scans. LY3002813 joins aducanumab and gantenerumab as the only immunotherapies that have been shown to reduce brain amyloid to date (see Mar 2015 news). Aducanumab had a considerable rate of ARIA-E and ARIA-E-related discontinuations at the higher doses.
“The significant reduction of PET signal, without showing any ARIA-E side effect, is a very important advance over other antibodies in development,” noted Hans-Ulrich Demuth, from the Fraunhofer Institute for Cell Therapy & Immunology, Leipzig, Germany. Demuth advises Probiodrug, a company developing anti-pyroglutamate antibodies and glutaminyl cyclase inhibitors to prevent formation of pyroglutamate Aβ, which is reportedly more aggregation-prone than the unmodified peptide (see Mar 2013 conference news). Indeed, Irizarry reported no cases of ARIA-E (aka vasogenic edema) in this small trial, but he did report two asymptomatic cases of ARIA-H (microhemorrhages). “It’s difficult to tell if these were related to the antibody since microhemorrhages are common in Alzheimer’s disease, but we will continue to monitor closely,” said Irizarry. Others still saw this as cause for concern.
Researchers were even more concerned that this antibody elicited strong immunogenicity. In the multiple-dose phase, six of the 37 patients developed an infusion reaction, such as chills, flushing, dizziness (postural hypotension), rash, and fever. Anti-drug antibodies turned up in the plasma of almost everyone treated. Dave Morgan, University of South Florida, Tampa, saw this as the end of the road, at least for this version of the antibody. “With seven of eight people mounting an immune response, this is basically dead,” he told Alzforum. “They could try to identify the epitope causing the immune response and modify it,” he suggested. Demuth agreed. He speculated that in designing an antibody with high avidity for pGluAβ, a new epitope was created that was highly immunogenic. Morgan said this was most likely in the variable region of the antibody, the business end that binds pGluAβ. Epitopes in antibody-variable regions, known as idiotypes, can, in some cases, elicit an immune response, he explained. Irizarry told Alzforum that his colleagues are characterizing the anti-drug antibodies, but said he does not know as yet the cause of the immunogenicity.
One problem with having an immune reaction is that it shortens the half-life of the therapeutic antibody, and indeed, Irizarry reported exactly that. At low doses, half the antibody was gone from the participants’ blood within four days, as opposed to the 21 days the researchers expected. Irizarry said he was very surprised at the clearance rates. “None of the preclinical work predicted either the immune response or the rapid clearance,” he said. He suspects the two are related, but noted that even after one low dose the antibody was cleared rapidly. “The clearance may not be fully explained by an immune reaction to the antibody,” he told Alzforum. “To understand the relationships with immunogenicity, immune safety, amyloid clearance, and pharmacokinetics, we have to take the therapy forward to another study,” he said. Irizarry said the company has begun a Phase 1b.
On the other hand, plaque-clearing antibodies might not have to be given long-term. They could help eliminate existing plaque, and its recurrence could then be kept in check with BACE inhibitors. Companies including Lilly are experimenting internally with combination regimens of antibodies and BACE inhibitors.—Tom Fagan
Refining Models of Amyloid Accumulation in Alzheimer’s Disease
The knowledge that Alzheimer’s disease begins with a preclinical phase that lasts for 20 years or more has focused researchers’ minds on prevention. However, they need to know more about how fast pathology progresses, how to predict who will develop the disease, and when a person’s symptoms might appear. Speakers at the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto, presented new pathology, imaging, and cerebrospinal fluid data that offered clues to these mysteries. Talks pinned down the rate of amyloid accumulation, highlighted a region where plaque formation might correlate with imminent cognitive decline, and clarified the temporal relationship between Aβ signals in the CSF and in the brain. Researchers also provided more evidence that inflammatory processes cause the brain to swell early in AD, which could complicate the interpretation of brain volume changes during disease progression and treatment. Ultimately, the new information will sharpen trial selection criteria and inform therapeutic strategies.
How Slowly Does Aβ Build Up?
Many current therapeutic strategies attempt to curb amyloid plaque buildup or clear existing deposits. However, researchers still do not know the rate of Aβ accumulation, or how much they need to remove to preserve brain health. Because everyone’s brain, healthy and otherwise, contains soluble Aβ, what constitutes abnormal levels of the peptide? The advent of amyloid PET may provide an answer to this last question, said Colin Masters of the University of Melbourne in Australia. Using PET, researchers have defined a cutoff between normal and abnormal Aβ levels, and since the majority of the Aβ in the brain occupies detergent-soluble or insoluble pools, both of which show up on PET scans, PET signal intensity should reflect total Aβ, Masters predicted.
Amyloid Predicts Decline.
Cognitively normal older people with amyloid in the striatum declined twice as fast as those without. [Courtesy of Bernard Hanseeuw.]
To relate PET scans to peptide levels, Masters and colleague Blaine Roberts at Melbourne compared PiB uptake to total Aβ measured in brain lysates from more than a dozen participants in the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL) who died shortly after undergoing the PET scan. As predicted, the researchers found a tight correlation between the PiB signal in the frontal cortex and the Aβ load in frontal cortex lysates. In people whose brains were at the threshold for amyloid positivity (an SUVR of 1.45 in this case) when they died, the researchers measured about 3.2 μg Aβ per gram of frontal cortex. In people who had a PiB scan typical of AD, with an SUVR of about 2.3, they found 13 μg Aβ per gram of frontal cortex.
To expand this analysis to the whole brain, Masters and Roberts used structural MRI to determine total volume—and by extrapolation, weight—of gray matter in each person’s brain, and multiplied it by the measured amounts of Aβ per gram of tissue. Assuming a similar concentration of Aβ throughout the brain, they calculated that the average PiB-negative brain contained 1.7 mg of Aβ, while AD brains held 6.5 mg. The difference, 4.8 mg, represents Aβ that accumulates in disease. Because it takes two decades or more for Aβ to accumulate in the brain, it likely accrues at a rate of about 28 ng/hour, the researchers calculated. This assumes that the rate of accumulation is a constant, which may not be the case, Masters noted.
Researchers at AAIC found the results intriguing. Clifford Jack of the Mayo Clinic in Rochester, Minnesota, was impressed by the calculation that 1.7 mg of brain amyloid could lie below the detection threshold for PET scans. The fact that this “invisible” Aβ makes up as much as one-fourth of the total in people with “heads full” of amyloid illustrates the limits of a PET scan’s sensitivity, he told Alzforum. Jack emphasized the therapeutic implications of knowing the Aβ accumulation rate. “The rate of reduction in amyloid deposition needed to achieve a meaningful effect could come right out of these calculations,” he suggested.
How does the 28ng/hr rate compare to other estimates? Previously, Randall Bateman and colleagues at Washington University, St. Louis, measured soluble Aβ production and clearance rates in the CSF of healthy volunteers and AD patients using stable isotope-linked kinetics (SILK). Bateman and colleagues found that healthy adults normally produce and clear about 580 ng total Aβ each hour, indicating no accumulation of Aβ. In late-onset AD patients, by contrast, the hourly clearance rate from CSF drops by about one-quarter, or 145 ng/hr, suggesting the peptide could be building up (see Jun 2006 news; Jul 2010 conference news; Potter et al., 2013). This does not necessarily mean that amount of Aβ piles up in the brain every hour, however, since other clearance routes exist besides CSF (see Sep 2015 news). The AIBL data, which measured brain deposition rather than peripheral clearance, suggest that only about 5 percent of the 580 ng/hr production of Aβ becomes trapped in amyloid plaques in people who develop AD.
Boosting the Prognostic Value of Amyloid Imaging
Researchers also want better biomarkers to tell them how close a person is to developing symptoms. Although amyloid accumulates for 20 years before symptoms appear, plaques by themselves are an insensitive marker of imminent decline. Striatal amyloid accumulation may better predict symptom onset than cortical amyloid does, argued Bernard Hanseeuw of Massachusetts General Hospital, Charlestown. Striatal plaques form at later Braak stages, and the presence of striatal, but not cortical, amyloid at autopsy correlates with a diagnosis of AD (see Thal et al., 2002; Beach et al., 2012).
To find out if striatal plaques could flag late preclinical stages, Hanseeuw analyzed brain imaging and cognitive data from 346 participants in the Harvard Aging Brain Study (HABS) and 1,087 in the Alzheimer's Disease Neuroimaging Initiative (ADNI). The whole group comprised 646 cognitively normal participants, 574 with mild cognitive impairment, and 213 with AD. In these cohorts, amyloid PET scans detected striatal amyloid only when cortical amyloid was high, in agreement with postmortem studies. High striatal amyloid also correlated with a high tau PET signal. Overall, Hanseeuw and colleagues found striatal plaques in 94 percent of HABS participants diagnosed with AD, but only 14 percent of the cognitively normal HABS group. The percentages were slightly lower in ADNI, at 68 and 8 percent, respectively. Hanseeuw noted that the PiB scans used in HABS might be more sensitive to striatal amyloid than the florbetapir scans used by ADNI, perhaps because the latter compound binds surrounding white matter more. In addition, ADNI contained a fraction of participants diagnosed with AD who never had the disease, or brain amyloid.
Overall, the data support the idea that those few cognitively normal elderly who had striatal amyloid might be further along in the disease process than those without, Hanseeuw believed. To test this, he studied the association between striatal amyloid and cognitive decline in just the cognitively normal participants. Over one to five years of follow-up, those with striatal plaques at baseline declined twice as fast on cognitive tests as those with cortical plaques only (see image above). People with striatal amyloid also lost more hippocampal volume over the course of the study. “Striatal amyloid could be used to identify cognitively normal elderly who are at the greatest risk of decline,” Hanseeuw claimed.
CSF Versus Imaging
While striatal amyloid may mark the late preclinical phase, Sebastian Palmqvist of Lund University, Sweden, made a case for CSF Aβ42 being one of the earliest preclinical markers of AD. Levels of this fluid biomarker drop as amyloid plaques accumulate in the brain, and this becomes apparent before amyloid PET scans turn positive, Palmqvist claimed. Previous cross-sectional studies had hinted at this, with some participants having low CSF Aβ42 in the absence of an amyloid PET signal, but it was unclear if all of these PET–negative individuals were on track for AD (see Fagan et al., 2009; Mattsson et al., 2015). Confusing matters further, at least one study reported finding a few people with positive amyloid scans and normal CSF Aβ42 (see Landau et al., 2013).
To observe the relationship of the two markers over time, Palmqvist and colleagues Niklas Mattsson and Oskar Hansson at Lund stratified 437 ADNI 2 participants according to whether they were positive or negative on each. For brain amyloid, they used florbetapir PET data, choosing a low, conservative cutoff of 0.79 SUVR for positivity. CSF Aβ42 levels in ADNI2 were determined by the AlzBio3 assay, with the standard cutoff of less than 192 ng/L indicating positivity. After excluding borderline individuals who were within 5 percent of the cutoffs, the researchers found no participants who were CSF–/PET+, but 26 people who were CSF+/PET–, i.e., people whose CSF Aβ levels were already abnormal but whose PET scans were not.
Most people in this group were cognitively normal and stable, Palmqvist said. They had no evidence of elevated CSF tau or hippocampal atrophy. They had also higher CSF Aβ42 levels than the PET+ group, suggesting they were at an earlier stage of disease. Reinforcing this, the PET+ group lost more hippocampal volume over the course of two years than the CSF+/PET– group, and their brain metabolism and memory dwindled more. Importantly, the CSF+/PET– group accumulated Aβ at the same rate (1.2 percent increase in PET scan SUVR per year) as the PET+ group, and three times faster than the CSF–/PET– group, suggesting both of the CSF+ groups were on the path to AD. Given this rate, it would take an average of seven to 10 years for a CSF+/PET– person to become amyloid-positive on a PET scan, Palmqvist calculated (see Palmqvist et al., 2016). Altogether, the findings suggest that CSF measurements can discern amyloid accumulation before PET scans do, and that people with low CSF Aβ generally go on to become amyloid-positive on PET as well, Palmqvist said.
The data engendered lively debate at AAIC. Some researchers speculated that the results illustrate the limitations of florbetapir, and that other tracers might pick up lower levels of accumulation. Palmqvist countered that he has found similar results with flutemetamol, albeit in a cross-sectional study. One audience member wondered if some people with low CSF Aβ42 could represent false positives, for example people who simply make less Aβ. Palmqvist considered this unlikely because they accumulated brain Aβ as fast as those in the CSF+/PET+ group.
Why would soluble Aβ fall so much sooner than it becomes visible as plaques in the brain? Perhaps CSF and PET ligands are measuring different processes, Palmqvist suggested. Some researchers have proposed that CSF Aβ42 drops as the peptide aggregates into prefibrillary deposits and diffuse plaques, while PET detects more mature neuritic plaques. The data argue for separating the CSF Aβ and amyloid PET curves in biomarker staging models, Palmqvist said (see Jan 2010 webinar; Feb 2013 conference news).
A Bigger Brain Can Be Bad
Other biomarkers change dynamically over the course of the disease as well, providing more clues to underlying processes. Christian Haass of Ludwig-Maximilians University, Munich, had earlier reported that levels of a soluble fragment of the microglial receptor TREM2 rise in the CSF in prodromal AD, but stabilize or even drop later on (see Jan 2016 news; Mar 2016 news). The sTREM2 peak may indicate an inflammatory response early in AD, Haass speculated.
At AAIC, Juan Domingo Gispert of the Barcelonaβeta Brain Research Center, Spain, expanded on this idea. Gispert and colleagues compared 45 cognitively normal controls without brain amyloid, 19 cognitively normal people with brain amyloid, 27 people with MCI due to AD, and 23 with mild AD. His was a cross-sectional study. Among cognitively normal people, CSF sTREM2 levels bore no relationship to brain volume. In the MCI group, however, high sTREM2 associated with relatively larger brain volume after accounting for the expected atrophy due to CSF p-tau levels, particularly in the medial and inferior temporal cortices and the precuneus. The AD group showed a trend in that direction as well. The researchers next looked at diffusion-weighted MRI, which measures water diffusivity in the brain and serves as a marker of edema. High sTREM2 went along with low diffusivity in the temporal lobes and precuneus, suggesting the sTREM2-related volume increase was due to inflammation that peaks at the MCI stage (see Gispert et al., 2016).
The data fit with the idea that in a certain phase of the disease process, neurodegeneration features inflammation and local swelling, Gispert said. Several trials of anti-amyloid antibodies have reported shrinking brain volume in the treatment group. This puzzled researchers at the time, because they usually think of brain shrinkage as indicating atrophy and worsening of the disease (see Jul 2004 conference news; Nov 2012 conference news; Apr 2013 conference news). The new data add more evidence for the presence of edema in AD, and reinforce the idea that a temporary drop in volume could be good under some circumstances (see also Aug 2016 conference news).—Madolyn Bowman Rogers
Homing in on Early Alzheimer’s Biomarkers: Does Connectivity Hold the Key?
In the quest to find biomarkers that detect presymptomatic Alzheimer’s disease, some researchers are turning to measures of brain connectivity. Many studies have reported that functional circuits become disorganized early in the disease process, sparking interest in their potential as preclinical biomarkers. At the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto, several speakers discussed specific imaging measures that appear to flag people at the highest risk for progressing to AD. They include a gray-matter measure of connectivity, deficits in the visual network, and imaging correlates of anosognosia, which is the lack of awareness of one’s own memory problems. In a well-received talk, David Jones of the Mayo Clinic in Rochester, Minnesota, argued that network disruptions may precede and trigger amyloid and tau pathology, although the mechanism remains unknown. He put forward a method for calculating the communication breakdown based on changes in connectivity within and between subsystems of the default mode network.
Overall, researchers expressed excitement about the potential of these measures. “By describing the brain as a network, regardless of the imaging modality used, we get a more comprehensive description of brain functioning than by looking at brain areas in isolation,” Betty Tijms of VU University Medical Center, Amsterdam, wrote to Alzforum. Connectivity changes often explain cognitive problems better than do simpler measures like atrophy or hypometabolism, researchers agreed. “Network measures are really promising. I think they’ll provide us with important mechanistic interpretations of how AD progresses,” said Prashanthi Vemuri, also of the Mayo Clinic in Rochester.
Patterns of Tau Pathology.
Tangles appeared in the working memory network (left) mostly when AD struck early in life, but lit up DMN regions (right) no matter when AD developed. [Courtesy of David Jones.]
Researchers cautioned that the study of connectivity is in its infancy. It remains unclear how best to evaluate brain circuitry. Studies are exploring different types of imaging and new ways to define brain organization, and more work is needed to discover which of these are most robust. “We have to be careful to acknowledge that these measures are probably not interchangeable. ‘Connectivity’ has become an umbrella term,” Renaud La Joie of the University of California, San Francisco, wrote to Alzforum.
Currently, researchers typically measure connectivity using resting-state functional MRI. This technology maps the regions of the brain where blood flow waxes and wanes in tandem, suggesting the areas form a functional network. During the preclinical stages of AD, fMRI connectivity falters in numerous brain networks (see Jul 2012 news; Aug 2013 news; Aug 2014 news). In particular, researchers have focused on disruption of the default mode network (DMN), which is active when a person is not focused on a mental task. The DMN is one of the first networks to accumulate Aβ deposits (see Mar 2004 news; Nov 2007 news; Feb 2009 news). However, studies have not yet sorted out how network disconnection relates to pathology, and which comes first.
Could Network Failure Trigger Protein Accumulation?
Jones believes that Alzheimer’s may start with network disruptions. He previously reported cross-sectional data from a cohort of 128 ADNI participants who ranged from cognitively normal to AD dementia. Everyone in the impaired groups had amyloid accumulation, indicating they were on the path to AD. By correlating connectivity with ADAS-cog scores, which rise as people become more impaired, Jones was able to infer how brain connections changed over the course of the disease. In cognitively normal ApoE4 carriers, connectivity within the posterior DMN subsystem sputtered in the absence of any detectable amyloid plaques or brain atrophy. This network disruption appeared to trigger enhanced connectivity between other DMN subsystems, perhaps as a compensatory response. Connections between subsystems were stronger in those with worse ADAS-Cog scores. The heightened connectivity between subsystems was associated with the presence of amyloid plaques throughout the affected networks. Jones hypothesized a cascading network failure model of AD, in which synaptic disruptions themselves kindle amyloid pathology, which may then accelerate tau tangles (see Jones et al., 2016).
At AAIC, Jones proposed using this network failure pattern as a biomarker of disease. He calculated a network failure quotient (NFQ), in which the numerator represents connectivity between DMN subsystems, and the denominator, connectivity within subsystems. Because the numerator increases and the denominator drops as AD advances, their ratio rises. NFQ goes up at the preclinical stage, and correlates with changes in numerous other biomarkers, including FDG PET, amyloid and tau PET, cortical thickness, and word-list learning, Jones said.
To see if there was a relationship between network failure and molecular pathology, Jones and colleagues compared NFQ scores to amyloid PiB PET scans and AV1451 tau scans in a cohort consisting of 177 cognitively normal participants, 12 people with mild cognitive impairment, and 29 with dementia. Participants were drawn from the Mayo Clinic Study of Aging and the Mayo Clinic Alzheimer’s Disease Research Center; all of those with cognitive impairment had positive PiB scans, hence would be called amnestic MCI.
The researchers used regression analysis to determine how much of the tau pathology in their cohort could be explained by network failure, and whether tangle accumulation was mediated by amyloid. They found that two-thirds of the tau signal could be explained by the presence of amyloid. This left one-third of tau signal that associated with NFQ scores independent of amyloid. The findings support the idea that network disruptions may somehow trigger both amyloid and tau pathology, although the mechanism is unknown, Jones said. “Global network failure may lead to amyloidosis, and after saturation of amyloidosis, that network failure may then accelerate pre-existing tauopathy,” he speculated to Alzforum.
Neurofibrillary tangle deposition in this cohort occurred in specific patterns that matched three functional brain networks: the visual network, the left executive-control network, and a memory network. The first two of these accumulated tangles mostly in people with young-onset dementia. Tangles in the memory network, on the other hand, cropped up no matter when disease started, and matched the typical tau deposition patterns seen in Braak staging. This memory network overlaps with the ventral DMN, involving regions such as the medial and inferior temporal lobes and the posterior cingulate (see image above).
In each of the three networks the researchers saw a low level of tau signal even in people without biomarkers of AD, in line with neuropathological findings from older adults. Once amyloid appeared, however, the tau signal jumped throughout the entire network, with no signs of an incremental advance. On the face of it, this seems to belie theories about the gradual spread of misfolded tau along network connections. “Amyloidosis seems to accelerate an existing network-related tau abnormality,” Jones concluded. Recent tau imaging studies report that amyloid frees age-related tau pathology in the medial temporal lobe to explode throughout the cortex (see Jul 2016 news; Aug 2016 news).
An enthusiastic AAIC audience peppered Jones with questions. “The concept of tau deposition happening throughout a network is really new. We’re very excited about it,” said Vemuri, who collaborates on the research. Others were intrigued by the potential of NFQ as a biomarker. “It will be interesting to study the robustness of this measure, and how it changes over time,” Tijms said.
Gray Matter Connectivity Predicts AD. Disorganized gray matter (colored regions) in people with low CSF Aβ42 indicative of preclinical AD. [Courtesy of Betty Tijms.]
Faltering Connectivity Highlights Early Disease
Other talks highlighted how specific measurements of disrupted connectivity might serve as biomarkers. Tijms described a unique way to measure functional brain networks. She evaluates three-dimensional spatial relationships between neighboring voxels in a structural MRI scan. These relationships capture brain characteristics such as cortical thickness, folding, and volume, she noted. She then applies mathematical theory to calculate brain connectivity from these data (see Tijms et al., 2012). “This is based on the idea that when brain areas are involved in similar cognitive functions, they may develop in a coordinated way,” she told Alzforum. Because this method determines connectivity based on a single MRI scan instead of a lengthy resting-state fMRI session, the data are quicker and easier to obtain and suffer less from movement artifacts, Tijms said. These connectivity patterns are less pronounced in AD brains, she noted. She previously reported a correlation between disrupted gray matter connectivity and low CSF Aβ42 in cognitively healthy older adults (see Tijms et al., 2016, and image above).
Tijms wondered if the measure could single out people most at risk of progressing to AD. She analyzed data from the Connectivity in Dementia (CODA) study. This subset of the Amsterdam Dementia Cohort comprises 62 participants with subjective cognitive complaints and 160 with mild cognitive impairment, with an average age of 68. All had abnormal baseline CSF Aβ42 indicative of plaque accumulation.
Over two years, 23 of the participants with subjective complaints and 99 of those with MCI became more impaired, as measured by CDR scores. Those who progressed had weaker gray-matter connectivity at baseline than those who stayed stable, particularly between the medial temporal lobe, precuneus, posterior cortex, and inferior frontal gyrus, Tijms said. Low connectivity values throughout the brain heightened the risk of progression threefold, while deficits in those particular regions accounted for a 2.5-fold increase. “Gray-matter connectivity might be useful to predict time to AD onset,” Tijms concluded.
Other researchers made the case that specific functional brain networks may have prognostic value. Joey Annette Contreras of Indiana University, Indianapolis, correlated functional connectivity changes, as seen with resting-state fMRI, with subjective cognitive complaints in a sample from the Indiana Memory and Aging Study. Participants included 13 cognitively normal people, 16 with subjective complaints, 22 with mild cognitive impairment, and eight with Alzheimer’s disease. The more self-complaints a person reported, as measured by the Cognitive Complaint Index, the lower their connectivity in several resting-state networks, including the DMN, visual, limbic, and frontoparietal networks (see Yeo et al., 2011). In addition, the more subjective complaints a person made, this time as described by either a trusted informant or themselves, the lower the connectivity in their visual network, Contreras reported. Impaired visual memory could indicate preclinical AD, she suggested. Visual memory deficits associate with hippocampal defects and have been used to detect decline in familial AD cases (see Mar 2011 news).
Other characteristics of AD patients may point to affected networks, as well. About 80 percent of people with Alzheimer’s are unaware of their memory problems (see Lindau and Bjork, 2015). Anosognosia can also occur at the MCI stage, although data are mixed on its prevalence there (see Kalbe et al., 2005; Wilson et al., 2015).
Anosognosia at prodromal stages predicts progression to AD, Catherine Munro of Massachusetts General Hospital, Charlestown, asserted at AAIC. Among 25 people with amnestic MCI who remained stable over two years, those who performed more poorly on cognitive tests at baseline were aware of that. However, in 11 people with aMCI who progressed to AD, worse memory performance at baseline was associated with fewer memory complaints—an indication of anosognosia. Moreover, their awareness of their memory problems faded over the course of the study. In this small sample, having anosognosia ballooned the risk of progression fivefold, Munro reported.
Patrizia Vannini, also of Mass. General, investigated the brain networks underlying this phenomenon in 31 people with amnestic MCI and 31 matched controls. Anosognosia correlated with weak connectivity in parts of the DMN, particularly between the precuneus and orbitofrontal cortex, as well as the inferior parietal lobes, she reported. Weak connectivity between the precuneus and orbitofrontal cortex was particularly evident in people who progressed to AD at follow-up. Anosognosia reflects a disconnection of functional networks, Vannini concluded. In addition, people with anosognosia had hypometabolism in the posterior cingulate and right hippocampus, as Munro found, as well.
Eric Salmon of the University of Liège, Belgium, presented similar results. He correlated anosognosia in 31 AD patients with low connectivity in the memory subnetwork of the DMN. This involved the medial temporal lobe, ventral posterior cingulate cortex, posterior inferior parietal lobes, and the lateral temporal cortex. Anosognosia also went along with hypometabolism in the ventral posterior cingulate, in agreement with the Mass. General study, as well as with prior work (see Perrotin et al., 2015). This pattern makes sense, Salmon said, because portions of this memory subnetwork are responsible for a person’s ability to ponder his or her own mental state and recall past experiences (see Summerfield et al., 2009).
While various proposed connectivity measures may hold promise, validation in larger cohorts is needed to pick out good biomarkers, Vemuri noted, and Tijms agreed. “We need to identify the most robust approach to describe brain connectivity that can detect reliable differences between patients and controls across different studies and imaging modalities,” she wrote to Alzforum.—Madolyn Bowman Rogers
Tijms BM, ten Kate M, Wink AM, Visser PJ, Ecay M, Clerigue M, Estanga A, Garcia Sebastian M, Izagirre A, Villanua J, Martinez Lage P, van der Flier WM, Scheltens P, Sanz Arigita E, Barkhof F.
Gray matter network disruptions and amyloid beta in cognitively normal adults.
Neurobiol Aging. 2016 Jan;37:154-60. Epub 2015 Oct 22
PubMed.
Amyloid and Neurodegeneration Have Different Underlying Genetics
When pursuing an elusive beast, hunters look for the traces it leaves behind as clues to its whereabouts. Geneticists are employing a similar method to hunt variants linked to Alzheimer’s disease, with changes in the brain representing the variants’ traces. By correlating biomarker changes with genetic factors, researchers gain clues to the mechanism of action of these genes. The method can also bump rare genes, or genes with small effects, above the line of genome-wide significance in genome-wide association studies (GWAS). At the Alzheimer’s Association International Conference 2016, held July 22-28 in Toronto, several scientists described the use of PET and MRI data to identify genes involved in pathology.
A common theme emerged when various groups reported finding distinct sets of factors that influenced amyloidosis versus tau degeneration. The findings imply that these processes have different underlying causes, researchers noted. Other talks homed in on specific genes involved in atrophy, in some cases analyzing known AD genes for associations, and in others looking for novel genes. To many researchers, the data reinforce that to prevent the progression of AD it will be important to treat not only factors that affect amyloid, but also those that affect neurodegeneration.
Amyloid and Atrophy March to Different Drummers
Previous data have long identified a disconnect between amyloid and atrophy. The regions affected by each form distinct, though overlapping, patterns in the brain. In addition, many older people have brain atrophy without amyloid accumulation (see Sep 2015 conference news; Sep 2015 news; Aug 2016 conference news).
Prashanthi Vemuri of the Mayo Clinic in Rochester, Minnesota, wondered if amyloid and atrophy might involve distinct risk and protective factors. To test this idea, she analyzed data from Mayo Clinic Study of Aging participants aged 70-90. The cohort comprised 713 cognitively healthy controls, 148 people with mild cognitive impairment, and 12 with AD dementia. Vemuri looked for demographic and health factors that associated with either global amyloid load as measured by PiB PET scans, or thickness of the entorhinal cortex and inferior and middle temporal cortices as seen with structural MRI.
For amyloidosis, as expected, older age and the presence of an ApoE4 allele heightened risk. Being a man, or having ApoE2, protected against plaques. However, little else affected amyloid deposition. The only other significant association Vemuri found was that high levels of cholesterol and other lipids in the blood at midlife increased risk. In contrast, many factors contributed to atrophy. Lifestyle choices such as smoking associated with brain shrinkage, as did numerous chronic diseases of aging, such as hypertension and diabetes. Vemuri combined 19 medical issues, including cardiovascular disease, diabetes, metabolic syndrome, obesity, and mental illness, into a single Multiple Chronic Conditions (MCC) score. MCC scores associated with atrophy. Men had higher MCC scores than women on average, and also lost more brain volume with age. Curiously, education and cognitive activity did not protect against either amyloid accumulation or atrophy in this cohort.
The data argue that Alzheimer’s progression is more complex than simply amyloidosis driving tangles that in turn drive atrophy, Vemuri said. Instead, different factors affect each process. She tweaked the common AD analogy that amyloid acts as the gun and tau the bullet by saying that amyloid is the gun and degeneration the bullet. The speed of the bullet varies, Vemuri believes, based on risk factors that have nothing to do with amyloid. Other AAIC talks addressed how genetic variants may underpin some of these factors.
How do tau tangles fit in? Neurodegeneration has often been thought of as synonymous with tangles, but tau PET imaging data has now made clear that the brain can shrink without any tangles present (see Aug 2016 conference news). To specifically compare risk factors for amyloidosis, for tangles, and for atrophy, Vemuri analyzed a smaller cohort of 326 cognitively normal participants who had undergone tau imaging with the tracer AV1451. She found that amyloidosis was the main factor driving tau pathology, in agreement with recent imaging studies (see Jul 2016 news; Aug 2016 conference news). In turn, tangles drove some atrophy. However, here, too, Vemuri calculated that MCC scores affected neurodegeneration independently of amyloid or tau deposits. “Non-AD pathways contribute significantly to AD-pattern neurodegeneration,” she concluded. She is currently investigating what those pathways might be.
Others found the data plausible. “It is not terribly surprising that other mechanisms, and perhaps those related to chronic illnesses, can lead to atrophy independent of amyloid. This implies that, in addition to anti-amyloid therapies, we need to look beyond amyloid for mechanistic pathways that lead to atrophy or tau pathology when developing new treatments for AD,” Adam Fleisher at Eli Lilly, Indianapolis, wrote to Alzforum.
Distinct Gene Sets For Amyloid and Atrophy
Some clues as to what those distinct pathways might be came from Michel Grothe of the German Center for Neurodegenerative Diseases (DZNE), Rostock. First, Grothe and colleagues delineated patterns of brain amyloid and atrophy in a cohort of 75 AD patients compared to 126 controls, using florbetapir PET and structural MRI data from the Alzheimer's Disease Neuroimaging Initiative (ADNI). This analysis provided a graduated pattern of how amyloid and atrophy susceptibility varied across the entire brain, Grothe said. He then compared these patterns to gene expression data from the Allen Brain Institute’s Atlas of the Adult Human Brain Transcriptome (see Hawrylycz et al., 2012). This database contains gene-expression profiles from six middle-aged people who died without brain pathology, and is distinguished by dense sampling from hundreds of regions across each brain. The researchers looked for functional gene sets whose regional expression patterns coincided with the graded patterns of amyloid deposition and atrophy they had found in the DZNE cohort.
They found distinct gene-expression differences in areas vulnerable to amyloid versus atrophy. The amyloid-prone regions of the control brains expressed low levels of genes involved in protein synthesis and mitochondrial respiration, while atrophy-prone areas expressed high levels of genes responsible for axon and dendrite growth and responses to extracellular signaling. For the atrophy pattern, some of the strongest individual gene associations within the overexpressed gene sets were with the tau gene MAPT and the tau kinases CDK5 and MAPK1/ERK2, Grothe told Alzforum.
These data suggest testable hypotheses, Grothe said. For example, perhaps the high rate of aerobic glycolysis that has been described for amyloid-vulnerable regions explains why mitochondrial respiration genes are scarce there (see Sep 2010 news). A relative lack of protein synthesis genes might lead to more protein-folding errors and accumulation of misfolded debris in amyloid-prone regions. Meanwhile, the high expression of dendrite growth genes in atrophy-susceptible regions suggests those areas have high synaptic plasticity, Grothe said. The medial temporal lobe is one of the most plastic neuronal systems in the human brain and also has the highest susceptibility to tau pathology (see Walhovd et al., 2016). Its high capacity for continued synaptic reorganization may come with the downside of a higher susceptibility to tau hyperphosphorylation and associated neurodegeneration as the brain ages, Grothe suggested.
The data complement a recent paper from researchers at the University of Cambridge who also correlated gene expression data from the Allen Brain Atlas with regions vulnerable to AD (see Aug 2016 news). The U.K. researchers took a slightly different approach. They focused on those areas known to accumulate tangles as described in the Braak staging scheme, and specifically looked for expression changes in genes such as chaperones and proteases that regulate Aβ or tau accumulation. They found high expression of genes that promote Aβ or tau production and aggregation, and low expression of genes that prevent it, in the vulnerable regions. On an individual gene level, they also found high expression levels of the MAPT gene in tau-susceptible regions, agreeing with Grothe’s data. Together, these studies highlight that the accumulation of plaques and tangles in specific regions may be due to underlying susceptibilities in the local cells, and that the cellular mechanisms conveying this susceptibility may differ for amyloid versus tau accumulation.
Andre Altmann of University College London presented additional data emphasizing the different risk factors for amyloidosis versus atrophy. Altmann and colleagues calculated a genome-wide polygenic score (GPS) for AD risk based on data from the International Genomics of Alzheimer’s Disease database of more than 54,000 participants. The score was based on the presence and number of alleles at up to 150,000 genetic loci across the entire genome, excluding ApoE, and included a weighting factor that took into account how much each variant increased risk. The researchers then applied this score to the ADNI cohort, looking for associations with amyloid or atrophy changes over two years.
In 844 participants who had undergone florbetapir PET, the researchers found no link between their polygenetic risk score and global amyloid accumulation, but did find a strong effect from the ApoE4 allele. In 953 people with FDG PET data, on the other hand, a high polygenic risk score predicted a decline in brain metabolism, mostly in people who had mild cognitive impairment at baseline. The effect was most pronounced in certain brain regions, such as the left temporal pole, and strongest in people who did not carry an ApoE4 allele. The effect of the combined genes was similar in magnitude to that of ApoE4 alone on amyloidosis. Overall, the data highlight that distinct drivers exist for amyloid deposition and atrophy, Altmann said. ApoE4 mainly drives amyloidosis, while many genes affect atrophy, he concluded.
A previous study reported that polygenic risk scores associated with both faster memory decline and higher amyloid load in the ADNI cohort (see Jul 2016 news). Other studies have tied GWAS hits to APP processing (see Apr 2015 conference news; Nov 2015 conference news). It may be that a person’s amyloid changes over two years are not pronounced enough to show an association with their GPS in this study, Altmann suggested.
Genes and Brain Volume
Other talks zoomed in on specific genes that affect atrophy as measured by brain volume. Tugce Duran and Liana Apostolova of the Indiana University School of Medicine, Indianapolis, investigated the associations of 27 variants from the top 20 Alzheimer risk genes with atrophy in the medial temporal lobe. The researchers used ADNI data from 441 cognitively normal controls, 764 people with mild cognitive impairment, and 294 with AD. They found different associations in each group. Among controls, only SLC24A4/RIN3—a gene that plays a role in lipid metabolism and has been associated with hypertension in African-Americans—correlated with atrophy (see Jul 2013 news). In the MCI group, SLC24A4 and the zinc finger gene ZCWPW1 turned up as significant. In people with AD, however, a different set of genes popped up: ABCA7, EPHA1, and INPP5D. The first of these associates with lipid metabolism and amyloid, the latter two with inflammation. “The influence of genes may be confined to specific disease stages,” Duran said. This may be because biomarker changes are limited to certain disease stages, Apostolova noted. “For example, one would expect amyloid-related genes to have an early effect and neurodegeneration genes to show a late effect,” she wrote to Alzforum.
Jake Vogel of McGill University, Montreal, turned up a different genetic association with atrophy. Working with Sylvia Villeneuve at McGill, he examined the relationship between atrophy and 15 GWAS SNPs that were present in the PREVENT-AD cohort. This Canadian study tracks biomarker changes in cognitively normal middle-aged people at risk of late-onset AD due to family history (see Ritchie and Ritchie, 2012). Vogel examined data from 271 participants averaging 62 years old, about one-third of whom carried an ApoE4 allele. Participants lost volume in several brain regions, including subregions of the default mode network (DMN), as they approached the age at which their parents had developed AD. These regions dwindled most in those who carried a harmful BIN1 allele, hence BIN1 may play a role in the DMN’s vulnerability, Vogel concluded. Previous studies have suggested both that BIN1 may bind tau and that it increases Aβ production, leaving its potential mechanism of action in the DMN obscure (see Aug 2012 conference news; Apr 2015 conference news; Nov 2015 conference news).
Rather than working with known genes, Marco Lorenzi of University College London used a joint model of brain atrophy and SNPs to find new genes. Lorenzi and colleagues compared more than one million SNPs to cortical and subcortical thinning in a cohort of 639 ADNI participants comprising healthy controls and AD patients. He found two distinct sets of genes that associated with volume changes in different brain regions. Cortical thinning in the hippocampus, amygdala, temporal and cingulate cortices associated with SNPs near or within the ADAM23, NAT2, ADAMTSL1, ANK3, NAV2, and CALCOCO1 genes. Many of these genes are involved in cell growth, adhesion, and axon guidance. Meanwhile, shrinking volume in the parahippocampal gyrus and subiculum was associated with variants near ADCY9, APOC1, APOE, PVRL2, and TOMM40, many of which relate to amyloid accumulation and nervous system development. In an independent ADNI MCI group, several of these SNPs discriminated between people who remained stable and those who progressed to AD, Lorenzi said. The findings help link brain atrophy to biological functions and suggest directions for future research, he added.—Madolyn Bowman Rogers
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, Dalley RA, Daly BD, Dang C, Datta S, Dee N, Dolbeare TA, Faber V, Feng D, Fowler DR, Goldy J, Gregor BW, Haradon Z, Haynor DR, Hohmann JG, Horvath S, Howard RE, Jeromin A, Jochim JM, Kinnunen M, Lau C, Lazarz ET, Lee C, Lemon TA, Li L, Li Y, Morris JA, Overly CC, Parker PD, Parry SE, Reding M, Royall JJ, Schulkin J, Sequeira PA, Slaughterbeck CR, Smith SC, Sodt AJ, Sunkin SM, Swanson BE, Vawter MP, Williams D, Wohnoutka P, Zielke HR, Geschwind DH, Hof PR, Smith SM, Koch C, Grant SG, Jones AR.
An anatomically comprehensive atlas of the adult human brain transcriptome.
Nature. 2012 Sep 20;489(7416):391-9.
PubMed.
To Know or Not to Know: Trial Participants Confront the Question
In the era of secondary prevention trials for Alzheimer’s disease, knowledge is power. It also may be unavoidable. As large international study cohorts and clinical trials seek to enroll participants in the earliest stages, they must rely on biomarkers—such as amyloid accumulation in the brain or possession of the ApoE4 allele—rather than outward cognitive symptoms. This means that inviting someone to join such a trial equates to disclosing their AD risk status to them. At a session dedicated to this topic at the Alzheimer’s Association International Conference, held July 22-28 in Toronto, researchers grappled with developing ethical and efficient procedures to break this news to participants. They also presented emerging findings on the psychological, cognitive, and social impacts of doing so. So far, results indicate that most people use the information positively—they may exercise more, eat healthier, or make plans for their future care. Still, others may contemplate ending their lives.
“Understanding how people react to these test results is as important as understanding their response to a drug,” said Jason Karlawish of the University of Pennsylvania in Philadelphia. This still will be true once a disease-modifying therapy hits the market. “The future practice will be ‘get a test, get a drug,’ and we need to understand the combined impact of both of those interventions,” he said.
Investigators have a responsibility to take great care how, when, and to whom they disclose biomarker or genotype information, said Krista Tromp of Erasmus Medical Center in Rotterdam, the Netherlands. Tromp and colleagues are working to create ethical methods of disclosure in the European Prevention of Alzheimer’s Disease (EPAD) consortium. “On our path to a cure, we must make sure to protect the people who are helping us get there—the study participants,” Tromp said.
The AAIC session provided updates on disclosure issues discussed at last year’s meeting (see Aug 2015 conference news).
Previous studies have suggested that among AD research cohorts, a majority of people want to know their genetic and/or biomarker status, and that they suffer no major psychological damage after they are told (see Ott et al., 2016; Lim et al., 2016; Jul 2009 news). However, Tromp noted that these studies were largely conducted on people with a family history of AD, who were familiar with the disease and less surprised if they tested positive for AD biomarkers. With the massive recruitment of thousands of potential participants into AD prevention trials that is starting now and will only grow in the next few years, Tromp said researchers will need to consider how people who are less versed in the language of AD may react.
How might people respond differently to learning they carry an ApoE4 allele compared to testing positive for amyloid? While the evidence is not there yet, Tromp speculated that more people may be spurred to make positive lifestyle by learning they have amyloid accumulation than by learning they have an ApoE4 genotype. “Amyloid accumulation is an ongoing biological process—something is really happening. ApoE4 genotype is just a status,” she told Alzforum. “People might feel they can act more on a biological process.”
If disclosing AD risk status to cognitively normal people is ethically tricky, sharing biomarker information with people who have mild cognitive impairment has its own challenges. In a recent study, Jennifer Lingler of the University of Pittsburgh and colleagues developed a procedure for disclosing amyloid scan results to people with MCI along with their caregivers. They found that clear explanations of the complex data were key, and that research participants expressed a desire to see their scans and receive follow-up calls to answer emerging questions (see Lingler et al., 2016). Lingler said that upon learning of a positive amyloid scan, many participants may express relief that there is an underlying explanation for their symptoms. Compared with cognitively normal people, those with MCI may feel greater urgency to arrange for the future, and the scans might help them to do that, Lingler told Alzforum.
Generation GeneMatch
At AAIC, Jessica Langbaum of Banner Alzheimer’s Institute in Phoenix outlined the disclosure procedures underway in the Alzheimer’s Prevention Initiative’s (API) Generation Study. This trial aims to enroll approximately 1,300 people who carry two copies of the ApoE4 allele to test an Aβ vaccine and a BACE inhibitor, both by Novartis.
One source for participants in the Generation Study is GeneMatch, a program run by the Alzheimer’s Prevention Registry. People interested in participating in AD research sign up for GeneMatch online, and then use a mail-in cheek swab kit to get tested for their ApoE genotype. Since its launch in November of last year (see Dec 2015 news), more than 7,000 people have joined GeneMatch, of whom around 5 percent carry two copies of the ApoE4 allele, Langbaum said. Importantly, however, they will only learn their ApoE genotype if and when they are invited to join a clinical study and expressly agree to disclosure. While Generation is currently the only trial recruiting participants from GeneMatch, Langbaum said other trial sponsors are planning to tap this resource as well. Beyond AD, researchers planning gene-based trials in frontotemporal dementias are watching GeneMatch closely as it gathers early experience with this approach.
To avoid a situation where simply being invited to the trial signals de facto disclosure, the Generation Study has developed a statistical algorithm to invite a mix of ApoE4 homozygotes, heterozygotes, and non-carriers from GeneMatch to undergo prescreening for the trial. After pre-test counseling and a psychological assessment, willing and qualified participants are informed of their ApoE4 genotype. Those who carry two copies then receive an invitation to the treatment portion of the trial. Langbaum said the researchers developed this two-stage system to shield people from learning of their high-risk genotype unless they are also given the opportunity join the trial, and also to allow scientists the opportunity to prescreen people for signs of depression or anxiety that might make disclosure hazardous. The set-up also affords the researchers a chance to study the effects of disclosure not only on ApoE4 homozygotes, but also on heterozygotes and non-carriers.
As of now, people invited for prescreening in the Generation Study undergo pre-disclosure counseling and disclosure in one of three ways, depending on the resources at each study site. At sites that have a genetic counselor on staff, participants discuss one-on-one what a positive or negative test result could mean. The counselor emphasizes that there are no guarantees—a positive result does not mean a person will develop AD for sure, just as a negative one doesn’t rule out the disease. Participants then receive their genotype information. However, a majority of sites have no genetic counselor. At those centers, this same information is given on site, with a phone call from Penn Telegenetic Services, a genetic counseling service at the University of Pennsylvania that cut its teeth disclosing genetic information related to cancer risk.
A third option—remote video conferencing conducted on-site—is being tested as part of a trial called CONNECT 4 APOE. Headed by Angela Bradbury at UPenn, CONNECT 4 APOE will compare how satisfied participants are with phone versus video conferencing. This, too, came out of the more established field of genetic disclosure of cancer risk. The researchers hope to enroll 3,000 participants into CONNECT 4 APOE as an investigator-initiated sub-study conducted in parallel with the Generation Study. So far 12 sites are participating in CONNECT 4 APOE, with others coming on board. Regardless of their ApoE genotype, all participants in the Generation Study will be monitored for a year following disclosure to assess the psychological, lifestyle, and social impact of learning this information.
So far, 28 people have been invited to the Generation Study through GeneMatch, 14 of whom accepted and are in various stages of screening. To date, those who have learned their ApoE results have reacted well to receiving this information, Langbaum said at AAIC.
Making sure to get disclosure procedures right in the context of a trial is really just a practice run for the future, when a disease-modifying treatment becomes available, said Pierre Tariot of Banner, who co-leads the Generation Study. “If a treatment works, implications are that every adult in the world will want to know their genotype, and we need to know how to do that,” Tariot said.
How is A4 Handling the Hot Potato?
The A4 trial, which aims to enroll 1,000 people with positive amyloid scans, is studying a face-to-face disclosure method developed by Karlawish and colleagues (see Harkins et al., 2015). In that trial, participants meet with clinicians prior to the scan to learn what the results may mean and to undergo psychological screening. They then take home an extensive brochure describing the basic science of amyloid and AD. After signing a consent form, they return to receive the scan and ultimately, its results.
At AAIC, Karlawish updated researchers on the Study of Knowledge and Reactions to Amyloid Testing (SOKRATES)-I, which is embedded within A4. The researchers are conducting two detailed interviews following the disclosure of scan results—one after four to six weeks, another after 12 months—on 50 participants with elevated amyloid and 30 people without elevated amyloid. So far, the researchers have finished both interviews for people with elevated amyloid, and the initial one for people with normal amyloid. The researchers asked participants to describe how the test results affected their lives, including how they changed their perception of how much time they have left, their own cognition, and their social and work relationships.
“Essentially, the use of biomarkers such as amyloid imaging has created a new disease state—one based on underlying pathophysiology rather than clinical symptoms,” Karlawish said. How people respond to existing in that new state, or being excluded from it, is the question studies such as SOKRATES-I seek to answer. Karlawish added that some people’s lives are so strongly based on their perception of their own cognitive strength that they may interpret emerging memory problems, or even just a positive amyloid scan, as threatening the core of their existence, despite their uncertain prognosis. For others, these early markers merely represent the beginning of a long journey with the disease.
Beyond SOKRATES-I, Karlawish and colleagues are conducting SOKRATES-II, which addresses similar questions for ApoE genotype disclosure in the Generation Study, and REVEAL-Scan, which will assess the cognitive effects of learning one’s amyloid status, much as the original REVEAL study had investigated ApoE genotype. Some researchers are concerned that learning of a positive amyloid status could affect a person’s cognition or perception of it, which in turn could alter trial results.
Disclosure in Countries Other than United States
For international studies, the disclosure field is further complicated by differing policies and cultures among participating countries. The European Prevention of Alzheimer’s Disease (EPAD) consortium is an example. Still in its recruitment phase, EPAD will serve as a source for participants in AD clinical trials, the earliest phases of which it will conduct itself.
The consortium recruits its participants from so-called “parent cohorts,” which are ongoing or completed regional and national studies on aged populations. Once invited by the investigator of their respective parent cohort to join the EPAD longitudinal cohort study (see Aug 2016 conference news), potential participants must agree to one day have their amyloid status disclosed to them. This sequence differs from the GeneMatch/Generation Study procedure, in which participants only agree to disclosure after they have been invited to a specific trial. After a potential EPAD participant has undergone prescreening and agreed to disclosure in the future, researchers measure CSF biomarkers, determine ApoE genotype, and administer cognitive tests. A subset of EPAD participants also undergo amyloid PET scans. With all this information in hand, EPAD trial sponsors can then invite specific participants who meet the risk profile they are looking for into the EPAD trial platform and randomize them to a specific trial arm. In essence, this means invitation to a trial will alert a potential participant of his or her risk status. That is why research participants in EPAD need to agree to learn their risk status at the start of their EPAD journey, Tromp said. Participants not invited to trials stay in the EPAD longitudinal cohort study tracking biomarkers and cognition; they can learn about their AD risk markers upon request.
“There will not be default disclosure of amyloid status just by entering the EPAD cohort; it only happens when a person is invited to a trial,” said Pieter-Jelle Visser of Maastricht University in the Netherlands, who coordinates recruitment of EPAD’s registry. “But if participants request this information, they have the right to know.”
While EPAD distributes educational materials on AD biomarkers to its participating centers across Europe, the precise methods for disclosure can vary between countries. Each has its own ethics review board. Tromp and colleagues are therefore also investigating the views on risk disclosure methods of ethics review boards. Tromp said that EPAD is working to honor these country-specific practices while maintaining as much consistency as possible in the disclosure process.
EPAD is also running the Approaches to Communication of Alzheimer’s Disease Risk study. ACAR comprises nine focus groups in Barcelona, Spain, and London. Researchers in each of these groups are assessing people’s attitudes about learning their risk for AD, either in terms of biomarker status, lifestyle factors, or genotype, as well as their expectations of the disclosure process. At AAIC, Tromp presented preliminary findings of the study, which is led by Richard Milne of the University of Cambridge in England. So far, the researchers have learned that most people do want to know their AD risk status, but that their willingness decreases when they learn about the uncertainty associated with current information. People also want advice on how to improve their odds of avoiding AD.
Drawing the Line
While researchers are striving to put disclosure schemes in place for the upcoming clinical trials, what about everyday patients who see their doctor with memory complaints? Do they also have the “right to know” their amyloid status? Not unless there is a bona fide medical reason, according to Brian Ott of Brown University in Providence, Rhode Island. Ott told Alzforum that in a survey he conducted on people in the Rhode Island Alzheimer Prevention Registry, 80 percent of participants indicated they would like to know their amyloid status and/or ApoE genotype (see Ott et al., 2016).
However, Ott pointed out that ordering these tests outside of a research setting—particularly for cognitively normal people—is a risky business. For one, the meaning of the results is not straightforward. People may live dementia-free for a decade or more after becoming amyloid-positive, and some ApoE4 carriers never develop AD. More worrisome, about 12 percent of people in Ott’s survey said they would contemplate ending their lives if given this information, despite the uncertain prognosis it would convey. Most of the people in Ott’s study wanted to learn their status for the purpose of participating in research. Attitudes about knowing one’s biomarker status may be different outside of research settings. But even in carefully managed prevention trials, occasionally people have had to be excluded because they expressed the possibility of suicidal thoughts if they were to learn of a positive amyloid scan, Karlawish told Alzforum.
John Morris of Washington University in St. Louis agreed that conducting amyloid PET scans or measuring CSF biomarkers in cognitively normal people in routine clinical settings is a ways off. “We just don’t know enough about what the results of biomarker tests portend, especially for people without memory problems,” he told Alzforum. Routine biomarker use could increase if results from the IDEAS study, which is measuring health and lifestyle outcomes of learning amyloid status in people who have clinical symptoms, indicate that gaining this knowledge leads to clinical benefits for recipients (see Apr 2016 news). This data would give health insurance providers a financial incentive to cover the scans for people with cognitive impairment and an otherwise uncertain diagnosis.
Karlawish added that presently, ordering a scan in the clinic in the absence of symptoms would be based on observational studies. This is shaky ground, he told Alzforum. Clinicians don’t do this now because they lack proof that amyloid scans predict disease, or that lifestyle interventions prevent disease. “Writing prescriptions based on observational data would be sloppy medicine, at best,” Karlawish said.
Of course, if an amyloid-targeted therapy is approved, clinicians will likely face an onslaught of people ready to head into the scanner.—Jessica Shugart

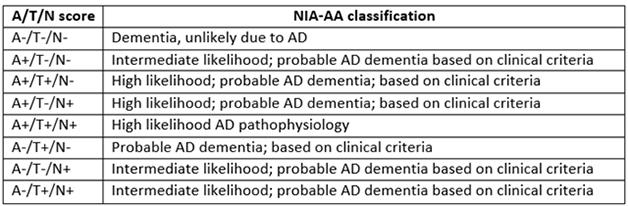






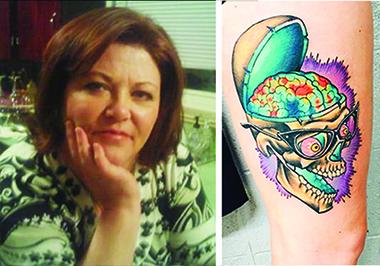





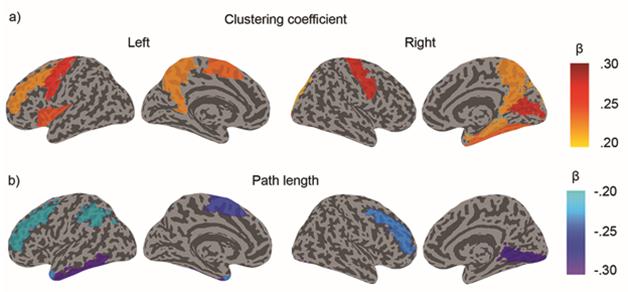

Comments
No Available Comments
Make a Comment
To make a comment you must login or register.