This year's Alzheimer's Association International Conference drew 4,000 attendees to Washington, D.C., where they soaked up everything from advances in human imaging and diagnostics to the latest clinical trial results. While the latter brought no surprises, scientists shared plenty of interesting preclinical data as well, among them a massive combination trial of two anti-Aβ therapies and research that shows that aerobic exercise preserves cognitive function even in people who are already impaired.
Massive Mouse Study Bolsters Rationale for Combination Therapy
Ron DeMattos and colleagues at Eli Lilly are challenging the field with extensive studies of combination therapies in mouse models of Alzheimer’s disease. At the Alzheimer's Association International Conference 2015, held July 18-23, DeMattos expanded a preclinical approach he had first described at AAIC in Copenhagen in 2014. To a packed room at the Walter E. Washington Convention Center in Washington, D.C., he reported the culmination of three years of work studying 700—yes, 700; that's no typo—mice given various combinations of Lilly's BACE inhibitor and anti-pyroglutamate Aβ antibody. "The take-away is that the work supports the rationale for combination therapy in the clinic," said DeMattos.
Pyrotechnics.
The anti pGluAβ antibody mE8 binds plaques in transgenic mice. [Image courtesy of Ronald DeMattos and Neuron.]
The wisdom of combination therapy has been hotly debated in recent years. Some, mostly in academia, argue that AD’s multiple pathologies necessitate a multipronged approach for a meaningful clinical benefit, and combination trials should start before the current batch of monotherapy trials read out (e.g., Apr 2015 conference news). Others counter that combination trials are a logistical nightmare and urge a go-slow approach (see Feb 2015 conference news; Mar 2014 conference news). Even starting combination trials of previously approved drugs is proving challenging (see Jun 2015 conference news).
Trying combinations of drugs in mice should be the first step, leaders in the field agree, but few have tried it. Among many possibilities for combination therapy, the Lilly researchers chose to focus on Aβ for now. They tested two drugs that could reduce amyloid deposits: the BACE inhibitor LY2811376 to turn off production of the peptide, and a humanized mouse monoclonal antibody (LY3002813) to the pyroglutamate form of N-terminal truncated Aβ to reduce existing plaques (see DeMattos et al., 2012). The pGluAβ antibody preferentially binds to dense-core plaques.
In Copenhagen last year, DeMattos reported that the combination of the two drugs worked better than either alone, knocking down plaques by 86 percent over four months when administered to PDAPP mice beginning when they were 18 months old. That study, in 180 mice, tested only one treatment duration and only one dose of each drug. In D.C., DeMattos shared data from two more recent studies, one that varied the doses of both drugs, and one that looked at the time course of combination therapy.
For the dose-response study, the researchers established 11 arms with around 28 PDAPP mice in each, covering a plethora of dose permutations. They included high (12.5 mg/Kg/week) or no antibody plus or minus high (30 mg/Kg/day), medium (3 mg/Kg/day), low (0.3 mg/Kg/day), or no BACE inhibitor. Medium- (4mg/K/week) and low-dose (1.5 mg/Kg/week) pGluAβ were combined with only the high dose of the BACE inhibitor. The study also had control arms for untreated and IgG2a-treated mice. The scientists treated the mice for four months beginning at 19 months of age, when plaques are well established in these animals. Total Aβ in guanidine HCl extracts measured by ELISA and immunohistochemistry served as end points.
For the high-dose combinations, the results matched those DeMattos reported last year. Antibody, BACE inhibitor, and the combination reduced total hippocampal Aβ by 30, 60, and 84 percent, respectively. For the BACE inhibitor, the dose response tightened when animals also received the antibody. That is, the 7 to 60 percent reductions in plaque load seen with increasing BACE inhibitor alone jumped to a 33 to 80 percent reduction in the presence of high-dose antibody as well.
Immunohistochemical analysis using the 3D6 anti-Aβ antibody paralleled the ELISA data. The high-dose BACE inhibitor strengthened the pGluAβ antibody dose response, such that 50, 63, and 84 percent of the Aβ was ablated by low, medium, and high doses of antibody, respectively. Low-dose antibody by itself removes about 30 percent of Aβ.
The longitudinal study was even larger. The researchers measured the effect of 4-, 8-, 12-, and 16-week treatments of high-dose BACE inhibitor and high-dose antibody either alone or in combination. Treatment began when the animals were 18 months old. Over the course of the 16 weeks, Aβ load as measured by ELISA slightly increased in controls, then reached a ceiling, which is very similar to what happens in AD patients, said DeMattos. In mice treated with the BACE inhibitor, the Aβ load decreased at four weeks, and continued to decrease slowly over the 16 weeks such that about half was removed. The antibody reduced Aβ by about 35 percent after four weeks and then it stabilized over the remaining 12 weeks. The important result, said DeMattos, was that on combination therapy, Aβ load fell faster over the 16 weeks than on either monotherapy alone, such that 80 percent was removed. The 16-week data in this longitudinal study lined up precisely with the four-month data from the previous two combination studies—all showing approximately 80 percent reduction in Aβ with the high doses of both drugs. "To repeat these experiments three times, over three years, with hundreds of animals, shows how robust this treatment and pharmacology are," said DeMattos.
What does might this mean for human trials? "Overall it fosters our confidence that combination therapy will result in more dramatic lowering of pre-existing plaque," said DeMattos. One upshot was that at medium and high doses of BACE inhibitor there was synergism with the antibody. "This tells us that when you have the two mechanisms engaged simultaneously, it promotes a feed-forward response resulting in more clearance of plaques," he said.
Others found this promising. "I have seen some of this data and find it very encouraging that combination therapies may have synergistic effects," wrote Reisa Sperling, Brigham and Women's Hospital, Boston. "I hope that other companies begin more animal studies and early safety work on combinations, including across mechanisms, such as anti-Aβ and anti-Tau." Other scientists noted that adding an antibody directed against plaques may allow researchers to go easy on the dose of the BACE inhibitor. Some BACE inhibitors on their own are able to reduce CSF Aβ levels down to about 80 percent, but because of BACE’s numerous reported substrates, some basic scientists have raised safety questions about doing so (see Nov 2014 news; Dec 2013 conference news).
DeMattos would not speculate on future mouse studies. He told Alzforum there is a lot more work to do to analyze the mice from the longitudinal study. The researchers have only processed the ELISA data, and still have to look at the histology, a large task with so many mouse brains to analyze.
Already the researchers know that plaque dynamics are complex. For example, plaques begin to dissolve with just the BACE inhibitor alone. DeMattos suggested that could be due to two ongoing processes, which are not mutually exclusive. First, as the inhibitor stops production of new Aβ, it may relieve overwhelmed clearance machinery, which can then work to remove existing Aβ deposits. Second, if free and plaque-bound Aβ are in equilibrium, then as the concentration of soluble Aβ falls in the brain, more may leech from the plaques. Add an antibody that reacts with a form of Aβ that mostly ends up in plaque cores, and the scenario becomes even more complicated. DeMattos said that it will be crucial to fully understand these dynamics. The longitudinal histology data will help with that, because it may reveal what happens to diffuse and dense-core plaques over time.
DeMattos said that besides providing proof of principle for combination therapy, the mouse study could inform the design of a human trial. The dose-response study will help clinicians establish rational choices for drug doses in humans, while data from the longitudinal study may predict when biomarker signals will change. "We will have an informed idea of when to do an amyloid PET scan, for example" said DeMattos. He cautioned, however, that all this will be contingent on what aspects of the animal model will hold true in people. Human plaques tend to be less soluble and the Aβ more truncated and modified. Paul Aisen, from the new Alzheimer's Therapeutic Research Institute, at University of Southern California, San Diego, co-chaired the session. He noted that it may be just as important to remove diffuse and dense-core plaques. "We have to take our insight and try to predict what will translate," said DeMattos.—Tom Fagan
TREM2 mutations raise one’s risk for Alzheimer’s disease—but which variants cause problems, and how? Researchers wrestling with these questions presented their latest results at the Alzheimer’s Association International Conference 2015, held July 18-23 in Washington, D.C. Many have been hampered by the extreme rarity of TREM2 variants, which meant they could only collect a handful of subjects for their work. Scientists have yet to come to agreement on whether TREM2 mutations can cause neurodegenerative conditions beyond Alzheimer’s, and how the gene contributes to neuropathology.
Microglia make TREM2 (red), which can be detected on the cell surface (green). [Courtesy of Konstantin Glebov and Patrick Wunderlich, University of Bonn.]
Mutations in TREM2 were discovered in people with frontotemporal lobar degeneration or Alzheimer’s disease in 2012 (see Oct 2012 news; Nov 2012 news). The gene has also been linked to ALS, leading scientists to wonder if it might be a general neurodegeneration risk factor (Feb 2014 news). However, the full picture of TREM2 and disease risk remains uncertain, in part due to small studies with conflicting results. According to a recent meta-analysis by Lars Bertram and Christina Lill of the University of Lübeck in Germany, published online April 30 in Alzheimer’s & Dementia, an arginine-47-histidine (R47H) substitution in the gene enlarges a person’s risk of Alzheimer’s by a factor of 2.71. The authors’ dataset included 24,086 AD cases, plus 2,673 people with FTLD, 8,311 with Parkinson’s, and 5,544 with ALS. They were unable to confirm a significant link between the R47H variant and FTD, ALS, or PD, though it remains possible that other TREM2 mutations contribute to those diseases.
Indeed, researchers have not completely catalogued the variety possible in TREM2 sequences, nor which variants are pathogenic. At AAIC, Alfredo Ramirez of the University of Bonn, Germany, reported preliminary data on a new variant likely involved in Alzheimer’s. Though he would not reveal the specific mutation, he told Alzforum it occurred in the extracellular portion of the protein. Ramirez and colleagues discovered the variant in two members of a Portuguese family diagnosed with an inherited form of AD, who had no APP or PS mutations and were ApoE4-negative. They could not detect the TREM2 variant in an unaffected relative or in 1,400 people with sporadic AD. Two other family members share the variant. One, who is 61, shows no signs of dementia, while the other, who is 50 years old, has had mild cognitive problems for 10 years.
Jochen Walter, also at the University of Bonn, investigated the function of the new variant. It ran faster on polyacrylamide gels, suggesting it was structurally different from the wild-type protein, but Ramirez said he detected no post-translational modification and he suspects the protein folds differently. They also examined shedding of the extracellular portion of TREM2, which occurs when microglia are active. When they expressed TREM2 constructs in HEK293 cells, the variant shed twice as much extracellular domain into the culture medium as did wild-type TREM2. In support of this, the researchers detected less of the variant on the cell surface.
TREM2 activation leads to internal cellular signaling that remodels the actin cytoskeleton and causes cells to round up, reducing their surface area. Since scientists have yet to find a ligand for TREM2, Konstantin Glebov and Patrick Wunderlich in Walter’s lab mimicked one by adding an anti-Myc antibody to COS cells expressing TREM2 with an N-terminal Myc tag. Surface area of cells expressing normal TREM2 modified in this way shrank on addition of the antibody, while it had no effect on cells producing the variant TREM2 with the N-terminal Myc tag. This ties in with heightened shedding of the variant, said Ramirez.
Amanda Heslegrave of University College London reported on TREM2 in the CSF. The receptor’s extracellular domain, shed from the microglia membrane after cleavage by γ-secretase, winds up in the spinal fluid. Scientists do not fully understand why TREM2 shedding occurs, said study co-author Henrik Zetterberg of the University of Gothenburg in Sweden. Might TREM2 in the CSF indicate something about the disease processes, or even serve as a biomarker?
Heslegrave and colleagues analyzed TREM2, Aβ, and tau in CSF from 34 people with AD who had a normal TREM2 genotype, and 20 cognitively normal controls. They struggled to obtain reproducible results from antibody-based assays for TREM2, so they switched to a mass-spectrometry method, quantifying a peptide unique to the TREM2 extracellular domain. They found the concentration of this peptide, VLVEVLADPLDHR, was higher in people with AD, and correlated with that of tau but not Aβ. Since CSF tau reflects injured neurons, the authors speculate that increased TREM2 shedding indicates that microglia are activated, yet ineffective at protecting the brain.
Zetterberg told Alzforum that TREM2 could be a potential biomarker for microglial activation, but probably would not be specific for AD. The researchers plan to examine TREM2 in longitudinal CSF samples to better understand how it changes with disease course, he said.
A separate study run by Christian Haass at Ludwig-Maximilians University in Munich used a TREM2 ELISA assay in more than 800 CSF samples from people who are cognitively normal, preclinical, prodromal or AD patients. This ongoing study also finds soluble TREM2 in CSF to be increased, particularly at the prodromal stage. The TREM2 elevation correlates with elevated CSF tau, indicating TREM2 could become a biomarker for an early inflammatory stage in AD pathogenesis.
In their paper, Lill and Bertram reported that TREM2 R47H carriers exhibited elevated CSF tau, corroborating the link between the two molecules.
Other researchers at the meeting investigated how known TREM2 variants affected Alzheimer’s onset and cognitive symptoms. Corinne Engelman of the University of Wisconsin School of Medicine and Public Health in Madison focused on R47H carriers. Engelman works with the longitudinal Wisconsin Registry for Alzheimer’s Prevention (WRAP) cohort. Most WRAP participants are too young to have developed dementia, but about two-thirds had a parent with the disease, and the data set includes clinical information from those participants. Among 1,200 of the cohort, Engelman discovered 10 carriers of R47H TREM2. Each had a family history of AD, though she has not checked yet to see if they themselves exhibited dementia symptoms. Their mothers' ages at disease onset averaged 66 years old—eight years younger than for affected mothers of non-carriers in this cohort. Without genetic data from the carriers’ parents, Engelman cannot be sure if any were also R47H carriers. Even so, her results are consistent with other work suggesting that TREM2 variants can speed up and worsen disease, Engelman said.
For fathers, however, onset occurred about six months earlier, which was not statistically significant. Engelman speculated that TREM2 variants may have a stronger effect in women, but she cautioned that her sample contained only four fathers, but seven mothers, meaning the difference may be due to chance. In six different tests of cognitive function, the 10 R47H carriers tended to perform slightly worse than non-carriers, but this difference was not statistically significant either.
Alexander Koppara, also at the University of Bonn, and Ramirez investigated the role of R47H and six other TREM2 variants in late-onset AD in the prospective AgeCoDe study (Luck et al., 2007). A 2014 paper had reported that in cases of early onset AD, R47H hastened the first symptoms by six years (Slattery et al., 2014), and one of the first studies to link TREM2 and AD indicated that R47H carriers fared worse, cognitively, than non-carriers in a cross-sectional cohort (Jonsson et al., 2013). Koppara found that among 10 carriers with dementia (eight female) and 270 matched non-carriers in AgeCoDe, TREM2 genotype made no difference to age of onset. They also examined Mini Mental State Examination (MMSE) scores and found that TREM2 mutation status did not affect the rate of decline.
In another poster, Klaus Fliessbach, also of the University of Bonn, reported a case study of a person with a threonine-96-lysine substitution in TREM2, which researchers suspected to be pathogenic. This person had cerebrospinal fluid (CSF) and PiB-PET imaging, and symptoms of the logopenic variant of primary progressive aphasia (lvPPA), which is characterized by difficultly retrieving words. Usually, people with lvPPA progress quickly, but this disease advanced slowly. Five years after the first symptoms, the patient was still able to live independently and scored 28 out of 30 on the MMSE. “The case is an example of an extremely benign clinical course in lvPPA,” the authors concluded. “We suggest that the investigation of genetics and other disease-modifying variables underlying the clinical variability in lvPPA progression is an important target for future research.”
The overall picture of TREM2 and AD remains murky for the time being, researchers told Alzforum. “The genetic findings are still too recent for us to have a firm and established idea of what TREM2 is doing,” Bertram said. “Clearly the immune system is involved, but it is not clear how.”—Amber Dance
Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J, Campbell T, Druyeh R, Mahoney CJ, Rohrer JD, Kenny J, Lowe J, Leung KK, Barnes J, Clegg SL, Blair M, Nicholas JM, Guerreiro RJ, Rowe JB, Ponto C, Zerr I, Kretzschmar H, Gambetti P, Crutch SJ, Warren JD, Rossor MN, Fox NC, Collinge J, Schott JM, Mead S.
R47H TREM2 variant increases risk of typical early-onset Alzheimer's disease but not of prion or frontotemporal dementia.
Alzheimers Dement. 2014 Nov;10(6):602-608.e4. Epub 2014 Aug 23
PubMed.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K.
Variant of TREM2 associated with the risk of Alzheimer's disease.
N Engl J Med. 2013 Jan 10;368(2):107-16. Epub 2012 Nov 14
PubMed.
New Data on Autosomal-Dominant Alzheimer’s Point to Early Fissures in the Brain’s Microarchitecture
The Alzheimer’s Association International Conference, held July 18 to 23 in Washington, D.C., showcased a growing interest in early brain changes beyond the established markers of amyloid, tau, and brain volume that anchor the diagrams of successive biomarker change in the pathogenesis of presymptomatic Alzheimer’s disease. What else changes in the brain? Could new measures fill in what happens in those long years between the first amyloid deposits and shrinkage? What other curves can scientists fit into the pathologic staging model of AD? “We want to detect changes earlier than volumetry does, before the neurons are gone,” said Natalie Ryan of University College London.
Something is Crumbling Early in White Matter
Conspicuously absent from the current staging diagram are white-matter hyperintensities (WMH). These bright areas on MRI scans have received plenty of research attention in recent years, but their role in Alzheimer’s is unclear. They are thought to represent damaged small vessels such as capillaries and arterioles, sometimes in connection with microbleeds, demyelination, or gliosis. Scientists typically think of them as a result of ischemic injury arising from a number of different underlying factors. There is debate about whether these lesions are truly a part of AD. To some, they are an independent comorbidity separate from Alzheimer’s core pathologies of plaques and tangles. To others, they predict the clinical onset and course of AD (e.g., Brickman et al., 2014; Tosto et al., 2015).
Researchers are exploring whether they can assess microdamage in gray matter by measuring how well water diffuses in brain areas vulnerable to Alzheimer’s disease. [Courtesy of Philip Weston and Natalie Ryan.]
Because age-related comorbidities make it difficult to study the emergence of WMH in late-onset Alzheimer’s disease (LOAD), Adam Brickman of Columbia University, New York, turned to the purer form of autosomal-dominant AD (ADAD). Mutation carriers do not have the hypertension, elevated cholesterol, or diabetes that heighten dementia risk by way of independently acquired cerebrovascular disease in old people. For this reason they enable scientists to dissect more cleanly which changes are genuinely part of the AD cascade itself.
At AAIC, Brickman reported an analysis of baseline scans from 299 DIAN participants, 184 of whom were mutation carriers. (One hundred of these people and their relatives traveled to Washington for a one-day workshop before the AAIC conference.) Seventy percent were still asymptomatic. The scans showed an inflection point six years before symptoms, when WMH became larger. What’s more, by using an estimated age at onset model for ADAD, Brickman was able to see that these lesions began to crop up in selected regions of the brain up to 22 years prior. In fact, WMH start to appear soon after amyloid deposits. Correlating the WMH with other biomarkers captured in the DIAN dataset suggests that amyloid deposits might cause them, not tau. “We observed a definitive relationship between WMH and amyloid markers of ADAD,” Brickman said.
“This greatly changes how we think about AD pathogenesis,” said Tammie Benzinger of Washington University, St. Louis. “We used to think about Alzheimer’s as a gray-matter disease, but there is something about these white-matter changes that is also a primary AD pathology. At least in ADAD, it is not a second hit.” When DIAN researchers first spotted WHM in this cohort in 2013, they realized that WMH were not due to independent diseases such as age-related hypertension; however, they still could have been a late step in the pathogenic process. Now, Brickman’s analysis suggests it is a primary step connected early on to amyloid.
The finding renews questions about amyloid-related imaging abnormalities, aka ARIA, in response to amyloid removal. The Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) will yield important information on that, Benzinger noted. If longitudinal observation shows that the WMH in the carriers progress over time, that might explain ARIA upon removal of amyloid in later stages of the pathogenic process. On the flip side, removing amyloid earlier, before the white-matter disease has progressed as much, might become possible without causing ARIA-H, Benzinger said. One issue to watch is whether the DIAN-TU trial of gantenerumab, an antibody that did cause ARIA in the SCarlet RoAD trial, causes less of it in these earlier-stage patients or not. Speaking in general terms, Bateman told DIAN families that the ongoing DIAN-TU trial thus far has had no safety concerns.
ARIAs are grouped into two types, E for edema and H for microhemorrhage. Microbleeds are a related manifestation of small-vessel disease, and they can be imaged with MRI, as well. Not only do microbleeds and white-matter hyperintensities tend to appear in the same DIAN participants, they also fall into a specific pattern depending on which presenilin 1 mutation a carrier has inherited, Nelly Joseph-Mathurin of Benzinger's lab reported at AAIC. Previous histological studies had suggested a curious dichotomy whereby people with PS1 mutations before codon 200 have worse parenchymal but milder vascular amyloid pathology, whereas people with mutations after codon 200 have milder parenchymal but worse vascular amyloid pathology. Would brain imaging bear out this genotype-phenotype relationship? Among 157 PS1 mutation carriers, it did. As a group, the carriers whose mutation was before codon 200 accumulated amyloid faster but were only one-third as likely to have microbleeds as the carriers of a PS1 mutation after codon 200, Joseph-Mathurin said.
Early Fissures in Gray Matter?
“Micro” was a recurrent theme in brain imaging at AAIC. Researchers are searching for ways to detect subtler changes that precede the wholesale contraction of the whole brain or the hippocampus as neurons die off in droves. For example, a pair of talks by University College London colleagues Phil Weston and Natalie Ryan showed how they are learning to capture finer presymptomatic changes by MRI. First, Weston adapted from LOAD to ADAD a method of detecting a specific regional signature of cortical thinning originally developed by Brad Dickerson at Massachusetts General Hospital. This work was not done within DIAN; rather, it drew on 43 mutation carriers and 42 non-carrying relatives from UCL’s longstanding observational cohort of ADAD families, some of whom are also in DIAN.
Weston divided the presymptomatic carriers into an early group of people in their 30s who were about a decade younger than their family’s expected age at onset and an older, late presymptomatic group of people in their 40s who were only three years away. Using longitudinal measurements of MRI and cognition, Weston found that a similar cortical signature as that in LOAD showed detectable thinning more than three years prior to symptom onset. Consisting of entorhinal, parietal, superior frontal, supramarginal cortex plus precuneus, this signature was able to distinguish presymptomatic carriers from healthy aging controls. Of these regions, the precuneus started to subtly thin out first, around nine years prior to onset. “Besides separating groups, this cortical signature was quite effective at identifying presymptomatic people who are likely to go on to develop symptoms,” Weston said.
Longitudinal imaging enables researchers to define a signature of cortical areas that begin to subtly thin out in vulnerable brain areas starting up to nine years before symptom onset. [Courtesy of Philip Weston and Natalie Ryan, UCL.]
Weston’s colleague Ryan picked up from there to see if the researchers could apply new MRI analyses to quantify tiny changes within the microarchitecture in the regions that would later thin out. Much like looking for hairline fissures that form inside a wall before the wall comes down, Ryan hoped to detect microchanges years earlier than outright macro-shrinkage of the width of cortex. To that end, she measured mean diffusivity in the same group of early presymptomatic and late presymptomatic mutation carriers. Mean diffusivity is measured with diffusion-weighted imaging, an advanced MRI technique that has been analyzed most widely as diffusion tensor imaging to capture changes in water diffusion in white matter. Rather than measure changes in the directed diffusion along fiber tracts, however, Weston and Ryan in this study analyzed the diffusion-weighted MRI data to quantify the average degree of diffusion in all directions in gray matter—basically measuring random Brownian motion as water molecules bump around within and between cells.
The London scientists acquired T1 and diffusion-weighted MRI of the ADAD cortical signature regions, and saw that mean diffusivity went up as people neared their expected age of symptom onset. This could be because cell and organelle membranes in those brain areas were breaking down as neurons started to degenerate, leaving fewer barriers to hinder diffusion. Intriguingly, mean diffusivity was somewhat lower in carriers at earlier presymptomatic stages than normal controls. Perhaps, Ryan speculated, this might be because prior to degeneration, glial proliferation and swelling of inflamed brain cells press them more closely together, hindering the diffusion of water. Weston and Ryan do not yet have amyloid scans in all those volunteers to see how the initial decrease and subsequent increase in water diffusion relates to the presence of amyloid deposits.
Ryan cautioned that this was a small, initial study mainly concerned with learning whether diffusion-weighted MRI might be a feasible way to capture presymptomatic processes in AD. Technical improvements, correlation with known biomarkers, and longitudinal observation in larger and independent samples still stand between this data and mean diffusivity becoming an established marker. An added caveat is that a number of different biological processes could underlie changes in diffusivity, as the measure itself is not tied to amyloid or tau pathology. That said, measuring microdamage might eventually offer an earlier marker of change than the atrophy curve that is part of the Alzheimer’s staging model.
What Does the Pathologist See?
Curiously, the suspected vascular micropathology that Brickman saw in the form of white-matter hyperintensities does not appear to develop into overt vascular damage of the sort that would show up in a postmortem exam. Also at AAIC, Nigel Cairns of Washington University, who runs the neuropathology core for both ADNI and DIAN, presented results from his direct comparison of 34 deceased AD patients in ADNI to 22 deceased relatives of DIAN families. Besides amyloid and tau pathology, LOAD and ADAD had in common that about half of the patients also had Lewy body pathology. But there were also clear differences. The amyloid and tau pathology were more intense in ADAD. Absent from these younger people’s brains were abundant comorbidities that marked the brains of people who had died of LOAD. TDP-43 proteinopathy, the tauopathy argyrophilic grain disease, hippocampal sclerosis, and cerebrovascular disease with infarcts were present in LOAD but absent in ADAD.
“From a pathological standpoint, ADAD is a purer form of the disease,” Cairns said. Cerebral amyloid angiopathy—where amyloid deposits lodge in blood vessel walls—is variable in a similar way between ADAD and LOAD, Cairns noted, with both types of AD having very severe and very mild cases of accompanying CAA. Microhemorrhages, too, are similar in ADAD and LOAD in that their number is comparable in symptomatic people of both types of AD. In that sense, both CAA and microhemorrhages appear not to be caused by independent diseases but to be an integral part of AD itself. —Gabrielle Strobel
Aducanumab, Solanezumab, Gantenerumab Data Lift Crenezumab, As Well
“Failures, repeated failures, are finger posts on the road to achievement. One fails forward toward success.” C.S. Lewis.
Flashed onto the projection screen by Roche’s Robert Lasser, the Irish novelist’s quote symbolized the “chin-up!” attitude that prevails among developers of anti-amyloid immunotherapy these days. The most anticipated news at this year’s Alzheimer’s Association International Conference were clinical trial updates on four antibodies that tackle various versions of the Aβ peptide—monomer, aggregated forms, or both. As it turned out, none of the data was momentous in its own right. By itself, each antibody’s performance was mixed, “meh,” or negative, respectively. Still, taken together, they fed a confidence across AAIC that Aβ immunotherapy may become a treatment, albeit one at only the very early stages.
Chief among the concerns is the side effect called amyloid-related imaging abnormality, ARIA. Clinicians are gaining more experience with it across the different antibodies currently in clinical trials. As they do, their attitude is evolving from tossing out an otherwise promising investigational therapeutic because of ARIA toward wanting to study and minimize it. “The problem is ARIA,” commented David Holtzman of Washington University, St. Louis, “There are challenges ahead managing this side effect, but I am optimistic that it looks like a solvable problem in the long run.” Philip Scheltens of VU Medical Center in Amsterdam said, “We need to be less risk-averse in Alzheimer’s disease. We should carefully dose up until side effects tell us to hold off. In cancer, we tell the patient: 'You will get nauseous from these drugs.'”
Taken on their own, none of the antibodies wowed the AAIC audience with new data. Aducanumab’s 6-milligram dose did not fit perfectly on all endpoints into the empty space waiting for it between the 3 mg and the 10 mg dose the way the audience—especially investors—had expected. Gantenerumab was flat-out negative on all endpoints in the SCarlet RoAD trial in prodromal Alzheimer’s disease, save for some blips in biomarker and subgroup analysis. Solanezumab came across as straining to prove disease modification for a small effect with what some called a geeky new statistical approach. Yet somehow, in the aggregate, the signs in the tea leaves, paired with consensus that the field’s measurement techniques are slowly improving, still added up to a positive vibe. By the end of AAIC, Roche had announced that it was moving into Phase 3 with gantenerumab and also crenezumab, the AC Immune antibody it is developing with Genentech (see Jul 2014 conference news). Aducanumab has begun enrolling for Phase 3, and solanezumab will finish dosing in its third Phase 3 trial next October.
Aducanumab: Just Shy of Sky-High Expectations Jeff Sevigny of Biogen showed new results from the Phase 1b trial of aducanumab, aka BIIB037, in people with prodromal AD. This presentation wrapped up the double-blinded portion of this study, and follows Sevigny’s presentation at the AD/PD meeting in Nice, France (see March 2015 conference news), of a 6 mg/kg dose and its placebo arm. Biogen had added this arm to the study when it became apparent that the 10 mg/kg dose was causing ARIA in a large proportion of patients. At AD/PD, Sevigny had only been able to show six-month data for this dose.
By one year, this dose had achieved a statistically significant reduction of brain amyloid as per florbetapir PET. This was widely seen at AAIC as the trial’s strongest, uncontested success. In exploratory analyses, the antibody also appeared to slow clinical progression as measured by the CDR-SB. The point value of this effect tracked with the dose-response relationship reported at AD/PD for the 1, 3 and 10 mg/kg doses. Specifically, Sevigny showed that the placebo group declined by 1.87 points, vis-à-vis 1.72 in the 1 mg/kg group, 1.37 in the 3 mg/kg group, 1.11 in the 6 mg/kg group, and 0.63 in the 10 mg/kg group. Except for the highest dose, each individual dose effect was not statistically significant on its own, but the overall dose-dependence of aducanumab’s effect on CDR-SB was significant in a test called linear trend of dose response, Sevigny said.
The blemish was the MMSE. There, the absolute value of the 6 mg dose (worsening by 1.96) did not fall between the 3 and 10 mg/kg doses but hovered nearer the 1 mg/kg dose. The point values were 2.81 reduction for placebo, 2.18 in the 1 mg/kg group, 0.7 in the 3 mg/kg group, and 0.56 in the 10 mg/kg group. The 3 and 10 mg/kg doses were close together and statistically significant, the 6 and 1 mg/kg doses were not significant. As with the CDR-SB, the linear test of dose response for overall dose dependence was significant, Sevigny reported.
The 6 mg/kg dose results were closely anticipated because of the expectations raised after Biogen’s AD/PD presentation. In defense, Sevigny told Alzforum, “We are talking about point estimates on scales that are being deployed in a Phase 1b study powered for PET imaging. As a drug developer and clinician, I find these results fantastic. Keep in mind what we can expect to see in this type of study with 30 people per active arm.”
The immediate stories by analysts and in the financial press were contradictory, and on July 24, when Biogen adjusted 2015 sales projections for its multiple sclerosis drug Tecfidera downward, the stock dropped. Alzheimer’s scientists largely shrugged off the market for now and focused on the new results. “The aducanumab data look good,” said Colin Masters of the University of Melbourne in Australia. “The new data look consistent with what Biogen have already reported. The fact that they got a signal across the measurements is encouraging for the field,” Holtzman concurred.
ARIA-E, a poorly understood kind of edema, was common in this trial. It occurred more often at higher doses and in people who carry the ApoE4 allele. In carriers, the incidence of ARIA-E climbed from 5 percent in the 1 and 3 mg/kg groups to 43 percent in the 6 and 55 percent in the 10 mg/kg group. In non-carriers, it still was 9 percent in the 3 mg/kg, 22 percent in the 6 mg/kg, and 17 percent in the 10 mg/kg group. A fraction of those with ARIA-E discontinued treatment; 56 percent continued, some at a lower dose. Patients had no further ARIA-Es after the first instance, Sevigny told Alzforum.
Of the ARIA-Es, 89 percent developed early in the course of treatment. “Since the AD/PD meeting, we have only had one additional case. Something happens biologically in a significant fraction of patients; if it does not happen early then it tends not to happen later, ” Sevigny said. A third of ARIA-E cases were symptomatic, and resolved, the remainder were only detected on the monitoring MRIs.
Other scientists were cautiously optimistic about what this might mean for the future. Several clinicians noted that it will be important for aducanumab’s future to learn how these patients fare in the long term. “The hope is we can learn to manage it. It depends on how severe the ARIA is clinically,” said David Knopman of the Mayo Clinic in Rochester, Minnesota.
Besides ARIA, headaches were common. Sevigny said they were unremarkable and resolved quickly. A few people developed antibodies against aducanumab, but these antibodies neither caused clinical symptoms nor reduced aducanumab blood levels, he told Alzforum.
Biogen has started two Phase 3 studies in a total of 2,700 mildly symptomatic patients in the United States and abroad. Both enroll ApoE4 non-carriers and carriers, the latter on a lower dose. Sevigny would not disclose the doses in Phase 3. About ARIA, he said that his company is focusing research on this phenomenon. “We are committed to being world experts in ARIA. It’s not like we say, ‘There is ARIA and we have to live with it.’ We are working on improving it,” Sevigny said.
Prior trials of prodromal AD used the free and cued selective reminding test, but struggled with high screen failure rates. Sevigny said patients would have to have measureable evidence of a cognitive deficit, but declined to say which memory test or exact screening procedure Biogen is using to speed up enrollment.
On a poster at AAIC, Yaming Hang of Biogen showed some pharmacokinetic characteristics of aducanumab. The higher a person’s blood aducanumab level, the stronger their amyloid removal in different brain regions of interest, regardless of ApoE genotype, Hang said. The distribution of the antibody in the body and its clearance depends on a person’s weight, and cumulative exposure in people who received multiple injections is linear, according to the poster. In pharmacology-speak, this implies to scientists that the antibody is “well-behaved,” Hang said.
Gantenerumab: Flatline Trial Shows Little Blips of Life
At AAIC, scientists picked up the pieces from gantenerumab’s setback last December, when a futility analysis halted the prodromal AD trial called SCarlet RoAD because the data thus far made it seem unlikely that the trial would show a treatment effect (see Dec 2014 news). Dosing stopped, but patients kept coming for assessments, and last month at AAIC, Lasser and Scheltens presented the trial’s clinical and biomarker results.
Lasser’s disappointment came through when he noted that a trial of this size engages thousands of people in dozens of countries—physicians, site staff, and patients and their partners. For SCarlet RoAD, 3,089 prospective participants were screened to enroll 799 people with a defined deficit on the FCSRT memory test and CSF evidence of Aβ deposition. They got monthly shots of either 105 or 225 mg of gantenerumab under the skin; ApoE4 carriers received the lower dose.
Lasser reported that, as with aducanumab, the main side effect of gantenerumab was ARIA. In most instances it caused no clinical symptoms, i.e., was evident only on the mandated safety MRIs. The incidence of ARIA-E and H ranged from 0.4 to 14 percent, rising both with gantenerumab dose and with the number of ApoE 4 alleles a patient carried.
Placebo and both gantenerumab dose groups were no different on the CDR-SB, MMSE , ADAS-Cog13, and FAQ outcome measures. “There was no efficacy,” Lasser and Scheltens both said explicitly. The researchers dug into the data to analyze subgroups in order to learn from the data, not to finesse a positive result from a negative trial, Scheltens emphasized to reporters in a media briefing. That analysis hinted that the patients whose disease progressed the fastest may have responded, though that made no difference in the overall outcome. Different progression rates from person to person, and the field’s inability to predict with any precision how quickly a given person will progress, are longstanding problems in Alzheimer’s disease trials. In this instance, the fast progressors—i.e., those whose hippocampal volume and CDR-SB performance declined the most over the duration of the trial—appeared to benefit, especially those whose serum levels of gantenerumab were high. “In fast progressors, we detected a concentration-dependent treatment effect on ADASCog and MMSE,” Lasser said. “This is a post hoc analysis, however.”
Looking at biomarkers, Scheltens spotted signs of biological activity in the trial’s wreckage. At AAIC, he reported that a florbetapir PET substudy showed that brain amyloid load dipped by about 5 percent in the high-dose group. This dose further reduced CSF total tau by 2.94 percent compared to an increase of 3.11 percent in those on placebo, and reduced CSF phospho-tau by 7.52 percent compared to a 2.62 percent increase in placebo. The point values for the mild dose fell in between. “This is the first antibody to show consistent improvement in target and downstream biomarkers of neurodegeneration. It seems to change something in the cascade,” Scheltens said.
This amounts to a negative clinical result in the face of an effect on amyloid and tau pathology. The audience question shot out immediately: “Does this mean the amyloid hypothesis is disproven? You have target engagement but no clinical benefit.” True, Scheltens conceded, but he suggested instead that the dose of gantenerumab in this trial was simply too low to make a clinical difference.
In preclinical studies, gantenerumab has performed similarly to aducanumab. The high dose in the SCarlet RoAD trial roughly compares to the 3 mg/kg dose in the aducanumab trial. Those SCarlet RoAD participants with the highest serum gantenerumab levels showed the most amyloid removal, and an overall comparison of the dose and pharmacokinetic relationship of both antibodies suggests that gantenerumab exposure in this trial was at the bottom of the range where aducanumab started showing efficacy. “I think it is right to continue evaluating gantenerumab,” Scheltens told Alzforum. Knopman and Holtzman agreed that the dose may have been too low.
Matthew Frosch of Massachusetts General Hospital, Boston, recommended digging deeper into the data of the PET substudy to learn if brain areas with ARIA showed more amyloid clearance than others.
Upping the gantenerumab dose would likely mean more side effects. “ARIA thus far has been self-limiting, without any long-term consequences,” Lasser said. For now, participants in the SCarlet RoAD trial can opt to take the antibody in open label at a higher dose, and future high-dose trials are being discussed with the EMA and FDA. This news was not lost on attentive participants in the DIAN-TU trial of gantenerumab (Aug 2015 news), who wondered in their social media group whether their dose could be increased, as well.
Solanezumab: What Does a Smidgen of Disease Modification Mean?
With this most advanced antibody against monomeric Aβ, the news at AAIC was different. Solanezumab, an antibody against Aβ’s midregion, is currently in its third Phase 3 trial. Rather than sharing data from it, Paul Aisen, University of Southern California, San Diego, presented a new statistical analysis of 3½-year treatment data from an open-label extension of the first two Phase 3 trials of solanezumab, called Expedition 1 and 2. “This is the first time the delayed-start method has been formally implemented in an AD trial,” Aisen said.
This far out, he reported, it is clear that people who chose open-label solanezumab after having been on placebo for the first 18 months benefitted, but they never caught up to fellow participants who had been on solanezumab from the get-go, i.e., 18 months longer. Hence, the claim goes, the antibody lastingly modifies the underlying disease process rather than just patching up its symptoms for a while, and for this reason should be started as early as possible. In a searching discussion session, however, regulatory scientists gave this delayed-start approach a cold shoulder. While complimenting the statistics, they mostly emphasized that achieving a large treatment effect is far more important than trying to prove that a small effect is disease-modifying.
The Expedition 1 and 2 trials were negative. Even so, a secondary analysis of the pooled mild subgroups, done by Lilly and also by the Alzheimer Disease Cooperative Study, both spotted an efficacy signal the scientists calculated to reflect 34 percent slower progression (Siemers et al., 2015; Oct 2012 news). In the present study, Lilly scientists implemented a trial design for showing disease modification that was proposed by former FDA scientist Paul Leber, who directed the agency’s Division of Neuro-Pharmacological Drug Products from 1981 to 1999 (Leber, 1996).
Called “delayed start,” it makes use of the commonly practiced open-label extensions, whereby everyone in the trial is offered drug after the placebo-controlled phase is over. A delayed-start study then analyzes whether the decline trajectories of the two groups merge over time (indicating symptomatic treatment) or stay parallel (indicating disease modification). The idea is that because people on placebo lose more neurons during the first 18 months than the people on drug, switching to drug at 18 months will start to slow down their neuronal loss at that point, but they will never catch up to the people who started taking the disease-modifying from the beginning.
The trouble with this design has always been that it is statistically complex, especially because more and more people drop out when trials drag on for years and comorbidities and advancing AD overwhelm the aging participant’s ability to come to the clinic for monthly shots and assessments. It has been tried once before, in the ADAGIO trial of rasagiline for Parkinson’s. There it was only a partial success, either because this transmitter-based drug truly has mixed effects, or because of the statistical challenges inherent in this design (Rascol et al. 2011). Conceptually, antibody therapeutics should give an either-or answer, Lilly’s Eric Siemers told Alzforum.
Of the solanezumab Expedition 1 and 2 participants, 95 percent chose open-label drug. Both patients and study staff continued to stay blinded to what patients had been on previously. Lilly’s statistician Hong Liu-Seifert developed a new way of analyzing the long-term data. It tests whether the treatment difference at the end of the placebo phase persists going forward or, in statistics lingo, stays “non-inferior.” At AAIC, Aisen presented data on up to two years of delayed-start treatment. In essence, the difference persisted on the ADAS-cog and the ADCS-ADL scales for a year. Over the course of the second year, as more people dropped out, statistical significance was gradually lost, but to the naked eye the lines of decline of the two groups stayed parallel. In other words, the people who had been on drug since day one continued to do somewhat better. By 3½ years, about half the people had dropped out, about equally in both the early and delayed-start groups.
“I was surprised that after two years of open-label we still see those hypothetical curves,” Siemers told Alzforum. He added that this additional data makes it more likely that solanezumab’s effect is real, not due to chance.
During an AAIC panel discussion of the delayed-start results, however, government regulators politely poured cold water on the effort to show disease modification for the Expedition 1 and 2 trials. Aisen moderated the discussion. Lisa LaVange directs biostatistics at the Food and Drug Administration. She said that while it would be wonderful to get disease modification, the treatment difference at the beginning of the delayed-start phase needs to be large enough to withstand the statistical complexity and the missing data problem of this type of trial. In essence, sponsors should show first that the difference is meaningful, and only then focus on showing that it is durable, LaVange said. Aisen noted that with disease modification, the clinical effect tends to grow over time, and asked if that lowers the bar for what is clinically meaningful at 18 months. “I don’t know. It makes no sense to me to show you are not inferior to something that is not meaningful to begin with,” Lavange said.
Rusty Katz, formerly of the FDA, acknowledged that Seifert’s approach addressed some of the statistical challenges that had bedeviled the ADAGIO trial, but he reminded the audience that Expedition 1 and 2 were negative trials. “We live and die by primary analysis. All subsequent analyses are suspect, and in this situation it is hard to know what the p values mean,” Katz said. Nick Kozauer of the FDA agreed that while disease modification is conceptually important, it should not be conflated with a big effect, “What’s really important is a meaningful effect,” Kozauer said.
Aisen next asked whether disease modification is important for drug approval. Billy Dunn of the FDA gave a clear answer. “Absolutely not. We work hard to dissuade sponsors from thinking that demonstrating disease modification in a modest fashion assumes some intrinsic importance over the pursuit of a dramatically large effect.” Dunn went on to explain that disease modification is intrinsically linked to the persistence of effect in the absence of drug. He agreed that the construct of disease modification is laudable. “But we should not take our eyes off the prize: a large treatment effect. I won’t comment on this particular data, but we are not dealing with a monster effect,” Dunn said.
In conversation, other clinicians agreed that they would prefer to see clinical meaningfulness on the primary outcome, get the drug to patients, and then figure out exactly how it works. But some biostatisticians greeted the delayed-start data more warmly as a sign that clinical trials methodology is improving. Take Suzanne Hendrix of Pentara Corporation, Salt Lake City. “I am excited that we can analyze this now. The data is clean, there is so little noise that we can see this small effect for 36 months,” Hendrix said. Being able to detect small effects means the field is not missing something that could be made to be meaningful with more work. Sometimes it is possible to build on a small effect by improving formulation, dosing, time of intervention, and especially by combining drugs (see Part 1).
“In many prior AD studies, we have been overwhelmed with variability and noise. Just look at spaghetti plots—they are all over the map. Even means plots often zigzag. It’s good to see smooth curves with tight error bars. It means that we now know better what we are seeing,” Hendrix said.
The ongoing Expedition 3 trial of solanezumab will add the same delayed-start analysis once its randomized phase concludes in October 2016. So far, dropout is low, at 8 percent, Siemers said. That may be because the participants have mild disease, but also because they know they have brain amyloid and that solanezumab looks to be quite safe.
The antibody crenezumab had no data presented at AAIC, but on the last day, word went around that its sponsor, Genentech, had decided to evaluate it in prodromal to mild AD in Phase 3 (see press release). The news came after a long period of deliberation following crenezumab’s Phase 2 results presented at AAIC 2014 (Jul 2014 conference news). Despite differences in IgG subclass and binding characteristics, crenezumab is widely considered to be clinically similar to solanezumab (see May 2015 news). With that, all of these four anti-Aβ antibodies remain standing in the long slog toward a new treatment for Alzheimer’s disease. —Gabrielle Strobel
Could Alzheimer’s disease be tempered by just by working up a sweat? Speakers at the Alzheimer's Association International Conference 2015, held July 18-23 in Washington, D.C., presented new evidence that regular aerobic exercise can help people in prodromal disease stages maintain their cognition, while for those with full-blown dementia it relieves neuropsychiatric symptoms. Some studies provided hints that exercise can also hone thinking at the dementia stage, but only if the participants reach moderate intensity heart rates during their workout. Exactly how exercise helps the brain is still not known, but several talks reported better cerebral blood flow and improved structural and functional connectivity in exercisers, and even some signs that six months or more of physical activity can slow pathology (see Part 2).
Researchers agreed that the duration and intensity of an exercise intervention are crucial to determining its effects. For aerobic exercise in particular, the field is standardizing methods and narrowing in on the appropriate dose to prescribe. Some believe supervised exercise classes could become part of the standard of care for people with cognitive problems. “Exercise is going to be an important adjunct to pharmacological treatment for patients with dementia,” Kristian Steen Frederiksen at the Danish Dementia Research Centre, Copenhagen, predicted to Alzforum. Overall, interest in this area seems to be growing, as evidenced by 11 talks and 23 posters on the topic at AAIC.
Researchers have few doubts now that exercise protects normal older adults against brain decline. Epidemiological studies suggest regular physical activity wards off future dementia (see Jul 2011 news and AlzRisk analysis). Supporting this, the most athletically fit older adults experience fewer age-related brain changes, and boast better cognition, than less-fit peers (see Nov 2011 conference news). In several small trials, cognitively normal older people who walked or did other aerobic exercise preserved or even increased their volume of brain gray matter compared with sedentary participants (see Sep 2008 news; Oct 2010 news; Feb 2011 news). In Finland, cognitively normal participants in the FINGER study, which comprised multiple lifestyle interventions including exercise, also notched cognitive gains (see Jul 2014 conference news).
Strengthening the Case for Benefits in the Cognitively Impaired
Can exercise also sharpen cognition in people who are already on the downhill slide? Emerging evidence, including several presentations at AAIC, indicates it can. Laura Baker of Wake Forest University Health Sciences, Winston-Salem, North Carolina, previously reported that aerobic exercise boosted executive function in a small cohort of cognitively impaired older adults in the Piedmont Aging, Cognition, and Exercise (PACE) study (see Baker et al., 2010). In Washington, Baker extended these findings to the larger Phase 2 PACE-2 study of 65 older adults with amnestic mild cognitive impairment and high blood sugar. This population is particularly vulnerable to further decline, she noted. Six months of moderate-intensity aerobic exercise, four times per week, enhanced executive performance over baseline measures on several tests. This contrasts with the continued decline seen in the control group, which took non-aerobic stretching classes.
Cognitive impairment can have many causes. In some people, it results from vascular issues, rather than AD pathology. Would exercise help this group? Teresa Liu-Ambrose at the University of British Columbia, Vancouver, has tested this hypothesis. In D.C., she described the PROMOTE study of 60 older adults with clinical diagnoses of mild vascular cognitive impairment. Participants also had evidence of subcortical white matter lesions by MRI (see Erkinjuntti and Rockwell, 2003). The 32 who exercised aerobically three times per week for six months preserved global cognitive function, as measured by the ADAS-Cog11, compared with controls who took an educational seminar once per month. This gain correlated with a drop in blood pressure, suggesting the improvement was due to better vascular health. The exercise did not protect against decline in executive function, which occurs in people with vascular cognitive impairment. However, the trial was powered to detect differences on the ADAS-Cog, and may not have been large enough to reveal an executive effect, Liu-Ambrose told Alzforum. She is following up with a larger, 12-month study in this population.
Do these promising findings in cognitively impaired people extend to those with full-blown dementia? Here the data are more equivocal. Frederiksen and Kristine Hoffman, also at the Danish Dementia Research Centre, presented findings from the Phase 3 ADEX study, a collaboration between eight memory clinics and seven research units in Denmark. In this 16-week trial, 200 people with moderate Alzheimer’s either worked out aerobically three times per week or performed their usual activities. By the end, the exercisers had become less depressed, less anxious, and less irritable than controls. Overall, exercisers saw no cognitive benefit compared to controls, but a subgroup of people who succeeded in working out at the intended intensity of 70 percent or more of maximum heart rate performed better on the Symbol-Digit Modalities test, a measure of executive function. In addition, the more exercise sessions they attended, the greater their improvement, implying a dose effect.
It’s All About that Dose: Finding the Right Prescription
Researchers have struggled to figure out how exercise should be dosed, a prerequisite for it to become a standard therapeutic intervention. For aerobic exercise, at least, the field is closing in on this, Baker said. Two crucial factors are the length of the intervention and the intensity. Several studies indicate a minimum of six months to produce cognitive improvement, Baker noted. At three months, she sees trends in her data, but nothing that reaches significance.
For intensity, data pinpoint moderate exercise as the most efficacious. This means that participants raise their heart rates to between 60 and 80 percent of functional capacity. This is calculated using a formula that takes into account the exercise intensity, resting heart rate, and maximum heart rate. A general rule of thumb is that a person's maximum heart rate per minute is 220 minus their age. For a 60-year-old, the desired zone would be around 125-145 beats per minute. While it is not yet clear why this level of exertion works best for the brain, Stephanie Schultz at the Wisconsin Alzheimer's Institute, Madison, noted that moderate aerobic exercise associated more closely with increased brain glucose metabolism than did vigorous workouts in the UW Fitness, Aging, and the Brain study. Brain glucose use drops during AD, which is thought to reflect ongoing neurodegeneration.
Baker noted several other factors important for standardizing trials of physical activity. For one, researchers must start with sedentary participants in order to see big effects, since fit participants may not have much room for improvement. Baker also stressed the importance of using an appropriate control group to account for the social and cognitive stimulation of participating in exercise sessions. In her trials, the control group takes a stretching class so that members receive the same social interactions as aerobic exercisers. She also instructs trainers to gradually ramp up the intensity of aerobic exercise over six weeks, so that participants do not become discouraged or find the class aversive and drop out. In her study, 92 percent of participants stuck with the intended program. “If we did not have the compliance we do, we would not see these effects,” she said.
On top of this, researchers said that there is a need to agree on which cognitive tests to use. Trials use a plethora of measures, making it difficult to compare them. Some researchers are adopting the NIH Cognitive Toolbox, a standardized and validated cognitive battery that is freely available online, Liu-Ambrose noted.
How close are researchers to delivering a prescription for exercise? Baker is enrolling participants for an 18-month aerobic exercise trial that she hopes will do just that. The Phase 3 EXERT trial will take place at 15 sites around the country and will start this fall. It will use the same parameters and methodology as her previous six-month trial, but will include some additional computerized cognitive outcome measures developed for prevention trials, to facilitate comparisons with other interventions (see Jun 2014 news; Dec 2014 conference news).
As in the PACE-2 study, exercise classes will be held at YMCAs. Baker’s team is working with YMCA leaders to develop a standardized program for supervised exercise that could be implemented across the country. Cognitively impaired people have special needs and cannot simply join a regular exercise class, researchers agreed. If the results from this trial are positive, ideally the intervention would be covered by Medicare for seniors with cognitive issues, Baker said.
Will people actually take such an exercise class? “We all know exercise is good for us, yet as a country, we still don’t exercise,” Baker noted. However, her experience in the PACE trials indicates that cognitive benefits motivate people more strongly than physical ones. “If you provide people who have cognitive impairment with scientific evidence that physical activity might slow their decline, they start exercising immediately,” she told Alzforum.—Madolyn Bowman Rogers
Can Exercise Slow the Progression of Alzheimer’s Pathology?
Part 2 of two.
Evidence for the cognitive benefits of exercise keeps growing, but researchers are still not sure how it helps the brain. At the Alzheimer's Association International Conference 2015, held July 18-23 in Washington, D.C., several speakers presented imaging data that addressed this question. Overall, the findings indicated that working out enhances vascular brain health and connectivity, implying a direct benefit to brain structure and function. Data were mixed on whether exercise slows the progression of underlying Alzheimer’s pathology, however. One six-month study of moderate aerobic exercise reported a drop in cerebrospinal fluid tau in cognitively impaired people, but a shorter intervention failed to budge brain amyloid in people with AD. In general, speakers agreed that the cognitive boost from exercise likely comes from diverse benefits on several different aspects of brain function, something that would be hard to match pharmacologically. “There are multiple pathways for how it affects cognitive health, and that would be hard to package into a single pill,” Teresa Liu-Ambrose at the University of British Columbia, Vancouver, told Alzforum.
Blood Flow Boost.
In people who exercised aerobically for six months, cerebral blood flow increased in the regions most vulnerable to aging and AD (prefrontal cortex, red; posterior parietal, blue; cingulate, green). [Courtesy of Laura Baker.]
One of the biggest questions is whether exercise can slow neurodegenerative processes. Laura Baker of Wake Forest University Health Sciences, Winston-Salem, North Carolina, provided some of the first evidence for this in her talk on the PACE-2 study. This trial enrolled 65 older adults with amnestic mild cognitive impairment and high blood-sugar levels. Half of them took six months of supervised, moderate-intensity aerobic exercise classes, while the rest attended low-intensity stretching classes. Researchers collected cerebrospinal fluid biomarkers at baseline and study completion.
CSF profiles changed in exercisers. With age, levels of CSF tau and phosphorylated tau normally rise, perhaps reflecting neuron death. In trial participants over 70, CSF p-tau levels instead fell with exercise, suggesting better brain health in this vulnerable population. CSF Aβ42 also varied between exercise and control groups, although those data are harder to interpret. This marker rises during normal aging, but begins to drop once amyloid starts depositing in the brain. In controls over 70, the researchers saw Aβ42 climb over the course of the study, whereas it remained stable in exercisers. Baker suggested that these data might represent a progression of aging in controls, but not exercisers. She plans to investigate this further in future studies by adding amyloid imaging to estimate where participants might be on the trajectory of amyloid deposition.
The results contrast with findings from the Danish Phase 3 ADEX study of exercise and AD. This 16-week trial enrolled 200 people with moderate Alzheimer’s, half of whom exercised aerobically three times per week. Exercise did not seem to slow pathology in this study. A subgroup of 34 participants who volunteered for amyloid imaging continued to accumulate brain amyloid at the same rate as the control group, Kristian Steen Frederiksen at the Danish Dementia Research Centre, Copenhagen, reported in D.C. Preliminary analysis of CSF from another 37 participants likewise revealed no change in Aβ42, according to a poster presented by Camilla Steen Jensen, also at the Danish Dementia Research Centre. However, Frederiksen noted that 16 weeks may not have been long enough to effect measurable changes on biomarkers. He pointed out that animal work and observational studies both provide encouraging evidence for the potential of physical activity to move biomarkers. “There’s still a lot of hope that exercise, even at the dementia stage, might modify disease,” he told Alzforum. The researchers are still analyzing biomarker and imaging data from this trial, and will continue to follow the exercise cohort to measure long-term effects of the intervention.
The Head-Heart Connection: Better Brain Vascular Health in Exercisers
How else might exercise yield cognitive benefits? One of the most obvious effects of exercise on the brain is through cerebral blood flow. Supporting this idea, Baker and colleagues recorded enhanced blood flow in the brain in the PACE-2 exercise group, specifically in prefrontal regions, where perfusion normally drops with age, and in posterior parietal regions, where flow ebbs during Alzheimer’s disease (see image above). Other studies have reported improved glucose metabolism in those regions as a result of exercise as well, she noted. Brain glucose metabolism wanes during AD. “Moderate-intensity aerobic exercise can attenuate the effects of aging and AD on brain function,” Baker concluded.
Other data adds to this hypothesis, including a study conducted by Elizabeth Boots, Stephanie Schultz, and Ozioma Okonkwo at the Wisconsin Alzheimer's Institute in Madison. They have measured fitness, brain structure, and cognitive function in 106 cognitively healthy older adults with an average age of 64 as part of the UW Fitness, Aging, and the Brain study. The participants also take part in the Wisconsin Registry for Alzheimer’s Prevention, a longitudinal study, and many have a family history of Alzheimer’s. In D.C., Boots reported that those with the greatest cardiorespiratory capacity, as judged by peak oxygen consumption, had greater cerebral blood flow in the angular gyri and right temporal cortex, regions implicated in AD. The fittest participants also did better on several tests of executive function. Moreover, less-fit participants accumulated more white matter lesions at older ages, indicating more wear and tear on the brain. By contrast, while the youngest of the fit participants had the same number of lesions as their less-fit counterparts, the number did not go up with age in the fitter volunteers. Exercise might attenuate this aging-related decline in brain health, Schultz suggested. The researchers are now conducting a six-month randomized controlled trial of aerobic exercise in this at-risk population. “It seems likely that what’s good for your heart is good for your brain,” Schultz noted.
With Exercise, Brain Connections Snap, Crackle, and Pop
Imaging studies delineate other alterations in brain structure and function in exercisers. In D.C., Liu-Ambrose reported data from the PROMOTE study of 60 older adults with mild vascular cognitive impairment. A subset of 21 participants underwent functional MRI while performing a task that measured selective attention. Those who had exercised for six months reacted faster and with greater accuracy than the controls. They also activated fewer brain regions, suggesting more efficient cognitive performance.
Structural MRI of another 30 PROMOTE participants found an increase in white matter and a drop in gray matter in the 16 exercisers. It is unclear why gray matter volume fell, but other studies have reported a drop in gray matter paired with better cognition after various treatments (see Jul 2004 conference news; Nov 2012 conference news). The volume loss may reflect amyloid and associated fluid moving out of the brain, or dampened inflammation, Liu-Ambrose speculated. In an upcoming study, she will add further imaging measures such as amyloid PET and analyze blood biomarkers of inflammation to get a better idea of what is going on. She will also investigate whether the proliferation of white matter lesions stops in exercisers.
In a poster, Rodrigo Dennis Perea, now at Massachusetts General Hospital, Boston, focused on the brain’s structural connections. While at the University of Kansas Alzheimer’s Disease Center, Fairway, Perea had found that AD patients with high cardiovascular fitness preserved white matter integrity better than their less-fit peers (see Perea et al., 2015). The causal relationship was unclear, however. To see if exercise could improve this white-matter integrity, he scanned 30 AD patients with structural and diffusion tensor imaging (DTI) MRI. The latter traces white-matter tracts. He took images before and after half of them completed six months of aerobic exercise. Exercisers developed greater connectivity between the thalamus and right cingulate gyrus, and less connectivity between the thalamus and left post-central gyrus, compared with controls. The data indicate that exercise can modify brain structure even in people with dementia, Perea noted. The findings complement Liu-Ambrose’s data on increased white matter after exercise, and other small studies tying exercise to greater functional connectivity and more efficient use of the brain (see, e.g., Rajab et al., 2014; Wang et al., 2015).
Wait, There’s More—Genes and Growth Factors
Exercise may also help the brain by pumping up production of brain-derived neurotrophic factor (BDNF), a key compound for neurogenesis, learning, and memory. The relationship between exercise and BDNF was first found in animal studies, but has also been reported in people (see May 2002 news; Cotman and Berchtold, 2002). Moreover, BDNF levels are low in AD (see May 2009 news). While BDNF may mediate some benefits of exercise, data presented at AAIC by Carla Nascimento of the Federal University of São Carlos, Brazil, suggest it does not explain the whole picture. A cohort of 47 cognitively impaired older adults took part in either an exercise intervention or normal care for 16 weeks. Eleven of the exercisers and 10 of the control group carried the Val66Met polymorphism in BDNF, which results in less secretion of the growth factor. Only people without the Met polymorphism gained a boost in peripheral BDNF after the exercise intervention, but all exercisers did better on a test of executive function. In addition, plasma levels of two inflammatory factors, TNF-α and IL-6, dropped in exercisers regardless of BDNF genotype (see Nascimento et al., 2015). The data imply that exercise can sharpen cognition and calm inflammation through mechanisms other than BDNF.
Exercise may even modify genetic risk. A subset of 50 participants in the UW Fitness, Aging, and the Brain study donated DNA, which the researchers used to genotype the AD risk genes ApoE, clusterin, and ABCA7. All three genes are involved in cholesterol metabolism. A combined risk score from the three genes associated with lower CSFAβ42 and a higher total tau/Aβ42 ratio, indicating worse pathology, in participants with poor fitness. In their fitter peers, this genetic risk was nearly abolished, with fluid biomarkers approaching the levels seen in people with no risk genes. “We think of genetic risk as something you can’t change, but this shows there are things one can do to protect against brain changes,” Schultz told Alzforum.
Despite all these clues, exactly how exercise supports cognition remains hazy. “That’s the million-dollar question,” Baker said.—Madolyn Bowman Rogers
As more scientists delve into the possibilities offered by imaging the second major protein pathology of Alzheimer’s disease, aggregates of tau, they are getting a close-up, live view of how this pathology relates to neurodegeneration and cognitive decline. At the Alzheimer's Association International Conference 2015, held July 18 to 23 in Washington, D.C., tau imaging data bolstered the idea that tau sits tight in the medial temporal lobe until Aβ builds up, then it spreads, wreaking havoc on cognition. New imaging data suggests that once it breaks loose, tau spreads through functional networks and impairs brain metabolism. What’s more, unlike Aβ, which appears diffusely throughout the brain, tau deposits seem to map closely onto regions where atrophy occurs and cognitive deficits originate. “Unlike amyloid imaging, tau PET seems to strongly correlate with cognition and clinical states,” said Gil Rabinovici, University of California, San Francisco. Researchers were also excited by a new tau ligand that appears to have better specificity and kinetics than its predecessors.
Roche Forges Ahead with New Tracer
Scientists collaborating with F. Hoffmann-La Roche in Basel, Switzerland, introduced the first human data on an up-and-coming tau PET ligand, [18F]RO6958948. Dean Wong and Hiroto Kuwabara from Johns Hopkins University in Baltimore presented two posters detailing preliminary data from a Phase 1 trial of this ligand and two others, [11C]RO6924963 and [11C]RO6931643. So far they have scanned seven young, healthy controls aged 25 to 38, and seven people with mild to moderate Alzheimer’s disease aged 64 to 86. Each person was scanned twice, one to two weeks apart, with two different tracers, so that the researchers could directly compare ligands.
Roche’s Lead Tau Tracer:
Ro6958948 binds areas expected to contain tau in a patient with AD. [Courtesy of Roche.]
Ro6931643 and Ro6958948 cleared quickly from controls and clearly differentiated them from AD patients, said Wong. These two tracers bound to frontal, temporal, parietal, and occipital cortices, fusiform and hippocampal gyri, and the entorhinal area in patients (see image at right). This pattern resembles that seen in Braak stage V/VI (Braak and Braak, 1991). Controls had weak but visible uptake in the dorsal cerebellum, which has been reported with other tau tracers. Subjects’ brains took up moderate amounts of Ro6931643, with a standard uptake value (SUV) of about 1.5 between 60 and 90 minutes. Ro6958948 entered the brain more readily, reaching an SUV of up to 3.5 the same time frame. Of the three compounds, Ro6924963 proved most problematic. It failed to enter the brain well, then stuck around in controls when it should have washed out quickly, Wong said.
Roche is now moving forward with Ro6958948. Scientists are examining its test-retest reliability by scanning five additional AD patients and five age-matched controls, two weeks apart, to see how binding varies. They will also scan out to 200 minutes to better determine tracer kinetics, and choose the best reference region—likely the pons or cerebellum.
How does Ro6958948 compare with other tau tracers in development? A head-to-head comparison has yet to be done, said Roche’s Edilio Borroni. Ro6958948 appears to have a better kinetic profile than one of the leading tau tracers, T807/AV1451, Borroni said. Binding of T807/AV1451 never stabilizes in the cortex during the scanning period. The tracer also has different kinetics in different brain regions (Feb 2015 news). By contrast, Ro6958948 uptake stabilizes quickly, and at relatively uniform times for all brain regions. These qualities could make it easier in longitudinal studies to detect subtle changes in tau load, said Robert Comley, also from Roche. In addition, researchers see fewer artifacts with Ro6958948. It does not label the choroid plexus, as does T807/AV1451, and the striatum takes up less Ro6958948 than it does THK5351 or T807/AV1451, said Comley.
Though more data are needed, these qualities may give Ro6958948 a competitive edge, said researchers at the conference. “This tracer is an interesting and promising one,” said Keith Johnson, Massachusetts General Hospital, Boston. “There’s been an evolution, a continuous improvement with amyloid tracers, and hopefully we’ll see that with tau, too,” he said.
“The work that they’ve done looks careful and thorough,” said Chet Mathis, University of Pittsburgh, who agreed that the early data make Ro6958948 look promising. It demonstrates a good tissue-to-plasma ratio and fast kinetics, he said, and the background washes out quickly in controls. He noted that the compound binds in places that contain no tau, but this off-target binding falls below that seen in THK5351 or T807/AV1451. This off-target binding is a common issue for tau tracers, said Bill Jagust, University of California, Berkeley. Figuring out what they are binding may help explain something about tau, he said.
Amyloid Triggers Tau Spread and Destruction
At AAIC, more evidence emerged to support the notion that Aβ accumulation causes tau to spread from the medial temporal lobe and wreak havoc (Feb 2015 news).
Michael Schöll, who works with Jagust, correlated T807/AV1451 uptake with cognition among 27 normal people, average age 79, from the Berkeley Aging Cohort Study. He found that ligand binding in the entorhinal cortex and hippocampus correlated with slightly poorer and deteriorating episodic memory. This fits with the primary age-related tauopathy (PART) reported almost universally in elderly people (Crary et al., 2014). However, in those who were also amyloid-positive, surrounding areas, such as the inferior and lateral temporal cortex, as well as lateral and medial frontal and parietal cortex, took up the tracer too, and the global cognitive score started to decline. “It’s seems that once you have amyloid, tau moves outside the medial temporal lobe and global cognition is affected,” Schöll told Alzforum. He cautioned that the results cannot say whether the tau spread or amyloid comes first, and noted that the small sample size and some uncertainties about what PET tracers bind limit the conclusions he can draw. “Now the questions are how this tau pattern develops, and what is making it spread into these particular regions,” said Jagust.
If tau does compromise cognition, could that manifest in other ways? Bernard Hanseeuw, from Johnson’s lab, tested if tau or amyloid deposition brought about hypometabolism in the parietal cortex, which occurs early in Alzheimer’s disease (Herholz 2010). Hanseeuw and colleagues performed PET using PiB, T807/AV1451, and FDG on 90 cognitively normal people, averaging age 74, from the Harvard Aging Brain Study. They found that tau accumulation in the entorhinal cortex associated with reduced metabolism in the parietal, regardless of amyloid. However, metabolism dropped even lower if amyloid had built up and tau had spread into the inferior temporal cortex. Hanseeuw said the results suggest that tau tangles in the entorhinal cortex cause some hypometabolism during normal aging. However, in Alzheimer’s disease, when amyloid accumulates and causes tau to spread into the inferior temporal cortex, the problem worsens. It also hints that the entorhinal and parietal cortices are functionally linked, and that tau affects that connection, he said. The group next plans to look at additional regions of hypometabolism, he told Alzforum.
Victor Villemagne of the University of Melbourne, Australia, commented that the findings could explain suspected non-amyloid pathology, or SNAP, which is defined by a negative Aβ biomarker in the presence of a marker of neurodegeneration, such as hypometabolism on an FDG-PET scan.
Disaster Strikes Wherever Tau Lands
Once outside the medial temporal lobe, tau seems to cause trouble wherever it lands. At AAIC, Daniel Schonhaut, who works with Rabinovici, expanded on the notion that tau builds up in brain regions thought to underlie clinical symptoms in patients with atypical forms of AD (Feb 2015 conference news). He compared performance on a variety of cognitive tests with T807/AV1451 scans from 19 patients with typical and atypical forms of AD, including posterior cortical atrophy, primary progressive aphasia, and corticobasal syndrome.
Schonhaut reported that AD patients who performed more poorly on memory tasks had more tau in the hippocampus and surrounding medial temporal areas, while patients with visuospatial deficits had more tau in occipital regions (see image below). Those with language difficulties, meanwhile, had more tau in left temporoparietal regions associated with language function. “The distribution patterns of tau we’re seeing with this PET tracer seem to underlie the clinical phenotype of our patients,” Schonhaut told Alzforum
Close Match: Areas of T807/AV1451 uptake align with regions associated with cognitive deficits. [Courtesy of the Rabinovici lab.]
“These data show nicely that there’s a relationship between where the tau is clinical symptomatology,” said Jagust. “We haven’t seen that with amyloid.” Rabinovici noted that these results corroborate findings from classic postmortem studies demonstrating that cognitive state correlates much more strongly with tangle than amyloid pathology. They also support tau as a therapeutic target, particularly in the symptomatic phase of AD, he told Alzforum.
Researchers are also finding that tau deposition aligns with areas of cortical thinning. Chenjie Xia, who works with Brad Dickerson at Massachusetts General Hospital in Boston, analyzed T807/AV1451 scans and structural MRI images of five patients, two with amnestic AD, and one each with posterior cortical atrophy, primary progressive aphasia, and corticobasal syndrome. In each case, the T807/AV1451 signal correlated with areas that atrophied the most. Those areas also corresponded with the expected areas of atrophy for each disease. For instance, the two patients with amnestic AD took up tracer in the bilateral parietal and medial temporal areas, which lost volume compared with those areas in controls. In the PCA patient with visual deficits, the tracer labeled atrophied occipital regions. In the PPA patient, T807/AV1451 bound in the left parietal lobe, and in the person with CBS, in the primary and association sensorimotor cortices, each time matching the pattern of cortical thinning. In all cases, the tau deposition correlated significantly with the areas of atrophy, but amyloid did not, Xia said.
How Does Tau Spread?
Though these results all point to tau’s destructive nature, none gives clues as to how tau is transported through the brain. The idea that it travels trans-synaptically has gained traction. Evidence from cell culture and animal models suggest as much, but scientists still question whether it spreads this way in humans (Mar 2013 conference news; Apr 2015 conference news). At AAIC, Joseph Seemiller, working with Alexander Drzezga, University of Cologne, Germany, claimed it does. They used imaging to correlate patterns of tau distribution with known connectivity maps of the brain.
By comparing T807/AV1451 images, the researchers subtracted the “normal” signal of 18 healthy controls from those of 18 AD patients to identify AD-specific regions of tauopathy. From these they found one spot, near the angular gyrus, where tau aggregated the most. Voxels closest and most functionally connected to this “tau max” accumulated more of the misfolded protein. This supports the idea that tau spreads outward from an epicenter and along functional networks, Seemiller told Alzforum.
To find out if that is the case, they compared the pattern of T807/AV1451 uptake to 14 known functional networks, including those for language, visuospatial, and auditory functioning. The posterior default mode network (DMN) strongly matched the pattern of ligand distribution. The precuneus, language, and higher visual networks correlated, too. The DMN is a site of early Aβ deposition in AD and activity there wanes early in disease, leading some to suggest the network may be particularly vulnerable to pathology (Feb 2009 news; Mar 2004 news).
Michael Greicius, Stanford University, California, told Alzforum that the findings suggest that tau spreads between functionally connected synapses in the human brain. “The idea that tau spreads through interconnected networks is appealing,” said Rabinovici. However, both scientists said more research is needed to prove it. It is unclear why tau would spread along predefined functional networks, said Seemiller. He said that direct evidence can only come from experiments at the molecular level.—Gwyneth Dickey Zakaib
CSF Aβ Assays Remain Fickle: Robots to the Rescue?
As amyloid fibrils hog Aβ in the brain, less ends up in the cerebrospinal fluid. Myriad studies suggest that this dearth correlates with a positive amyloid PET scan. However, the use of CSF Aβ as diagnostic tool has been stymied by lab-to-lab and day-to-day variability in the immunoassays used to quantify the peptide. Though an international consortium has tried to standardize assays and reduce this variability to acceptable levels, it still prevails to some degree. Could fully automated assay systems be the answer? At this year’s Alzheimer’s Association International Conference (AAIC), held July 18-23 in Washington, D.C., researchers reported that two of these systems, which require users to do little more than unscrew a tube and push a button, yield highly reproducible results. If given the nod by regulatory agencies, these devices could pop up in clinics and serve as both selection and monitoring tools in clinical trials. The jury is still out on whether the platforms will edge amyloid PET as a diagnostic, or academic researchers will ditch their traditional CSF assays for automated ones. Some experts suggest that much may depend on price and who controls the reimbursement purse strings. Approval of a treatment that slows or stops AD could be a game-changer, as it would unleash a flood of older adults looking for a diagnostic test, and that in turn might be enough to tip the balance toward simpler and cheaper CSF analysis, they say.
Two kinds of variability plague the CSF measurements. On one hand, assays developed by different companies give different results. On the other, there is variability from run to run in any given assay. The Alzheimer’s Association funded the Quality Control Initiative in 2009 to attack the latter problem (see Nov 2009 news). Headed by Kaj Blennow at the University of Gothenburg in Sweden, the initiative periodically sends out CSF samples to member labs, which use their assay of choice to measure levels of Aβ42, phospho-tau (p-tau), and/or total tau. The researchers then attempt to weed out the sources of variability and standardize protocols. In 2012, the variability hovered around 25 percent (see Aug 2012 news).
At AAIC, Blennow reported that the variability hasn’t budged much in the last three years. It runs at 18 percent to 20 percent for several popular tests, including immunoassays from Meso Scale Discovery (MSD) and INNO-BIA AlzBio3 from Fujirebio (formerly Innogenetics), which was used in the Alzheimer’s Disease Neuroimagining Initiative 2 (ADNI2) and ADNI GO studies. To be fair, ADNI researchers have reduced this variability to around 10 percent by streamlining their protocols between study centers and trading human hands for pipet robots in some cases (see Figurski et al., 2012; Kang et al., 2015). However, the variability is not quite up to snuff for diagnostic standards, which ideally would fall between 2 percent and 4 percent, Blennow said. Michal Figurski of the University of Pennsylvania attributed the remaining variability to changes in lab temperature and humidity, as well as inconsistency in reagents from the manufacturer. “I do not know whether automated systems will change that. In principle they have the potential to do so; in practice, we’ll see,” Figurski told Alzforum.
Responding to this call for consistency, a handful of in vitro diagnostic companies have put forward fully automated systems that require nothing more from the user than plopping the sample tube into a machine. At AAIC, Blennow presented data from Roche Diagnostics’ Elecsys® immunoassays for Aβ42, p-tau, and tau that run on the Cobas E601 instrument. Blennow reported a variability of 0.6 to 3.5 percent for Aβ assays run at four different labs in the United States and Europe, which tested three different immunoassay lots twice a day for five days. This falls well within the realm of diagnostic acceptability, he said.
Roche Diagnostics is seeking FDA approval for the automated CSF biomarker tests as an in vitro diagnostic (IVD). The company has nearly completed the technical portion of the approval process, which ensures that the assay is consistent and reliable, explained Tobias Bittner of Roche Diagnostics in Penzberg, Germany. It now faces the more difficult hurdle of convincing the FDA that the test can be used as a clinical indicator of amyloid burden. Access to well-characterized clinical samples is key for this, and Roche is vying for its biomarker assay to be selected for the upcoming ADNI3 study.
In a poster presentation, Sandra Pereson from Fujirebio in Ghent, Belgium, summarized results from that company’s automated system, the Lumipulse. Its assay can measure CSF Aβ42 up to 4,500 pg/mL and total tau up to 2,500 pg/mL, and its in-house reproducibility for CSF Aβ42 was similar to Roche’s Cobas system. Fujirebio is also competing for the prized ADNI3 spot. The company plans to market this assay and device in Europe and the United States in addition to Japan, Pereson told Alzforum.
Assuming an automated system is approved, how will that change the landscape of biomarker testing? Bittner said the Roche Diagnostics platform is already being used under an Investigational Use Only (IUO) status for clinical trial patient selection. Roche is currently using the instrument in a Phase 3 trial of its monoclonal antibody gantenerumab in people with mild dementia due to Alzheimer’s disease. The company is also seeking the wider designation of a stand-alone IVD, which would allow physicians and other pharma companies to purchase the assays off the shelf for use in trials. As of now, they can use these Roche Diagnostics assays only through collaboration with the company.
Whether academic labs will adopt one of the automated platforms will depend on how much they will use it and why, said Henrik Zetterberg of the University of Gothenberg in Sweden. In addition to measuring Aβ, tau, and p-tau, researchers can use the instrument to quantify other potential markers, such as neuroinflammatory or cardiovascular-related proteins. Labs that process very few samples may have less incentive to automate. Zetterberg added that the current variability in tests such as AlzBio3 may still be acceptable for many research uses, but that as the cost of fully automated systems drops, more labs may jump aboard. Another factor is time. Blennow pointed out that while it only takes 20 minutes to run an assay on a fully automated machine, it takes two days before a standard ELISA returns a result.
The automated instruments could make a splash as diagnostic tools, since they are already generally used in clinical history labs at hospitals for a wide variety of diagnostic tests, Blennow said. They can run one patient sample at a time and pop out results so quickly that the clinician will have direct feedback to support the diagnostic decision. However, although CSF analysis is much cheaper than a PET scan, which costs several thousand dollars, some doctors in the United States still may prefer the latter to performing a lumbar puncture. Blennow thinks that may change once a treatment that slows the disease is approved, because then insurers will reimburse for the most cost-effective diagnostic test, especially given that CSF Aβ42 and amyloid PET give the same diagnostic accuracy. While payers currently cover either test, the Centers for Medicare and Medicaid Services (CMS) is reimbursing for PET in an exploratory study (see Apr 2015 news).
Most experts agree that CSF biomarker tests will never fully supplant amyloid PET, as the two measure two different things. Fresh data presented by Niklas Mattsson of Lund University in Sweden bore this out. While his findings strengthened the concordance between the two measures, showing that CSF Aβ, as measured by the AlzBio3 test, predicted amyloid PET positivity 98 percent of the time, he also found that a small number of patients in ADNI2 had pathological CSF Aβ42 levels but appeared in the normal range on amyloid PET. These patients subsequently tested positive in repeat amyloid-PET scans, suggesting that CSF Aβ detects earlier stages of disease.
Despite their similarities, each test has its unique benefits, Mattsson said. Unlike CSF tests, PET reflects the distribution of amyloid throughout the brain, and the scans may be particularly relevant in monitoring the effects of treatments aimed at fibrillar amyloid. They also don’t cause the headaches associated with lumbar puncture, especially in younger patients. On the other hand, CSF biomarker tests have a leg up on PET scans in that they can test multiple markers at once, and even measure the effects of treatments such as BACE inhibitors on APP processing.
While having automated systems, as well as streamlining assay techniques, will improve variability between runs of the same assay, researchers still need a way to harmonize findings between different assays. To this end, researchers led by Zetterberg and Blennow, in collaboration with the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC), have whipped up a master reference material made of pooled human CSF samples that contain either low, medium, or high levels of Aβ42, confirmed by mass spectrometry (see Leinenbach et al., 2014). Once a giant batch is made by the Institute for Reference Materials and Measurements (IRMM), assay vendors will use this material to calibrate their kits.
At a pre-meeting on the eve of AAIC, members of the Alzheimer’s Association-backed Global Biomarker Standards Consortium met to discuss progress on development of the reference materials. While the Aβ42 lot is well on its way to completion, researchers are now working out strategies for similar batches of tau and p-tau. These decisions, including which isoforms of tau to use as a reference material, are set to be made at meetings in the fall, Zetterberg said, and after that researchers can start making and validating the material.—Jessica Shugart
Does Brain Development in Childhood Set the Stage for Dementia?
It’s clear by now that in dementia, a disease mostly of old age, trouble starts decades earlier—but how early? Well—how about in the womb? Data presented at the Alzheimer's Association International Conference, 2015, held July 18-23 in Washington, D.C., lend some support to this radical idea. Scientists presented epidemiological, imaging, and even genetic studies that drew associations between early brain development and some atypical forms of Alzheimer's disease.
They also grappled with what those associations mean. “Might how your brain is wired in early stages influence how it degenerates later in life? That’s an extremely interesting concept,” said Jonathan Schott, University College London. Zachary Miller, from the University of California, San Francisco, stressed that early developmental patterns are not necessarily risk factors for later disease. "A more parsimonious explanation is that if you are going to get some form of dementia, then where it presents first, or the ‘locus of least resistance,’ might be determined by how your brain has developed," he told Alzforum. Whatever the cause and effect, researchers agreed that studying how learning disabilities and brain development influence later dementia could yield valuable insights into the disease processes, and even provide an opportunity for very early intervention.
Researchers have suspected for some time that there is a connection between learning problems and dementia. In 2008, researchers led by Marsel Mesulam at Northwestern University, Chicago, reported that people with learning disabilities, particularly dyslexia, were over-represented among patients with primary progressive aphasia (PPA), an atypical form of Alzheimer's disease characterized by language problems (see Feb 2008 news). Miller has studied this relationship, correlating learning disabilities with different forms of PPA and with posterior cortical atrophy (PCA), a form of dementia centered in the visuospatial cortex. He outlined in D.C. that screening 198 people in the PPA cohort at the UCSF Memory and Aging Center uncovered a connection between learning disability and the logopenic variant of PPA (lvPPA), but not with the semantic PPA or non-fluent variants (see Miller et al., 2013). Twelve of 48 people in the lvPPA group reported a history of learning disability, a prevalence as much as five times higher than that in the general population. Dementia started earlier in those 12 volunteers, hinting at particularly aggressive disease. Au contraire, MRI scans revealed their brain atrophy was much more restricted than in PPA patients with no history of learning problems, suggesting that neurodegeneration was more focal (see image below). In keeping with this idea, the learning disability/lvPPA patients scored higher on tests of global cognition than PPA patients in general. Miller's work suggests that early learning patterns might subtly influence the course of sub-types of dementia.
Deconstructing Aphasia.
Logopenic variant PPA patients with a history of learning disability have more focal atrophy (right). [Courtesy of Zachary Miller and Oxford University Press.]
Most of the patients with learning disability/PPA had childhood dyslexia, which predominantly causes reading difficulties. Could other learning disabilities, such as dyscalculia, or trouble with math, also herald PPA or some other kind of dementia?
Dyscalculia manifests as problems with numerosity, aka the ability to guess "more," as in which pile of acorns is bigger. The skill is conserved across most of the animal kingdom, said Miller. Without this basic math, animals would make poor decisions, e.g., squirrels might not survive the winter. In children, dyscalculia is the most common non-language learning disability. It can be inherited and has been linked to differences in brain structure and cortical activation patterns (see Kucian et al., 2006). Unlike language, which originates from the left side of the brain, the location of mathematical abilities is challenging to pin down. Symbolic mathematics typically localize to the left side, but non-verbal mathematical abilities tend to be rooted to the right side. Would people with dyscalculia be more likely to have neurodegeneration in the right brain hemisphere?
That's exactly what Miller found. In a UCSF cohort of 95 PCA patients, 17 had learning disabilities. Eleven of these had mathematical or visuospatial problems rather than language difficulties—a rate twice that in the general population. Interestingly, PCA patients with dyscalculia had atrophy predominantly in the right side of the brain, and much less in left side, compared with PCA patients without a history of learning disability. And similarly to the lvPPA patients with dyslexia, PCA patients with dyscalculia scored better on the MMSE and global cognition tests than did PCA patients without learning disabilities. Miller proposed that as for PPA, there may be sub-types of PCA with a more focal atrophy that might be distinguishable by brain imaging and natural history. Such subtypes might open a window onto the origin and progression of disease pathology. "The learning-disability groups may help us understand what is happening more generally in dementia," said Miller. Gil Rabinovici, who works with Miller at UCSF, agreed. "Trying to understand the mechanism that drives heterogeneity in dementia is incredibly important," he said. "There are fundamental clues among the rare variants that may teach us about vulnerability in general."
Researchers at the meeting asked if these results meant that people with learning disabilities were at greater risk for dementia. Miller said the available data is insufficient to draw that conclusion. He favored a "two-hit hypothesis" of how a person is built plus how he or she lives as determining overall risk. Others wondered why the atrophy in the cases with prior disability was so focal. Miller was unsure, but wondered whether it might be related to the structure of the brain in those regions or to some barrier to the spread of toxic tau.
Could genetic variants that cause or influence learning disabilities also predispose to dementia? Miller said there is as yet no clear evidence for this question, but it needs to be investigated. In his presentation, Schott described a variant that might do just that.
Schott led an international collaboration to carry out the most extensive genome-wide association study on PCA to date. Because the disease is so rare, the previously largest genetic analysis relied on about 80 patients. Schott and colleagues almost quadrupled that, obtaining DNA from 302 patients at 11 centers in Europe, North America, and Australia. "This might seem small for a genetics study, but it’s huge for PCA," said Schott. He credits the Atypical Alzheimer’s Disease and Associated Syndromes professional interest area organized through the Alzheimer's Association with helping pull together interested parties.
Patients recruited were 59 years old on average at symptom onset, which is typical for PCA. CSF, amyloid PET, or autopsy data was available for 82 of the volunteers; it was consistent with a diagnosis of Alzheimer's disease.
The researchers first looked in these DNA samples for genetic variants known to associate with AD. They confirmed that ApoE and CR1 variants were risk factors, though the odds ratio for ApoE was lower than for typical, late-onset AD. The researchers also detected nominal risk for ABCA7 and BIN1. In the GWAS, they identified three novel loci near the genes CNTNAP5, FAM46A, and SEMA3C that met genome-wide statistical significance. CNTNAP5 codes for a transmembrane protein involved in cell adhesion and signaling; FAM46A is expressed in the retina and has been linked to retinitis pigmentosa (see Barragán et al., 2008); and SEMA3C regulates axonal guidance in the developing brain.
Schott emphasized that these results are preliminary and need to be replicated. That said, he noted that FAM46A bears detailed study given the visual problems that plague patients with PCA, such as poor depth perception and difficulty tracking objects that may be related to reading difficulties. He is also intrigued by the notion that a gene involved in brain development—SEMA3C—could be linked to dementia. This gene codes for semaphorin-3C, one of a family of proteins that guide developing axons to the proper location. It is expressed in the visual cortex and the hippocampus, particularly near cholinergic afferents, said Schott. Interestingly, in a separate study SEMA3C emerged among 100 genes that supported connectivity among different brain networks, while SEMA3A, another semaphorin, was linked to control of axonal growth cones by β-secretase (see June 2015 news). "The possibility that subtle differences in brain development might influence the development of specific forms of dementia in later life is intriguing," said Schott. "If true, then this adds another dimension for study," he said. Schott noted that such developmental risks might get drowned out in studies of AD, but that by studying selective phenotypic variants of dementia, researchers might be able to lay bare those relationships.
What might vulnerabilities rooted in early life mean for the later treatment of dementia? "We could have 50 years lead time if we utilize the education system to identify learning disability," suggested Miller. In fact, a study in Sweden bears this out. At AAIC Serhiy Dekhtyar from the Karolinska Institute in Stockholm reported a link between test scores in primary school and dementia incidence later in life. Researchers in Sweden have followed 7,500 adults, age 65 and older, in the Uppsala Birth Cohort Study, for more than 20 years. Dekhtyar and colleagues correlated data on incident dementia with a variety of historical markers, including primary school grades, education level, and complexity of their occupation. The study is unique, said Dekhtyar, in having robust data on patient history. Though people in the cohort were born between 1915 and 1929, and school test records were archived and accessible. Dekhtyar obtained educational and occupational data from a population census taken around 1980, and dementia diagnoses from the national patient and cause-of-death register.
At AAIC, Dekhtyar reported that those who were in the lowest quintile for test scores at around age 10 had a 20 percent greater risk of developing dementia later in life. Interestingly, the association held even if the child went on to have many years of higher education or work in an occupation of higher complexity, both of which increase cognitive reserve. "It could be that the foundation for cognitive reserve begins very early in life," said Dekhtyar.
Dekhtyar noted that the Swedish registries tend to underestimate dementia diagnoses. To test if that might have biased the results, Hui-Xin Wang, also from the Karolinska, conducted a similar analysis on data from the Kungsholmen Study in Stockholm, which uses extensive clinical and neuropsychological assessment to diagnose AD. Among 440 men and women over age 75, those who had been in the lowest quintile for school grades at age 9-10 were 50 percent likelier to be diagnosed with dementia during the nine-year follow-up period. Neither study looked at specific types of dementia, but Dekhtyar said that data could be extracted from the records and agreed it would be interesting to see if test scores correlated with PPA, PCA, or other subtypes of dementia
While the Swedish studies did not specifically account for learning disabilities, Miller said they underscored the importance of a healthy brain in early life. "We need to think about lifespan neurology," said Miller. "It's important to understand people's entire life history. That influences who they are and their later vulnerabilities," he said.—Tom Fagan
At AAIC, Researchers Debate Neurogranin's Measure as a Marker
Last month in Washington, D.C., while U.S. political pundits eyed presidential hopefuls, researchers at the Alzheimer’s Association International Conference (AAIC) were scrutinizing neurogranin, a synaptic protein found in cerebrospinal fluid. At the meeting, held July 18-23, new data corroborated and extended earlier reports that neurogranin levels climb in the CSF of people with AD as well as in those with mild cognitive impairment, sparking enthusiasm about the protein's potential as a diagnostic and prognostic biomarker of AD.
AD research has been helped enormously by the availability of biomarkers for amyloid accumulation and neurodegeneration, and many hope a marker for synapse loss might provide additional information on the pathological progression of this disease. Neurogranin first surfaced as a potential biomarker a few years ago, when Kaj Blennow and colleagues at the University of Gothenburg, Sweden, analyzed CSF samples by western blot and found higher levels of the protein in AD patients than in healthy controls (Thorsell et al., 2010). More recently, aided by the development of new quantitative immunoassays, Blennow, Eugeen Vanmechelen at ADx NeuroSciences, Ghent, Belgium, and others, observed an uptick of neurogranin in the CSF of people with mild cognitive impairment (de Vos et al., 2015). Moreover, high neurogranin levels in MCI subjects predicted progression to AD (see Jan 2015 news).
Neurogranin, a post-synaptic protein enriched in dendritic spines, is believed to leak out into CSF as synapses in the brain degenerate. “The assumption has been that it reflects passive neurodegeneration,” said Anne Fagan, Washington University, St. Louis. “But we do not know yet if what we are seeing reflects neurodegeneration or some other mechanism.”
At AAIC, Fagan shared data from the largest cohort assessed for neurogranin thus far, more than 300 people with AD and healthy controls from WashU. Using a neurogranin immunoassay developed at WashU by Jack Ladenson, Fagan's team observed high levels of CSF neurogranin in people with various degrees of cognitive impairment. Compared with healthy controls, people with very mild AD (CDR 0.5) and those with mild and moderate dementia (CDR 1 and 2, respectively) had more neurogranin in their CSF (2,020 pg/mL in mild AD versus 1,470 pg/mL in controls). Furthermore, among healthy controls (CDR 0), high baseline levels of neurogranin nearly tripled the risk of progressing to cognitive impairment within two to three years. Risk jumped to 11-fold in those who also had low CSF Aβ42.
"It's important to note that these were cognitively normal folks," Fagan said. "These were not people with very mild AD that progressed to dementia, but those with a CDR of zero at baseline who progressed to AD dementia within the follow-up period."
These results suggest that neurogranin levels in CSF may start to rise very early in the disease process, prior to cognitive symptoms. This is consistent with postmortem studies showing that synapse loss is well underway by the time individuals develop MCI (Scheff et al., 2007).
Although the trajectory of CSF neurogranin awaits thorough longitudinal characterization, at AAIC researchers received their first glimpse into neurogranin’s behavior over time. Charlotte Teunissen, VU University Medical Center, Amsterdam, tested CSF in 163 participants from the Amsterdam Dementia cohort. This study included people with AD (n=65), MCI (n=61), as well as a cognitively normal group (n=37), the latter comprised mainly of individuals with subjective memory complaints (n=31). All participants provided two CSF samples an average of two years apart and were followed clinically for about four years. In keeping with other studies, baseline neurogranin levels were higher in the AD and MCI groups than in the cognitively normal group. High baseline neurogranin nearly doubled the risk of progressing from MCI to AD, comparable to the predictive power of core biomarkers such as Aβ42, total tau, and phospho-tau181.
Neurogranin concentrations were surprisingly stable over time within the AD and MCI groups. Compared to concentrations at baseline, Teunissen’s team saw no significant increase in samples collected two years later. In contrast, neurogranin levels were higher at follow-up in the group with subjective memory complaints, increasing from an average of 1712 pg/mL at baseline to 2015 pg/mL two years later. Teunissen believes that the higher concentrations at follow-up in those with subjective memory complaints may be an early sign of pathology. Subjective memory complaint is a known risk factor for AD and 10 of the 31 participants who entered the study with subjective complaints progressed to MCI or dementia within four years (Sep 2014 news; Rönnlund et al., 2015).
Tapping into brain pathology. Proteins in CSF may represent different pathological processes in the brain. CSF neurogranin is thought to reflect synaptic loss. [Courtesty of Kaj Blennow.]
As a putative indicator of synaptic loss, would CSF neurogranin correlate with a decrease in brain volume or other signs of atrophy? Several groups are now investigating this possibility. At AAIC, Harald Hampel and colleagues at the Pierre and Marie Curie University, Paris, reported that levels of neurogranin in CSF negatively correlated with total white-matter volume in the brain, but had no association with hippocampal volume. In this pilot study, first author Simone Lista analyzed a cohort of 10 AD patients and 12 people with prodromal AD. “These preliminary results suggest neurogranin is a promising biomarker reflecting atrophy of brain white matter in AD dementia,” Lista wrote to Alzforum.
A study in press by Erik Portelius and colleagues at the University of Gothenburg, Sweden, bolsters the correlation between CSF neurogranin and structural brain changes. "We found significant associations between high CSF neurogranin at baseline and increased rate of hippocampal atrophy, as well as reductions in cortical glucose metabolism,” Portelius wrote to Alzforum. “There is now overwhelming evidence supporting CSF neurogranin as a biomarker for synaptic pathology in Alzheimer's disease.”
Synaptic loss occurs in many neurodegenerative diseases, raising the question of whether CSF neurogranin creeps up in diseases other than AD. At AAIC, Henrietta (Henny) Wellington, a student at University College London, reported that elevated neurogranin is specific for AD. Working with Henrik Zetterberg at the University of Gothenburg, she measured neurogranin in the CSF of more than 300 participants with a variety of neurodegenerative diagnoses. CSF neurogranin in people with frontotemporal dementia, dementia with Lewy bodies, Parkinson’s disease, progressive supranuclear palsy, or multiple system atrophy was similar to CSF neurogranin in controls. It was elevated only in the AD group.
Wellington, who won the best poster presenter in the Diagnosis and Prognosis theme, attributes neurogranin’s specificity for AD to both its pattern of expression in the brain as well as the magnitude of synaptic loss in the AD brain. “Neurogranin is enriched in brain regions highly affected by AD, such as the hippocampus and cerebral cortex,” she said, “And, in AD there is more widespread synaptic loss, not restricted to one or two specific areas, as in PD.”
AAIC attendees found the new neurogranin results encouraging. However, some wondered what, if anything, neurogranin adds to the existing biomarker toolkit, noting that established biomarkers like CSF Aβ42 and total tau are also elevated in AD patients compared with healthy people and associate with progression from MCI to AD (see, for example, Hansson et al., 2006; Fagan et al., 2007). Multiple studies have now shown that levels of CSF neurogranin in AD patients strongly correlate with levels of total tau in CSF, a finding further corroborated in the WashU and Amsterdam Dementia cohorts, according to Fagan and Teunissen. “Given the correlation with total tau, it will be interesting to see if neurogranin can add additional information,” said Markus Otto, University of Ulm, Germany, who was not involved in the studies.
Blennow pointed out that levels of phospho-tau also correlate very well with total tau in AD patients and controls, but nevertheless the two markers provide distinct information about tangle-load and neurodegeneration, respectively. “So, even if neurogranin and total tau show similar increases in AD, they may tell us different things or could be used in different ways,” he said.
Given that synapse loss correlates better with cognitive decline than tau or amyloid pathology (Terry et al., 1991), one possible future application for CSF neurogranin is as a surrogate endpoint in clinical trials. This may seem a distant goal, but at AAIC, Diane Stephenson, co-director of the Coalition Against Major Diseases (CAMD), announced that CAMD had recently singled out CSF neurogranin as a promising fluid biomarker for use in clinical trials.
“When we surveyed the biomarker landscape, neurogranin, along with tau PET, rose to the top as having the potential to meaningfully advance drug development,” said Stephenson. CAMD is an initiative of the Tucson, Arizona-based Critical Path Institute (C-Path), which brings together industry and academic scientists to speed therapy development for neurodegenerative disease, in part by promoting standardized assays and protocols. Last May, CAMD recommended CSF neurogranin, along with tau PET, to the FDA in response to an open request for biomarker nominations. According to Stephenson, “The purpose of the recommendation was to get everyone on the same page so that when these markers are ready for prime time they can be employed in pivotal clinical trials.”
In the meantime, there is much to be learned about neurogranin, starting with what exactly is being measured in CSF. The majority of studies thus far have used ELISAs to detect neurogranin, but this type of assay does not distinguish between full-length proteins and peptide fragments. Last year, a mass spectrometry study indicated that the vast majority of neurogranin in CSF comprised not intact protein, but C-terminal fragments of varying lengths. One fragment in particular, amino acids 48-76, was enriched in CSF of AD patients compared with healthy controls (Kvartsberg et al., 2014). At AAIC, Hlin Kvartsberg, a student at the University of Gothenburg, compared neurogranin peptides in CSF with those found in brain tissue, specifically in parietal lobe samples from 10 AD and 10 control cases. Using hybrid immunoaffinity mass spectrometry (HI-MS), a meld of antibody-based purification and MS, she detected more than 70 neurogranin peptides in the brain. The reason for this heterogeneity is unknown. “Our theory is that as synapses degenerate, the protein becomes available to a variety of proteases in the brain,” said Kvartsberg.
Kvartsberg had also previously characterized neurogranin peptides in plasma and found a very different set of peptides than she observed in the brain and CSF. She said that the plasma signal likely derives from peripheral sources of neurogranin, such as blood cells, and that plasma neurogranin is not associated with AD (Kvartsberg et al., 2015; de Vos et al., 2015).
“There appears to be two separate neurogranin pools with only very minor or no exchange between them,” said Kvartsberg. “A CNS pool, comprising brain and CSF neurogranin with very similar peptide profiles, and a peripheral pool, as measured in plasma.”
Very little is known about the life cycle of neurogranin in the brain, including how it is metabolized, and why the protein is detectable in CSF. “We now need to understand the mechanism of release of neurogranin and determine if it is in fact reflecting neurodegeneration and dendritic loss, or perhaps more subtle synaptic dysfunction, or even a change in the regular turnover of the protein,” said Zetterberg.
A plethora of peptides. A variety of C-terminal fragments, but no intact neurogranin, can be detected in CSF. Four peptides (green) are also found in brain extracts. The 48-76 peptide (asterisk), is common to both, and is present at higher concentrations in the CSF of AD patients. [Courtesy of Hlin Kvartsberg.]
In today’s biomarker field, neurogranin is not the only synaptic protein in the running. The presynaptic protein SNAP-25 is also elevated in the CSF of AD patients (Brinkmalm et al., 2014). At AAIC, Fagan reported higher SNAP-25 CSF levels in people with AD compared with healthy controls, among a cohort from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). CSF SNAP-25 also ran higher in MCI subjects, particularly those classified as amyloid-positive due to low CSF Aβ42. Fagan noted these data are preliminary and from a comparatively small number of individuals (152), but nevertheless indicate that disruptions to synaptic integrity are detectable very early in the disease process.
“A year ago, when I was asked to prepare this talk about synaptic biomarkers, I thought it was going to be a five-minute presentation,” Fagan said, “But in the meantime we and others have done a lot of work on candidate markers such as neurogranin and they are shaping up to be very interesting.”—Kelly Dakin
How Do You Communicate Alzheimer’s Risk in the Age of Prevention?
Part 1 of two.
With the growing use of biomarkers, researchers can now identify cognitively normal people who are at elevated risk for Alzheimer’s disease. This has enabled secondary prevention studies such as the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) trial. To participate in this type of study, however, people typically need to learn about their risk factors. It is not feasible to keep participants blinded on this point, as that would necessitate including many more people, ballooning the cost of these already-expensive undertakings, researchers said. Hence clinicians are now grappling with how best to disclose risk information to cognitively healthy people without causing undue despair. They also want to monitor how the information affects people’s psychological well-being and cognitive function in the long term.
At the Alzheimer's Association International Conference 2015, held last month in Washington, D.C., speakers detailed innovative ways in which the A4 trial and the Alzheimer’s Prevention Initiative’s ApoE4 trial are approaching the issue, the former revealing brain amyloid imaging results and the latter ApoE genotype (see Dec 2014 conference news). Researchers are drawing inspiration from the breast cancer field, where communicating genetic risk has become standard practice, noted Angela Bradbury of the University of Pennsylvania in Philadelphia, and from the AD REVEAL studies that disclosed ApoE status to participants. However, AD researchers face logistical barriers in scaling up these previous disclosure models to communicate risk to several thousand people within the setting of a large trial. To accomplish this, the API ApoE4 study will make extensive use of remote technologies such as the Internet, telephone, and videoconferencing (see Part 2 of this story).
“We’re starting to think about providing risk information and empowering participants to make decisions based on this information,” Jessica Langbaum of Banner Alzheimer’s Institute in Phoenix told Alzforum. “I think in the coming years we’ll see more and more studies that disclose.”
Little data is yet available to determine how well these new protocols work, but preliminary results from small studies suggest that most people enrolled in trials handle knowing their risk well, speakers said. Surveys also reveal a growing desire for that knowledge, particularly if that qualifies the person for a treatment trial. Overall, the talks showcased a field moving toward an era of widespread risk disclosure.
At the same time, speakers acknowledged looming societal unknowns, such as whether knowledge of AD risk could lead to insurance or other types of discrimination. Federal law prohibits employers and health insurers from discriminating against people based on genotype, but this protection does not extend to amyloid-imaging results. Likewise, disability, life, and long-term-care insurers are not bound by the same federal protections (see Aug 6 conference news; Jun 2008 news). Long-term care is particularly important, since many people at risk of AD purchase such coverage, speakers said, adding that this is fast becoming a pressing issue for legislators to resolve.
Laying the Groundwork for Disclosure Trials
Cancer researchers have forged this path already. Twenty years ago, after the identification of variants in the BRCA1 and BRCA2 genes that magnify the risk of breast cancer, clinicians struggled with the issue of how to tell women about their genetic status. “We were concerned about their ability to understand complex genetic information, and the potential for distress when facing tough decisions about prophylactic surgery,” Bradbury told Alzforum. Bradbury is an expert in the ethics of breast cancer risk disclosure.
In the classic genetic counseling model, people who may carry risk variants first meet with a counselor face-to-face to learn about the limitations of testing and give consent to the procedure. They return for a second meeting to hear the results. Genetic counselors assess their mood and anxiety, and follow up later by phone to see how they are coping with the information. Providers in the cancer field developed several strategies for communicating technical genetic information, including using visual aids and providing only a small amount of information at a time to avoid overwhelming people, Langbaum noted. Studies found that most women who went through the process did well over time and used the information to improve their health, Bradbury said (see, e.g., Lynch et al., 2006; Bosch et al., 2012; Borreani et al., 2014).
Alzheimer’s raises different issues, however. Chief among these is that people with increased risk of the disease have no access to treatments that would slash their odds of getting sick. Many researchers worry that knowledge of brain amyloid or ApoE status would be deeply distressing to cognitively healthy, middle-aged people, perhaps lowering their quality of life or leading to rash decisions. Even so, surveys indicate that an increasing number of people want to know their AD risk.
At AAIC, Joshua Grill of the University of California, Irvine, presented a poster describing attitudes toward genetic testing among 80 participants in the Dominantly Inherited Alzheimer’s Network (DIAN). For these people, who may have inherited a familial AD gene, a positive test result means they are destined to develop the disease. Nonetheless, 55 percent wanted to know their genetic status. Among those who did not, almost three-quarters said they would change their mind if knowing their status gave them access to a clinical trial. All but one person said they would get testing and join such a trial if they were assured of getting active drugs during an open-label extension study (see Grill et al., 2015). “That is a big jump in interest from previous surveys,” Grill told the audience.
Anecdotal reports suggest that more people at risk for familial disease are choosing to get tested now than ever before. Many of these people recently shared their stories at a July 18 DIAN meeting in Washington, D.C. (see Aug 2015 conference news).
Attitudes appear similar among people at risk for late-onset AD. Brian Ott of Brown University, Providence, Rhode Island, presented data from 158 participants in the Rhode Island Alzheimer’s Prevention Registry. In this cohort, three-quarters had at least one parent with AD, and half cared for someone with the disease. Only 15 percent already knew their own ApoE or amyloid-imaging results, but of those who did not, 80 percent said they wanted to find out. The most common reasons they cited were to participate in research and plan for their future. Likewise, a recent survey of 4,036 people enrolled in the Alzheimer’s Prevention Registry found that 89 percent of them wanted either genetic or biomarker testing, Langbaum noted. Ninety percent said they would seek a healthier lifestyle if test results were positive. Ominously, 12 percent reported they might consider suicide (see Caselli et al., 2014; Caselli et al., 2015).
This latter finding dovetails with experiences from oncology. Although most patients do well after finding out they have a genetic risk for breast cancer, there are also subgroups of people who find the information very distressing and will struggle, Bradbury said. As the Alzheimer’s field moves toward incorporating disclosure into trials, it will be important to identify these subgroups and develop protocols to minimize psychological risk for them, Bradbury noted.
The Next Frontier: Disclosure of Amyloid Status
With the Food and Drug Administration’s approval in recent years of three amyloid imaging tracers, cognitively normal people now have the option to find out if they are accumulating amyloid deposits in their brain. Large prospective studies have linked brain amyloid to a greater risk of cognitive decline within a few years (see Dec 2014 conference news). The field is grappling with the issue of whether to tell cognitively normal adults their amyloid status, and if so, how and under what circumstances (see Feb 2012 conference news).
In D.C., David Johnson of the University of Kansas, Lawrence, presented preliminary data that suggested many people can handle this information well. As part of the APEX exercise study at his institution, 85 cognitively normal participants went through a counseling protocol similar to that used in genetics, and learned their brain amyloid status. In this cohort, 25 people had positive amyloid scans. Researchers followed up with participants six weeks and six months after disclosure. Six weeks after testing, clinical measures of anxiety and depression stayed stable, and low, in all groups; however, people with a positive scan did report feeling more upset, sad, anxious, or worried. In nearly all cases, participants listed these feelings as being “very rare” or “infrequent,” and the frequency declined to nearly baseline measures by six months. The researchers saw no change in memory performance, or in how participants rated their own memory. People with positive scans did report a greater intent to exercise than those with negative results, however.
In a similar vein, Michael Pontecorvo of Avid Radiopharmaceuticals, Philadelphia, updated the audience on a trial that examines the health outcomes of amyloid imaging (see Aug 2015 conference news). Patients with memory complaints who were randomized to the disclosure arm of the study did not demonstrate any significant psychological distress upon learning the news, he said. Researchers saw no changes in measures of anxiety, depression, or psychotropic drug use.
More data may soon be available. The A4 trial enrolls cognitively normal people between 65 and 85 who have a positive amyloid scan. With an enrollment goal of 1,150, trial investigators will have to inform large numbers of people of their amyloid status. Researchers formed a group of six amyloid imaging experts and six genetic-testing experts to develop a protocol for this (see Harkins et al., 2015). Jason Karlawish of UPenn, who leads the A4 disclosure study, said that this group incorporated lessons from REVEAL in this work.
As in genetic-disclosure models, participants first meet with A4 personnel for an educational session. There, they learn about the science behind amyloid imaging and what a positive or negative result means in terms of risk. For A4, these meetings will be face-to-face. People who decide to proceed sign a consent form. Then the investigator assesses each participant’s motivation for getting the scan, as well as their understanding of the process, and administers standard psychological tests for anxiety and depression. If no concerns surface, they schedule the scan. Some days after the scan, the participant returns for a face-to-face disclosure session, accompanied by a friend or family member if desired. The investigator again assesses the participant’s mood and willingness to receive the results, then discloses the findings using scripted language. Study personnel follow up by phone within three days to see how participants are coping. While no data from the A4 study is yet available, about 200 people have now gone through the procedure, and the process appears to be working well, Jeffrey Burns of the University of Kansas, Kansas City, told the audience in D.C.
A4 researchers also look at the long-term impact of disclosure. The companion Study of Knowledge and Reactions to Amyloid Testing (SOKRATES) will track people in the A4 study from just after disclosure to nine to 12 months later, and compare them to people in the adjunct observational LEARN study who have discovered that they do not have brain amyloid. Participants will complete measures of mood and cognition, and estimate how much time they think they have left to live. “The goal is to understand how people incorporate information about amyloid status into their sense of self,” Karlawish told Alzforum. Researchers will also investigate how the knowledge affects participants’ social relationships, and whether they are subject to stigma (see Sep 2012 news).
Overall, these studies are paving the way toward a future when physicians might routinely disclose brain amyloid status as part of clinical care, Karlawish noted. Thus, researchers need to find the best ways for physicians to communicate and interpret these results for patients. But Karlawish believes the issue is bigger than that. “We also need to make sure that learning this information has value to patients,” he said. Other speakers noted that once an effective Alzheimer’s treatment becomes available, people may demand to know their amyloid status. However, Karlawish pointed out that even the existence of treatment options might not mitigate the stress of such knowledge. Rather, a potential treatment would introduce uncertain risk-benefit calculations to the equation, such as how safe the treatment is and how much it delays disease, without changing the fundamental reality of dealing with impending Alzheimer’s disease. “There may be an existential burden to learning that one’s brain is at risk for, or in the process of, failing,” Karlawish suggested.—Madolyn Bowman Rogers
Testing a New Model for Disclosure of ApoE4 Status
Part 2 of two.
With Alzheimer’s secondary prevention trials now underway, the question of how best to tell people about their AD risk has arrived. The ongoing Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) trial uses a protocol similar to traditional genetic counseling methods to inform approximately 1,000 people about their brain amyloid status (see Part 1 of this story); however, the soon-to-start Alzheimer’s Prevention Initiative’s ApoE4 trial focuses on genetic risk for late-onset AD and will therefore draw on an even larger pool of potential participants. Faced with the challenge of finding and communicating risk to many thousands of people, API researchers have had to tailor classic in-person disclosure models making greater use of technology such as computers and telephones. At the Alzheimer's Association International Conference 2015, held last month in Washington, D.C., Jessica Langbaum of Banner Alzheimer’s Institute in Phoenix detailed the new protocol. She noted that the API trial will provide the first large-scale test of whether such remote methods can work as well as established, face-to-face genetic counseling protocols.
The established protocols first proved themselves in the cancer field, where genetic counselors are more plentiful than at Alzheimer’s clinics. Nonetheless, in D.C., Angela Bradbury of the University of Pennsylvania, Philadelphia, pointed out that breast cancer researchers, as well, have since moved toward remote disclosure. Few studies have compared the outcomes against the older methods, she stressed. “We don’t know the impact of these alternative delivery models,” she told Alzforum. A handful of studies have reported no difference in anxiety or depression among participants counseled about their cancer risk remotely versus face-to-face, but fewer people chose to proceed with genetic testing when the initial meeting with a counselor occurred remotely (see Hilgart et al., 2012; Schwartz et al., 2014; Kinney et al., 2014). Bradbury is collaborating with API researchers to help test such newer methods.
Traditional disclosure methods have a good track record in AD. The Risk Evaluation and Education for Alzheimer’s Disease (REVEAL) study led by Robert Green of Brigham and Women’s Hospital, Boston, reported that middle-aged people informed of their ApoE genotype handled the information without undue distress or long-term negative effects (see Jun 2007 webinar; Jul 2009 news). Subsequent REVEAL studies have examined additional issues, such as whether the information is more disturbing to people who already have mild cognitive impairment and thus higher risk of progression. The results were similarly positive (see Aug 2012 conference news).
In D.C., Scott Roberts of the University of Michigan, Ann Arbor, updated the audience on what REVEAL says about ApoE4 homozygotes, who inherit about 12 times the risk of AD that non-carriers do. Researchers pooled data from the 28 homozygotes who took part in the first three REVEAL studies. As a group, they reported worrying about the test results more than heterozygotes in the first few weeks after disclosure, but clinical measures of depression and anxiety did not budge. In addition, people with two copies of ApoE4 were more likely than heterozygotes to report that they made lifestyle changes in response to learning the news, such as improving their diet, taking vitamins or supplements, or starting an exercise program.
Will High-Tech Disclosures Work?
The REVEAL studies had limitations, however. The number of ApoE4 homozygotes was very small, and participants were a highly educated group who had chosen to learn their status, so the studies may not reflect the general population. Clinicians at Banner expect to get more definitive data on the effects of disclosure soon. They are preparing to enroll 1,300 people who carry two copies of the ApoE4 allele for the first API ApoE4 prevention trial (see Jul 2014 conference news). Potential participants must learn their ApoE genotype to be considered for the trial. Some may already know it through testing done at a memory clinic, through a direct-to-consumer service, or at a cardiac clinic (the latter because ApoE4 boosts the risk of coronary artery disease). But for the majority who do not, Banner offers the API ApoE4 Genetic Testing and Counseling Program. This sub-study will examine the impact of disclosure and the best way to go about it, Langbaum said. To compare the effect of disclosure on different genotypes, the study will include ApoE4 homozygotes, heterozygotes, and non-carriers, although only the former are candidates for the intervention trial.
First, the researchers need to find potential participants. Only 1 to 2 percent of the population carry two copies of ApoE4, so trial personnel will need to genotype upward of 60,000 people to identify enough carriers. Researchers will reach some people through memory clinics, but most potential participants will come from the Alzheimer’s Prevention Registry. This online registry currently has more than 135,000 members, who have signed up to receive information about Alzheimer’s research and future trials. After the Genetic Testing and Counseling Program receives regulatory approval, which may occur this fall, registry members between the ages of 55 and 75 will receive an invitation to donate DNA, Langbaum said.
Those who express interest will be asked to first complete a brief, interactive module about genetic testing online or over the phone with a health educator at Banner. “We recognize that not everybody has Internet access or wants to do this online,” Langbaum said. The module will inform participants that Banner will use the genotyping results to match people to future trials, but it will not disclose the findings to participants. It would not be practical or ethical to try to disclose genetic status to everyone, Langbaum noted.
People who choose to proceed will sign a consent form and then receive a cheek-swab kit through the mail. They can collect their own DNA at home, then mail it to a central lab for analysis. Langbaum noted that her team encountered unexpected hurdles while setting up this process. Because many states consider DNA testing to be a practice of medicine, a doctor’s approval is required to mail samples across some state lines. To comply with regulations, API contracted with medical providers in those states to review orders that come in through the website and approve the cheek-swab kits. “There are other initiatives looking at doing cheek swabs by mail, so this experience can be a lesson for all,” Langbaum told Alzforum.
After genotyping, API will invite about 3,000 DNA donors in the United States, and additional participants overseas, to learn their ApoE status. In Europe, participants will go to a clinic to meet face-to-face with genetic counselors to get their results, but U.S. participants will be told either by phone or videoconference from a centralized group based at the University of Pennsylvania. This format will allow researchers to directly compare the effects of disclosure by phone, videoconference, or in person. “No study has yet compared all three. This is really novel,” Langbaum said. She noted that this protocol modifies the traditional genetic-testing model used in oncology, since participants will receive pre-test counseling remotely, take part in genotyping through the mail, and in many cases learn their results via technology. “We need to think about ways to scale up disclosure to a real-world delivery model,” she said.
After disclosure, study personnel will follow up with participants by phone within two to seven days to see how they are coping with the information. Additional follow-ups will occur at six weeks, six months, and 12 months. Researchers will administer a battery of psychological tests, including for anxiety and depression, as well as surveys adopted from REVEAL that assess attitudes and knowledge about genetic testing. The surveys will measure how well people remember the results of testing, as well as how satisfied they are with the process. The researchers also want to explore the effects of testing on family members, since a positive result implies that relatives may be carriers as well. “We need to think about these issues as we move forward to a time when more and more people may learn their genotype,” Langbaum said. She noted that so far, the people she has spoken to seem very interested in learning their results. “They want to know they are at increased risk before embarking on such a long trial,” she said.
Researchers will watch for cognitive effects of disclosure as well, including the “stereotype threat” whereby a negative perception becomes a self-fulfilling prophecy. For example, a recent study of 144 cognitively normal older adults found that those who knew they carried an ApoE4 allele rated their own memory more poorly and performed worse on tests of verbal memory than ApoE4 carriers who did not know their genotype (see Lineweaver et al., 2013). In a separate follow-up study, API researchers will invite participants from the disclosure study to complete online cognitive assessments and answer questions about how they think they are doing mentally. For controls, the study will enroll some people who donated DNA but did not learn their genotype. Researchers will examine whether ApoE4 carriers who learned their genotype perform differently than those who did not, or have more concerns about cognition, and whether this effect varies with the number of copies of ApoE4.
“We want to make sure that genetic disclosure is done in the best way to try to minimize any negative impact,” Langbaum said.—Madolyn Bowman Rogers
Amyloid scans have allowed researchers to follow Alzheimer’s pathology as it develops in the human brain, but they are still working out whether and how this technology will help patients. At the Alzheimer’s Association International Conference 2015, held July 18 to 23 in Washington, D.C., scientists presented results of three recent trials that examined whether scans led to changes in patient diagnoses, influenced how doctors managed their disease, or improved health outcomes. These studies aim to help doctors and insurers define which patient groups will benefit the most. “We want to be very cautious in how we use these expensive instruments and tease out the populations that really need imaging,” said Philip Scheltens, VU University Medical Center, Amsterdam.
The largest of the studies comes from Philadelphia-based Avid Pharmaceuticals, owned by Eli Lilly and Company in Indianapolis. Avid makes the amyloid ligand florbetapir, aka Amyvid. Company scientists completed a randomized Phase 4 study of positron emission tomography (PET) scans with the tracer to test short-term consequences on diagnosis and patient management, as well as longer-term health outcomes. Avid’s Michael Pontecorvo presented results at AAIC. The trial enrolled 618 patients aged 50 to 91, with an average age of 73, from France, Italy, and the United States. All had a diagnosis of mild cognitive impairment or dementia, but physicians were less than 85 percent certain that AD pathology underlay their symptoms. Doctors first gave a working diagnosis and outlined would-be plans for treatment and further testing in the absence of a scan. Then, all patients were scanned for brain amyloid (see image below).
Scans in the Clinic.
Amyvid PET scans distinguished between brains with high (left) and low (right) amyloid. [Courtesy of Eli Lilly & Co. and Avid Radiopharmaceuticals.]
For 308 of them, participating physicians received results right away and some used them to modify their action plan. The researchers compared outcomes in that patient group with outcomes from 310 patients who served as a control group—this "delayed feedback" group got their scan results a year later. The researchers wanted to know if outcomes would change with or without knowledge of amyloid status.
Three months after scanning, the participating physicians recorded for the researchers any modifications they had made to their original diagnoses and any further tests or treatments they had ordered or prescribed. Doctors who got scan results immediately changed their diagnosis for a third of patients, most often to fit a scan that countered their original thinking. For patients who initially had a non-AD diagnosis but a positive scan, scan results changed the doctor’s mind 92 percent of the time. Conversely, doctors switched to a non-AD diagnosis in 81.5 percent of cases considered AD at the outset when the scan was negative. In the delayed-feedback control group, physicians altered only about 6 percent of diagnoses, for reasons other than amyloid PET scan results.
Doctors altered their treatment strategy in 68 percent of patients in the experimental group, versus 56 percent in the control group, a statistically significant difference. These numbers were driven mostly by prescription of cholinesterase inhibitors: In the group that got scan results immediately, physicians began prescribing these drugs to people with positive scans and discontinued them in those with negative scans. In the delayed-feedback group, doctors prescribed more of these drugs to everyone.
Patients returned for one-year follow-up appointments so doctors could assess change in cognitive status, health outcomes, mood, function, or quality of life. There was no difference between groups. The study lacked the power to discern small or rare effects, Pontecorvo said. He added that the company found no evidence that disclosing amyloid status to patients or their caregivers led to higher rates of depression, anxiety, or drug use (see Aug 2015 conference news).
“The study shows how amyloid imaging is likely to change clinical practice,” said Glenda Halliday, Neuroscience Research Australia, Randwick, New South Wales, who co-chaired the session with Matthew Frosch, Massachusetts General Hospital, Boston. Frosch cautioned against weaknesses in the data. For instance, Pontecorvo did not collect data on what the alternative diagnoses had been, or reasons for maintaining disease management that ran counter to scan results.
“This study demonstrates that in diagnostically uncertain cases, amyloid PET can help refine diagnosis and guide clinical management,” said Gil Rabinovici, University of California, San Francisco. It also suggests that amyloid PET disclosure is safe in a clinical setting, he said. Rabinovici leads the IDEAS study, a separate effort to examine benefits of amyloid scans in Americans 65 years and older (Apr 2015 news). He was surprised by the high rate of management change in the control group. It could be explained by clinicians not knowing whether scan feedback would be immediate or delayed when they completed the pre-PET management plan, he said, so they may have held off on final management recommendations hoping they could incorporate amyloid status into their decision. “In IDEAS, we will compare disease management that assumes amyloid PET will not be accessible with management that occurs after an amyloid scan,” he told Alzforum. “This design may better isolate the impact of the scan.”
A different study, from Scheltens’ lab, tested the utility of amyloid scans in patients with an earlier age at onset of disease. Marissa Zwan pointed out that scans give valuable information in younger people, because they are less likely to have age-related amyloid deposits in their brain, have fewer comorbidities, and more likely to have a form of dementia that is harder to diagnose because of overlapping clinical phenotypes, for example frontotemporal dementias.
Patients were eligible to take part if their neurologist said they were less than 90 percent certain of the cause of their dementia after a standard clinical workup. This included a neurological and psychiatric screen, as well as a magnetic resonance imaging scan of the brain. The study included 211 patients with an unclear, atypical presentation of dementia, who ranged from 45 to 70 years old. Of those, 145 were tentatively diagnosed with AD, 66 with a non-AD dementia. All got a flutemetamol PET scan, and the results were given to the clinician, who then re-evaluated the diagnosis and revised their measure of confidence and management plan.
Eighteen percent of patients originally thought to have AD had negative brain amyloid scans. This led their clinician to diagnose another disease, such as frontotemporal dementia or dementia with Lewy bodies (DLB). Twenty-three percent of those with a non-AD diagnosis had a positive scan. This led the clinician to pronounce AD as the underlying cause, though in one case they pinpointed DLB. Doctor confidence increased for 86 percent of patients. The doctors changed how they managed disease for 37 percent of the patients, again driven mostly by medication changes. Patients initially labeled AD whose scans were negative went on for more diagnostic tests, probably to seek evidence for an alternative cause of the dementia, said Zwan.
“This confirms that amyloid PET imaging does have an impact on clinical diagnosis and patient management,” said co-author Femke Bouwman. Zwan agreed, adding, “It’s really important to determine in which patients the use of this technology will add value.” She pointed out that while appropriate-use criteria have been proposed (Johnson et al., 2013), they still need to be validated by evidence from the field, especially in a memory clinic setting.
Scheltens noted that the IDEAS study enrolls only people 65 and older—the minimum age for Medicare coverage—and so will miss out on testing a younger population that could gain more from a scan. Rabinovici agreed that examining younger patients is important, saying he and colleagues are brainstorming about a parallel study to do that.
Robert Laforce, Hôpital de l'Enfant-Jésus, Quebec, presented similar results from a small study of 25 subjects. He used F18-labelled NAV4694 (see Oct 2014 news) to test how amyloid PET affects diagnosis and clinician confidence. Laforce also assessed the impact on caregivers. Participating patients were younger than 65, with an average age of 59. For nine of them, physicians changed their diagnosis after the scan: six to a non-AD diagnosis and three to AD. Overall diagnostic confidence rose from 63 percent to 88 percent. Post-scan treatment plans changed in 18 patients, including changes in medication or referral to other research projects. Treatment with cholinesterase inhibitors began in everyone with a positive scan who had taken none before, whereas most recipients with a negative scan stopped taking them.
Assessing caregiver impact proved less straightforward. Caregivers reported less anxiety and depression, but didn’t feel strongly one way or the other about the value of the scan. Laforce interprets this to mean that people appreciate a more certain diagnosis but are generally unhappy that their loved one has developed a degenerative disease. One audience member noted that the scientists did not assess caregiver status prior to the scan. Bouwman explained the researchers could have missed an important change as a result. She said that future studies by other groups should likewise add a focus on caregivers.
While the percentage benefit varies some, these studies suggest that the most eligible patient groups for a scan are those for whom doctors are uncertain of an Alzheimer’s diagnosis, and that in this group, diagnosis and treatment may change. Whether insurers will decide to cover amyloid PET is unclear. For that decision, in the United States at least, the Centers for Medicare & Medicaid Services will look to the IDEAS study to see if scans lead to better outcomes. IDEAS will enroll 18,500 participants and is powered to detect whether short-term changes in management translate to better long-term outcomes.—Gwyneth Dickey Zakaib
Ossenkoppele R, Prins ND, Pijnenburg YA, Lemstra AW, van der Flier WM, Adriaanse SF, Windhorst AD, Handels RL, Wolfs CA, Aalten P, Verhey FR, Verbeek MM, van Buchem MA, Hoekstra OS, Lammertsma AA, Scheltens P, van Berckel BN.
Impact of molecular imaging on the diagnostic process in a memory clinic.
Alzheimers Dement. 2012 Nov 16;
PubMed.
Estrogen Therapy Could Hold Back Alzheimer’s, Shrink the Brain?
The hormone estrogen has a long and fitful history in Alzheimer’s research, and at the recent Alzheimer’s Association International Conference, scientists grappled anew with its possible impact on amyloid-β deposition, brain structure, and risk. Perhaps unsurprisingly, mixed messages emerged at the meeting, held July 18-23 in Washington, D.C. On a positive note, a large epidemiological study found that estrogen given early in menopause reduced Alzheimer’s risk, and results from a small cohort indicated that early treatment with the hormone may have slowed amyloid accumulation. However, another study found that early treatment with the hormone shrank the brain. After many years of study and a batch of new data, the jury is still out on whether hormone replacement therapy is good or bad for the brain, and researchers agreed that the nature of estrogen’s effects likely come down to who is being treated, and when.
Women take supplemental estrogen to ease symptoms of menopause, which include hot flashes and insomnia. In model systems, estrogen has been reported to protect neurons and reduce the production of Aβ. Given that two-thirds of people with Alzheimer’s are women, researchers have wondered whether flagging estrogen was partly to blame, and if supplemental hormones could help. The answer is far from simple. The Women’s Health Initiative Memory Study (WHIMS) showed that hormone therapy increased the risk of dementia when initiated after age 65, but subsequent studies reported that the hormones seemed harmless or helped prevent dementia when taken during early menopause (see May 2003 news; Shao et al., 2012; Maki et al., 2011; Jun 2013 news). From this, the “critical window" hypothesis was born, which posits that early treatment with the hormone may be beneficial, while later treatment could be harmful. Roberta Diaz-Brinton, at the University of Southern California in Los Angeles, expanded on that with the “healthy cell bias of estrogen action” hypothesis, which claims that healthy neurons respond positively to estrogen, but that sickly neurons, which are more likely to be present in older age, respond negatively.
At AAIC, researchers discussed several batches of new human data. For one, a long-term, Finnish epidemiology study offered support for estrogen’s positive effects on cognition. More than 13,000 participants, who were aged 47 to 56 at the initiation of the Kuopio Osteoporosis Risk factor and Prevention (OSTPRE) study, answered questions about their use of hormone therapy as well as other health and lifestyle factors every five years from 1989 until 2009. Almost 9,000 women completed the study. In addition, researchers drew from prescription registries to corroborate the questionnaire data. They found that women who used some form of hormone therapy for more than 10 years halved their risk of Alzheimer’s disease. The study was led by Bushra Imtiaz of the University of Eastern Finland in Kuopio.
These OSTPRE results appear the opposite of the WHIMS finding that hormone therapy doubled dementia risk when it was started after age 65. However, Imtiaz told Alzforum that women in her study reported starting hormone therapy at an average age of 52, more than a decade younger than the women in the WHIMS trial. Thus she concluded that the findings supported the critical-window hypothesis.
The multicenter Kronos Early Estrogen Prevention Study (KEEPS) trial was designed in part to test the critical-window hypothesis. More than 700 participants in this study underwent four years of placebo or hormone therapy, either in the form of oral conjugated equine estrogens (CEE) or 17-β estradiol administered through a transdermal patch. Both estrogen treatments were accompanied by progesterone, which prevents the harmful uterine growth that can occur when estrogen is administered alone. KEEPS started all treatments between five months and three years after menopause onset (see Wharton et al., 2013). Recently published findings from KEEPS indicated that estrogen therapy did not alter cognition in women during the trial or within more than two years of follow-up (see Gleason et al., 2015).
In Washington, KEEPS leader Kejal Kantarci of the Mayo Clinic in Rochester, Minnesota, presented structural MRI data from a subset of 95 KEEPS participants who had scans at the beginning and end of the four-year treatment phase of the trial. Gray matter volume, as measured by expansion of the ventricles, fell in volunteers taking CEE compared to placebo. Transdermal estrogen had no effect on brain volume. Women who started their CEE treatment later in menopause had the largest ventricular volume expansion. The researchers are continuing to measure brain volume changes in this group of women longitudinally.
The findings, though from a small cohort of scanned women, were both fascinating and disturbing, Diaz-Brinton commented after the talk. However, neither of the treatment groups displayed signs of cognitive decline compared to placebo. Kantarci said the brain changes may be transient. She hopes to address this, as well as whether the structural changes ultimately have cognitive consequences later in life, in follow-up studies on this cohort. It is also important to note that use of transdermal estrogen did not significantly reduce brain volume as seen with CEE, she said.
Further evidence that estrogen can shrink the brain came from the WHIMS trial. Christina Hugenschmidt of Wake Forest School of Medicine in Winston-Salem, North Carolina, in collaboration with Diaz-Brinton, analyzed brain MRI scans of 1,400 women in the WHIMS trial more than two years after that study concluded. Half of them returned for an additional scan about five years after that. Hugenschmidt reported that during that five-year period, brain volume shrank by a decrement of -18.6 mL in women on estrogen therapy who were also diabetic. For women without diabetes, brain volume only decreased slightly (decrement of -0.4 mL) with estrogen treatment. The women in this study had initiated hormone therapy when they were all at least 65 years old. The researchers suggested that the results, published July 10 in Neurology, supported the healthy-cell-bias hypothesis, because the therapy had the strongest neurodegenerative effects on women with underlying metabolic problems. However, clearly future studies about the effects of estrogen in the diabetic brain will be needed to strengthen that support, Hugenschmidt said.
Val Lowe, also from the Rochester Mayo, reported on amyloid burden in a small subset of KEEPS participants. The researchers recruited 17 women who took CEE, 21 who took estradiol via the transdermal patch, and 30 women from the placebo arm, to undergo amyloid PET scans three years after the four-year treatment phase concluded. These women did not have a baseline PET scan at the beginning of the study. Women who had taken transdermal estradiol had lower levels of amyloid than the placebo group at this time. However, this finding was only statistically significant for ApoE4 carriers. In contrast, women who had taken CEE had comparable amyloid burden to controls three years after treatment.
The findings from this small pilot study mesh with animal studies, which have implicated estrogen in the reduction of Aβ production. CEE may not have affected amyloid because it contains a mix of non-human estrogens, including estrone, Kantarci said. That ApoE4 carriers appear to have responded more robustly to the transdermal treatment could reflect the fact that without treatment, they accumulate amyloid at a faster rate than non-carriers and thus had a bigger response to treatment, she said. Kantarci emphasized that the small numbers of participants in the study preclude any strong conclusions. She plans to test if the apparent dip in amyloid correlates with a reduction in AD risk.
Most researchers seem to accept the idea that a critical window exists during which estrogen treatment is most likely to be beneficial. “The question is, when is that window open, and when is it closed?” said Brinton. “When women have menopausal symptoms, we think that’s a good indication that it’s open.” Brinton proposed that menopausal symptoms are a sign of the ongoing bioenergetic transition in the brain that is caused by loss of estrogen (see Brinton et al., 2015). Estrogen promotes glucose metabolism in the brain, and loss of the hormone triggers the brain to go into starvation mode, which ultimately leads to compensation through metabolism of small molecules called ketone bodies. (Not to be confused with protein aggregates, ketone bodies are derivatives of fatty acids.) This ketogenic shift occurs in aging female mice as well as in AD mouse models (see Yao et al., 2010; Ding et al., 2013). Once the brain successfully starts using ketone bodies as a supplemental fuel, Brinton proposes, menopausal symptoms fade.
An extension of Brinton’s hypotheses would be that as long as estrogen therapy is started before the menopausal transition is complete, it could be continued indefinitely. However, currently physicians only recommend women take the therapy for a few years after menopause onset, then taper off. This is in part due to concerns of elevated breast cancer risk in some women with continued estrogen use. A form of estrogen that is only active in the brain could help solve that problem. One such therapy is currently in preclinical studies (see Aug 2015 news).—Jessica Shugart
Suspected Non-Alzheimer Pathophysiology: It’s Not Exactly a Snap
Suspected non-Alzheimer's pathophysiology has puzzled the field since researchers coined the term three years ago. SNAP describes cognitively normal older adults who have one of several markers of neurodegeneration but test negative for brain amyloid and have not been diagnosed with a specific neurodegenerative disorder (Jack et al., 2012). While it seems clear that SNAP is not a preclinical stage of AD, scientists are having a hard time deciding what it is. At this year's Alzheimer’s Association International Conference, held July in Washington, D.C, SNAP was a focus of presentation and debate. Some researchers reported that SNAP might correlate with other morbidities such as cerebrovascular disease. Others believe SNAP progresses very slowly or not at all, and in some cases might reflect normal aging. Most agreed that the definition of aging versus neurodegeneration might have to change, because the biomarkers used to measure degeneration each reflect slightly different processes. "While we know that some degree of brain atrophy occurs with age, we don’t really know what aging is and we really need to understand that better," said Bill Jagust, University of California, Berkeley.
SNAP has been documented in several large studies, including the Mayo Clinic Study of Aging, the Berkeley Aging Cohort, the Alzheimer's Disease Neuroimaging Initiative, the Alzheimer's Disease Research Center at Washington University, St. Louis, and others. Consistently, up to a quarter of cognitively normal older adults in these cohorts show signs of neurodegeneration on FDG PET scans, on structural MRIs of the hippocampus, or by CSF tau, while falling below the threshold for a positive amyloid scan in the brain. In this way, SNAP posed a challenge to the National Institute on Aging–Alzheimer's Association (NIA-AA) proposed diagnostic criteria for preclinical AD. They classify people into sequential stages of having: no abnormality (stage 0); amyloid (stage 1); amyloid and evidence of neurodegeneration (stage 2); or amyloid, neurodegeneration, plus subtle cognitive/behavioral decline (stage 3, Sperling et al., 2011). What causes SNAP, and what does it mean for people who have it?
SNAP Is Not Progressive.
As measured by the cognitive battery of AIBL, people with SNAP (blue) start out slightly worse than controls (green), but stay stable over the course of six years. The yellow and red groups represent preclinical Alzheimer’s. [Courtesy of Samantha Burnham, University of Melbourne, Australia.]
Researchers are beginning to refine the clinical picture of SNAP and follow it longitudinally. At AAIC, Beth Mormino, Massachusetts General Hospital, Charlestown, reported on SNAP in the Harvard Aging Brain Study. As with other cohorts, a quarter of the 242 people enrolled fell into this category. At an average age of 76, they tended to be older than cognitive normals who tested negative for neurodegeneration, and less likely to carry an ApoE4 risk allele than normals who were positive for amyloid. This was true in other cohorts, as well. Mormino tested for several morbidities that might explain the neurodegeneration markers in the SNAP group, but found no correlation with cardiovascular disease risk factors such as high body-mass index or white-matter hyperintensities (WMH) in the brain. Likewise there was no correlation to tau tangles in various regions of the brain, including in the medial-temporal lobe, the entorhinal cortex, and parahippocampal gyrus. Mormino found that people with SNAP and age-matched controls who test negative for both amyloid and markers of neurodegeneration (i.e., stage 0 for AD) similarly take up the neurofibrillary tangle tracer T-807 in the brain. "At that age, tangles in the medial temporal lobe of the brain are prevalent, even in people at stage 0," Mormino said. "While there could be tau pathology in SNAP, it does not seem specific to it," she added.
The findings suggest that SNAP is not the same as PART, aka primary age-related tauopathy, which was evoked to describe tangle pathology in the absence of Aβ pathology (see Nov 2014 news).
Melissa Murray, Mayo Clinic, Jacksonville, Florida, agreed that tau pathology does not fully explain SNAP. Murray autopsied 65 people who had taken part in the Mayo Clinic Study of Aging or enrolled in the Mayo ADRC. During life, 11 had been diagnosed with SNAP based on glucose hypometabolism in FDG PET scans. At death, none of them fulfilled NIA-AA pathological criteria for Alzheimer's disease, supporting the SNAP designation. What explains the neurodegeneration marker? Postmortem pathology revealed that one person had had hippocampal sclerosis, four argyrophilic grain disease, and six fulfilled criteria for PART.
Murray’s data supports the idea that tau pathology alone cannot explain all SNAP cases. In fact, seven of these 11 had moderate to severe arteriosclerosis and white-matter disease, she said, though it is not clear how these pathological findings relate to SNAP. The timing of the autopsies complicates analysis of this data. The volunteers were 83 years old, on average, when they had their FDG PET scan, and 87 when they died. Of the 11 cases, five came to autopsy within three years of the initial SNAP diagnosis, but the delay was longer for the other six so some of the pathology could have developed after the scan.
Previous analysis of the Mayo cohort had reported that patterns of WMHIs in SNAP tracked more closely with those in stage 2/3 preclinical AD than those in stage 0/1. The incidence of hypertension and diabetes were also similar in SNAP and stage 2/3 AD, and higher than in the stage 0/1 (see Knopman et al., 2012). These findings indicate that neurodegeneration in cognitively normal individuals may be related to vascular pathology, said Miranka Wirth, Inserm, Caen, France, who is now at Charité University Medicine in Berlin.
To explore this relationship, Wirth compared hippocampal volume, glucose metabolism, and regional cortical atrophy with white-matter lesions in people enrolled in the Berkeley Aging Cohort. In people without brain amyloid, the size of the white-matter lesions correlated with thinning of the cortex; intriguingly, however, their cortex thinned out in regions that do not typically atrophy in AD. Conversely, in people with brain amyloid, white-matter lesions exacerbated cortical thinning in those areas of the brain that typically accumulate Aβ. This is a finding Sylvia Villeneuve had previously reported while working with Wirth at Bill Jagust's lab (Villeneuve et al., 2014).
Cerebrovascular disease also appears more common in SNAP cases than in controls in the Australian Imaging, Biomarkers, and Lifestyle (AIBL) study. In Washington, Samantha Burnham from the Commonwealth Scientific and Industrial Research Organisation (CSIRO) in Melbourne, Australia, reported among that among 132 AIBL participants diagnosed with SNAP, 36 percent tested positive for WMHIs, a much higher proportion than in normal elderly controls.
Taken together, the SNAP versus AD patterns of imaging and fluid markers suggest that vascular factors can accelerate neurodegeneration in vulnerable brain regions, which vary depending on the presence of a disease-specific pathology such as Aβ.
Does SNAP Get Worse?
Based on current data, it appears that cognition in people with SNAP deteriorates much more slowly, if at all, than in those who have both brain amyloid and neurodegeneration. In keeping with this idea, Mormino reported at AAIC that the practice effect in the Harvard Aging Brain Study, aka the ability of cognitively normal people to score better on repeat testing, diminishes in those with SNAP, much like it does in people at stage 1 AD. At a group level, about 15 percent of people with SNAP declined to CDR 0.5 over an average of two years. Mormino said that is about the same rate as people with stage 1 AD, but much slower than people with stage 2. Researchers at the Mayo Clinic and at Washington University have seen similar rates of decline, she said. More recently, Mormino measured change in specific cognitive domains. Executive function and memory declined in SNAP at about the same rate as in people with stage 1 AD. "The pattern is always the same," said Mormino.
In the AIBL study, people with SNAP decline even more slowly, Burnham reported. She followed 545 cognitively normal healthy controls, testing them with the Preclinical Alzheimer's Cognitive Composite, a battery that measures executive function, episodic memory, and global cognition (see June 2014 news). The 132 people designated as SNAP had slightly poorer scores at baseline, but maintained their cognition over the next seven years, just like controls who tested negative for both amyloid and markers of neurodegeneration (see image above). "SNAPs do not decline and behave over time like the Aβ-negative/neurodegeneration-negative group," said Burnham.
If not cognition, does neurodegeneration worsen in SNAP? Not much, apparently. At an AAIC imaging preconference, Pierrick Bourgeat from CSIRO, Brisbane, Australia, reported atrophy rates among 320 people enrolled in AIBL. This dataset has a full eight years of follow-up. Hippocampal volume fell fastest in 11 people who tested positive for both amyloid and neurodegeneration (stage 2). In 48 people who had evidence of amyloid but none of neurodegeneration, the hippocampus shrank more slowly. In contrast, in the 55 SNAP cases, hippocampal volume behaved quite differently. It started out low, but did not shrink over the course of eight years.
Bourgeat reported the same pattern for temporal and parietal cortex atrophy, with fast decline in the group with both pathology markers and slowest in SNAP. Other cortical regions trended the same way. Likewise, ventricular volumes expand fastest in people with amyloid/neurodegeneration markers and slowest in SNAP.
The upshot of all this, said Bourgeat, is that SNAP follows a wholly different trajectory to Alzheimer's disease (see image below).
More than a Snapshot.
Over eight years, there was no atrophy in people with SNAP. [Courtesy of Pierrick Bourgeat, University of Melbourne, Australia.]
Then what in the world is SNAP? In his talk, Jagust cautioned that the construct is artificial. He noted that scientists apply a bimodal distribution for amyloid, classifying it as either positive or negative based on a specific cut-off, but treat hippocampal volume and glucose metabolism as continuous measures. "It is clear to me that SNAP is a biomarker construct that classifies people based on thresholds for Aβ and biomarkers that have no natural cut point," Jagust said. "A lot may depend on thresholds that are arbitrarily chosen."
On top of that, biomarkers of neurodegeneration, be they hypometabolism, atrophy, or neuronal damage reflected as CSF tau, are not necessarily equivalent. They all measure slightly different things and only modestly correlate with each another, noted David Knopman, Mayo Clinic, Rochester, Minnesota. "The field could get itself into trouble if investigators are not appropriately cautious about specifying which neurodegeneration biomarker they are referring to." In fact, Brian Gordon and colleagues at WashU reported that among 212 normal older adults (mean age 67), CSF tau and hippocampal volume correlated poorly, agreeing only 57 percent of the time. "Different people have either one or the other marker. They are not equivalent," said Gordon. Because high CSF tau and low hippocampal volume are not concordant, i.e., they don't always occur in the same person, it is not clear that they reflect AD pathology, he said. By comparison, amyloid PET and CSF Aβ42 are highly concordant, Gordon said.
This raises the question of exactly what causes the positive neurodegeneration marker in SNAP to begin with. "A lot of different biological processes can lead to abnormal hippocampal volume or abnormal CSF tau," said Gordon. "Unlike amyloid, those processes are not AD specific." The markers might reflect underlying cardiovascular disease, argyrophilic grain disease, hippocampal sclerosis, diabetes, stress, or some yet-to-be-discovered problem. Jagust noted that a certain amount of atrophy occurs with age (see Jun 2013 news).
In short, scientists don't know what causes the neurodegenerative signal in SNAP. Victor Villemagne, University of Melbourne, believes it may be unrelated to AD. "For me, the SNAP findings demonstrate that there is no 'no-Aβ' pathway to AD. You might have some degree of cognitive impairment and 'AD-like' neurodegeneration, but if Aβ is not present you have a completely different progression over time compared to when it is present, suggesting two completely different underlying pathophysiologies." With SNAP, we can now identify people who definitely do not belong in AD therapeutic trials, he added. Vice versa, many scientists agreed that AD therapy trials may want to refrain from using structural MRI as an inclusion criterion. If an AD trial enrolls patients due to their low baseline hippocampal volume, then it may end up with SNAP participants who do not have AD and will barely progress during the duration of the trial.
If SNAP does not progress over the course of eight years, can it be called neurodegeneration? "There is some reason for the abnormalities, but it is not progressive neurodegeneration," said Gordon. Jagust agreed that having a small hippocampus may be unrelated to a progressive disease and recommended that SNAP be reconsidered. Jagust suggested incorporating evidence of neurodegeneration into SNAP. Categorizing subjects by longitudinal change would undoubtedly alter the proportion of people who are called SNAP and may be a better, and more specific, index of brain degeneration, Jagust said.
"We do know that if you have neurodegeneration plus Aβ in the brain, then you are in trouble," said Jagust. "The key question is can you get neurodegeneration without Aβ? That's the fundamental question. We need to follow people longitudinally to answer it."—Tom Fagan

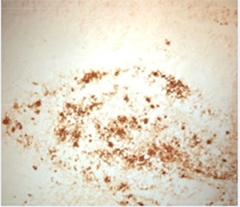
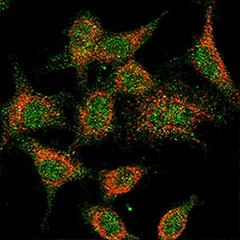


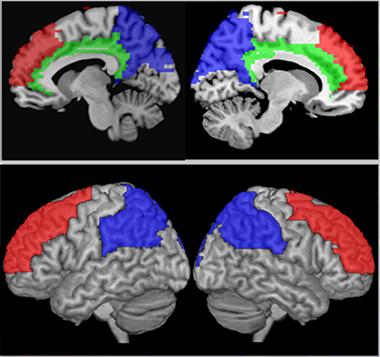
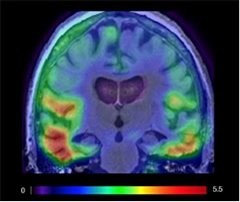
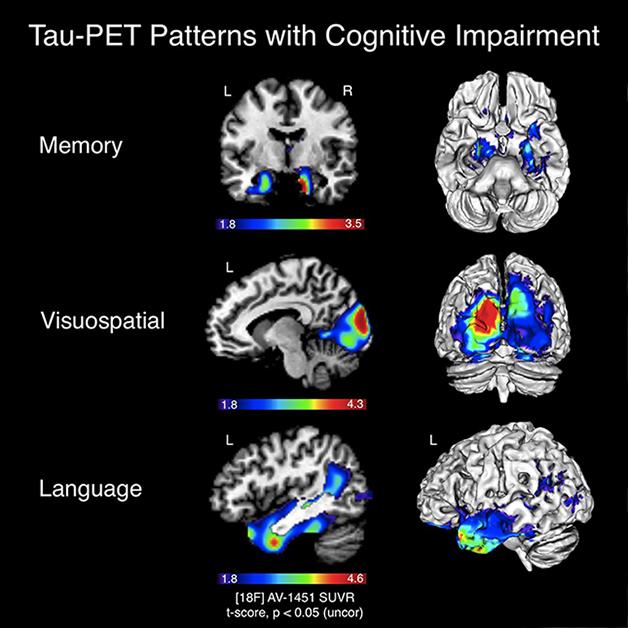
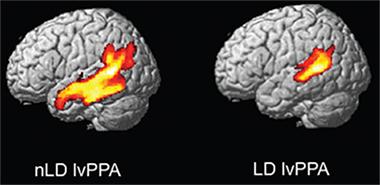

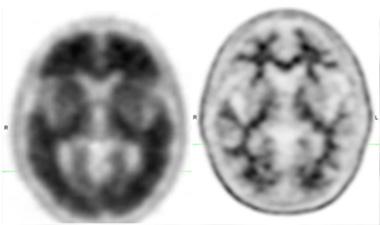

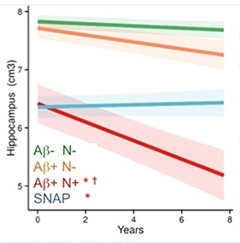
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.