A new meeting in the calendar, Tau2020, covered the biology and pathology of all tauopathies, both primary and secondary. Some of the 650 attendees saw it as an inflection point in tau research, getting researchers to think more broadly and bringing their collective knowledge to bear on a common problem. Highlights included a new staging scheme for tau progression in AD, new ligands for primary tauopathies, cryoEM structures for α-synuclein, and a receptor that facilitates spread of mutant tau in mouse models.
Tau2020: Meeting for Tauopathies Debuts Genetic Variants
Tau2020, a new international conference on all things—you guessed it—tau, unfolded in Washington, D.C, February 12 to13. This open meeting grew out gatherings of the Tau Consortium held in previous years, which were funded by the Rainwater Charitable Foundation and were closed to the public. Tau2020 was larger, drawing 650 researchers from 21 countries, and was co-organized by RCF, the Alzheimer’s Association and CurePSP. Unlike other conferences, Tau2020 covered all tauopathies, including Alzheimer’s disease, Progressive Supranuclear Palsy (PSP), frontotemporal dementias, and Pick’s disease. “I see this meeting as an inflection point in tau biology,” GilRabinovici, University of San Francisco, told Alzforum. “It brought together researchers studying AD with those studying primary tauopathies and got them thinking outside their own silos.”
The Rainwater foundation has poured $100 million into tau research since its founder Richard Rainwater was diagnosed with PSP in 2009. “Tau2020 grew from a desire to partner with other groups committed to sharing ideas and moving the science forward,” said Diana Kerwin from the Kerwin Research Center, a private clinic in Dallas. Kerwin was Rainwater’s geriatrician before his passing in 2015. She co-led the Tau2020 program committee with Peter Davies, Albert Einstein College of Medicine, Manhasset, New York.
The meeting touched on most aspects of tau biology, from genetics and protein structure to toxicity, biomarkers, imaging, and therapeutics. An all-plenary format and plenty of time for panel discussions ensured lively debate. Scientists presented new data at this meeting. There were high-resolution cryoEM structures of α-synuclein, a newly discovered receptor for tau that might facilitate cell-to-cell transmission, PET pinpointing a region of proposed earliest tau deposition in AD, and human data on how well PET ligands image tau in non-AD tauopathies.
The biggest genetic news came courtesy of Edwin Jabbari and Huw Morris, University College London. They described a collaborative search for variants that influence PSP progression. To their surprise, their one hit landed near LRRK2, broadening this gene’s rap sheet beyond Parkinson’s disease.
Previously Jabbari and Morris had identified polymorphisms near the TRIM11/17 locus that associate with clinical subtypes of PSP (Nov 2018 conference news). These phenotypes include Richardson syndrome and the even less common, non-Richardson forms of PSP, such as PSP-Parkinsonism (PSP-P) and pure akinesia with gait freezing (PAGF).
Of those, Richardson syndrome progresses fastest. Its median duration is six to seven years, while people with PSP-P and PAGF generally survive for around nine and 13 years, respectively. Researchers are trying to develop therapeutics to slow progression of PSP. Morris wondered if genetic variants that influence progression might point to drug targets.
To find out, Jabbari and colleagues ran a survival genome-wide association study (GWAS) of a combined 1,001 people from a clinical and a postmortem cohort. The former came from the Progressive Supranuclear Palsy-Cortico-Basal Syndrome-Multiple System Atrophy (PROSPECT) study in the U.K.; the postmortem samples came from brain banks in the U.K., U.S., and Germany. PROSPECT analyzes brains upon death, hence more than 90 percent of people in both cohorts had a neuropathology-confirmed diagnosis.
None of the previously identified PSP risk genes, including TRIM11/17, MAPT, MOBP, EIF2AK3, and STX6, correlated with progression. That said, in the survival GWAS, one significant association emerged, at rs2242367. It slowed progression a bit. At this locus GG, AG, and AA genotypes correlate with 7.7, 6.9, and 6.6 years survival, respectively.
How might rs2242367 slow PSP? The SNP sits in intron 3 of the gene SLC2A13. It also lies only190 Kb away from rs76904798, a common variant near the LRKK2 gene that has been linked to Parkinson’s disease. Does rs2242367 affect SLC2A13 or is it a proxy for some co-inherited LRRK2 variant? The authors found no coding variants that fit either bill. Therefore, to figure out how this genetic polymorphism influences progression of PSP, the authors looked to gene expression. They searched databases that match expression changes of specific genes with variants, aka expression quantitative trait loci (eQTL), that correlate with those changes.
Jabbari turned to two eQTL datasets—the eQTLGEN whole-blood dataset of 32,000 samples, and a BRAINEAC eQTL dataset of about 120 samples. The former comprises data from 37 different projects worldwide, including the Framingham Heart and the Rotterdam Studies. BRAINEAC, aka, the U.K. Brain Expression Consortium, studies expression regulation in up to 12 different brain regions sampled from people who died without any sign of neurodegenerative disease. Jabbari discovered that, in eQTLGEN, rs2242367 alleles correlated with expression of two long, noncoding RNAs and also with expression of LRRK2, but not SLC2A13. Among 120 samples in the BRAINEAC eQTL dataset, there also was a correlation with LRRK2 expression, but it fell short of statistical significance. Among BRAINEAC samples of frontal cortex, putamen, and substantia nigra, the AG and GG genotypes, which correlated with longer survival, trended toward reduced LRRK2 expression. Whether the long coding RNAs may play any role in disease needs to be determined. “We still have a lot of work to do to understand this data,” said Morris.
Which is more important for a person’s PSP risk then, the blood or the brain? Morris said his study lacked power to detect significant expression differences in the brain, but he thinks changes there might be more important. However, he did not discount peripheral mechanisms. The variant could affect blood-derived cells that enter the brain, or remotely affect microglia in the brain. Jabbari told Alzforum that he plans to analyze single-cell eQTL data to see if it points to LRRK2 expression in specific brain cells.
This new data pleased Mark Cookson, National Institute on Aging, Bethesda, Maryland, who collaborated on the study and co-authored the paper now uploaded to bioRxiv. Cookson believes it could speed up clinical development of LRRK2 kinase inhibitors. “I think this [variant] is a big deal,” he told Alzforum. “If it can be confirmed, then it opens up LRRK2 kinase inhibitors to a broader population of patients, including some with very short survival. This would make clinical trial readouts shorter than looking for slower progression rates in PD,” he said. Cookson believes this could be a “game changer” if such a trial finds a sponsor.
Pfizer, Merck, Glaxo-SmithKline, Genentech, and Biogen have all LRRK2 programs. Denali Therapeutics has LRRK2 kinase inhibitor in Phase 1 clinical trial for Parkinson’s disease. Jabbari told Alzforum that while he is gathering data for replication studies, he has also already contacted companies to collaborate on PSP trials.
Tau sub haplotypes and primary tauopathies
Exactly how LRRK2 might influence progression in PSP remains to be seen. Come to think of it, researchers do not even fully understand how variants in the tau gene itself confer risk. In her talk, Alison Goate, Icahn School of Medicine at Mount Sinai, New York, reviewed the complexity of the MAPT locus at chromosome 17.
Goate showed how an inversion of a large DNA fragment at 17q21.31, which spans MAPT and multiple other genes, gives rise to the two major haplotypes H1 and H2 (Baker et al., 1999; Stefansson et al., 2005). People who carry H1 are at higher risk for both PD and PSP even though each disease has distinct clinical symptoms and neuropathology. How can this be? Kathryn Bowles, a postdoc in Goate’s lab, found that different sub-haplotypes hold the answer.
Researchers know that a sub-haplotype called H1c associates with PSP but not with PD. However, no one has mapped the association beyond the single nucleotide polymorphisms that tags H1c (Pittman et al., 2005; Hoglinger et al., 2011). In D.C., Goate showed that broader structural differences complicate this association. Namely, people with PSP have a different linkage disequilibrium pattern near the MAPT locus than do people with PD. In other words, PSP families inherit a structurally different version of this region of chromosome 17. “This suggests altered recombination or gene conversion in the MAPT 1 regulatory region,” said Goate. Gene conversion is where one copy of a DNA sequence is replaced by a homologous sequence.
How would such a structural difference manifest in disease risk? Because Bowles found no evidence that MAPT intron 1 variants associate with altered expression of tau, or of any other gene in the 17q21.31 locus, she wondered if the variants cause more dramatic changes, namely, in the way DNA is packaged. She used a technique called Assay for Transposase-Accessible Chromatin (ATAC-Seq), to test whether the chromatin near this locus is open and active, or closed. Bowles tested this in neurons induced from iPSC cells that were derived from people with the H1c haplotype. In these neurons, chromatin was open near SNPs that associate with PSP. The same was true in induced microglia, but not astrocytes.
In contrast, researchers know of no linkage disequilibrium shenanigans associated with Parkinson’s. That said, three H1 sub-haplotypes Bowles designated H1.1, H1.2, and H1.3, associated with PD in two independent case-control datasets. They correlated with expression of the astrocyte gene LRRC37A, but here the story gets even more complicated. Each of these sub-haplotypes can be further divided based on co-inherited SNPs, and some of those smaller haplotypes turned out to be the risk or protective variants. For example, H1.1b and H1.1e increased risk of PD, whereas H1.1c was protective.
All told, the genetic data suggest that the 17q locus has different kinds of association with PSP than with PD. In the former, altered chromatin in neurons and microglia increase risk in some way yet to be determined. In the latter, sub-haplotypes seem to modulate expression of LRRC37A, which encodes a cell membrane protein of unknown function. Gene ontology and pathway analysis predict it may function in chemotaxis, cell migration, and/or fatty acid metabolism.
As for PSP, Bowles can’t say for sure whether the risk comes from neurons or glia, but she leans toward the latter. “I think the argument for there being a larger, or at least unique, effect in microglia is quite compelling,” she said. “In the human brain ATAC-seq data, there are peaks in this region only in the NeuN-negative cells, nothing in the NeuN-positive cells. That points more to glia than to neurons.”—Tom Fagan
Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, Desnica N, Hicks A, Gylfason A, Gudbjartsson DF, Jonsdottir GM, Sainz J, Agnarsson K, Birgisdottir B, Ghosh S, Olafsdottir A, Cazier JB, Kristjansson K, Frigge ML, Thorgeirsson TE, Gulcher JR, Kong A, Stefansson K.
A common inversion under selection in Europeans.
Nat Genet. 2005 Feb;37(2):129-37. Epub 2005 Jan 16
PubMed.
Behold the First Human α-Synuclein CryoEM Fibril Structure
The first high-resolution cryoEM structures of pathological α-synuclein aggregates were unveiled at Tau2020, a new conference held Feb 12 and 13 in Washington, D.C. (see Part 1 of this series). Michel Goedert, MRC Laboratory of Molecular Biology, Cambridge, U.K., presented an assembly that looks quite distinct from the core protofibrils found in tau neurofibrillary tangles or amyloid-β plaques. What’s more, two different protofibrils make up the core of α-synuclein filaments, rendering it asymmetric. Scientists hope this information will finally help them score compounds that will make good ligands for α-synuclein PET imaging, which have thus far eluded them.
Goedert revealed the structure while accepting the Rainwater Prize, a $250,000 award for outstanding innovation in neurodegenerative research. The Rainwater Charitable Foundation co-sponsored the conference. Patrick Hsu, University of California, Berkeley, received a $150,000 innovative early career scientist award for correcting defective tau splicing in MAPT-mutant neurons with CRISPR RNA editing (Oct 2019 news). On March 4, Rainwater and the Alzheimer’s Association announced eight new research grants, totaling nearly $4 million, as part of their Tau Pipeline Enabling Program. The awards go to drug discovery programs at private and academic labs in the U.S. and Europe.
Type I α-Synuclein. Type I α-synuclein fibrils comprise two different protofibrils. PF-IA (yellow) folds from amino acids 14 to 94. Its N-terminus (left) forms a cross β-sheet hairpin that continues into an L shape. Starting at amino acid 45, the C-terminus (right) forms three stacked L shapes. PF-IB (orange) comprises amino acids 21 to 99, and similarly stacks three L-shaped layers at its C-terminus (left). [Courtesy Schweighauser et al., 2020, under Creative Commons license.]
In collaboration with Masato Hasegawa at the Tokyo Metropolitan Institute of Medical Science, Goedert, co-senior author Sjors Scheres from the MRC, and colleagues isolated α-synuclein fibrils from five people who had had multiple system atrophy. MSA is a synucleinopathy that mostly affects glial cells. In his speech, Goedert noted that, as is the case for tau, human brain-derived structures are markedly different than those of recombinant α-synuclein. “This casts doubt on a lot of the work done using recombinant molecules,” he emphasized. “We see again in the case of synuclein that recombinant proteins do not reflect the pathology in the brain.”
In their paper uploaded to bioRxiv on February 6, co-first authors Manuel Schweighauser, Yang Shi, and colleagues describe two different forms of fibril present at different ratios in these five people. The three who had died after nine or ten years with MSA had predominantly Type I fibrils. Type II fibrils predominated in the brains of the other two, who had lived with MSA for 18 and 19 years, respectively. This hints that fibril structure may have something to do with how quickly MSA leads to death, though more evidence is needed to be sure, the authors say.
Complicating matters, the scientists found different fibrils in different brain regions. In one case, Type I fibrils marked the putamen, Type II fibrils the cerebellum. Another case had mostly Type I in the putamen, Type II in the frontal cortex.
The researchers resolved the structure of Type I fibril cores from one patient to a resolution of 2.6 Ångstroms (see image above). One protofibril, PF-IA, encompassed amino acids glycine 14 to phenylalanine 94. PF-IB was slightly shorter, running from lysine 21 to glutamine 99. Both contained extensive β-sheet structure—12 strands in PF-IA and 10 in PF-IB. Both have a β-sheet hairpin at the N terminus and a three-layered, L-shaped β-sheet motif at the C terminus. PF-IA also has a long straight section connecting the two ends; PF-IB does not. The arrangement of the three-layer L differs between the two, particularly in how the innermost layer packs against the middle layer after glycine 86.
Type II α-Synuclein. Protofibril IIA (light purple) adopts almost the same shape as PF-IA (see image above), but features a wider cavity between the inner and central layers of its three-layered C-terminal motif. PF-IIB (dark purple) is 15 amino acids shorter than PF-IB. It comes in two forms. PF-IIB1 forms nearly the identical structure at its C terminus as does PF-1B. PF-IIB2 assumes a slightly different backbone shape. [Courtesy Schweighauser et al., 2020, under Creative Commons license.]
Type II fibrils from one donor were solved to a resolution of 3.1 Ångstroms. PF-IIA resembled PF-IA, extending from glycine 14 to phenylalanine 94 with the same basic folds. However, the three-layered motif at the C-terminus packed together slightly differently, opening a cavity between the inner and central layers near glutamic acid E61. It also has 12 β strands.
PF-IIB turned out to be much smaller than PF-IB. It starts at glycine 36 rather than lysine 21, and has but nine β strands. PF-IIB exists in two distinct forms as well. PF-IIB1 is nearly identical to PF-IB, but PF-IIB2’s backbone is shaped differently between amino acids 81 and 90 (see image above).
A and B Type protofilaments abut to form the Type I and II fibril cores.
What’s With the Cavities?
In a curious nod to tau structures from people with corticobasal degeneration (CBD) and chronic traumatic encephalopathy (CTE), the center of the asymmetrical α-synuclein structure features a small cavity that contains an unknown molecule unattached to the protein (Feb 2020 news).
Schweighauser and colleagues believe this molecule, or molecules, is something other than a protein. It is almost certainly negatively charged, to counter a net positive charge of four lysines that surround the cavity. It may stabilize the structure and even promote assembly, though that remains to be determined. In Type I filaments, the slightly bigger cavity seems to contain additional smaller molecules. Other molecular entities appear on the fringes of the protofilament cores; they may represent extensions of the synuclein polypeptide chain.
Because of the height of the amino acid side chains, and also because the protofibrils tilt away from the fibril axis, each protofibril monomer contacts three monomers in the opposing protofilament. For example, PF-IA’s N-terminus connects to the C-terminus of the PF-IB one rung up the chain, while PF-IA’s C-terminus connects to the N-terminus of the PF-IB one down the chain (see image below).
This is different than in tau filaments. There, each protofilament only contacts its opposing counterpart. The interface between the synuclein monomers is larger, stretching for 25 amino acids, in comparison to at most six amino acids for tau fibrils. “This is bound to have an impact on seeded aggregation, as it results in differences in enthalpic and entropic gains when a new molecule is incorporated into the filament,” the authors wrote.
Protofibril Troika. Each synuclein protofilament monomer touches three in the opposing chain. The N terminus of the green PF-IB contacts the C terminus of the faded red PF-IA one up the chain. The C terminus of the green PF-IB binds the N terminus of the faded blue PF-IA one down the chain. [Courtesy Schweighauser et al., 2020, under Creative Commons license.]
This information is fine and good, but does it help scientists understand why mutations in α-synuclein cause disease? Yes. For example, either a glycine-to-aspartic acid mutation at amino acid 51, or an alanine-to-glutamic acid switch at 53 lead to familial Parkinson’s disease. Both residue 51 and 53 lie at an interface between the two protofilaments. The filament structure can accommodate either the aspartic acid or glutamic acid side chains. Each will add one extra negative charge per protofibril, or two for each rung of the fibril structure, reducing the net charge in the cavity by two. This might affect the non-protein mystery molecule in the cavity; this idea, however, requires more work.
Researchers are also banking on cryoEM for clues toward PET imaging ligands. They have sought suitable compounds for years, but failed to find any that bind with high specificity and affinity. Robert Mach, at University of Pennsylvania, Philadelphia, leads a new collaboration to model compounds that might bind known α-synuclein structures. So far, scientists have based their screening on steady-state NMR and cryoEM of recombinant synuclein. But in-silico modelling is only as good as the structural information it uses.
“This is why these new cryoEM structures are so incredibly important,” Mach told Alzforum. “They differ from the NMR and the four to five cryoEM structures of recombinant synuclein solved to date. It does not take a very large change to dramatically affect binding sites,” he said. While some lab-made types contain the three-layered L shape, most are symmetrical, containing only one, or sometimes two identical protofilaments. None of the prior structures have the long N-terminal arms. “We hope the structures of α-synuclein filaments from MSA will help to design PET ligands that are specific for assembled α-synuclein,” Goedert told Alzforum.
What about fibrils from other α-synucleinopathies, such as PD or dementia with Lewy bodies (DLB)? Will they each have their own unique structures, just as different tauopathies do? It seems likely.
Goedert and colleagues tried to solve cryoEM structures for α-synuclein fibrils isolated from three people who had had neuropathologically confirmed DLB. Unlike the MSA fibrils, the DLB ones were thinner and did not twist, as has been previously described (Spillantini et al., 1998; Tarutani et al., 2018). The lack of a twist meant the researchers were unable thus far to determine a three-dimensional structure. However, based on two-dimensional analysis, Goedert and Scheres concluded that DLB and MSA protofibrils are distinct.—Tom Fagan
New at Tau2020: PET Detects First Traces of Tangles in Rhinal Cortex
Tau2020, the first of what may become a regular “pan-tau” conference (see Part 1 of this series), got off to a brisk start February 12–13 in Washington, D.C. Researchers who focus on primary tauopathies, such as progressive supranuclear palsy, rubbed shoulders with others who study secondary tauopathies, such as Alzheimer’s disease (AD).
As with any tau conference these days, PET imaging featured prominently. The session showed how unequal the field of tauopathy imaging has been of late. AD researchers have reasonably well-characterized ligands that bind AD neurofibrillary tangles with respectable specificity and sensitivity, and they use them in both natural history studies and clinical trials. Alas, those ligands work poorly in the PSPs and CBDs of the world. In D.C., scientists described newer tracers, such as APN-1607 and CBD2115. How well these will perform remains to be seen (see Part 4 of this series).
AD researchers are using PET to figure out how the disease causes cognitive decline. In D.C., Keith Johnson of Massachusetts General Hospital, Boston, reviewed data from the Harvard Aging Brain Study (HABS) that correlated longitudinal change in amyloid plaques, neurofibrillary tangles, and cognition. This data fits with the field’s majority view of a sequential model of progression, whereby baseline amyloid load drives both plaque and tangle accumulation, and the latter drives cognitive decline (Jun 2019 news; Feb 2020 conference news).
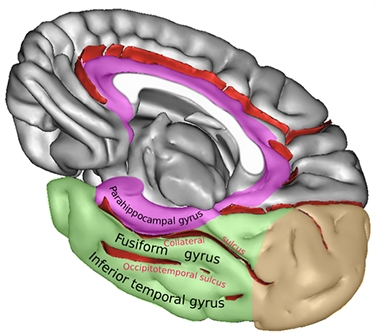
Rhinal Sulcus. The rhinal sulcus comprises the anterior section of the collateral sulcus. It separates the entorhinal part of the parahippocampal gyrus from the fusiform gyrus. [Courtesy of Sebastian23, Creative Commons license.]
But where in the brain does tau accumulation start? In D.C., Johnson suggested that, at least as visible by PET, it may be in a speck of brain he calls the “rhinal cortex.” The rhinal cortex isn’t a histopathologically defined region, said Johnson. Rather, it is a small area surrounding the temporal lobe’s rhinal sulcus and is delineated purely based on the resolution of PET imaging. The rhinal sulcus, which some consider an extension of the collateral sulcus, is the fissure separating the entorhinal cortex from the temporal pole.
Using flortaucipir PET imaging in a cross-sectional study of healthy controls and older people from the A4/LEARN clinical trial, Justin Sanchez in Johnson’s lab found that tangles begin to accumulate in this rhinal cortex in people who have only a low amyloid burden. Once amyloid starts to build, then the spread of tangles takes off in the temporal lobe. As the disease progresses, tangles invade the precuneus.
Based on this data—and on Gaussian mixture modelling, which finds sub-distributions among larger probabilistic outcomes—Sanchez then identified four tau PET staging thresholds. Called Tp0 to Tp3, they reflect no specific flortaucipir uptake, binding in the rhinal cortex, binding in rhinal cortex plus inferior temporal lobe, and binding in rhinal cortex, inferior temporal lobe, and precuneus, respectively.
This staging trajectory replicated in 104 volunteers in HABS, said Johnson. The tau stages correlated with Aβ burden, he added, noting that the scheme could become a tool to stage early tau pathology in living people.
Will this staging scheme distinguish age-related from disease-related tau accumulation? Johnson is trying to address this question. Alas, it is confounded not only by pathology, but also by resilience, genetic, and other factors. “We may be able to transform the data to have some diagnostic value, but that will be hard to do,” he said.
Is the rhinal cortex ground zero for tau deposition? Maybe not quite. Histopathology suggests that it begins even earlier, even deeper in the brain, i.e., in the locus coeruleus (Braak et al., 2011). Alas, this has been difficult to detect with current tau tracers. Therefore, Heidi Jacobs in Johnson’s lab decided to probe the locus coeruleus by measuring how it held up as tau pathology progressed.
She measured its size with MRI and correlated that data with flortaucipir binding in the cortex. In D.C., Johnson reported that the integrity of the locus coeruleus correlated inversely with tangle burden in the entorhinal and medial prefrontal cortex. This supports the idea that a flagging locus coeruleus may be a harbinger of AD neuropathology.—Tom Fagan
Scientists studying the non-Alzheimer’s disease tauopathies have struggled to find tracers that will bind the particular forms of tau that predominate in those diseases. At Tau2020, held February 12–13 in Washington, D.C., Gil Rabinovici, University of California, San Francisco, reviewed in vivo and autoradiography data of human tissue.
It showed that flortaucipir bound far more weakly to the straight and twisted filaments of the 4R tau found in progressive supranuclear palsy (PSP) and corticobasal disease (CBD), or to the 3R versions found in Pick’s disease, than it does to the 3R/4R paired helical fragments found in AD (Aug 2016 conference news; Sep 2018 news). While flortaucipir does bind to regions that accumulate tangles in primary tauopathies, such as the globus pallidus in PSP, the signal is obscure because off-target binding in healthy controls also occurs in those same regions. In toto, flortaucipir is not useful in these diseases, Rabinovici said.
New autopsy data from 19 people who had flortaucipir PET scans during life supported this. The cases ranged from 34 to 76 years old, with an average of 30 months between the scan and autopsy. Eight were diagnosed with AD, four with PSP, two each had CBD or frontotemporal dementia (FTLD) caused by a tau mutation, and one each had had FTLD-FUS, FTLD-TDP43, or argyrophilic grain disease. David Soleimani-Meigooni in Rabinovici’s lab found excellent PET-to-tau immunohistochemistry correlations among the eight AD patients, who were all at Braak stage VI.
Autopsy Versus Scan. Comparing tangle immunohistochemistry after death (bottom rows) with flortaucipir scans during life (top rows) shows tight correlation in AD and modest to poor correlation in certain primary tauopathies, such as PSP and CBD, as well as other FTLD and AGD. [Courtesy of Gil Rabinovici.]
As predicted from in vivo imaging, autopsy correlations were modest to poor for the primary tauopathies. While their scans had shown more flortaucipir retention in the brain than in normal controls, it was still much less than in AD, and the binding did not always correlate with tangle pathology. For example, non tau-mutation FTLD cases had bound tracer in frontal white matter and basal ganglia, but no tau showed up there on autopsy. The data corroborate that flortaucipir’s specificity and sensitivity for detecting tau in primary tauopathies is low, said Rabinovici.
CTE Tau. PET imaging of one coronal slice of the brain of a former American football player showing extensive flortaucipir binding. [Courtesy William Mantyh, UCSF.]
The same seems true for a case of chronic traumatic encephalopathy (CTE), a sporadic tauopathy. Working with Rabinovici, William Mantyh at UCSF compared autopsy data from a former professional football player in the U.S., who had had a flortaucipir PET scan 52 months before he died (Mantyh et al., 2020). Postmortem pathology indicated stage 4 CTE with limbic argyrophilic grain disease, limbic-predominant TDP43 encephalopathy, and Braak stage III neurofibrillary tangles. Strong flortaucipir binding mapped to regions of high tau pathology in his left fusiform and inferior temporal gyri and in frontal white matter, and yet, other regions that lit up strongly in the scan, such as the basal ganglia, thalamus, motor cortex, and calcarine cortex, had low tau burden on autopsy.
Overall, the correlation between tau PET and pathology at the regional level was weak. “Flortaucipir does not seem to be a great tracer for CTE, at least based on this case,” said Rabinovici.
What about other tau tracers, then? Radiography data suggest that MK6240, RO948, and JNJ067 also are not specific and sensitive enough in primary tauopathies. On the other hand, PBB3 and Life Molecular Imaging’s (formerly Piramal) PI-2620 show promise, said Rabinovici. He reported recent in vivo data showing that 48 people with PSP bound much more PI-2620 in the globus pallidus, subthalamic nucleus, substantia nigra, and dentate nucleus than did 10 healthy controls.
The PBB3 derivative PM-PBB3 is being developed by Taiwan-based Aprinoia Therapeutics under the name APN-1607. Richard Margolin from Aprinoia claimed it is the only tracer to demonstrate age-associated increase in signal in a 4R tau mouse model.
About 265 people have been scanned with APN-1607 so far, including 36 subjects in a Phase 1 study that enrolled 12 healthy controls, 12 AD, three FTD, three PSP, three CBS, and three people with vascular cognitive impairment. In D.C., Margolin said that uptake in AD patients matches the known pathological distribution of tau. In PSP, the tracer bound much more strongly in the globus pallidus, subcortical white matter, and midbrain.
Binding correlated with the PSP Rating Scale, a first for a PSP tracer, said Margolin. Controls retain some APN-1607 in the meninges and choroid plexus, which is a problem with other PET tracers, as well. APN-1607 does not bind to monoamine oxidase or melanin (see also Part 3 of this series).
Tsuneya Ikezu at Boston University considers APN-1607 a tracer to watch, because it binds to all forms of tau. Rabinovici was more cautious. He noted that APN-1607’s dissociation constant (Kd), which reflects binding strength, for AD and PSP tangles is a bit weak at 4 and 6.4 nM, respectively. By comparison, the Kds for PI-2620 and MK6240 are 2.1 and 0.73 for AD tangles. Rabinovici also believes the off-target binding might still be an issue. APN-1607 is unstable under ambient light, where it isomerizes, making it more difficult to use. Rabinovici thinks better tau tracers are still needed.
One new candidate for 4R tau might be CBD-2115, which is being developed by Samuel Svensson and colleagues at CBD Solutions in Sweden. Rabinovici showed that this compound bound more than twice as strongly to frontal gyrus extracts from CBD patients than did MK6240 or flortaucipir, but he said no scans of people have been done yet with this compound.
Rabinovici is collaborating with Chet Mathis at University of Pittsburgh Medical Center, to develop new 4R tau ligands. They will exploit the different cryoEM structures of tau fibrils identified by Michel Goedert, Sjors Scheres and colleagues at the MRC in Cambridge, England. These investigators have solved structures of four different forms of tau fibril isolated from people with AD, Pick’s disease, CTE and, most recently, CBD (see Feb 2020 news).
Under a new U19 “center without walls” NINDS grant, Mathis, together with Robert Mach and colleagues at the University of Pennsylvania, Philadelphia, will run in silico modelling experiments to screen thousands of compounds in search for some that bind specifically to the unique groves formed by each of these fibrils, then whittle down the hits to candidates for clinical testing. “The other approach will be to take existing tau tracers, model how they bind to different tau forms, and see if we can tweak them to improve their binding properties,” said Rabinovici.
In parallel, Mach and E. James Petersson, also at UPenn, will use a similar strategy to screen for compounds that might make good PET ligands for α-synuclein. Mach collaborates with John Karanicolas, Fox Chase Cancer Center, who has developed in silico methods to find small molecules that bind to pockets on protein surfaces. Starting with the three-dimensional structure of those pockets, the Karanicolas method virtually “adds” 1 Ångstrom diameter spheres until the pocket is filled. He then uses the configuration of these spheres as an exemplar to find compounds that assume a similar shape (Johnson and Karanicolas, 2016).
Mach, Petersson, and colleagues have used this exemplar method to screen compound libraries for small molecules that fit sites suggested by solid-state NMR structures of α-synuclein determined by Chad Rienstra and colleagues at the University of Illinois, Urbana-Champaign (Tuttle et al., 2016).
Screening was extremely fast. Mach told Alzforum that within a few weeks they had whittled 3.5 million compounds down to a handful of candidates, ultimately identifying a compound that has 7nM affinity for α-synuclein and fivefold selectivity over binding to Aβ. “This is not great, but it’s a lot better than what we had before,” he said. Mach thinks he can improve that further using cryoEM structures of two different synuclein fibrils isolated from patients who had multiple-system atrophy. Goedert showed these structures at Tau20202 (see Part 2 of this series). “We are now using these structures to build new exemplars for further screening,” said Mach.—Tom Fagan
Evidence now seems overwhelming that toxic forms of tau spread from cell to cell, causing the characteristic progression of tauopathies such as Alzheimer’s and frontotemporal dementia. But how does tau get from one cell to another? At Tau2020, a new conference held February 12 and13 in Washington, D.C., Ken Kosik, University of California, Santa Barbara, reported that tau slips into cultured human neurons by binding LDL-receptor-related protein 1 (LRP1), a cell surface receptor. In mice, knocking down LRP1 expression shut down the spread of injected mutant tau. “LRP1 is a master regulator of tau uptake and spread,” Kosik told the audience.
LRP1 popped up as a hit when Jennifer Rauch, a postdoc in Kosik’s lab, used a CRISPR-based screen to search for receptors of various forms of tau, including oligomers and fibrils.
Previously, Rauch had developed a cell-based assay for tau uptake based on a fluorescently labeled tau reporter. In essence, she added labeled tau into cell culture medium, then used flow cytometry to measure its incorporation into the cells over time. This confirmed what others had reported, namely that heparin sulfate proteoglycans (HSPGs) seem to mediate tau uptake (Rauch et al., 2018; Apr 2015 news). However, even after blocking HSPGs, some tau still entered cells, suggesting there are other receptors.
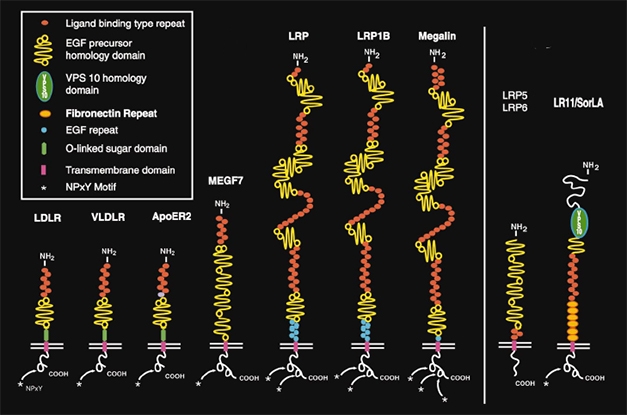
LDLR Family. LRP1 belongs to the LDL receptor family (left). These, and more distantly related cell surface receptors (right), bind a wide range of proteins and play important roles in cholesterol metabolism, endocytosis, and cell signaling in the brain. [Courtesy Joachim Herz, University of Texas Southwestern Medical Center, Dallas, and Neuron.]
To cast a wider net, Rauch adopted a candidate approach. She used the same assay with cells she had denuded of various cell-surface receptors. Specific guide RNAs and CRISPR ensured that H4 neuroglioma cells lacked various members of the LDL receptor family.
Rauch found that knockdown of LRP1 totally abolished tau uptake. In contrast, ridding the cells of LRP1 homologs, including LDLR, LRP1B, LRP2, LRP5, LRP8, and VLDLR, had no effect. Without LRP1, the H4 cells were unable to absorb monomers, oligomers, and fibrils of tau. Likewise, when Rauch knocked down LRP1 in neurons derived from human induced pluripotent stem cells, they also no longer endocytosed tau. “The results were dramatic,” said Kosik.
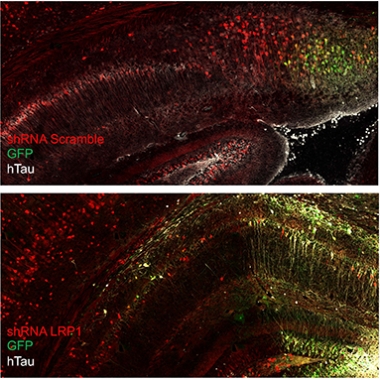
Don’t Spread ’Em. In the mouse brain, human mutant tau (white) can spread from donor cells (green) to recipient cells. Knocking down LRP1 (bottom) arrests the spread, restricting human tau to donor cells. [Courtesy Jenny Rauch, UCSB.]
Cultured cells are well and good, but does LRP1 mediate cell-to-cell transmission of tau in the brain? To find out, Rauch used a mouse model developed by Susanne Wegmann. When at Brad Hyman’s lab at Massachusetts General Hospital, Charlestown, Wegmann wanted to distinguish tau made in cells from tau taken up by cells. She engineered an adeno-associated virus encoding a chimera of enhanced green fluorescent protein coupled via a 2A peptide domain to human P301L mutant tau. The linker undergoes self-proteolysis, releasing tau from eGFP once the chimera is expressed. Hence, tau “donor” cells show up by their GFP fluorescence, and “recipient” cells are marked by tau immunohistochemistry but no GFP fluorescence (Wegmann et al., 2019).
Rauch used this AAV reporter to track the spread of tau. First, though, she injected, retro-orbitally, 6-week-old wild-type mice with a short-hairpin RNA to knock down expression of LRP1 in their brains. Retro-orbital injections into the sinuses is commonly used instead of tail vein injections for systemic delivery of a variety of agents. Two weeks later, she injected the AAV, intracerebroventricularly, into the brain. Three weeks after that, she killed the mice and looked for human tau throughout their brains. In mice injected with a scrambled shRNA and normal LRP1expression, human P301L tau lit up all through the hippocampus and connected cortical areas. In mice treated with the LRP1 shRNA, human tau was restricted to a tiny focus of eGFP fluorescence at the AAV injection site. In short, knocking down LRP1 had prevented the spread of the mutant tau.
Kosik does not know what type of brain cells tau entered. The cells bound MC1, an antibody that recognizes a conformation of tau found in neurofibrillary tangles.
How does LRP1 bind tau? The receptor is one of the largest in the LDLR family, having four cysteine-rich ligand binding domains. It is well-known and promiscuous, binding more than 40 different ligands, including α2 macroglobulin, amyloid precursor protein, Aβ, and ApoE (Holtzman et al., 1995; Fuentealba et al., 2010). It wears many hats, being important for cholesterol homeostasis, endocytosis, and cell signaling through its many ligands.
To figure out which LRP1 domain might hook tau, Rauch made mini receptors, each having only one of its normally four binding domains. Cells expressing ligand binding domain IV had highest affinity for tau. Domain IV binds receptor-associated protein (RAP), a common ligand for most LDLR family members. Looking at specific tau regions, Rauch found that tau’s microtubule binding domains bound to LRP binding domain IV, while tau’s N-terminal bound to ligand binding domain II.
Crystal structures of RAP bound to LRP1 have been solved, noted Kosik, and they are guiding scientists with regard to how tau binds LRP1. For example, lysines on RAP bind to aspartic acid residues on LRP1 through salt bridges. Tau may bind similarly, since capping tau lysines with a chemical called sulfo-NHS-acetate completely abolished endocytosis via LRP1.
Researchers at the Tau2020 meeting were excited by the findings. They asked how LRP1 fits with HSPG’s role in tauopathies. Might both bind tau simultaneously, either in a ternary complex, or even a quaternary one with ApoE? Kosik said this remains to be sorted out. He speculated that HSPGs act as catch-alls, scooping various molecules out of the extracellular space and bringing them closer to the cell surface, where more specific interactions take place.
As for ApoE, it, like RAP, seems to suppress endocytosis of tau, at least in cell assays. All isoforms of ApoE behave similarly in this regard. Kosik noted that LRP1 binds ApoE in a region where the protective Christchurch mutation is located (Nov 2019 news). “It would be worth looking into how this mutation might affect ApoE4-LRP binding and tau dynamics,” said Kosik.—Tom Fagan






Comments
No Available Comments
Make a Comment
To make a comment you must login or register.