Shape of a Hug: How the Embrace of a Therapeutic Aβ Antibody Really Matters
Quick Links
One must know oneself and one's enemy to prevail in battle, according to the legendary militarist Sun Tzu. The first shot in the war against amyloid was fired long ago, but researchers are still getting to know which species of Aβ pose a threat and are fashioning their antibody weaponry accordingly.
Take a recent crystal structure of solanezumab bound to a fragment of Aβ. Published in the April 16 issue of Scientific Reports, it showed that in the antibody’s clutches, Aβ exhibited a unique configuration that was part helix, part extended coil. The study's authors, Luke Miles and collaborators at St. Vincent’s Institute for Medical Research in Fitzroy, Australia, noted that the amino acids in solanezumab that interacted with Aβ had nearly the same sequence as those in crenezumab. This suggested that the two antibodies bind Aβ in the same way, despite reports that the antibodies bind different forms of Aβ. A patent application on Biogen’s BIIB037/aducanumab antibody disclosed that it binds a linear stretch of Aβ. More broadly, researchers believe that the precise spatial details of how Aβ antibodies in clinical development engage their antigen may correlate with which ones succeed in therapeutic trials, both with regard to amyloid reduction and minimal collateral activity such as vasogenic edema. Binding experiments and a slowly growing portfolio of crystal structures are beginning to build this knowledge base.
Aβ Antibodies—What’s in That Pocket?
Each of the clinical anti-Aβ antibodies target specific parts of the infamous peptide. For example, solanezumab and crenezumab bind Aβ’s midsection, while bapineuzumab, gantenerumab, and aducanumab recognize the N-terminal region. In turn, the antibodies have been reported to preferentially recognize different Aβ complexes. Solanezumab has a penchant for soluble monomers, while crenezumab recognizes monomers, oligomers, and aggregates. The N-terminal antibodies recognize primarily aggregates, though some are reported to recognize soluble forms as well. Researchers speculate that these patterns reflect the exposure of Aβ’s N-terminus to solution in monomeric, aggregated, and plaque-deposited forms (Zago et al., 2012). In contrast, Aβ’s midsection is masked when it forms aggregates but exposed in monomers. The antibodies further differ by their affinity for their respective targets.

Five antibodies bind different epitopes and conformations of Aβ.
In 2012, Miles started to investigate how therapeutic antibodies engage Aβ. “We believe that developing structure-activity relationships for the immunotherapies will be informative in understanding why one antibody works and another does not,” Miles wrote to Alzforum. His group first reported that bapineuzumab bound the N-terminus of Aβ in a helical form, offering a possible explanation for why the antibody also recognizes aggregates (see Feb 2013 news).
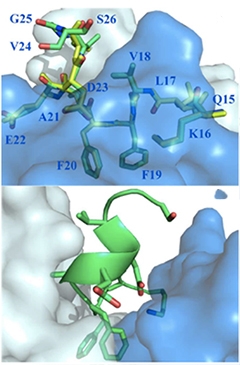
Hybrid Hug.
Solanezumab heavy (blue) and light (white) chains cradle Aβ. Side chains of phenylalanines F19 and F20 bore into the core of the binding pocket (top). Residues C-terminal to F20 snap into a helix (bottom).
To address the structural relationship between solanezumab and Aβ, first author Gabriella Crespi and colleagues crystallized the Fab portion of solanezumab (containing the Aβ binding region) with Aβ12-28—the portion of the peptide reportedly recognized by the antibody. X-ray analysis resolved Aβ residues 16-26 complexed with the antibody fragment. In this structure, the antibody made extensive contacts with the side chains of Aβ residues lysine 16, phenylalanine 19, phenylalanine 20, and aspartate 23, as well as with the Aβ peptide backbone. The researchers found the two phenylalanine side chains wedged deep within the antibody; their dive into the antibody’s folds consumed nearly half of the 960A2 surface area with which the antibody and Aβ interacted.
In the antibody binding pocket, residues 16 to18 assumed an extended coil configuration, lying flat over the solanezumab surface. Phenylalanine residues 19-20 projected down into the pocket. Residues 21-26 formed a helix held together by hydrogen bonds between Aβ residues. The researchers described the structure as an intermediate between a previously reported crystallographic β-sheet and a helical structure reported in NMR studies. They designated residues 16-20 (KLVFF) as the core epitope required for antibody specificity, and the helical region as affinity enhancer. They predicted that the core epitope would become masked in the context of Aβ oligomers, which could explain why solanezumab binds only monomers.
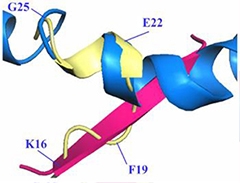
Two in One?
The shape of Aβ in the grip of solanezumab (yellow) is proposed to be a hybrid of previously reported β-sheet (pink) and helical structure (blue). A hairpin turn separates the two structural domains.
In a separate argument, Miles and colleagues claim that solanezumab recognizes hundreds of other proteins, a subset of which share the KLVFF motif. Researchers at Eli Lilly dispute this (see comment by DeMattos and colleagues, below, and Siemers et al., 2014).
While it is unclear what this hybrid Aβ structure represents, Colin Masters of the University of Melbourne, Australia, speculated that it could be an intermediate between monomeric and oligomeric forms. Miles agreed, writing, “The structure suggests to me that the conformation solanezumab recognizes is on a pathway to oligomerization.”
Next, Crespi and colleagues compared the binding pockets of a recombinant Fab fragment of solanezumab with that of crenezumab. The sequences are identical bar two residues—serine 33 and phenylalanine 36 in solanezumab are tyrosine and glycine, respectively, in crenezumab. Using homology modelling, the researchers determined that these changes would not affect the shape of the Aβ-antibody complex, and predicted that crenezumab mimics solanezumab folding and binds the same Aβ conformation. That said, exchange of the serine in solanezumab to tyrosine in crenezumab would remove a hydrogen bond between crenezumab and Aβ, while the second amino acid difference (glycine instead of phenylalanine) would lessen the hydrophobic interaction between the crenezumab and Aβ phenylalanines 19 and 20. Crespi suggested that these differences could explain why solanezumab binds Aβ with picomolar affinity, while crenezumab has a weaker, nanomolar affinity.
Unlike solanezumab, crenezumab is reported to bind aggregated forms of Aβ (Adolfsson et al., 2012). Miles suggested that perhaps solanezumab does as well, but this binding to aggregates may have gone unnoticed amid its high affinity for monomers. Ron Black, who headed Pfizer’s Phase 3 bapineuzumab trial, offered the same hypothesis.
How do the N-terminal antibodies compare? Researchers led by Guriqbal Basi, then at Elan, published a crystal structure showing 3D6, the murine parent of bapineuzumab, binds Aβ1-7 as an α-helix (see Jan 2009 conference news; Feinberg et al., 2014). Later, Miles solved a crystal structure that shows bapineuzumab binding a unique helix at the Aβ N-terminus, though he does not know which form of Aβ assumes this configuration (see Crespi et al., 2014).
In marked contrast, Biogen's patent application discloses that BIIB037/aducanumab binds residues 2 to 9 of Aβ in an extended coil conformation. Glutamate 3, phenylalanine 4, arginine 5, and histidine 6 all make contact with the antibody, with the phenylalanine and histidine being buried deep in the binding pocket. Epitope mapping identified amino acids 3-6 as the primary epitope for aducanumab.

Extended Coil.
Some antibodies to the Aβ N-terminal bind a linear structure (yellow). [Image courtesy of Basi et al. Copyright © 2010, American Society for Biochemistry & Molecular Biology.]
Other monoclonal antibodies targeting Aβ at the N-terminus, for example 12A11 and others, also bind this linear Aβ structure and have sequence similarities with aducanumab in the binding region (see image at right and Basi et al., 2010). As for gantenerumab, it recognizes a linear conformation in Aβ as well, but its epitope comprises residues 1-11. Moreover, this antigen is bound by antibody in reverse orientation compared with the crystal structure of Aβ as reported for 12A11 (Feinberg et al., 2014). Furthermore, the complementarity determining regions of gantenerumab are different from those of aducanumab and the 12A11 family (see Bohrmann et al., 2012).
Epitope May Make a Difference in the Clinic
Are those just arcane details? Or does the atom-by-atom way each antibody binds Aβ influence the outcome of therapeutic trials? Some similarities in past trials hint as much, scientists say. Both the mid-region binding solanezumab and crenezumab had overall negative results, but sub-group and post-hoc analyses hinted at a benefit in mild AD (see Aug 2012 news; Oct 2012 news; July 2014 conference news). Both produced low rates of amyloid-related imaging abnormalities with brain edema (ARIA-E).
These similarities could arise from binding specificity and the extent of inflammation triggered, Masters suggested. Because solanezumab ignores plaques deposited in the brain, it largely avoids arousing the microglia-triggered inflammation associated with plaque clearance and ARIA-E. Crenezumab binds aggregated Aβ in addition to monomer. This would suggest that it triggers more ARIA-E, but whereas solanezumab is an IgG1 antibody, crenezumab’s IgG4 subclass may provide an explanation for the lower ARIA-E rate observed with crenezumab (see Jul 2012 news).
Trial results from N-terminal antibodies also share some commonalities. Bapineuzumab, gantenerumab, and aducanumab all reduced amyloid burden and provoked ARIA-E, in keeping with the idea that mobilizing plaque-bound Aβ activates microglia. But there were stark differences too. High ARIA-E sans cognitive benefit sunk bapineuzumab in 2012, and a Phase 3 trial of gantenerumab ended because the drug failed a futility analysis for lack of a cognitive benefit (see Dec 2014 news). In contrast, interim data from an ongoing Phase 1b trial showed aducanumab nearly eliminated brain amyloid with hints of a cognitive benefit.
Where do the differences come from? Masters speculated that the extended conformations recognized by aducanumab and gantenerumab could represent fibrillar Aβ species rather than the helical structure recognized by bapineuzumab. Furthermore, aducanumab was derived from a natural Aβ-specific antibody from an older person. This suggests this antibody recognizes an Aβ conformation that truly exists in the human brain and has clinical relevance, Masters said. Bapineuzumab and gantenerumab were raised against synthetic and phage-displayed peptides, respectively, and then humanized.
Even with a growing collection of antibody-Aβ structures in hand, it remains unclear whether targeting monomeric, oligomeric, or fibrillar forms of Aβ is the right strategy for treating AD, and which molecular interactions will prove to work best.—Jessica Shugart
References
Therapeutics Citations
News Citations
- Packed in Crystals, Bapineuzumab-Aβ Give Up Secrets
- Eibsee: Antibody Binding Crystal Clear; New Vaccine in the Mix
- Phase 3 Solanezumab Trials "Fail"—Is There a Silver Lining?
- The Solanezumab Benefit: Oh, So Small, But Probably Real
- Crenezumab Disappoints in Phase 2, Researchers Remain Hopeful
- A Close Look at Passive Immunotherapy Newbie, Crenezumab
- End of the RoAD for Gantenerumab? Roche Declares Prodromal Alzheimer’s Trial Futile
Paper Citations
- Zago W, Buttini M, Comery TA, Nishioka C, Gardai SJ, Seubert P, Games D, Bard F, Schenk D, Kinney GG. Neutralization of soluble, synaptotoxic amyloid β species by antibodies is epitope specific. J Neurosci. 2012 Feb 22;32(8):2696-702. PubMed.
- Siemers E, Dean RA, DeMattos RB, Hutton ML, Blennow K, Shaw LM, Holtzman DM. Anti-Aβ antibody target engagement: commentary regarding Watt et al. Acta Neuropathol 127:803-810 (2014). Acta Neuropathol. 2014 Oct;128(4):609-10. Epub 2014 Aug 14 PubMed.
- Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, Watts RJ. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci. 2012 Jul 11;32(28):9677-89. PubMed.
- Feinberg H, Saldanha JW, Diep L, Goel A, Widom A, Veldman GM, Weis WI, Schenk D, Basi GS. Crystal structure reveals conservation of amyloid-β conformation recognized by 3D6 following humanization to bapineuzumab. Alzheimers Res Ther. 2014;6(3):31. Epub 2014 Jun 2 PubMed.
- Crespi GA, Ascher DB, Parker MW, Miles LA. Crystallization and preliminary X-ray diffraction analysis of the Fab portion of the Alzheimer's disease immunotherapy candidate bapineuzumab complexed with amyloid-β. Acta Crystallogr F Struct Biol Commun. 2014 Mar;70(Pt 3):374-7. Epub 2014 Feb 20 PubMed.
- Basi GS, Feinberg H, Oshidari F, Anderson J, Barbour R, Baker J, Comery TA, Diep L, Gill D, Johnson-Wood K, Goel A, Grantcharova K, Lee M, Li J, Partridge A, Griswold-Prenner I, Piot N, Walker D, Widom A, Pangalos MN, Seubert P, Jacobsen JS, Schenk D, Weis WI. Structural correlates of antibodies associated with acute reversal of amyloid beta-related behavioral deficits in a mouse model of Alzheimer disease. J Biol Chem. 2010 Jan 29;285(5):3417-27. PubMed.
- Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, Rothe C, Urban M, Bardroff M, Winter M, Nordstedt C, Loetscher H. Gantenerumab: A Novel Human Anti-Aβ Antibody Demonstrates Sustained Cerebral Amyloid-β Binding and Elicits Cell-Mediated Removal of Human Amyloid-β. J Alzheimers Dis. 2012;28(1):49-69. PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Crespi GA, Hermans SJ, Parker MW, Miles LA. Molecular basis for mid-region amyloid-β capture by leading Alzheimer's disease immunotherapies. Sci Rep. 2015 Apr 16;5:9649. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Eli Lilly and Company
Eli Lilly and Company
Acumen Pharmaceuticals
Crespi and colleagues performed X-ray crystallography studies on two central domain-targeting anti-Aβ antibodies in order to identify structural interactions of the antibodies with the Aβ peptide (Crespi et al., 2015). Based upon the patent literature, the group developed their own “bio-similar” versions of solanezumab and crenezumab and subsequently investigated X-ray structural relationships in the presence of the Aβ fragment comprising amino acids 12 to 28. Their X-ray structural studies demonstrated that the antibody they produced corresponding to solanezumab bound to an Aβ epitope comprising amino acids 16 to 26, a finding that is consistent with our own internal epitope-mapping studies. Also, our epitope-mapping data indicates that the KLVFF domain (amino acids 16 to 20) by itself is not capable of binding to solanezumab by Biacore analyses. Of additional interest was the group’s finding that the Aβ peptide bound by the antibody had a central domain helical element that is quite different from the β-sheet structure found in oligomerized forms of the peptide. Our own studies have demonstrated the selectivity of solanezumab for soluble monomeric forms of the Aβ peptide (DeMattos et al., 2001; Racke et al., 2005). Our viewpoints diverge when the authors speculate in the discussion that the current structural work may explain their previously published observation of potential cross-reactivity of solanezumab and crenezumab to a number of proteins (Watt et al., 2014).
We believe it is important to highlight that there were a number of technical issues with the author's earlier manuscript (Watt et al., 2014), especially as it pertains to the potential identification of cross-reacting proteins. These perspectives were highlighted in a letter to the editor by Lilly colleagues and several academic thought leaders (Siemers et al., 2014). To truly characterize a protein signature as “cross-reacting” from immunoprecipitation (IP) pull-down followed by MS/MS experiments on plasma, a matrix that has an immensely high protein concentration of 35 to 50 g/L, multiple rigorous controls and orthogonal biochemical approaches are necessary (binding assays, Biacore affinity measures, occupancy measures, and selectivity assays). Without rigorous orthogonal approaches, it is impossible to determine whether the signature was specific or merely representative of normal non-specific binding that would be expected for any globular protein. For example, would the authors believe the binding should be described as “cross-reacting” if they were to discover that it only represented a tiny fraction of the plasma pool (i.e. < 0.1 percent)? The specific binding of solanezumab results in an approximately 1000-fold increase in plasma Aβ (DeMattos et al., 2001). Indeed, our laboratory has recently analyzed non-specific plasma protein binding to other purified antibodies and we learned that hundreds, if not thousands of proteins can be detected in the IP-pull down experiments. Importantly, we analyzed these proteins by parallel approaches, such as SDS-PAGE followed by silver stain, where we determined that these non-specific protein interactions amounted to extremely low mass levels as compared to their concentration in plasma. Also, Crespi and colleagues conclude that the KLVFF homology is underlying the potential cross-reactivity (Crespi et al., 2015); however, our own epitope-mapping experiments in conjunction with Biacore analyses show that the KLVFF sequence is not capable of binding to solanezumab by itself.
Furthermore, in the publications by Watt et al. and Crespi et al., the authors speculate that binding to the cross-reacting plasma proteins might result in a lack of target engagement of the central domain antibodies to plasma Aβ, and that if cognitive benefit is observed in current clinical studies, the effect could actually be due to unknown pharmacology linked to the cross-reacting proteins. To address the question of Aβ target engagement, in our subsequent letter to the editor we referenced an extensive literature from over two decades of work from multiple groups using multiple orthogonal biochemical approaches (multiple ELISA formats including multiple clinically validated assays, Western blotting, RIAs, acid urea gels, and MALDI mass spec) that demonstrate that the central domain anti-Aβ antibodies engage soluble Aβ in plasma and CSF (DeMattos et al., 2001; Racke et al., 2005; Lachno et al., 2013; Siemers et al., 2010; DeMattos et al., 2002; DeMattos et al., 2002; Dodart et al., 2002; Yamada et al., 2009; Seubert et al., 2008; Bard et al., 2003; Seubert et al., 1992; Adolfsson et al., 2012; Doody et al., 2014; Farlow et al., 2012). These orthogonal approaches are more sensitive and quantitative than the SELDI methods employed by this group. Regarding the suggestion that binding to cross-reacting proteins leads to novel unknown pharmacology, we believe this hypothesis is premature, as their MS/MS experiments to explore this question did not adequately control for normal non-specific protein binding. Prior to this type of assertion, it would be prudent to perform the orthogonal biochemical approaches listed above. Moreover this hypothesis is inconsistent with the historical data for solanezumab referenced above.
The clinical experience with solanezumab to date is consistent with a lack of meaningful binding to other proteins. Over 130,000 infusions of solanezumab have been administered to nearly 4,000 patients with AD, for durations in some cases over five years. The Phase 1, 2, and 3 clinical studies have documented robust target engagement (plasma and CSF) with good tolerability (Siemers et al., 2010; Doody et al., 2014; Farlow et al., 2012). The EXPEDITION 3 trial is now fully enrolled and the last patient visit is anticipated to occur around October 2016. If the EXPEDITION 3 trial confirms the slowing of cognitive and functional decline in mild AD that appeared to be present in EXPEDITION and EXPEDITION 2 pooled data (Carlson et al., 2013), and also confirms what appears thus far to be good tolerability, these findings will support the amyloid cascade hypothesis and the targeting of soluble Aβ as one therapeutic avenue for the treatment of Alzheimer’s disease.
Eli Lilly's Michael Hutton, chief science officer, neurodegeneration, and Jirong Lu, senior research fellow, contributed to this comment.
References:
Crespi GA, Hermans SJ, Parker MW, Miles LA. Molecular basis for mid-region amyloid-β capture by leading Alzheimer's disease immunotherapies. Sci Rep. 2015 Apr 16;5:9649. PubMed.
DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001 Jul 17;98(15):8850-5. Epub 2001 Jul 3 PubMed.
Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, Demattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005 Jan 19;25(3):629-36. PubMed.
Watt AD, Crespi GA, Down RA, Ascher DB, Gunn A, Perez KA, McLean CA, Villemagne VL, Parker MW, Barnham KJ, Miles LA. Do current therapeutic anti-Aβ antibodies for Alzheimer's disease engage the target?. Acta Neuropathol. 2014 Jun;127(6):803-10. Epub 2014 May 7 PubMed.
Siemers E, Dean RA, DeMattos RB, Hutton ML, Blennow K, Shaw LM, Holtzman DM. Anti-Aβ antibody target engagement: commentary regarding Watt et al. Acta Neuropathol 127:803-810 (2014). Acta Neuropathol. 2014 Oct;128(4):609-10. Epub 2014 Aug 14 PubMed.
Lachno DR, Evert BA, Vanderstichele H, Robertson M, Demattos RB, Konrad RJ, Talbot JA, Racke MM, Dean RA. Validation of Assays for Measurement of Amyloid-β Peptides in Cerebrospinal Fluid and Plasma Specimens from Patients with Alzheimer's Disease Treated with Solanezumab. J Alzheimers Dis. 2013 Jan 1;34(4):897-910. PubMed.
Siemers ER, Friedrich S, Dean RA, Gonzales CR, Farlow MR, Paul SM, Demattos RB. Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease. Clin Neuropharmacol. 2010 Mar-Apr;33(2):67-73. PubMed.
Demattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002 Apr;81(2):229-36. PubMed.
Demattos RB, O'Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2002 Aug 6;99(16):10843-8. PubMed.
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002 May;5(5):452-7. PubMed.
Yamada K, Yabuki C, Seubert P, Schenk D, Hori Y, Ohtsuki S, Terasaki T, Hashimoto T, Iwatsubo T. Abeta immunotherapy: intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci. 2009 Sep 9;29(36):11393-8. PubMed.
Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, Kling K, Schenk D, Johnson-Wood K, Schroeter S, Gill D, Jacobsen JS, Pangalos M, Basi G, Games D. Antibody capture of soluble Abeta does not reduce cortical Abeta amyloidosis in the PDAPP mouse. Neurodegener Dis. 2008;5(2):65-71. PubMed.
Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, Guido T, Hoenow K, Hu K, Johnson-Wood K, Khan K, Kholodenko D, Lee C, Lee M, Motter R, Nguyen M, Reed A, Schenk D, Tang P, Vasquez N, Seubert P, Yednock T. Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer's disease-like neuropathology. Proc Natl Acad Sci U S A. 2003 Feb 18;100(4):2023-8. Epub 2003 Feb 3 PubMed.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000 Aug;6(8):916-9. PubMed.
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992 Sep 24;359(6393):325-7. PubMed.
Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, Watts RJ. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci. 2012 Jul 11;32(28):9677-89. PubMed.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R, Alzheimer's Disease Cooperative Study Steering Committee, Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014 Jan 23;370(4):311-21. PubMed.
Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, Friedrich S, Dean RA, Gonzales C, Sethuraman G, Demattos RB, Mohs R, Paul SM, Siemers ER. Safety and biomarker effects of solanezumab in patients with Alzheimer's disease. Alzheimers Dement. 2012 Jul;8(4):261-71. PubMed.
Carlson C, Sundell K, Estergard W, Raskin J, Liu-Seifert H, Case M, Sethuraman G, Chen Y, Henley D, DeMattos R, Siemers E. Efficacy of solanezumab in patients with mild or moderate Alzheimer's disease: Pooled analyses findings from two phase III studies. Alzheimer’s Dement. 2013 Jul;9 (4 Suppl):P139.
Certara
Besides specific interaction details of the antibody with the amyloid peptide, one also has to consider the larger environment of Aβ synthesis, breakdown, forward and backward aggregation parameters, antibody-mediated microglia-dependent clearance, and impact on cognitive functioning through interaction with glutamate (Wang et al., 2013) and other neurotransmitter system such as the α7 nAchR (Lee et al., 2014).
Based on human SILK data, Aβ1-40 is synthesized faster and cleared more slowly than Aβ1-42 (Potter et al., 2013). Together with biochemical observations on the aggregation rates of these two amyloid peptides (Garai and Frieden, 2013), this results in a complex relationship between monomers, oligomers, and aggregated forms of both peptides. Assuming also that oligomers might be formed from breakdown of plaques, it is clear that many non-linear processes play a role and that the clinical outcome might depend upon the specific epitopes and their affinities of the antibodies.
Recently we showed that the choice of the epitope for the antibody does matter, both in terms of biomarkers (CSF Aβ levels) and functional outcome (effect on ADAS-Cog). Our approach was a mechanism-based Quantitative Systems Pharmacology and humanized computer model, constrained by existing clinical data on amyloid modulation in AD patients based on integration of biological, biochemical and clinical domain expertise (Geerts 2014).
References:
Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, Mawuenyega K, Blazey T, Goate A, Chott R, Yarasheski KE, Holtzman DM, Morris JC, Benzinger TL, Bateman RJ. Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med. 2013 Jun 12;5(189):189ra77. PubMed.
Garai K, Frieden C. Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ. Proc Natl Acad Sci U S A. 2013 Feb 26;110(9):3321-6. PubMed.
Wang Y, Zhou TH, Zhi Z, Barakat A, Hlatky L, Querfurth H. Multiple effects of β-amyloid on single excitatory synaptic connections in the PFC. Front Cell Neurosci. 2013;7:129. Epub 2013 Sep 3 PubMed.
Lee L, Kosuri P, Arancio O. Picomolar Amyloid-β Peptides Enhance Spontaneous Astrocyte Calcium Transients. J Alzheimers Dis. 2014 Jan 1;38(1):49-62. PubMed.
Geerts H. Using in silico mechanistic disease modeling to address the effect of amyloid beta manipulation on cognitive clinical readouts. Alzheimers Dement. 2011 Jul; 7(4 Suppl),S774–5.
Make a Comment
To make a comment you must login or register.