PET imaging is adding a new layer of understanding to scientists’ concept of Alzheimer’s disease pathogenesis and progression. At the 14th Human Amyloid Imaging conference, speakers detailed nuances in the relationship between plaques, tangles, and cognitive decline. They also described nascent efforts to tie pathology in specific brain regions to specific behavioral and cognitive symptoms. While HAI featured new data on the pros and cons of various tau tracers in development, the synaptic tracer UCB-J jumped to the fore. It appears to detect synapse loss throughout AD cortex, as well as in other neurodegenerative diseases such as frontotemporal dementia and progressive supranuclear palsy.
PET Tracer Detects Synapse Loss Across Alzheimer’s Brain
Part 1 of a 2-part story.
A PET tracer that lights up synapses is able to detect a loss of connectivity across the Alzheimer’s brain, according to new research presented at the Human Amyloid Imaging conference, held January 15–17 in Miami, Florida. This new evidence of widespread loss in cortical regions broadly extends previous hints that the tracer, UCB-J, picks up synaptic loss in the hippocampi of AD patients. It holds out the prospect of live imaging of a phenomenon that neuropathologists have been describing for many years. “For the first time, we’re seeing with UCB-J PET in vivo what we expected to see based on the autopsy studies,” Christopher van Dyck of Yale University, New Haven, Connecticut, the senior author of the research, told Alzforum. In this study, lower UCB-J uptake also correlated with worse cognitive performance.
Other presentations at HAI linked decreased UCB-J signal to more plaques and tangles as seen by PET. Overall, the emerging data strengthen the case that this tracer could be used to help diagnose Alzheimer’s disease, and perhaps even detect a slowing of synapse loss with treatment, researchers said. However, so far, few people have been scanned, and scientists are repeating the findings in larger cohorts. They are also beginning to collect longitudinal data and scan people at presymptomatic disease stages to find out when synapses start to disappear.
Broadly speaking, studies increasingly combine imaging with multiple tracers, and these emerging data are helping scientists stage which biomarker changes follow others in the long process of Alzheimer’s pathogenesis. The same methods are shedding light on other neurodegenerative diseases. Some researchers in Miami reported decreased UCB-J uptake in frontotemporal dementia, Parkinson’s disease, and progressive supranuclear palsy, as well (see Part 2 of this series).
Developed by the Belgian pharma company UCB and first tested at Yale, UCB-J binds to the presynaptic vesicle protein SV2A (Jul 2016 news). In an initial study of 10 AD patients and 11 controls, van Dyck and colleagues found that patients took up less UCB-J than did controls in their hippocampus, though they were unable to see a difference in cortical regions (Dec 2017 conference news; Aug 2018 conference news).
More Amyloid, Fewer Synapses. In healthy people (left), the synaptic PET signal (top) is high, while amyloid PET (bottom) is low; in people with AD (right), the opposite is true. [Courtesy of Adam Mecca and Chris van Dyck.]
Dwindling Synaptic Signal Flags Alzheimer’s Disease
In Miami, Adam Mecca of Yale described how the methodology for analyzing the UCB-J signal had improved. Instead of using the centrum semiovale, a patch of white matter above the lateral ventricles, as the reference region, the researchers turned to the cerebellum. There, tracer uptake was less variable from person to person, lowering noise and sharpening the signal. Using this method, Mecca and colleagues scanned 19 cognitively normal controls, 14 people with mild cognitive impairment due to AD, and 20 with AD dementia.
In every brain region examined, amyloid-positive people bound less tracer than did controls (see image above, part A). The differences were biggest in the hippocampus and entorhinal cortex, but were statistically significant across the board. After correcting for brain atrophy, the signal remained significant in the hippocampus and amygdala, as well as in the entorhinal, lateral temporal, parietal, pericentral, and prefrontal cortex.
The uncorrected signal probably represents the total number of synapses in the region, while the corrected one indicates synaptic density in the remaining tissue; both are potentially interesting, van Dyck told Alzforum. Overall, the findings match the pattern of synaptic loss seen in postmortem AD brain.
And UCB-J binding mattered. More signal tracked with better scores on the CDR-SB and on a measure of episodic memory. This association between tracer uptake and cognition was robust across the whole cohort, though within the MCI and AD groups alone, it was weak. Possibly, synapse numbers vary enough between individuals to obscure disease effects in such small cohorts, van Dyck suggested. More sensitive cognitive measures may be needed to pick up the earliest effects of a modest loss in connectivity. The early studies that first described the close relationship between synaptic loss and cognitive decline were done in postmortem samples from people at advanced stages of dementia, van Dyck noted (e.g. Terry et al., 1991; DeKosky et al., 1996). Longitudinal data in larger cohorts will clarify the issue.
Other research at HAI hinted at a link between synapse loss and cognitive impairment in general. In Miami, Alexandra DiFilippo of the University of Wisconsin-Madison, presented preliminary data from UCB-J scans of participants recruited from the UW Alzheimer’s Disease Research Center. Working with Sterling Johnson, Barbara Bendlin, and Bradley Christian, DiFilippo has scanned 15 study participants so far with UCB-J, the tau tracer MK-6240, and PiB PET. Eleven of them were amyloid-negative controls, one was amyloid-positive but cognitively normal, and three were amyloid-negative but cognitively impaired. Compared with controls, the amyloid-positive person had a slight drop in UCB-J uptake in his or her hippocampus and inferior parietal and medial frontal lobes. The impaired participants had larger declines in hippocampus, parahippocampus, and entorhinal cortex. The synaptic losses in these cases of non-AD dementia suggest that this tracer could be useful for tracking many neurodegenerative conditions, DiFilippo noted.
The researchers are now scanning more participants, hoping to enroll 120. Ideally, 30 of them will have MCI, 30 AD dementia, DiFilippo said. All participants will be scanned at two time points. UW-Madison is partnering with the Yale group on synaptic imaging grants and harmonizing their methodology so they can pool data.
However, as usual in emerging AD research, things are not cut-and-dried. A recent autoradiography study of seven AD cases and seven controls reported no difference in UCB-J binding in postmortem samples (Metaxas et al., 2019). In Miami, some researchers noted that they, too, saw no large difference between AD and control UCB-J scans. However, others in Miami reported excellent results from quantitative autoradiography. The discrepancies may reflect methodological differences, such as choice of reference region.
“The past three years have been spent in method development and confirming that the tools we’ve been using do reflect the underlying biological reality,” noted Eugenii (Ilan) Rabiner of King’s College and Invicro, an imaging services company based in London. With better tools in hand, researchers can begin to ask broader questions about how synapse loss changes in aging and disease, Rabiner added.
Plaques, Tangles, Synapses: A Complicated Trio
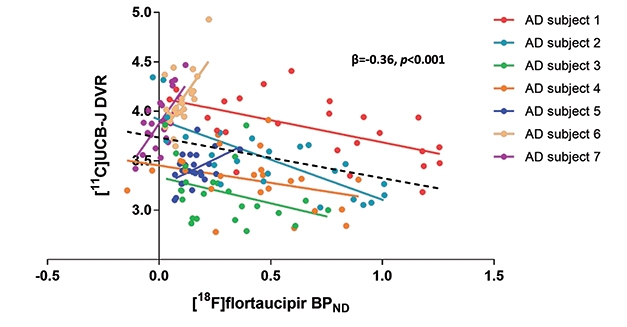
How do changes in SV2A correlate with other markers of Alzheimer’s disease? Preliminary data suggest but a rough correspondence with plaque. At HAI, Ryan O’Dell in the Yale group reported that, among 19 controls, 14 people with MCI due to AD, and 24 with AD dementia, higher PiB PET signal correlated with lower UCB-J signal (see image above, part B). However, this relationship was driven by the difference between the AD groups and controls. Within the MCI due to AD and AD dementia groups alone, amyloid PET did not relate to synaptic PET. In fact, medial temporal cortex, the cortical region with the least fibrillar amyloid, had the greatest drop in UCB-J uptake compared with controls. Perhaps this is because only healthy neurons can make amyloid, and the profound neuron death in this region lowers both amyloid production and synapse number, van Dyck speculated.
Tangles appear to correspond more closely with synapses. Mecca compared flortaucipir to UCB-J uptake in 10 cognitively normal controls and 10 with AD; half of whom were at the MCI, half at the dementia stage. In all brain regions examined, people with AD had more flortaucipir uptake than controls, and in most of those regions, they also had fewer synapses. Again, the decrease in UCB-J was most profound in hippocampus and entorhinal cortex. Tangles in EC correlated with fewer synapses in the hippocampus, which would be expected if tauopathy attacked neurons that project from the EC to the hippocampus. In future work, the Yale group will switch from flortaucipir to Merck’s tau tracer MK-6240, in order to harmonize their data with the UW study.
Stage-Specific Response? A spaghetti plot of seven participants shows a negative correlation between tangles and synapses in four people with high neocortical tau pathology, but positive in three people with low neocortical tau. [Courtesy of Bart van Berckel and Emma Coomans.]
Other groups are also correlating UCB-J with AD biomarkers. At HAI, Emma Coomans of Vrije University, Netherlands, showed data on UCB-J and flortaucipir scans in seven amyloid-positive AD patients. Working with Bart van Berckel, she found that within the group, higher flortaucipir uptake came with lower UCB-J uptake, similar to the results from Yale. However, when looking at individuals, the researchers saw a dichotomous response. In four people who had high flortaucipir signal in their neocortices, the more tangles they had in a given region, the lower the synaptic signal was in that region. The other three people had low neocortical tangles; in them, more tangles in a given brain region associated with a higher synaptic signal there (see image above).
Coomans suggested that this may be because high synaptic density in a region might, at first, actually attract and facilitate tangle pathology as it travels through synaptically connected circuits. On the other hand, it could reflect compensatory synaptic upregulation at early stages of disease, followed by synapse loss later. Some early studies have reported larger synapses in AD brain, as well as axonal sprouting and formation of new synapses (Scheff et al., 1990; Dekosky and Scheff, 1990; Cotman and Anderson, 1988). To see if their results hold up, the researchers will scan 30 people and follow them over time. They will also compare UCB-J imaging to magnetoencephalography, a different measure of synaptic activity.
Researchers are eager to start scanning people at presymptomatic stages. Synapse loss may be the earliest marker of degeneration, before atrophy, van Dyck believes. How early does it start? Can it be detected before cognitive decline manifests on tests? And if synapses vanish that early, might the UCB-J signal correlate better with amyloid accumulation at that stage than later on?
In cell culture, Aβ oligomers damage dendritic spines. Pedro Rosa-Neto, McGill University, Montreal, believes that amyloid may be the culprit behind synaptic loss at preclinical stages, while tangles may kill synapses later. “Amyloid is not necessarily associated with a change in cognition, but it is associated with a change in synaptic markers,” Rosa-Neto told Alzforum. Alternatively, van Dyck speculated that Aβ oligomers and pathological forms of tau might interact at early disease stages to harm synapses.—Madolyn Bowman Rogers
Multimodal Imaging of Neurodegenerative Diseases Links Pathology and Cellular Dysfunction
Part 2 of a 2-part story
Scientists are increasingly optimistic that PET tracer UCB-J, which recognizes a presynaptic vesicle protein, can reliably detect synapse loss in the Alzheimer’s brain (see Part 1 of this series). And it’s not just for Alzheimer’s. Synapses are vulnerable to protein pathologies besides plaques and tangles, and the tracer may become broadly useful for detecting the earliest signs of degeneration in numerous brain disorders, reported scientists at the Human Amyloid Imaging Conference, held January 15–17 in Miami. To investigate how disease progresses in these disorders, researchers are combining multiple PET tracers and fluid biomarkers in observational cohort studies.
Beyond Alzheimer’s: Synapse Loss Marks Neurodegeneration Eugenii (Ilan) Rabiner of King’s College and Invicro, an imaging services company based in London, leads MIND MAPS, a London-based public-private consortium that characterizes new imaging markers across several diseases. In Miami, Rabiner and Roger Gunn, at Imperial College and Invicro, reported preliminary data from 31 healthy volunteers, 12 Alzheimer’s patients, 12 Parkinson’s patients, and six with frontotemporal dementia, all of whom underwent scans with three different tracers.
One is the synapse marker UCB-J. One is BCPP-EF, a tracer that detects mitochondrial complex 1 as a measure of cellular energy generation (Harada et al., 2013; Tsukada, 2014). The third is SA4503, a tracer that binds the sigma-1 receptor (Mansur et al., 2020). This receptor regulates calcium signaling between the endoplasmic reticulum and mitochondria. It maintains neuronal homeostasis, is considered to be a marker of cellular stress, and has been a drug development target (see Edonerpic, Blarcamesine, Nuedexta). All participants are invited to return for follow-up scans after one year, and longitudinal scans are ongoing.
In AD and FTD, uptake of both the synapse and the mitochondria marker was down one-tenth to one-third across multiple brain regions, reaching a maximum drop of 40 percent in the hippocampus. This correlated with declining MMSE scores. AD patients also had some increases in SA4503 uptake. The Parkinson’s patients, who currently have early stage disease, had only modest declines in the synapse and mitochondrial markers, and no change in SA4503. The PD cohort has completed follow-up, but did not progress either clinically or by biomarkers in this time frame.
Jonathan Rohrer and Mica Clarke, University College London, lead the FTD portion of this study. It is still recruiting participants, and aiming for 12 people. The six enrolled so far all have behavioral variant FTD. Compared with 17 healthy controls, they had a marked loss of UCB-J uptake, particularly in frontotemporal regions, but also in parietal and the cingulate cortex and in subcortical regions such as hippocampus and amygdala. BCPP-EF binding was down in most of the same regions, as well. Some participants had a distinct pattern, with a more global decrease in uptake of both tracers across the brain. Overall, the magnitude of the loss was similar to that in AD patients, and perhaps more consistent across the cohort, Rabiner noted. The drop-off in tracer uptake exceeded atrophy in a given region, hinting that synapses die before the tissue shrinks.
“The magnitude of the change was quite surprising to me, especially when you compare it to flortaucipir change in FTD, which isn’t very large and doesn’t show much presymptomatically,” Rohrer told Alzforum. He noted that this FTD cohort comes from GENFI, i.e., it has familial forms of FTD due to mutations in tau, progranulin, or C9ORF72. Eventually, the researchers hope to have enough participants to characterize differences between these variants.
Rabiner speculated that mitochondrial dysfunction might precipitate synaptic loss, although he has not yet seen any sign that low BCPP-EF uptake precedes changes in UCB-J. Intriguingly, Rosa-Neto and Tatsuhiro Terada at McGill reported in Miami that increased tau tracer uptake in the entorhinal cortices of AD patients correlates with lower uptake of BCPP-EF there, hinting that tangles could precede mitochondrial failure.
Meanwhile, in a preprint on medRxiv, researchers led by James Rowe and Negin Holland at the University of Cambridge report lower UCB-J uptake in two sporadic primary tauopathies, progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). In 10 people with PSP and 12 with CBD, UCB-J uptake was down as much as 50 percent in numerous cortical and subcortical regions, including frontal, parietal, temporal, and occipital cortex, and hippocampus, insula, and amygdala. This decrease correlated with disease severity (Holland et al., 2020).
Another recent study from the U.K., led by Oliver Howes at the MRC London Institute of Medical Sciences, describes lower UCB-J binding in the frontal and anterior cingulate cortices of 18 middle-aged people with schizophrenia compared with 18 controls. They report a large effect size, with a Cohen’s d of 0.8 to 0.9 (Onwordi et al., 2020).
The Next Generation
One drawback of UCB-J is that it can only be labeled with C11, limiting its use to facilities that have a cyclotron on site. Scientists at Yale and Invicro have developed an 18F-labelled version of UCB-J, originally called MNI-1126/SDM8 and recently renamed SynVesT1 (Patel et al., 2019; Li et al., 2019; Constantinescu et al., 2019). Pedro Rosa-Neto of McGill University, Montreal, is testing its usefulness.
Rosa-Neto runs the Translational Biomarkers of Aging and Dementia (TRIAD) study at McGill. This longitudinal project enrolls 504 participants ranging from cognitively healthy to MCI, AD dementia, and atypical dementia. Each of these volunteers agreed to slide into the scanner for amyloid PET with AZD4694, tau PET with MK-6240, plus MRI. In addition, one-third will be scanned with SynVesT1 PET, another third with the inflammation marker PBR28, and the final third with martinostat, a marker for epigenetic dysregulation.
“The idea is to create a benchmark system for comparing new biomarkers,” Rosa-Neto told Alzforum.
Besides PET, the TRIAD group also investigates fluid biomarkers. Andrea Benedet and Nesrine Rahmouni of McGill presented some of these data at HAI, reporting, for example, that neurogranin levels in cerebrospinal fluid correlate with amyloid and tau tracer uptake in temporal, parietal, and occipital cortex. This substudy comprised 99 healthy controls, 36 people with MCI, and 23 with AD. The rise in CSF neurogranin was apparent already at the MCI stage.
Neurogranin is a marker of synaptic damage, again linking synapse loss to AD pathology at early disease stages, but there is no PET tracer for it. There is, however, a fluid assay for SV2A (Heurling et al., 2019). Rosa-Neto will try to correlate CSF levels of the protein with the SynVesT1 PET signal.—Madolyn Bowman Rogers
As the field of Alzheimer’s and related dementias is trying to go all-in on detecting—and targeting—tau pathology, scientists are still grappling with exactly how to deploy PET imaging in the effort. A still-new, and still-limited set of tools, existing tau tracers vary quite a bit in terms of their sensitivity, off-target binding, and exactly which form of tau they recognize. The 14th Human Amyloid Imaging conference, held January 15–17 in Miami, brought together researchers for the latest characterization of these agents, as well as for some contrast-and-compare data sharing. The conference was organized by Keith Johnson of Massachusetts General Hospital, Boston, William Jagust of the University of California, Berkeley, William Klunk and Chester Mathis of the University of Pittsburgh, and Maria Carrillo of the Alzheimer’s Association.
One upshot from a diverse array of presentations: PI-2620 and APN-1607 might recognize the four-repeat (4R) aggregates of tau that are found in non-AD tauopathies such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). Another: Flortaucipir off-target binding may be a bit more extensive than previously thought. Physical activity during the tracer uptake period seemed to lower nonspecific binding, however, suggesting one way to manage this. Finally: Some researchers argued that MK-6240 is more sensitive than other tracers, and two new tracers—JNJ-067 and SNFT-1—made their entré. Read below for a summary.
Do We Have a First Tracer For 4R Tau?
Four current tracers—flortaucipir, MK-6240, RO-948, and GTP1—detect, with high specificity, the paired helical filaments of 3/4R tau that are present in the Alzheimer’s brain, but they bind weakly, if at all, to the 4R tau fibrils that mark PSP and CBD (Sep 2016 conference news; Dec 2016 conference news). Initially, scientists were disappointed that tau PET might not work in non-AD tauopathies, but appreciated starting out with tracers that distinguished between different types of tauopathy (Sep 2018 news). Now, they appear to be getting a tool for 4R tau, after all, in Life Molecular Imaging’s PI-2620. In Miami, Matthias Brendel of University Hospital Munich noted that in autoradiography studies, the tracer binds to the globus pallidus and putamen in sections from PSP brains, but not controls.
Brendel and colleagues scanned 60 clinically diagnosed PSP patients and 10 controls at three centers in Germany, one in Australia, and one in the United States. The former comprised 40 people with classic PSP symptoms, aka Richardson’s syndrome, and 20 with atypical symptoms. Four-R tau predominates more in Richardson’s syndrome than in atypical PSP (Williams et al., 2005), and PI-2620 retention reflected this. People with PSP-RS had elevated uptake in all brain regions affected by this disease: the globus pallidus, putamen, subthalamic nucleus, substantia nigra, and dentate nucleus. The atypical PSP group had elevated signal only in the globus pallidus and subthalamic nucleus, and their signal was less intense than in PSP-RS brains. Overall, a semiquantitative read of PI-2620 scans distinguished PSP-RS cases from controls with a sensitivity and specificity of 85 and 83 percent, respectively. For atypical PSP, sensitivity and specificity were 60 and 83 percent, respectively. The findings suggest this tracer could aid PSP diagnosis, Brendel suggested.
In a separate study of 10 controls and 32 people with corticobasal degeneration, patients again had elevated tracer signal in the globus pallidus, putamen, subthalamic nucleus, and dentate nucleus, with a trend for higher binding in the frontal cortex. Dentate nucleus binding in these patients tracked with how severe their symptoms were, and cortical binding with how long they had been symptomatic. For 23 of the CBD patients, their scans were positive by visual read alone, suggesting the tracer might be readily adapted for diagnosing this disorder, as well.
More Inclusive Tracer? PI-2620 binds to 4R aggregates in the tauopathies PSP (second row) and CBD (third row), and to paired helical filaments of tau in AD (bottom row), while having little background in healthy controls (top row). [Courtesy of Roesler et al., Progress in Neurobiology.]
While PI-2620 binds both 3/4R and 4R tau aggregates, it does so with different kinetics, Brendel reported in Miami. He compared tracer uptake in 14 amyloid-positive participants, 14 with suspected CBD, and 15 with suspected PSP. The latter two groups bound less tracer in cortex and cleared it faster than the amyloid-positive people. The data imply that PI-2620 binds with lower affinity to 4R than 3/4R tau. This difference could help distinguish these two types of tau pathology in imaging studies, Brendel believes. This is relevant, because patients with a clinical diagnosis of CBD can have either type of tau aggregate.
Lest it seem progress was easy or straightforward, some data presented in Miami cast doubt on PI-2620’s ability to detect 4R tau. Cinthya Agüero at Massachusetts General Hospital, Boston, compared binding of flortaucipir, MK-6240, and PI-2620 in the same set of postmortem samples. She reported that all three bound strongly to tangles in AD brain, but not in non-AD tauopathies such as Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, and chronic traumatic encephalopathy. What gives? Brendel noted that the Boston group used cryopreserved tissue sections, while his group used paraffin embedding. In preliminary experiments on cryopreserved sections from PSP brains, Brendel, too, sees a weaker PI-2620 signal than in paraffin sections. Thus, this could be a methodological difference, he said.
Flortaucipir Off Target
At this stage in tau PET research, one of the bigger worries with detecting 3/4R tau, i.e. Alzheimer’s tangles, is off-target binding that may confound the interpretation of a person’s scan. Just how serious a problem is it? Flortaucipir, the only one of the first-generation tau tracers still standing, binds nonspecifically to basal ganglia and choroid plexus. Choroid-plexus binding is problematic because its signal could bleed into the hippocampus lying just below it, potentially inflating the estimation of AD pathology.
In Miami, Shaney Flores of Washington University in St. Louis reported that, in the hands of the WashU group, flortaucipir binds to the skull, as well. Among 216 amyloid-negative and 97 amyloid-positive participants, about 16 percent had high flortaucipir uptake in the skull. This binding was most pronounced in inferior occipital and temporal regions. It did not correlate with the intensity of other off-target binding, suggesting it recognizes a unique target. Inferior temporal regions are an early site of tau accumulation in AD. In the WashU cohort, most of those with skull uptake did not have amyloid pathology, suggesting the signal could potentially lead to a false-positive AD diagnosis.
In fact, off-target binding to the skull or meninges might explain some gender differences that were previously reported with flortaucipir scans, said Davneet Minhas of the University of Pittsburgh. The UPitt group examined flortaucipir scans from 162 people scanned there, plus 169 ADNI participants. They were cognitively healthy or had MCI. In both cohorts, women had higher uptake than men around the perimeter of the brain but not in the brain, possibly clouding comparisons of tau pathology between the sexes. Several studies had previously reported higher flortaucipir uptake in women (Feb 2019 news; Nov 2019 news).
Now for some good news: It appears off-target binding can be held at bay by physical activity in the time between a person’s injection and scan, i.e., while the tracer is traveling into the brain and finding its targets there. According to Hoon-Ki (Paul) Min of the Rochester Mayo Clinic in Minnesota, among a cohort of 330 people scanned with flortaucipir, those who reported napping during this 80-minute uptake period had higher binding to bone and meninges than those who remained active. The more sleep a person reported getting, the higher this off-target binding was. On the other hand, participants who walked around during this period had less uptake in these regions, as well as less off-target binding in basal ganglia, than those who stayed sedentary.
Scientists in Sweden flagged an additional type of off-target binding. Antoine Leuzy of Lund University, Malmö, directly compared flortaucipir and RO-948 uptake in the same three people with semantic variant primary progressive aphasia (svPPA), a form of FTD distinguished by TDP-43 pathology rather than tau tangles. Flortaucipir lit up the lower temporal lobe, whereas RO-948 did not. Which molecule in the temporal lobe might generate flortaucipir’s off-target signal is a mystery. A previous study found little evidence for flortaucipir binding TDP-43 (Smith et al., 2019). One possibility is that flortaucipir is binding to MAO-B, which is expressed by reactive astrocytes, Leuzy noted. “In common with most next-generation tau tracers, RO-948 seems to have a better off-target binding profile and broader dynamic range than flortaucipir,” he wrote to Alzforum (Smith et al., 2020).
Meanwhile, Milos Ikonomovic of UPitt led a postmortem study that correlated the flortaucipir PET signal with region-matched ELISA concentrations of total tau and tau phosphorylated at Ser396, Ser199, or Thr231. He did this in autopsy brains from three people who had had Alzheimer’s dementia and one cognitively normal person with AD pathology. Flortaucipir PET correlated with the ratio of pSer396 to total tau and pSer199 to total tau in the three people who had been impaired, but not in the one who had died while still cognitively normal. In one brain from a cognitively impaired person with severe cortical tangle pathology, flortaucipir PET also correlated with the ratio of pThr231 to total tau.
Broader Range. MK-6240 (top) generates a higher signal than flortaucipir (bottom) in the same AD patients. [Courtesy of the University of Pittsburgh PPG/ADRC.]
MK-6240, the Sensitive Guy Among the Lot?
With so many tau tracers being studied, which one is best? It may depend on what exactly you need it for. MK-6240 detects tau tangles present in AD brain with an affinity of 0.3 nM, which is considered high. Some HAI presentations suggested that MK-6240 is particularly sensitive to low levels of tangles, and thus might be suitable for early detection.
Brian Lopresti of UPitt directly compared flortaucipir and MK-6240 uptake in the same five people with AD, one with MCI, and nine controls. They evaluated six brain regions per person, for 90 total reads. In this study, MK-6240 had twice the dynamic range of flortaucipir in target regions, while background uptake in cerebellum was similar for both tracers. Even so, readers interpreted flortaucipir and MK-6240 scans similarly, making the same diagnostic call in 13 of the 15 people. Where the calls were discordant in specific brain regions, flortaucipir was more often read as positive than MK-6240, perhaps due to noise. The two tracers had distinct patterns of off-target binding, with flortaucipir lighting up striatum and choroid plexus, MK-6240 meninges. Both had some binding to bone.
How problematic is the meningeal binding? In Miami, Justin Sanchez of Mass. General reported that about half of clinically normal people have some extra-cerebral MK-6240 uptake that forms a halo around the brain in scans. This signal closely apposes the entorhinal cortex, a region of early tangle accumulation, with a proximity near the limit of spatial resolution. That said, this off-target signal can be distinguished from cortical uptake, Sanchez said. The trick? An analytic method that examines cortical surface projections, rather than volume-based regions of interest.
Sanchez further noted that MK-6240’s on-target cortical signal correlates with age and amyloid load, whereas its extra-cerebral signal does not. The two types of uptake follow different time courses; the on-target signal plateaus, while the extra-cerebral signal continues to rise over two hours. “We are optimistic about the ability of the surface projection method to distinguish tau-like from non-tau-like signals, and to optimize our sampling of cortical MK-6240 signal,” Sanchez wrote to Alzforum.
Firoza Lussier of McGill University in Verdun, Canada, examined MK-6240 uptake in 12 people with a familial AD mutation and in 11 noncarriers. Most of the carriers had a PS1 mutation; six of them were symptomatic. In carriers, higher tracer uptake correlated with how close a person was to his or her estimated year of onset (EYO). Regionally, this correlation was significant in the entorhinal cortex, right posterior cingulate, and precuneus. The MK-6240 signal could be detected as many as 10 years before EYO, hinting that this tracer might allow for early detection. In DIAN, which thus far has reported results with flortaucipir, the tau signal became apparent some years later, closer to a carrier’s EYO (Aug 2017 conference news).
Finally, Zhizhen Zeng of Merck reported that MK-6240 can detect tangles in Parkinson’s disease. In postmortem sections from 36 PD brains, binding of MK-6240 to the substantia nigra, caudate, and putamen matched immunostaining with antibody AT8, which recognizes paired helical filaments of tau. In all, 18 of 22 substantia nigra samples and nine of 13 caudate and putamen samples bound MK-6240. Off-target binding was limited to cells containing neuromelanin, Zeng reported. About half of Parkinson’s patients have notable tangle accumulation at autopsy, but flortaucipir shows little signal in these patients (Irwin and Hurtig, 2018; Smith et al., 2018; Hansen et al., 2020). “The results of this study show that there is significant tau pathology in the striatum and substantia nigra in almost all of the postmortem PD brains we sampled,” Eric Hostetler of Merck wrote to Alzforum. He noted that this has implications for development of an α-synuclein tracer, suggesting that that ligand will have to be selective for α-synuclein over tau tangles.
Low Background. The new tracer JNJ-067 distinguishes cleanly between control (left) and Alzheimer’s (right) brains. [Courtesy of Invicro, a Konica Minolta company.]
Meet the New Tau Tracers
Additional tau tracers are in development. In preclinical studies, Janssen’s JNJ-067 showed higher affinity than flortaucipir for tangles (Apr 2017 conference news), and in Miami, Janssen’s Mark Schmidt presented new data from scans of five amyloid-positive Alzheimer’s patients and five healthy age-matched controls. Four of the AD patients bound the tracer in their cortices. Controls had negative scans, except one person, who had focal hotspots in the occipital cortex around shotgun pellets lodged there from an old hunting accident.
JNJ-067 is in a Phase 1 trial conducted at the University of California, Berkeley, and Suzanne Baker of Lawrence Berkeley National Laboratory, California, reported preliminary findings at HAI. So far, four healthy controls, three people with AD, three with amyloid-positive MCI, and three with progressive supranuclear palsy have been scanned. Baker noted that JNJ-067 appears “cleaner” than flortaucipir, without off-target binding to choroid plexus and less affinity for MAO-B. Unlike other second-generation tau tracers, it does not appear to bind meninges. However, JNJ-067 did display off-target binding in midbrain, basal ganglia, and white matter, Baker reported.
In AD patients, JNJ-067 lit up the entorhinal cortex and widespread cortical regions, as expected. This on-target signal was higher in people whose MMSE scores were lower, but it was a generally weaker signal than seen with MK-6240 and PI-2620, Baker said. Schmidt agreed that JNJ-067 has a similar dynamic range to flortaucipir, smaller than that of MK-6240.
One MCI participant had focal cortical uptake of JNJ-067, but none had any signal in entorhinal cortex. Baker noted that one-third of ADNI MCI participants have no tracer uptake in this early cortical region, either, so it is too early to draw conclusions about the ability of this new tracer to detect MCI. So far, JNJ-067 has not labeled any brain areas in PSP patients. Most current tau tracers detect only 3/4R tau, because they were selected for binding to AD brain tissue.
In regions with high tauopathy burden, JNJ-067’s kinetics are slow, reaching steady state in about two hours. This is common with high-affinity tracers, Schmidt said, and is true for flortaucipir and MK-6240, as well. Janssen plans to make the tracer available via a royalty-free license to academic and industry groups, Schmidt said. At the moment, it is being produced at Invicro and Berkeley.
The first-generation tau tracer THK-5351, developed at Tohoku University in Sendai, Japan, was felled by nonspecific binding to the enzyme MAO-B (Apr 2017 conference news). Since then, researchers there have identified a new candidate, THK-5562, now renamed SNFT-1. In Miami, Ryuichi Harada of Tohoku reported that SNFT-1 selectively binds tau aggregates over other proteins, including Aβ, α-synuclein, TDP-43, MAO-A, and MAO-B. In autoradiography of postmortem human brain sections, SNFT-1 had a strong signal in cortex but not basal ganglia. The tracer quickly entered control mouse brain after intravenous injection, and washed out quickly, too. Researchers are now optimizing radiosynthesis for clinical study, Harada wrote to Alzforum.
Meanwhile, Aprinoia Therapeutics in Taipei, Taiwan, is developing a derivative of the discontinued tau tracer PBB3. Initially known as PM-PBB3 or MNI-958, the derivative is now called APN-1607 and is in Phase 1 in Taiwan. The compound is light-sensitive. This complicates its production and use, but this can be overcome with longer wavelength, i.e. red/yellow, lighting.
In Miami, Ing-Tsung Hsiao of Chang Gung University in Taoyuan, Taiwan, reported preliminary data comparing APN-1607 with THK-5351, not to its own predecessor PBB3. Unlike THK-5351, APN-1607 did not light up basal ganglia or thalamus in healthy controls or AD patients. The new tracer appeared to more faithfully reflect pathology than the older one, Hsiao showed. In 10 AD patients, APN-1607 uptake varied more than THK-5351 did between brain regions expected to have high and low tangle load, and it matched Braak staging. In five controls, meanwhile, APN-1607 showed but a low, uniform background signal, without the hot spots of off-target binding seen with THK-5351. The findings suggest APN-1607 would have more diagnostic power than THK-5351, Hsiao noted. APN-1607 does not bind MAO-A or MAO-B in competition assays. In a separate study of three people who had had a stroke, the new tracer, unlike THK-5351, did not bind to ischemic lesions.
Finally, APN-1607 may recognize 4R forms of tau. In Miami, Richard Margolin of Aprinoia reported that the tracer’s signal matched 4R histopathology in the tau mouse model rTg4510. So far, researchers have scanned 45 people with PSP and nine with CBD with APN-1607. Binding patterns matched the expected distribution of tangles and correlated with the clinical severity of PSP, Margolin said. Likewise, in 40 people with AD, APN-1607 uptake showed the expected pattern of binding and correlated with disease severity. Uptake in 75 healthy controls scanned so far has been low. APN-1607 does not bind meninges, but does bind to choroid plexus.—Madolyn Bowman Rogers
Can PET Match Up Areas of Protein Deposit With Alzheimer’s Symptoms?
Even though, overall, Alzheimer’s disease robs all people it afflicts of mind and memory, it progresses differently in different people. Its specific symptoms, their sequence, and how fast they worsen can vary quite a bit from one person to another. This used to be a tough nut to crack for researchers, but now, by combining data from multiple PET tracers in longitudinal study cohorts, they are beginning to decipher which regional pathologies provoke particular disease manifestations. The Human Amyloid Imaging conference, held January 15–17 in Miami, showcased the latest findings tying tangles, or plaques, to particular behavioral and cognitive impairments. Intriguingly, links between pathology and subtle symptoms emerged even in people who were still cognitively healthy. The data suggest that multitracer, and also multimodal, brain imaging may eventually allow researchers to predict how disease will progress in a given person.
Scientists’ budding ability to parse which regional pathologies and cellular processes underlie particular symptoms will grow as new tracers come onto the scene.
At HAI alone, scientists introduced first-in-human or late preclinical data for tracers recognizing α-synuclein, the α7 nicotinic receptor, the muscarinic cholinergic receptor, microtubules, activated astrocytes, and cyclooxygenase enzymes that mark inflammation.
Regional Tangles and Behavior. Tau PET signal in highlighted brain regions correlates with mild behavioral impairment in 91 cognitively impaired older people. [Courtesy of Firoza Lussier, Serge Gauthier, and Pedro Rosa-Neto.]
Do Regional Tangles Cause Psychiatric Symptoms?
Several speakers tied behavioral symptoms to tangle accumulation in specific brain regions. Jennifer Gatchel and Bernard Hanseeuw of Massachusetts General Hospital, Boston, presented data from 252 participants in the Harvard Aging Brain Study, mean age 73. At the outset, all were cognitively healthy and not depressed. That said, those among them who at baseline had more tangles in their entorhinal or inferior temporal cortices on flortaucipir scans gradually worsened on the Geriatric Depression Scale over an average of six years of follow-up. Their baseline cortical amyloid had no effect on depression.
A separate, albeit cross-sectional, study confirms a link between tangles and depression. Beau Ances of Washington University in St. Louis examined the relationship between tau pathology and depression in 301 cognitively normal participants seen at the Knight Alzheimer’s Disease Research Center. Those whose flortaucipir signal was elevated across the brain were twice as likely to be depressed as were their peers with fewer tangles. In this group, too, amyloid plaques were not associated with depression.
Ances noted that 42 percent of depressed people in this study had tangles but no plaques, i.e., suspected non-AD pathology (SNAP). Perhaps mood disorders like depression characterize SNAP, he speculated. Oddly, antidepressant use correlated with an even higher risk of depression in this study. This could be because depressed people are more likely to be prescribed antidepressants. However, Ances noted that some participants were on antidepressants for other reasons. He believes the data hint at a deleterious role for antidepressants in people who are tau-positive, but noted that larger and longitudinal studies are needed to confirm this.
Other scientists linked tangles to psychiatric symptoms more generally. Cécile Tissot and Pedro Rosa-Neto of McGill University in Verdun, Canada, compared AZD4694 amyloid PET and MK-6240 tau PET to the global score on the neuropsychiatric inventory questionnaire (NPI-Q) in 35 people with mild cognitive impairment and 28 people with AD dementia. Tangle pathology, particularly in the precuneus, hippocampal formation, and frontal cortex, correlated with worse scores on the NPI-Q. Amyloid PET and volumetric MRI did not.
Besides depression, the NPI-Q measures delusions, hallucinations, agitation, disinhibition, and sleep problems. The researchers reported seeing links between those symptoms and regional tangles. Delusions correlated with tracer uptake in the ventromedial prefrontal cortex and cuneus, hallucinations with uptake in the cuneus. This makes sense, the authors noted, because the cuneus is involved in vision, and the ventromedial prefrontal cortex in inhibition. For its part, agitation correlated with tracer signal in the frontal cortex and precuneus, parts of the default mode network. Disinhibition correlated with uptake in the precuneus, dorsomedial prefrontal cortex, and temporal poles, areas involved in emotional processing. Unsurprisingly, motor disturbances were seen in people with tangles in the motor cortex. Getting up at night, or too much daytime napping, correlated with having tangles in the ventrolateral prefrontal cortex, another area involved in inhibition and, indeed, in sleep and memory consolidation (e.g., Cowan et al., 2020).
Firoza Lussier, from the same McGill team, linked mild behavioral impairment (MBI) in older adults to their tangle burden. The clinical term MBI recently has been proposed as denoting a cluster of symptoms, including mood changes, lack of motivation, weak impulse control, and socially inappropriate behavior, that precede or go along with cognitive decline. MBI is assessed via a 34-item checklist (Ismail et al., 2017).
Lussier analyzed 91 cognitively impaired participants in the McGill Translational Biomarkers of Aging and Dementia (TRIAD) cohort, who were on average 71 years old. A person’s total MBI score associated with his or her having tangles in the orbitofrontal cortex, posterior cingulate cortex, precuneus, cuneus, and lateral temporal lobes (see image above). In 94 cognitively healthy controls, by contrast, there was little MK-6240 uptake, or MBI.
While most talks linked tau pathology to behavior, a separate study from Tissot suggests plaques can play some role, too. She assessed 37 cognitively healthy, 18 MCI, and seven AD participants in the TRIAD study. All underwent AZD4694 amyloid PET, as well as scans with the PET tracer PBR28, which binds mitochondrial translocator protein and acts as a marker of inflammation (Wang et al., 2009; Donat et al., 2018). Uptake of both tracers in the mediofrontal cortex and the anterior nucleus of the thalamus picked out people who scored high on the Apathy Inventory. The mediofrontal cortex helps regulate behavior, while the thalamus relays communication between brain regions. The findings suggest that plaques and inflammation together may make a person apathetic, the researchers said. Apathy is the most common behavioral symptom of AD, and gets worse as the disease becomes more severe.
The Pattern of Plaques or Tangles Predicts Cognitive Impairment
Other research links regional pathology to particular cognitive deficits. Michelle Farrell and Reisa Sperling of Massachusetts General Hospital reported at HAI that low levels of plaque accumulation suffice to precipitate subtle cognitive deficits in people who are not measurably impaired. In 67 cognitively normal participants in the Harvard Aging Brain Study who initially had a low amyloid load by PiB PET, plaque accumulation over four years predicted subsequent declines on the Buschke Selective Reminding Test and the Digit Symbol Substitution Test over the next two years. These tests measure executive function and processing speed.
In this cohort, tangles alone, as seen by flortaucipir PET, did not affect decline. However, in participants whose baseline amyloid load was high—1.22 SUVR or more—a flortaucipir signal in the inferior temporal cortex did presage subsequent decline on the Selective Reminding Test. The findings strengthen previous data suggesting that amyloid by itself can trigger subtle cognitive problems early in disease, but that tangles are responsible for decline later on.
A sensitive and quick new cognitive test detects early damage wrought by both plaques and tangles. Danielle Mayblyum and Emma Thibault of Mass. General, working with Keith Johnson and Dorene Rentz, evaluated 52 cognitively healthy and nine cognitively impaired participants with the digital clock drawing test (Dec 2017 conference news). The more PiB a person retained in his or her frontal, lateral temporal, and retrosplenial cortices, the worse they performed on this test. MK-6240 signal in entorhinal and inferior temporal cortex, amygdala, and hippocampus also correlated with worse performance, suggesting that both pathologies blunt the skills required for this task.
What are the earliest cognitive consequences of having tau tangles? Working with Elizabeth Mormino of Stanford University School of Medicine, Tyler Toueg examined PI-2620 tau PET scans in 39 participants in the Stanford Memory and Aging Study. Of these, 30 were cognitively healthy. Nine had subtle deficits, which the scientists defined as scoring more than 1.5 standard deviations below the population mean on a neuropsych test, or having a family member who expressed concerns about his or her memory. These nine were roughly equivalent to stage 2 AD patients as defined by recent NIA-AA recommendations, i.e., they have some cognitive decline even though they still score in the normal range on tests (Apr 2018 news).
In Toueg’s study, the nine had more tau deposition in their medial temporal lobes, including the entorhinal cortex, hippocampus, and amygdala, than did the cognitively normal group. The data suggest that tangle accumulation in these regions can precipitate cognitive decline. As expected, seven patients from the Stanford Memory Disorders Clinic who had amyloid-positive amnestic MCI or AD dementia had even more tracer uptake in these regions.
There seem to be no hard, dichotomous switches in AD pathogenesis, however. Everything slowly creeps up on the unsuspecting brain. Case in point: Even among cognitively healthy people, a higher tangle burden in the hippocampus accounts for subtle memory deficits, reported Alexandra Trelle, who leads the Stanford Aging and Memory Study. In an overlapping group of 36 cognitively healthy study volunteers, Trelle found that more hippocampal tangles meant worse performance on tests of associative memory and mnemonic discrimination. Hippocampal tangles also came with low levels of CSF Aβ42, a presymptomatic marker of AD.
In this cohort, having tangles only in the entorhinal cortex did not correlate with test scores, nor did CSF Aβ42 and p-tau. Entorhinal tangles precede hippocampal tangles in Braak staging. The findings suggest that tangles start to dull memory once they reach the hippocampus, and also that tau tracer uptake in the hippocampus might be a more sensitive early marker of decline than CSF, the authors noted.
Tangles in a given region can also predict future cognitive decline. Adam Martersteck and Emily Rogalski of Northwestern University Feinberg School of Medicine in Chicago examined 17 people with primary progressive aphasia due to Alzheimer’s disease. Aphasia—problems understanding or expressing speech—results from damage to language centers, such as Broca’s and Wernicke’s areas, in the left hemisphere. At baseline, both tangle burden and cortical thinning in these regions correlated with difficulties on the Boston Naming Test, in which participants name objects from line drawings. In the eight participants who took the test again a year later, however, only the baseline tangle load correlated with decline. Tau PET better predicts decline than does atrophy, these authors concluded. In other words, tangles are an early marker of decline, and atrophy a later marker.
It’s not just objects. Apparently tangles also mess with one’s ability to remember names. Victoria Tennant and William Jagust of UC Berkeley tested 85 cognitively healthy people enrolled in the Berkeley Aging Cohort Study on the Northwestern Famous Faces task. It asks participants to name famous people when shown photos of their faces. The more entorhinal cortical flortaucipir signal a participant had, the worse he or she did. Thinning of the fusiform gyrus also correlated with naming difficulties.
Overall, research in multiple independent groups is converging on the point that tangles in the entorhinal cortex, one of the first regions affected in AD, can already harm cognition.
But wait. David Berron and Oskar Hansson at Lund University, Sweden, implicated an even earlier region. Before appearing in the entorhinal cortex, tangles pop up in the transentorhinal cortex, also known as area 35. From there, they spread to the entorhinal cortex and from there, to the hippocampus.
Berron examined RO-948 tau PET scans in 322 cognitively healthy participants in BioFINDER2. Of these, 241 were amyloid-negative, 81 amyloid-positive. Among the amyloid-positive group, tracer uptake in area 35 correlated with atrophy there, as well as in the anterior hippocampus. The findings suggest that tangles can have both local and remote effects on atrophy, the authors noted.
Area 35 tangles, but not cortical thickness, matched up with poor delayed word recall. Regression analysis showed that this effect was partially mediated by anterior hippocampal atrophy, which explained about 19 percent of the variance in memory. Intriguingly, in 107 amyloid-positive people with MCI, hippocampal atrophy explained 91 percent of the variance in delayed recall. The data imply that other disease mechanisms besides atrophy are at work in very early stages of AD.
On the face of it, this seems to contradict Trelle’s finding that tangles have to reach the hippocampus to harm memory. The studies used different tracers, PI-2620 versus RO-948, and focused on different regions of early tangle accumulation, entorhinal cortex versus area 35, so some of the difference could be methodological. But also, these are early days for the approach of tying symptoms to regional pathologies by PET. Further research will make clear which findings are robust and repeatable, and shed more light on how local pathology leads to functional loss.—Madolyn Bowman Rogers
Ismail Z, Agüera-Ortiz L, Brodaty H, Cieslak A, Cummings J, Fischer CE, Gauthier S, Geda YE, Herrmann N, Kanji J, Lanctôt KL, Miller DS, Mortby ME, Onyike CU, Rosenberg PB, Smith EE, Smith GS, Sultzer DL, Lyketsos C, NPS Professional Interest Area of the International Society of to Advance Alzheimer’s Research and Treatment (NPS-PIA of ISTAART).
The Mild Behavioral Impairment Checklist (MBI-C): A Rating Scale for Neuropsychiatric Symptoms in Pre-Dementia Populations.
J Alzheimers Dis. 2017;56(3):929-938.
PubMed.
How Much Amyloid Will Kick Off Tangles, and Decline?
As more research groups are conducting brain imaging in Alzheimer’s disease, separately and on different continents, their data nonetheless converge into a confirmation overall of the disease’s basic progression pathway: plaques, then tangles, then cognitive decline (Perrin and Holtzman, 2009). Large longitudinal imaging studies—and lots and lots of correlation exercises with fluid biomarker and clinical measures—are now adding heft to the idea. Scientists are calculating thresholds of amyloid and tau accumulation associated with progression. They are pinpointing the brain regions involved and parsing how each pathology relates to cognitive decline. At the Human Amyloid Imaging conference, held January 15–17 in Miami, several groups reported that tangles spread beyond the medial temporal lobe only after plaques have pretty much carpeted the cortex, in agreement with prior work.
“The data at HAI reinforce previous models of Alzheimer’s progression predicted by Cliff Jack and others,” noted Michael Devous of Avid Radiopharmaceuticals in Philadelphia (Dec 2011 news). “Now we have quantitative data to support those ideas,” Devous said.
Alas, in AD research, nothing is ever simple and unanimous. Studies disagreed on the exact threshold level of amyloid needed, and some said inflammation needs to interact with amyloid for it to spark tangles. Finally, researchers quantified how strongly plaque and tangles predict cognitive decline. There, too, overall convergence was tempered by the nuance that not all amyloid-positive people with cognitive impairment have a high tangle load, hinting at other factors at work. In part, some seeming discrepancies could be because different groups set thresholds differently, use their own quantification methods, or simply slice and dice large data sets differently for their own analyses.
Widespread Tangles Correlate With Impairment. Among cognitively healthy people with amyloid plaques (left), only a few accumulate tangles outside the medial temporal lobe (darker colors indicate higher percentage of people with pathology); in the cognitively impaired group (right), the percentage is much higher. [Courtesy of Susan Landau.]
Plaques Drive Tangles
Previous imaging studies have shown that the spread of amyloid through the cortex triggers tangles to break out of their previous confinement within the medial temporal region (Mar 2016 news; Aug 2016 news). How much amyloid does it take to unleash tangles? In Miami, Keith Johnson of Massachusetts General Hospital gave an answer. He analyzed a cohort of 131 participants in the Harvard Aging Brain Study (HABS) whose average age was 75. All but eight were cognitively healthy at baseline. Participants were followed longitudinally with PiB amyloid and flortaucipir tau scans for up to four timepoints over a median span of 2.5 years.
A regression analysis of the change in plaque and tangles found that amyloid load needed to reach a threshold of 1.1 to 1.2 SUVR for tau deposition to take off. The higher a person’s baseline amyloid load, the faster both plaques and tangles accumulated. Fast progression of both pathologies correlated with greater cognitive decline on the PACC cognitive composite over two years, in line with previous HABS findings (Jun 2019 news). The PACC is sensitive to change in preclinical populations (Jun 2014 news).
David Knopman and colleagues from the Mayo Clinic in Rochester, Minnesota, independently confirmed that tangles spread only when amyloid is high, but calculated a different threshold value. They stratified 175 cognitively healthy participants in the Mayo Clinic Study of Aging into people who were amyloid-negative, with PiB PET loads below 22 centiloids; those above the threshold, from 22 to 67; and a group whose amyloid load exceeded 67 centiloids. The researchers then examined change over about two years in flortaucipir PET signal in these people’s entorhinal, inferior temporal, and lateral parietal cortex, as well as in a composite of regions typically affected in AD.
At baseline, all participants with amyloid levels above background also had an elevated tau PET signal in most regions. However, only those whose amyloid was above 67 centiloids at baseline accumulated tangles over the next two years, mostly in the inferior temporal cortex and the AD composite regions. Notably, 67 centiloids corresponds to a PiB PET SUVR of 2.0—much higher than the amyloid threshold at which the Harvard group found tangle buildup. Knopman noted that the higher cutoff fits with previous data showing that cognitive impairment occurs later in disease, at high amyloid loads, and closely corresponds to tangle spread.
One implication of Knopman’s finding is that tau PET would not be a practical outcome measure in therapy trials in preclinical AD populations unless participants’ plaque burden was very high. Knopman calculated that if participants’ baseline amyloid burden was below 67 centiloids, a trial would need to enroll 2,000 to 4,000 people to see a 25 percent slowing on tau PET progression. With participants above a cutoff of 67 centiloids, 500 participants would suffice.
Another study took a regional, rather than quantitative, approach to address the question of how much amyloid is needed to kick off tangle spread. Hazal Ozlen and Sylvia Villeneuve of the Douglas Mental Health University Institute in Montréal compared NAV4694 amyloid PET, flortaucipir tau PET, and cognitive performance in 129 participants in the PREVENT-AD cohort. All were cognitively healthy, with a family history of AD. The authors had previously defined seven cortical regions that accumulate plaques early in disease: precuneus, posterior cingulate, rostral anterior cingulate, medial orbitofrontal, rostral middle frontal, superior frontal, and inferior parietal cortex (Villeneuve et al., 2015). In this cohort, 81 participants had no amyloid tracer uptake in these regions, while 28 did in some of the regions—most commonly the precuneus and posterior cingulate—and 20 had uptake in all seven regions.
Compared with the 81 amyloid-negative controls, the 28 people with regional amyloid had an elevated tau signal in entorhinal cortex and middle temporal gyrus. Participants with widespread plaques across all seven regions, on the other hand, also had widespread tangles in the inferior temporal cortex, amygdala, and fusiform, lingual and parahippocampal gyrus. Over up to seven years of follow-up, only the group with widespread amyloid declined on a test of delayed memory in the RBANS. In addition, only the widespread amyloid group reported subjective complaints about their cognition. “Widespread amyloid is needed for tau to get outside of the temporal lobe and cause cognitive impairment,” Villeneuve wrote to Alzforum.
In all of these cognitively healthy cohorts, tangle accumulation precipitated cognitive decline. Another study examined a population that already was cognitively impaired. In cross-sectional data, tangles were not essential for memory impairment. Susan Landau of the University of California, Berkeley, analyzed data from 709 ADNI participants with baseline flortaucipir and either florbetapir or florbetaben amyloid scans. Among them, 315 had positive amyloid scans; 142 of them were cognitively healthy, 173 impaired.
Landau’s work agreed with that of the other groups in that it was primarily the amyloid-positive participants who had accumulated tangles outside the medial temporal lobe. But then the researchers used 1.4 SUVR as the threshold for regional tau positivity, because a previous flortaucipir imaging study had identified that as the best cutoff for discriminating cognitively impaired people from amyloid-negative controls (Maass et al., 2017). And they found that, among the 315 amyloid-positive ADNI participants, the tau signal exceeded this threshold in regions outside the medial temporal lobe in 40 percent of the cognitively healthy, and 60 percent of the impaired (see image above). While low tau signal was frequent in participants who otherwise seemed to be on the AD pathway, the low-tau individuals were clinically indistinguishable from those with high tau.
“People were interested in the fact that there is such a high proportion, up to 40 percent, of amyloid-positive, impaired subjects with low tau,” Landau told Alzforum. “This is likely due to comorbid pathology, but is not fully understood.” In Miami, Julie Schneider of Rush University in Chicago noted that co-morbidities in AD brain include Lewy bodies, TDP-43 deposits, and vascular disease. In some studies, vascular pathology contributes to one-third of dementia cases, Schneider said.
As has been reported in other studies, in Landau’s ADNI analysis, a higher tau PET signal correlated with lower scores on the PACC. The findings reinforce that amyloid pathology drives tangles to spread beyond the medial temporal lobe, and that tangle buildup greater than 1.4 SUVR causes impairment, Landau said.
Other recent ADNI analyses agree with this. In a preprint on medRxiv, researchers led by Rik Ossenkoppele of Vrije University in Amsterdam report that, among 730 non-demented ADNI participants, only those who were amyloid-positive on baseline PET scans accumulated tangles over the next five years. Those who were amyloid-positive by CSF Aβ42, but negative by PET, did not (Reimand et al., 2020). This is in keeping with the idea that CSF changes before PET (Aug 2016 conference news).
Partners in Crime. In people with amyloid plaques (right), but not low-plaque controls (left), the combination of plaques and activated microglia in the indicated brain regions associates with tangle deposition there. [Courtesy of Pedro Rosa-Neto.]
How do plaques drive tangles? Perhaps through inflammation. Min Su Kang and Pedro Rosa-Neto of McGill University presented cross-sectional data from 95 participants in McGill’s Translational Biomarkers of Aging and Dementia (TRIAD) cohort. The majority were cognitively healthy, 18 had MCI, 10 Alzheimer’s dementia. All underwent scans with the amyloid tracer NAV4694, tau tracer MK-6240, and microglial activation PET tracer PBR28. Among the 35 amyloid-positive participants, amyloid load and microglial activation synergistically associated with tangles in precuneus, entorhinal cortex, basolateral temporal cortex, and medial frontal cortex, these authors claim, suggesting that these two pathologies together might spur tau pathology (see image above). In turn, tangle load correlated with worse performance on the MOCA, MMSE, and CDR-SB.
Similarly, Paul Edison of Imperial College London reported elevated PBR28 uptake in the medial and lateral temporal lobes of 18 people with AD compared with 16 healthy controls. The PBR28 signal correlated with amyloid PET in the temporal and occipital lobes, and with tau PET in temporal, parietal, and occipital lobes. In 30 people with MCI, on the other hand, the PBR28 signal was no higher than in controls. These findings, too, support a link between amyloidosis, microglial activation, and tangles in AD.
Pathology and Progression
On the question of how plaques and tangles relate to neurodegeneration and progression to dementia, new data from the Australian Imaging Biomarkers and Lifestyle Study of Ageing (AIBL) underscore the finding that high amyloid is required for a person to get worse. In Miami, Chris Rowe of the University of Melbourne, Australia, described a longitudinal analysis of 534 cognitively healthy people. At baseline, 73 percent of them had little amyloid, below 26 centiloids; 10 percent had moderate accumulation, defined as 26 to 50 centiloids; 14 percent were high, at 51 to 100; and 3 percent had more than 100 centiloids.
Over three years, 57 of the 534 participants progressed to MCI. That risk correlated strongly with their baseline amyloid load: People below 26 centiloid did not progress; those with moderate accumulation had a hazard ratio of 3.2; those with high loads, a ratio of 7; and for those with very high loads, risk of progression stood at a whopping 11.4. The steepness of cognitive decline echoed these numbers, with moderate baseline load associated with a slide on the PACC of 0.07 standard deviations per year, high loads with 0.32, and very high loads with 1.38.
Unlike ADNI, in AIBL the picture was similar for people who started the study already impaired. Among 125 people with mild cognitive impairment, 31 percent started out with no amyloid accumulation, 14 percent with moderate, 34 percent with high, and 20 percent with very high accumulation. Sixty people, almost half the cohort, developed dementia over three years. Two-thirds of them had high or very high amyloid loads; one-third, moderate. Those with moderate baseline amyloid declined by 0.1 per year on the PACC, those with more than 50 centiloid by 0.2 per year. “Greater than 50 centiloids of Aβ substantially increases the risk of clinical progression and rate of cognitive decline,” Rowe said.
Tangles, even more than plaques, are closely linked to progression and degeneration. Leonardo Iaccarino of the University of California, San Francisco, described a study of 51 amyloid-positive people with MCI or dementia. In them, flortaucipir binding in medial frontal, anterior temporal, and insular cortex correlated with the neuronal injury marker CSF NfL. Flortaucipir uptake in the left temporoparietal lobe correlated with the inflammation marker CSF YKL40, though only in older participants. NfL correlated with CSF total tau but not with CSF Aβ42. Thus, these markers of injury and inflammation seem to reflect tangles more than plaques, the researchers concluded.
For this reason, tangles as seen by tau PET may make a good progression marker. Lilly’s Devous reported on 717 amyloid-positive people with MCI or dementia. The data were pooled from three in-house flortaucipir studies and a flortaucipir add-on to the solanezumab Expedition 3 trial. First, the researchers dichotomized flortaucipir scans into those that had an advanced tauopathy pattern, with uptake in parietal or frontal regions, and those that did not. The former had nearly double the risk of cognitive decline on the MMSE, ADAS, CDR-SB, and FAQ over the next 18 months.
The upshot was the same when the researchers evaluated tau pathology quantitatively using MUBADA, Lilly’s in-house measure for how extensive tangles are in the brain. They deemed a person’s neocortical flortaucipir signal positive if it was 2.5 standard deviations above the average value seen in healthy young controls. Nearly all MUBADA-positive participants were at Braak stage V or VI, and above a brain-wide cutoff of 1.1 SUVR on flortaucipir scans. Once again, the group defined in this way was most likely to decline on the four cognitive tests.
“You need to have tau pathology beyond the temporal lobe in order to have cognitive progression over 18 months,” Devous concluded.
Braak V or VI means a lot of tangles. Can tau PET pinpoint people earlier on? To find out, the Lilly team derived a second statistical measure, called Etau. It corresponds to tangle accumulation in mesial and inferior temporal regions, roughly Braak stage IV. In this same cohort, only 15 people were positive on Etau but not on MUBADA. And their cognition? They had some decline on the same four tests, midway between that of tau-negative and MUBADA-positive participants, suggesting that Etau might pick out an earlier AD population (Feb 2020 conference news).
That tangles in these regions harm cognition fits with the neuropathology literature and other studies. “We are excited about the ability of tau imaging to inform us of the risk of progression, which is otherwise hard to predict,” Devous told Alzforum. In toto, researchers believe that these kinds of quantitative data build a nuanced understanding of disease progression. This will inform how better to select trial participants, and to detect the effects of therapeutic intervention.—Madolyn Bowman Rogers







Comments
No Available Comments
Make a Comment
To make a comment you must login or register.