With a Shot of Adrenaline, Amyloid-β Sparks Tau Cascade
Quick Links
Aβ oligomers may set off tau phosphorylation by hijacking the adrenergic system, according to a study published January 15 in Science Translational Medicine. Researchers led by Qin Wang at the University of Alabama in Birmingham reported that Aβ oligomers bind to the α2A-adrenergic receptor. This not only sensitized the receptor to its ligand, norepinephrine, but also rejiggered the downstream signaling cascade to include activation of a new substrate, GSK-3β, a kinase that phosphorylates tau. The researchers reported that blocking α2AAR signaling in mouse models of amyloidosis effectively nipped Aβ toxicity in the bud—dousing neuroinflammation, reducing tau hyperphosphorylation, and even preventing memory loss. All this occurs at one hundredth the concentration of Aβ needed to induce GSK-3β in cells lacking α2AAR.
- In mice, Aβ oligomers bind α2A adrenergic receptors.
- This triggers GSK-3β activation, and tau phosphorylation.
- Blocking α2AARs reduced p-tau, Aβ plaques, and memory loss.
“This elegant study provides a possible pathway to explain the interaction between Aβ and hyperphosphorylated tau in the pathogenesis and progression of Alzheimer’s disease,” commented Lea Grinberg of the University of California, San Francisco. “Modulation of this pathway has the potential to slow down AD progression, if the same mechanism proves to be relevant to AD in humans.”
Produced by neurons in the locus coeruleus (LC)—a tiny kernel nestled deep within the brainstem—the neurotransmitter norepinephrine sharpens attention, arousal, cognition, and responses to stress. NE orchestrates all this by binding to a variety of adrenergic receptors expressed in the LC and elsewhere in the brain. In AD, LC neurons are among the first afflicted by tau pathology and start to wither early in the disease (Braak et al., 2011; Weinshenker, 2018; Kelly et al., 2017).
Researchers have attributed agitation, aggression, and psychosis in people with AD to surviving LC neurons that overcompensate for the loss of their brethren by ramping up adrenergic signaling (Herrmann et al., 2004). Damage to the LC may accelerate AD progression, since previous studies have demonstrated that loss of LC neurons exacerbates Aβ deposition, neuroinflammation, and memory loss in mice (Dec 2010 webinar).
First authors Fang Zhang and Mary Gannon investigated the potential role of one subfamily of adrenergic receptors in AD—α2AAR. These G-protein-coupled receptors are expressed in several organs, including the brain. Agonists trigger a range of responses including sleepiness, low blood pressure, and improved cognition (Cottingham et al., 2011).
To discern if α2AAR signaling went haywire in AD, the researchers measured α2AAR signaling in membrane extracts from postmortem brain samples of 15 people with AD versus 15 controls. In the former, α2AAR were abnormally sensitive, as gauged by the amount of NE required to set off G protein activation. Sifting through the National Alzheimer’s Coordinating Center database, Zhang and colleagues also found that the α2AAR agonist clonidine worsened cognition in people with dementia. This hypertension drug did not tax cognition in healthy people. Together, the findings hinted that hyperactive α2AAR receptors might exacerbate AD.
The researchers next looked for α2AAR shenanigans in mice. They found that it took about 20-fold less NE to stimulate α2AAR signaling—as measured by G protein activation—in cells from APP/PS1 mice than in cells from non-transgenic mice, despite similar receptor levels. Cells from APP knock-ins also had slightly more sensitive receptors than controls. Similarly, adding synthetic Aβ42 oligomers to cell membrane preparations from wild-type mice boosted sensitivity of the receptor to NE, as did extracts of soluble matter from AD brain. Together, the findings suggested that Aβ oligomers somehow lower the threshold for NE activation of α2AAR signaling.
To find out how Aβ boosts NE signaling, the researchers ran biochemical, genetic, and structural experiments. They found that Aβ oligomers bound to an allosteric site on the α2AA receptor, distinct from its NE binding site. This changed the signaling cascade, leading to phosphorylation of new target proteins. From a kinase screen, the researchers found that one was GSK-3β.
Not only did Aβ enhance NE-mediated G protein activation, NE signaling enhanced Aβ activation of GSK3β as well. When added with clonidine to cultured neurons, just 20 nM Aβ oligomers activated GSK3β—less than 1 percent of the Aβ concentration previously reported to activate GSK3β (Jo et al., 2011; Kirouac et al., 2017). In the absence of clonidine, Zhang found no activation of GSK-3β by up to 100 nM Aβ. These findings suggest that α2AAR signaling dramatically enhanced the potency of Aβ to set off GSK-3β activation and tau phosphorylation, at least in vitro.
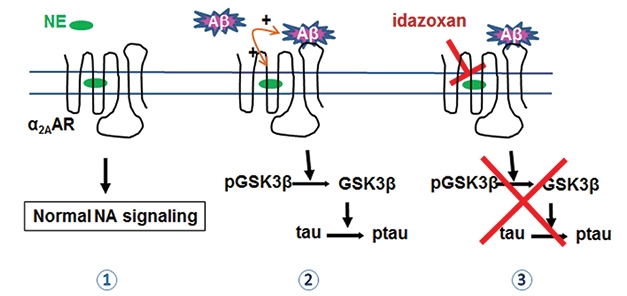
Adrenergic Hijacking? Norepinephrine (NE) activates normal α2AAR signaling (1). Aβ redirects α2AAR to activate GSK-3β, which phosphorylates tau (2). Idazoxan, an α2AAR antagonist, blocks the cascade (3). [Courtesy of Zhang et al., Science Translational Medicine, 2020.]
Next, the researchers injected Aβ oligomers into the brains of non-transgenic mice, with or without blocking GSK-3β or α2AARs. On their own, Aβ oligomers activated GSK-3β, resulting in tau hyperphosphorylation in the hippocampus. In the presence of lithium, a GSK-3β inhibitor, or idazoxan, an α2AAR blocker, both GSK-3β activation and tau hyperphosphorylation were curbed. The findings implicated the adrenergic pathway in the instigation of tau hyperphosphorylation by Aβ oligomers.
Would blocking α2AAR signaling stave off disease in mice with amyloidosis? Indeed, treating plaque-ridden 7.5-month-old APP/PS1 mice with idazoxan for eight weeks reversed GSK-3β hyperactivation and lowered the burden of hyperphosphorylated tau “pretangles,” i.e., phospho-tau clusters in Aβ plaque-laden areas. These mice do not develop the fibrillar tau tangles observed in people with AD. Idazoxan-treated mice had roughly 30 percent less phospho-tau per area of Aβ than control mice did (see image below). The α2AAR blocker also curbed Aβ plaque burden, and lowered the density of Iba1-positive microglia, suggesting it reduced neuroinflammation. The treatment had similar effects in APP knock-in animals.
Finally, the researchers found that idazoxan treatment spared APP/PS1 mice from spatial memory loss. In APP knock-ins, which are slow to retreat to dark, hidden areas when placed in more environs, idazoxan restored their reticence.

Tau Blockade. Clusters of phosphorylated tau (red) comingle with Aβ plaques (green) in control APP/PS1 mice. Idazoxan reduces phospho-tau clusters. [Courtesy of Zhang et al., 2020.]
Wang told Alzforum that the findings not only mechanistically link Aβ to tau pathology, but also support the idea that adrenergic signaling promotes Aβ accumulation. This is in keeping with a previous study that linked GSK-3β activation to Aβ production, as well as one that found adrenergic signaling disrupts proper trafficking of APP by sorting receptors (Chen et al., 2014; Ly et al., 2013).
To Wang’s mind, the potency of Aβ oligomers in activating GSK3β via the adrenergic cascade implies that tau pathology starts early in AD, perhaps even before amyloid shows up on PET scans. This is in keeping with recent CSF biomarker data that detected an uptick in p-tau at the earliest stages of AD (Aug 2019 news).
Virgil Muresan of Rutgers University, Newark, New Jersey, proposed more than a decade ago that the Aβ cascade starts out in the LC. He wrote that the findings explain why the region is also host to the earliest accumulation of tau (Muresan and Muresan, 2008). “The paper does not address what is the primary event that initiates AD, i.e., where the initial pool of Aβ oligomers—even infinitesimal in amount—comes from,” Muresan added, though his previous studies suggest that LC neurons release copious amounts of Aβ oligomers.
The low concentration of Aβ oligomers required to set the tau cascade in motion could explain why Aβ-targeted therapies have so far failed to dramatically slow cognitive decline in people with AD, Wang said. Immunotherapies would be hard-pressed to lower Aβ concentrations into the nanomolar range required to prevent Aβ oligomerization, especially since Aβ commonly reaches micromolar levels in the AD brain. Wang believes adding an α2AAR blocker such as idazoxan might make Aβ-targeted therapies more effective. Idazoxan has been tested in clinical trials for depression, schizophrenia, and progressive supranuclear palsy (Litman et al., 1996; Ghika et al., 1991).
“This study represents an important advance in the field, particularly given the rigor of the experiments,” commented David Weinshenker of Emory University School of Medicine in Atlanta. “Moreover, identification of the α2AAR as a mediator of Aβ oligomer-triggered tau pathology provides a new target for Alzheimer’s disease pharmacotherapies.” However, Weinshenker cautioned that α2AAR antagonists block all α2AAR transmission throughout the body and have cardiovascular and anxiogenic effects. More detailed structural knowledge of Aβo-α2AAR binding could aid in the development of therapeutic molecules to block the interaction between the two molecules, while leaving normal α2AAR signaling intact, he said. Wang is running screens in search of just such a compound.
Khalid Iqbal of the New York State Institute for Basic Research in Staten Island wondered how to reconcile the study’s findings with the fact that about 30 percent of aged people have significant brain amyloid accumulation, but no clinical symptoms of AD. “Either there is some mechanism overriding that reported in this study in the human brain, or the findings are limited to transgenic mouse models used,” he wrote.—Jessica Shugart
References
Webinar Citations
Research Models Citations
News Citations
Paper Citations
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011 Nov;70(11):960-9. PubMed.
- Weinshenker D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018 Apr;41(4):211-223. Epub 2018 Feb 20 PubMed.
- Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol Commun. 2017 Jan 21;5(1):8. PubMed.
- Herrmann N, Lanctôt KL, Khan LR. The role of norepinephrine in the behavioral and psychological symptoms of dementia. J Neuropsychiatry Clin Neurosci. 2004 Summer;16(3):261-76. PubMed.
- Cottingham C, Chen H, Chen Y, Peng Y, Wang Q. Genetic variations of α(2)-adrenergic receptors illuminate the diversity of receptor functions. Curr Top Membr. 2011;67:161-90. PubMed.
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, Cho K. Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat Neurosci. 2011 May;14(5):545-7. PubMed.
- Kirouac L, Rajic AJ, Cribbs DH, Padmanabhan J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer's Disease. eNeuro. 2017 Mar-Apr;4(2) Epub 2017 Mar 27 PubMed.
- Chen Y, Peng Y, Che P, Gannon M, Liu Y, Li L, Bu G, van Groen T, Jiao K, Wang Q. α(2A) adrenergic receptor promotes amyloidogenesis through disrupting APP-SorLA interaction. Proc Natl Acad Sci U S A. 2014 Dec 2;111(48):17296-301. Epub 2014 Nov 17 PubMed.
- Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013 Jan 2;123(1):224-35. PubMed.
- Muresan Z, Muresan V. Seeding neuritic plaques from the distance: a possible role for brainstem neurons in the development of Alzheimer's disease pathology. Neurodegener Dis. 2008;5(3-4):250-3. Epub 2008 Mar 6 PubMed.
- Litman RE, Su TP, Potter WZ, Hong WW, Pickar D. Idazoxan and response to typical neuroleptics in treatment-resistant schizophrenia. Comparison with the atypical neuroleptic, clozapine. Br J Psychiatry. 1996 May;168(5):571-9. PubMed.
- Ghika J, Tennis M, Hoffman E, Schoenfeld D, Growdon J. Idazoxan treatment in progressive supranuclear palsy. Neurology. 1991 Jul;41(7):986-91. PubMed.
Further Reading
Papers
- Raskind MA, Peskind ER. Neurobiologic bases of noncognitive behavioral problems in Alzheimer disease. Alzheimer Dis Assoc Disord. 1994;8 Suppl 3:54-60. PubMed.
- Olivieri P, Lagarde J, Lehericy S, Valabrègue R, Michel A, Macé P, Caillé F, Gervais P, Bottlaender M, Sarazin M. Early alteration of the locus coeruleus in phenotypic variants of Alzheimer's disease. Ann Clin Transl Neurol. 2019 Jul;6(7):1345-1351. Epub 2019 Jun 23 PubMed.
- Muresan Z, Muresan V. Neuritic deposits of amyloid-beta peptide in a subpopulation of central nervous system-derived neuronal cells. Mol Cell Biol. 2006 Jul;26(13):4982-97. PubMed.
Primary Papers
- Zhang F, Gannon M, Chen Y, Yan S, Zhang S, Feng W, Tao J, Sha B, Liu Z, Saito T, Saido T, Keene CD, Jiao K, Roberson ED, Xu H, Wang Q. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci Transl Med. 2020 Jan 15;12(526) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Emory University
We have known for some time that Aβ and tau interact with each other and that Aβ can trigger and exacerbate tau pathology, but the molecular mechanisms have not been fully defined. The present study shows that Aβ oligomers (Aβo) can act as a positive allosteric modulator of α2A-adrenergic receptors (α2AAR), and that α2AAR signaling increases tau hyperphosphorylation via inappropriate engagement of the protein kinase GSK3β. This study represents an important advance in the field, particularly given the rigor of the experiments, which included cell culture and transgenic mouse models as well as postmortem human brain tissue, controls for the specificity of the Aβo-α2AAR interaction, and effects on cognition across species. Moreover, identification of the α2AAR as a mediator of Aβo-triggered tau pathology provides a new target for Alzheimer’s disease pharmacotherapies.
These findings have important implications for both the basic neurobiology underlying Alzheimer’s disease as well as potential treatments, and suggest new directions for future research. For example, although Aβ-based transgenic mice show some tau hyperphosphorylation, wild-type mouse tau is relatively resistant to aggregation and toxicity, and it would be informative to assess the consequences of Aβo-α2AAR signaling on tau pathology in other models that accumulate bona-fide neurofibrillary tangles.
Noradrenergic neurons of the brainstem locus coeruleus are among the first to display tau pathology during the early progression of Alzheimer’s and degenerate later in the disease, in part due to the effects of norepinephrine metabolism on tau cleavage and aggregation (Braak et al., 2011; Weinshenker, 2018; Kang et al., 2020). Given the high expression of α2AAR in the locus coeruleus, the current study suggests that Aβo-enhanced α2AAR signaling may also contribute to the selective vulnerability of this nucleus, which would necessitate experiments specifically examining locus coeruleus neurons. Although the authors show that α2AAR antagonists can prevent Aβo-induced GSK3β activation, tau phosphorylation, and cognitive deficits in mice, these drugs are not ideal for clinical use because they block all endogenous α2AAR transmission throughout the body and have cardiovascular and anxiogenic effects.
More detailed knowledge of the structural basis mediating the Aβo-α2AAR interaction via the use of x-ray crystallography, cryo-electron microscopy, and/or in silico modeling could inform the development of therapeutic molecules that specifically interfere with Aβo-α2AAR binding without impairing endogenous norepinephrine signaling.
References:
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011 Nov;70(11):960-9. PubMed.
Weinshenker D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018 Apr;41(4):211-223. Epub 2018 Feb 20 PubMed.
Kang SS, Liu X, Ahn EH, Xiang J, Manfredsson FP, Yang X, Luo HR, Liles LC, Weinshenker D, Ye K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J Clin Invest. 2020 Jan 2;130(1):422-437. PubMed.
University of California, San Francisco
The paper shows a series of logically linked experiments using human brain tissue, wild-type mice, two lines of mice producing excessive Aβ, and neuronal cultures. It demonstrates that Aβ oligomers can bind allosterically to α2AAR receptors of the norepinephrine signaling system, which redirects norepinephrine-induced α2AAR signaling to activate Glycogen synthase kinase 3 (GSK3β), known to phosphorylate tau and increase tau phosphorylation. Blockage of the proposed pathway via mutation of the Aβ oligomer allosteric binding site to α2AAR, agents to block α2AAR receptors, or GSK3 activation and others, prevent the cascade and also reverse behavioral phenotypes in animal experiments.
This elegant study provides a possible pathway to explain the interaction between Aβ and hyperphosphorylated tau via α2AAR in the pathogenesis and progression of AD. Modulation of this pathway has the potential to slow down AD progression if the same mechanism proves to be relevant to AD in humans.
AD-type misfolded tau first aggregates in selected subcortical neurons, followed by the trans-entorhinal and entorhinal cortices. This early tau accumulation often precedes Aβ deposits, thus constituting the first evidence of AD in humans. The distribution of AD-tau aggregates is the best correlate to neuronal loss and clinical symptoms in AD.
Interestingly, neuropathological and radiological evidence suggest that AD-tau changes are likely to remain restricted to subcortical and limbic areas and only produce mild symptomatology until Aβ starts aggregating as plaques, first in the neocortex and later in limbic and subcortical regions. The mechanisms underlying the interaction between Aβ and AD-tau that, in turn, modulate AD-tau spread to the neocortex, remain mostly elusive.
This paper goes beyond providing a possible explanation underlying Aβ and AD-tau interaction, but supports the importance of norepinephrine dysfunction to AD pathogenesis. Degeneration of the pontine locus coeruleus (LC), the main seat of norepinephrine-producing neurons in the brain, is an early and integral part of AD pathophysiology. The LC is one of the first brain regions to accumulate tau-AD, already at stage 0 of neurofibrillary pathology proposed by Braak and Braak. By late AD stages, only 20 percent of the original neurons survive in the LC.
Interestingly, an early effect of LC accumulation of AD-tau is an increase in norepinephrine release and an increase in norepinephrine receptors in remote areas. Although this paper suggests a feed-forward mechanism in which Aβ oligomers make α2AAR more responsive to norepinephrine, it does not answer the question of what first caused these receptors to overreact or how early this happens in the relation to increasing levels of Aβ oligomers.
It would be interesting to learn more about temporal LC changes, if any, in mouse models used in this study. Would it be plausible that early LC dysfunction causes changes in α2AAR that, in turn, will enter a vicious cycle with Aβ oligomers perpetuating disease progression? Also, given the robustness of the experiments, it would be essential to understand how the proposed pathway complements changes in neuroinflammatory pathways mediated by LC, or a recently reported pathway showing that the norepinephrine metabolite DOPEGAL may directly trigger tau phosphorylation.
Finally, changes in beta-adrenergic signaling also have been reported in AD and Down syndrome, and it is worth looking into how both mechanisms interact. In summary, this paper sheds light into the mechanisms of AD acceleration and spreading and supports that modulating neurotransmitters systems may modify AD progression, rather than only target symptoms.
Rutgers - New Jersey Medical School
Different regions of the brain are differentially affected by plaque and tangle pathology, with the cerebral cortex and hippocampus being the primary targets. Subcortical regions are also affected. The noradrenergic locus coeruleus (LC) is involved in the control of complex behavior, including arousal, alertness, as well as cognitive and endocrine functions. As such, the LC is a target in many psychiatric and neurodegenerative disorders, including Alzheimer’s disease (AD). In fact, the LC is largely affected by cell death early in AD. However, since this brain region mostly lacks neuritic plaques, the prevailing view was that neuronal loss in the LC is caused by the cortical Aβ, which “poisons” the projections of brainstem cells (German et al., 2005).
Subsequent studies provided some evidence that the degenerating LC neurons may deprive brain regions supplied by their axons of norepinephrine, and promote AD pathogenesis in the brain regions that are primarily affected by Aβ and Tau pathology: the cortical areas, the hippocampus, the entorhinal cortex, and the frontal cortex (Heneka et al., 2006; O'Neil et al., 2007).
At about the same time, we proposed that the Aβ pathology in AD could actually begin in the LC, with the production of Aβ oligomers (Aβo) inside the LC neurons, and their transport throughout the entire brain via axonal transport (Muresan and Muresan, 2006; Muresan and Muresan, 2008). Our hypothesis envisioned that the pathogenic Aβo would be released from the axon terminals in the AD vulnerable brain regions, but not in the LC itself. This would explain the lack of Aβ accumulation, and lack of plaque formation, in the LC.
Several years after we published our hypothesis, Braak and Del Tredici also arrived at the conclusion that the LC could be the site where the pathology in AD begins—and from where it spreads throughout the brain—based on tau pathology. Their studies have shown that AT8-immunoreactive abnormal tau aggregates (pretangles) develop within proximal axons of noradrenergic LC projection neurons in the absence of both tau lesions in the trans-entorhinal region as well as cortical Aβ pathology (Braak and Del Tredici, 2011; Braak and Del Tredici, 2012). Thus, LC neurons appear to be particularly vulnerable to tau pathology. Why this would be the case remained unknown. Zhang et al. now provide a possible explanation.
This paper addresses the mechanism of preferential accumulation of AT8-immunoreactive abnormal tau aggregates in LC neurons, in the AD brain. Using mouse models of AD, AD brain samples, and data mining, the authors find that this tau pathology is initiated by low amounts of Aβo, which alter norepinephrine signaling, and lead to tau phosphorylation by activating GSK3β. The activation of the GSK3β/tau cascade requires direct binding of Aβo to α2AAR. Unexpectedly, this binding does not block binding of norepinephrine to α2AAR; moreover, activation of the pathogenic mechanism absolutely requires binding of norepinephrine to its receptor (simultaneously with Aβo, which binds to an allosteric site).
Zhang et al.’s paper is important for several reasons, some of which are listed below. We also list some issues that remain unsolved.
References:
German DC, Nelson O, Liang F, Liang CL, Games D. The PDAPP mouse model of Alzheimer's disease: locus coeruleus neuronal shrinkage. J Comp Neurol. 2005 Nov 28;492(4):469-76. PubMed.
Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006 Feb 1;26(5):1343-54. PubMed.
O'Neil JN, Mouton PR, Tizabi Y, Ottinger MA, Lei DL, Ingram DK, Manaye KF. Catecholaminergic neuronal loss in locus coeruleus of aged female dtg APP/PS1 mice. J Chem Neuroanat. 2007 Nov;34(3-4):102-7. PubMed.
Muresan Z, Muresan V. Neuritic deposits of amyloid-beta peptide in a subpopulation of central nervous system-derived neuronal cells. Mol Cell Biol. 2006 Jul;26(13):4982-97. PubMed.
Muresan Z, Muresan V. Seeding neuritic plaques from the distance: a possible role for brainstem neurons in the development of Alzheimer's disease pathology. Neurodegener Dis. 2008;5(3-4):250-3. Epub 2008 Mar 6 PubMed.
Braak H, Del Tredici K. Alzheimer's pathogenesis: is there neuron-to-neuron propagation?. Acta Neuropathol. 2011 May;121(5):589-95. PubMed.
Braak H, Del Tredici K. Where, when, and in what form does sporadic Alzheimer's disease begin?. Curr Opin Neurol. 2012 Dec;25(6):708-14. PubMed.
Make a Comment
To make a comment you must login or register.