International Symposium Puts PSP/CBD on the Map
Quick Links
Progressive supranuclear palsy and corticobasal degeneration are rare, poorly understood tauopathies with a broad array of symptoms and subtypes. What causes them? How to prevent, diagnose, track, and treat them? On October 25 and 26, 220 scientists gathered at the Royal College of Physicians in London to tackle these questions. Organized by Alex Klein of CurePSP in New York and Kate Arkell of the PSPA in Milton Keynes, U.K., the first PSP & CBD International Research Symposium drew researchers from fields of basic science to neuropathology. The meeting was opened by the Duchess of Gloucester, a longtime patron of PSPA.
The meeting marked a coming together of minds and gave these rare diseases their own voice in the dementia and movement disorders community. “Just a few years back we’d have been lucky to get 15 people at a PSP meeting,” said John Hardy, University College London. Klein noted, “It was exciting to have key opinion leaders of the field so focused on PSP/CBD, which often gets drowned out by discussion of more common dementias, such as Alzheimer’s.” Jeff Friedman, who is on the board of CurePSP and manages the PSP Genetics Consortium, agreed. “The quality of the science and the collaborative nature of the meeting showed how dynamic the field has become. It was encouraging,” he told Alzforum.
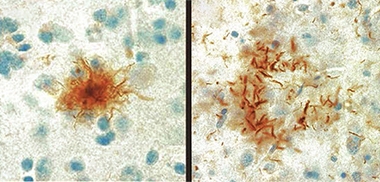
Tufts and Plaques.
Astrocytic tufts (left) and astrocytic plaques (right) of tau, are the pathological hallmarks of PSP and CBD, respectively. [Courtesy of Ruth Lamb, University College London.]
PSP and CBD typically become symptomatic in a person’s early 60s. They can progress quite fast, leading to death within three to six years after diagnosis, or six to nine years after symptoms start. Symptoms are similar in PSP and CBD, making the diseases difficult to distinguish clinically. Patients typically have trouble controlling movement, speech, swallowing, vision, and thought processes. Memory and cognition are often affected.
Postmortem, PSP is marked by so-called tufted astrocytes, which are caused by intracellular accumulation of tau in the soma of these glia. CBD also has astrocytic tau aggregates, but these look morphologically different, sit near the cells’ end processes, and are called astrocytic plaques. Neurons in PSP and CBD can also accumulate tau. In CBD, tau aggregates tend to be in cortical areas and basal ganglia, whereas in PSP they are in the basal ganglia and midbrain. Physicians tend to mistake CBD in particular for Alzheimer’s disease, and amyloid PET or CSF testing is often needed to get the diagnosis right. Unlike frontotemporal dementia, no tau mutations have been linked to PSP/CBD. Both diseases are rare, affecting fewer than five people per 100,000.
Presenters used the London conference as an opportunity to review ongoing research efforts, from the basic biology of tau to immunotherapy trials, from PSP/CBD brain banks to patient registries, from imaging and fluid markers to genetics. The latter had new data on offer, as well.
Genetics Points Toward Pathways
Scientists are starting to tap into the genetic variants shared by people with PSP or CBD. The largest PSP genome-wide association study to date, led by Ulrich Müller at Germany’s Justus-Liebig University, Giessen, and Gerard Schellenberg at the University of Pennsylvania School of Medicine, Philadelphia, genotyped about 2,000 cases in two phases, each with 1,000 cases. First author Günter Höglinger and colleagues identified risk variants in the STX6, EIF2AK3, and MOBP genes, which encode syntaxin 6, eukaryotic translation initiation factor 2-a kinase, and myelin-associated oligodendrocyte basic protein. The GWAS also confirmed that loci near the MAPT gene for tau, including the known rs242557 single-nucleotide polymorphism (SNP), associate with PSP (Jun 2011 news on Höglinger et al., 2011).
A smaller CBD GWAS led by Schellenberg and Dennis Dickson at the Mayo Clinic in Jacksonville, Florida, genotyped 219 cases (Kouri et al., 2015). It bubbled up the same SNPs near MOBP (rs1768208) and tau (rs242557), plus variants near the long noncoding RNA gene KIF13B-1 (rs643472) and near SOS1 (rs963731). The latter encodes son of sevenless homologue 1, a potential tau phosphatase.
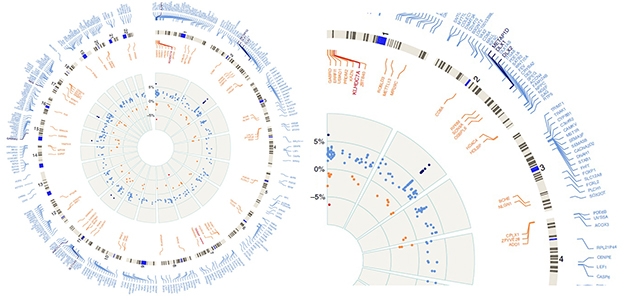
Epigenetic Changes. Circular plot, enlarged slice show hyper- (blue) and hypomethylated (red) regions of the whole genome by numbered chromosomes (outer gray circle). Inner gray circle represents percentage of up (to 5 percent) or down (to -5 percent) methylation relative to controls. The DLX1 locus on chromosome 2 is the most highly methylated in PSP, as represented by black dots on inner circle showing greater than 5 percent change in methylation status. [Courtesy of Günter Höglinger and Nature Communications.]
In London, researchers reported recent hits. Edwin Jabbari and Huw Morris at University College London took a phenotype approach to hunt for genetic variants that specifically associate with subtypes of PSP. They compared cases of Richardson syndrome against all other forms. This most common form of PSP was first described by Clifford Richardson at Sunnybrook Hospital in Toronto in 1955, with further cases identified by John Steele at the same institution (Steele et al., 1964). People with PSP-RS develop a rigid gaze, parkinsonian movement difficulties, and dementia. Because their eyes tend to be fixed straight ahead, people with PSP are prone to trip over pets, rugs, and furniture, putting them at great risk of falls. Their disease progresses more rapidly than non-Richardson PSP. It also has a higher tau pathology burden, especially in deep brain areas such as the subthalamic nucleus, globus pallidus, substantia nigra, and the pons.
Jabbari compared GWAS data from 367 PSP-RS cases to those of 130 non-RS cases. He found 27 SNPS, all in the q42.13 region of chromosome 1, and all inherited together, i.e., in linkage disequilibrium with each other. Of the 27, Jabbari believes rs564309 to be the lead candidate for a PSP variant. It lies between exons 3 and 4 of the gene for tripartite motif-containing protein 11 (TRIM11), which encodes a RING R3 ligase that activates the proteasome. Two of the other three genes residing in this locus are unlikely candidates for PSP genes, agreed Morris. One, obscurin, is expressed mainly in skeletal muscle, while Histone H3 is barely detectable in the brain. The fourth gene there, TRIM17, encodes another RING R3 ligase that shares 60 percent identity with TRIM11. Like its homolog, TRIM17 is expressed across the brain, and predominantly in the cerebellum. It is unclear which TRIM might be the link to PSP. It may be both, Morris said, since they seem to be co-regulated.
How do they increase risk? Jabbari suspects tau. He cited work suggesting that tau scuppers the 26S proteasome (Dec 2015 news), and said both TRIM 11 and 17 prime the proteasome to degrade proteins.
Deepening his confidence in a link between TRIMs and PSP-RS, Jabbari found variants that were in linkage disequilibrium with rs564309 in 100 well-characterized cases from Phase 1 of the Muller/Schellenberg GWAS. These findings appeared in the October Annals of Neurology (Jabbari et al., 2018).
Another potential risk variant emerged from whole-exome sequencing (WES). Schellenberg described WES data from 750 people with PSP. It identified the rs17662853 variant in the KANSL1 gene, which lies close to MAPT. KANSL1 encodes a component of a histone acetylation complex that regulates expression of up to 400 genes, said Schellenberg. A different mutation that introduces a premature stop codon in KANSL1 causes the neurodevelopmental cognition syndrome Koolen-de Vries.
Schellenberg has been unable to pinpoint how KANSL1 variants affect PSP, but is examining expression quantitative trait loci (eQTL) possibly regulated by the variant. Thousands of single-nucleotide polymorphisms in that region of the genome complicate the analysis. Schellenberg said KANSL1 SNPs don’t affect expression of tau, but might affect expression of nearby long noncoding RNAs. That would jibe with a recent report that PD variants in the MAPT region boost expression of transcribed non-coding elements, or TNEs, near KANSL1 (Oct 2018 news).
For his part, Höglinger investigates epigenetic regulation, which remains understudied in PSP. Samples of frontal cortices revealed 1,118 sites in the genome that are hypermethylated among 94 PSP patients relative to 88 controls. He also found 530 hypomethylated sites. Together, they cover some 450 protein-coding genes, but not MAPT.
The strongest signal came from a region encoding the DLX1 homeobox transcription factor. In PSP patients, this locus was highly methylated at multiple sites (see image above). This did not affect DLX1 expression but, lo and behold, reduced expression of a DLX1 antisense transcript. That, in turn, upregulated translation of DLX1. In keeping with this, the researchers found less DLX1AS in the brains of PSP patients relative to controls, and more DLX1 protein. Friedman complimented this approach. “It shows there are additional technical modalities we can use with existing sample sets, to pull out more data and identify potential targets,” he said.
What would having more DLX1 do? In neuroblastoma cells and Ntera-2 cells, which are an embryonal carcinoma line akin to neural precursors, Axel Weber and colleagues in Höglinger’s lab found that, at least in these cells, overexpressing the protein reduced tau levels. This is counterintuitive for a tauopathy. Höglinger thinks DLX1 may contribute to PSP by disrupting neuronal function. DLX1 appears to regulate tau expression indirectly by controlling expression of other genes, including those involved in Wnt and GABA signaling pathways. That might spell trouble for neurons. This work was published July 26 in Nature Communications.
Looking to future genetics studies, UCL’s Hardy called for more GWAS and challenged funders to support them. “Yes, maybe GWAS is so '90s and a bit boring, but we need more pieces to the jigsaw puzzle,” he said, referring to biological pathways that might be uncovered by new GWAS hits.
Taking his plea further, Hardy suggested that GWAS be done routinely and noncompetitively. This could be achieved by taking samples from all well-phenotyped cases as they come to clinics and running one large GWAS per year. “Since GWAS are now incredibly cheap, at about $60 a pop, they should be systematically run as samples are gathered by brain banks and even during clinical trials,” he said. Friedman agreed, saying the current GWAS are a good start but more are needed. Lawrence Golbe, Rutgers University, New Jersey, told Alzforum that Hardy’s idea holds merit provided the samples are well-characterized. “That could certainly work,” he said. “You’d have to get everybody to send their sample to a central lab and make sure the diagnostic quality was good. One thing that kills a GWAS is a few false positives.” On the up side, the positive predictive value for diagnosis of PSP-RS now exceeds 95 percent since clinicians have gotten adept at recognizing the disorder.
Meanwhile, the PSP Genetics Consortium plans to sequence at least 2,000 whole genomes. As outlined by Friedman, this project started as a collaboration among 11 investigators once they realized they had about 2,000 autopsy-verified cases between their groups, and another 100–200 accruing annually. Co-funded by CurePSP, the Tau Consortium, and NIH, and now expanded to 13 investigators, the project will sequence up to 3,000 PSP whole genomes and do GWAS on any new cases. Friedman hopes this work will find new risk alleles, targets, and pathways involved in pathology. It could also help stratify responders in clinical trials based on genotype, and identify protective alleles or mechanisms that delay disease onset. The project is committed to data sharing and joint analysis.—Tom Fagan
References
News Citations
- GWAS Fingers Tau and Other Genes for Parkinsonian Tauopathy
- Protecting Proteasomes from Toxic Tau Keeps Mice Sharp
- Noncoding RNAs Evince World of Gene Regulation in Dopaminergic Neurons
Paper Citations
- Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, PSP Genetics Study Group, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Müller U, Schellenberg GD. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011 Jun 19;43(7):699-705. PubMed.
- Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, Baker M, Finch NC, Yoon H, Kim J, Fujioka S, McLean CA, Ghetti B, Spina S, Cantwell LB, Farlow MR, Grafman J, Huey ED, Ryung Han M, Beecher S, Geller ET, Kretzschmar HA, Roeber S, Gearing M, Juncos JL, Vonsattel JP, Van Deerlin VM, Grossman M, Hurtig HI, Gross RG, Arnold SE, Trojanowski JQ, Lee VM, Wenning GK, White CL, Höglinger GU, Müller U, Devlin B, Golbe LI, Crook J, Parisi JE, Boeve BF, Josephs KA, Wszolek ZK, Uitti RJ, Graff-Radford NR, Litvan I, Younkin SG, Wang LS, Ertekin-Taner N, Rademakers R, Hakonarsen H, Schellenberg GD, Dickson DW. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015 Jun 16;6:7247. PubMed.
- STEELE JC, RICHARDSON JC, OLSZEWSKI J. PROGRESSIVE SUPRANUCLEAR PALSY. A HETEROGENEOUS DEGENERATION INVOLVING THE BRAIN STEM, BASAL GANGLIA AND CEREBELLUM WITH VERTICAL GAZE AND PSEUDOBULBAR PALSY, NUCHAL DYSTONIA AND DEMENTIA. Arch Neurol. 1964 Apr;10:333-59. PubMed.
- Jabbari E, Woodside J, Tan MM, Shoai M, Pittman A, Ferrari R, Mok KY, Zhang D, Reynolds RH, de Silva R, Grimm MJ, Respondek G, Müller U, Al-Sarraj S, Gentleman SM, Lees AJ, Warner TT, Hardy J, Revesz T, Höglinger GU, Holton JL, Ryten M, Morris HR. Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Ann Neurol. 2018 Oct;84(4):485-496. Epub 2018 Sep 15 PubMed.
- Weber A, Schwarz SC, Tost J, Trümbach D, Winter P, Busato F, Tacik P, Windhorst AC, Fagny M, Arzberger T, McLean C, van Swieten JC, Schwarz J, Vogt Weisenhorn D, Wurst W, Adhikary T, Dickson DW, Höglinger GU, Müller U. Epigenome-wide DNA methylation profiling in Progressive Supranuclear Palsy reveals major changes at DLX1. Nat Commun. 2018 Jul 26;9(1):2929. PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.