When the anti-Aβ protofibril antibody Ban2401 was first shown to reduce amyloid deposition in a Phase 2b clinical trial, researchers worried. Was its otherwise exciting cognitive benefit just an artifact of an imbalance of APOE4 carriers between the treatment and placebo groups, caused by regulatory changes during the trial? At the11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, investigators allayed some of those concerns. Their subgroup analysis by APOE genotype indicated that the drug effect was likely real. An early look at biomarkers suggests they are adding up toward a treatment effect on Alzheimer’s pathophysiology.
Bump in the Road or Disaster? BACE Inhibitors Worsen Cognition
Currently, hopes for an Alzheimer’s prevention therapy hang on BACE inhibitors that squelch generation of Aβ peptides. When these compounds failed to slow cognitive decline in symptomatic patients, researchers argued that they might work at earlier disease stages, before neurodegeneration has become entrenched. Alas, at the 11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, Spain, researchers at Merck shook the field with the stunning announcement that in a Phase 3 trial, people with prodromal AD who took the inhibitor verubecestat scored worse on cognitive tests than those on placebo. Michael Egan of Merck reported that the effect was small, but significant. The cognitive deficit appeared at the earliest time point and did not grow over time, nor did it appear to correlate with increased neurodegeneration.
What’s worse, other data suggest this may be a general property of BACE inhibitors. In Barcelona, researchers from Janssen and Lilly presented similar data on their compounds. Participants on all three inhibitors also had more neuropsychiatric symptoms and lost more brain volume. The troubling findings did not hold across the board, however. Biogen and Eisai reported a cognitive benefit in a small trial of its inhibitor elenbecestat, and no sign of neuropsychiatric symptoms or increased atrophy.
At CTAD, at least, researchers appeared to take this latest gut punch in stride. The mood at the conference was one of resolve rather than discouragement. Academic scientists said they want to figure out the mechanism behind the adverse effects, if they are reversible, and whether they are specific to certain patient populations or stages of disease. Many thought the data implied a stable synaptic deficit rather a speeding up of pathology. Because the cognitive loss appeared to be dose-dependent, scientists suggested that lower doses, or an intermittent dosing schedule, might alleviate the problem. Notably, no one at CTAD said the new data threw the amyloid hypothesis in doubt.
“I think there is a safe dose for BACE inhibition. We just need to find it,” said Robert Vassar of Northwestern University in Chicago. Paul Aisen of the University of Southern California, San Diego, agreed. “I believe BACE inhibition may still be an important part of our armamentarium against AD. It remains our leading candidate for primary prevention.”
Class Effect? Three Different Inhibitors Knock Cognition
In 2017, Merck halted its Phase 3 EPOCH study of verubecestat in patients with mild to moderate AD, after an interim analysis forecast that it would fall short. Subsequent analysis confirmed that participants on drug declined cognitively and functionally at the same rate as controls (Feb 2017 news; Dec 2017 conference news; May 2018 news). In February 2018, Merck conceded that the prodromal AD trial, APECS, appeared futile, too, and halted dosing (Feb 2018 news).
In Barcelona, the field got its first detailed look at APECS data. This trial enrolled 1,454 people with subjective memory decline and a positive amyloid PET scan. They had an average MMSE of 26 and age of 72. In a planned two-year trial, 484 people took 40 mg of verubecestat, 485 took 12 mg, and 485 took placebo. Although the trial ended a year early, many participants had completed the full dosing schedule by February 2018.
On the primary outcome measure, the CDR-SB, the 40 mg group scored worse than controls at nearly every time point from 13 weeks to 104 weeks. The effect was small, with a Cohen’s d of 0.2 mean difference divided by standard deviation between the treatment and placebo groups. However, it was significant at the 5 percent level, i.e. p<0.5. How about the lower dose? The 12 mg group also performed below controls, although for them the difference only reached significance at the 52 and 78 week time points.
Treatment groups did worse on the ADAS-Cog13 as well, scoring about one to 1.5 points higher than controls from 13 weeks on. On this test battery, the difference between drug and control groups shrank over time, with the 12 mg group no longer significantly different from placebo starting at 78 weeks.
Did the cognitive decrement matter to people’s lives? Participants taking verubecestat scored worse on the ADCS-ADL measure. The functional deficit appeared a little later than the cognitive one, starting at week 39. Both treatment groups progressed to dementia faster than did controls, at a rate of 25 percent per year instead of 20. Participants on drug also had more anxiety, depression, and disturbed sleep than those on placebo.
Is this just verubecestat, or a class effect? Ominously, other data at Barcelona pointed to the latter. Gary Romano of Janssen presented preliminary data from the Phase 2/3 EARLY trial of atabecestat, which was halted during its recruitment phase due to liver toxicity (May 2018 news). EARLY targeted a preclinical population, with participants having a CDR of zero and confirmed amyloid positivity by PET or cerebrospinal fluid. The 557 participants took atabecestat for no more than 18 months; more than half of them for only three. Equal numbers of participants received placebo, 5 mg, or 25 mg.
In this trial, only the high-dose group had a statistically significant cognitive decrement. They scored about 1 point worse than controls on the PACC cognitive composite at six and 12 months, and about four points worse on the RBANS at three months. This group started with 183 participants at baseline, but only 110 completed the three-month assessment, 66 the six-month, and 28 the 12-month. For later time points, the number of participants was even smaller, and findings not statistically significant. Breaking the data down by cognitive domain, the deficits appeared mainly memory, Romano said. The researchers saw no differences in ADCS-ADL in this study, but they did pick up more depression, anxiety, and sleep or dream-related problems.
Meanwhile, John Sims of Lilly said that findings for lanabecestat look similar to those for verubecestat, although he showed no data. Lilly recently pulled the plug on lanabecestat after futility analysis suggested no benefit in symptomatic disease (Jun 2018 news).
“I think we will find a BACE inhibitor class issue,” Sims said. Egan agreed that the weight of evidence now points toward an on-target, BACE-related mechanism. How dire this news is for BACE inhibitors, and whether all patient populations will be equally affected, are unanswered questions at this point.
Based on the evidence to date, it appears cognitive dampening mainly occurs early on in disease, at prodromal stages. While mild AD patients in the verubecestat EPOCH trial showed a trend toward cognitive worsening, it was not consistent. In Barcelona, Eli Lilly’s Albert Lo presented similar data on the company’s newest investigational BACE inhibitor, LY3202626. Its Phase 2 NAVIGATE trial was terminated after an interim futility analysis gave it little chance of success. NAVIGATE had enrolled 316 amyloid-positive people with mild AD whose average MMSE was 23; randomizing 133 to placebo, 55 to 3 mg, and 128 to 12 mg of drug. Researchers prioritized the higher dose as enrollment went along, so the 3 mg group consisted of those who had started earlier and had the longest exposure to drug. The researchers saw no clear effect on cognition, though there were hints of a deficit in the 3 mg group at 24 weeks on the ADAS-Cog13 and at 52 weeks on the MMSE.
Overall, researchers were unsure why BACE inhibitors would cause more harm early in disease than later. Bruce Albala of Eisai in Woodcliff Lake, New Jersey, noted that people at prodromal stages have a wider cognitive range, perhaps allowing researchers to detect subtle deficits.
What Do the Exceptions Mean?
Other data in Barcelona suggest that not all BACE inhibitors follow this pattern. In a panel discussion, Ana Graf of Novartis noted that the company's BACE inhibitor CNP520, which it develops in collaboration with Amgen, exhibited no ill effects in a small dose-finding study of elderly controls (Aug 2017 conference news). About 60 participants took varying doses of drug for three months, with no deficits apparent on monthly computerized cognitive tests. The participants had no increase in neuropsychiatric symptoms either, though they did report vivid dreams. Graf suggested that some BACE inhibitors may not cause cognitive effects because of differences in their mechanisms of action.
Take elenbecestat, for example. Shau Yu Lynch of Eisai reported sharply divergent data for this BACE inhibitor, which selectively inhibits BACE1 over BACE2, unlike most others in development. Yu Lynch claimed that elenbecestat slows cognitive decline. She reported a small, 18-month Phase 2 trial, in which 70 participants took either placebo or 5, 15, or 50 mg of drug. The groups started out evenly balanced, but people on the lower doses were switched to 50 mg as the trial proceeded. Half had mild, the rest prodromal AD. Treatment groups declined one-third less on the CDR-SB and ADCOMS than the placebo group did, with their separation beginning to show at about six months. No neuropsychiatric symptoms cropped up in this study, though people on drug had more nightmares (May 2017 conference news; Jun 2018 news).
Egan cautioned, however, that subtle cognitive deficits might not be detectable in trials with sample sizes as small as 60 or 70. Only a highly powered trial can detect the slight worsening seen with verubecestat, he said. The Merck trials represent the largest data sets for BACE inhibitors to date.
Mechanism Is a Mystery
What do these drugs do to memory? Discussion in Barcelona quickly turned to synapses. BACE1 loiters in presynaptic terminals. In mice, high doses of BACE inhibitor interfere with synaptic plasticity and memory formation (Nov 2014 news). “An early separation of curves that does not widen is consistent with a synaptic effect,” Aisen noted.
Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases in Munich nominated three of the more than 40 known BACE1 substrates as potential culprits in the synapse. Seizure protein 6 (SEZ6) helps maintain dendritic spine density and long-term potentiation, with BACE1 knockout mice impaired in both (Oct 2016 conference news). Perhaps SEZ6 effects manifest as cognitive deficits or neuropsychiatric symptoms in people, Lichtenthaler speculated. Another candidate, close homolog of L1 (CHL1), guides axons to their targets. Vassar and colleagues recently knocked out BACE1 in adult mice and saw stunted growth of mossy fiber axons in the hippocampi (Sep 2018 news). Finally, neuregulin helps maintain myelination and muscle spindles; its absence during development causes seizures (Jul 2013 news; Dec 2013 conference news). Lichtenthaler suggested probing these proteins first to find answers.
Alternatively, it could be that inhibiting cleavage of APP itself causes problems, Lichtenthaler added. Suppressing BACE1 has been reported to promote an alternate cleavage pathway, in which η-secretase snips APP to create a synaptotoxic Aη fragment (Aug 2015 news), hence buildup of this fragment after BACE1 inhibition could theoretically blunt synaptic transmission. Lastly, older work suggests that Aβ itself stimulates presynaptic release, and that too little of it causes harm (Nov 2009 news).
Could blocking of BACE2 be to blame? Most current inhibitors act on both BACE1 and BACE2. BACE2 is expressed in the brain and more so under conditions of inflammation. Even so, Lichtenthaler thought the enzyme’s normally low levels in brain make it an unlikely culprit (Mar 2018 news).
Some clues to what may be happening in the brain came from MRI data. In the APECS verubecestat trial, hippocampal volume shrank about half a percentage point more in the 40 mg treatment group than in the others. The difference appeared at 13 weeks, and held steady at 104. Likewise, people who took any dose of LY3202626 lost more volume over a year in the hippocampus, medial temporal lobe, and parietal regions than controls did. In contrast, in the elenbecestat study, hippocampal volume declined equally on drug and placebo.
This volume loss puzzled researchers. Some speculated it could represent a loss of dendritic spines or mossy fiber axons, in keeping with mouse data. Another recent study claims BACE inhibition can squelch hippocampal neurogenesis, which might also affect volume (Aug 2018 news).
As expected from previous mouse and human studies, amyloid load dipped slightly with BACE inhibitor treatment (Jan 2016 news). On the highest dose of verubecestat, the PET signal fell by 0.04 SUVR, a statistically significant difference. Lilly reported a similar drop in people taking LY3202626, although it did not reach significance in this smaller study. Janssen did not report amyloid PET findings, while Eisai and Biogen claimed a drop of about 25 centiloid in people taking elenbecestat, on a par with reductions seen after a year of anti-amyloid antibody treatment (Aug 2018 conference news).
The cognitive deficits seen with BACE inhibitors do not appear to come with increased neurodegeneration. None of these trials reported any change in tau PET or CSF tau or NfL relative to placebo. Although loss of gray matter increased on drug, this MRI finding did not correlate with cognitive scores, Egan said. “It looks like the effect is independent of AD progression,” Eric Reiman of Banner Alzheimer’s Institute in Phoenix told Alzforum.
What Now For BACE Inhibitors?
Researchers at CTAD emphasized that this is not the end of the road for BACE inhibitors. One crucial question is whether a person’s cognition rebounds after he or she stops taking the inhibitor. Egan said APECS was not designed to address this, but Merck is doing exploratory analyses now. Romano presented preliminary data implying the deficit might be reversible. In a single cognitive assessment done after atabecestat treatment stopped, scores rose slightly for people who had been on drug. However, Romano cautioned that the number of participants at this time point was small, about 36 per group. Lichtenthaler noted that in mice, spine density fully recovers after withdrawing BACE inhibitor.
Many researchers suggested lowering the dose of BACE inhibitors and starting treatment earlier. Vassar said that in mice, BACE1 activity has to be curtailed by 90 percent to cut Aβ levels in half. All BACE inhibitor trials to date suppress Aβ production from 50 to 90 percent, suggesting they shut off BACE1 almost completely. This may be far more stringent than needed, Vassar suggested. Eric McDade of Washington University in St. Louis agreed, noting that the protective APP mutation lowers plasma Aβ40 only 28 percent (Martiskainen et al., 2017). Vassar and Lichtenthaler both recommended slashing Aβ production by no more than half; others suggested trying an intermittent dosing schedule to give other BACE1 substrates a chance to recover.
Lichtenthaler proposed measuring levels of the BACE1 substrates SEZ6, CHL1, and neuregulin, as well as Aη, in stored CSF samples from inhibitor trials to find out if changes in any of these proteins correlate with cognitive effects. If so, the proteins could be used as biomarkers to monitor dosing and find safe levels of inhibition, he said. Aisen concurred that these ideas have merit. “We have the tools to move forward,” he said.
In the meantime, some inhibitor trials will continue. Johan Luthman of Eisai said they have confidence in their program and will carry on. Novartis and Amgen’s CNP520 program will also remain active, although Pierre Tariot of Banner noted that they will need to see more data before deciding whether to alter their plans for CNP520 dosing in the ongoing API Generation trial of ApoE4 homozygotes. Egan said Merck is not yet taking a public position on verubecestat’s future, noting they will take findings from other inhibitors into account before making a decision.
McDade and colleagues at WashU have been planning to use BACE inhibitors in the first primary prevention trial for AD in the DIAN cohort (Aug 2017 conference news). McDade believes this strategy is still viable. Because the cognitive deficits appear quickly, it will be possible to adjust dosing to achieve a safe level, he said. He plans to use an adaptive design, start with a low dose of inhibitor, and titrate up. Since all BACE inhibitors so far produce vivid dreams and sleep disturbances, researchers can monitor sleep quality as a readout for side effects, he added. McDade compared the challenge to that of balancing safety and efficacy during chemotherapy.
What do AD patients think? Offering some perspective, Tariot noted that families in the verubecestat trial are begging the scientists not to throw in the towel on BACE inhibitor research. The magnitude of the cognitive deficit pales in comparison to cognitive side effects from chemotherapy, and to the terror of AD, trial participants tell him. Maria Carrillo of the Alzheimer’s Association observed that the first vaccine trials also had frightening side effects, but ARIA has now proven to be manageable. “Patients are counting on us to be bold,” she said.—Madolyn Bowman Rogers
Second Look at BAN2401 Data Still Positive, Despite Snafu
Researchers got a fuller view of the data coming out of the Phase 2b trial of the anti-Aβ BAN2401 antibody at the 11th Clinical Trials on Alzheimer Disease conference, which convened in Barcelona, Spain, from October 24–27. Eisai’s Chad Swanson presented a preplanned subgroup analysis in response to criticism that arose in the wake of a regulator-induced hiccup in this Bayesian adaptive trial, which had led to an imbalance in the number of APOE4 carriers between treatment and placebo arms. According to the new analysis, the imbalance was not the reason for the treatment benefit on cognition that was first reported last July. On the contrary, Swanson said, it may have led the trial to underestimate the antibody’s effect on the cognitive endpoints. Changes in cerebrospinal fluid biomarkers of tau, synaptic function, and neurodegeneration also moved in a positive direction in the treatment groups, suggesting the antibody may moderate underlying pathology. Ditto for amyloid PET.
Jeffrey Cummings, Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, told the audience that the subgroup analysis should allay worries that the drug effect was driven by the uneven allocation of APOE4 carriers. To Cummings’ mind, the finding that results were consistent across all clinical measures and biomarkers supports the conclusion that the treatment effect is real. Some members of the audience shared his optimism. Others did not, noting that the highest dose group of 161 participants included just 48 APOE4 carriers, of whom only 10 completed the study. Only a truly randomized trial will properly test BAN2401, they maintained.
According to data presented this past summer at AAIC, at the highest doses tested, this anti-Aβ protofibril antibody being jointly developed by BioArctic, Eisai, and Biogen slashed brain amyloid and slowed cognitive decline in people with MCI due to Alzheimer’s disease or mild Alzheimer’s dementia (Jul 2018 conference news). There was a fly in the ointment, however. Because of a change in study design midway through the trial, the high-dose treatment group had far fewer APOE4 carriers than the placebo group, rendering the comparison suspect. At AAIC, commenters asked for the data broken down by APOE genotype, and at CTAD, Swanson delivered.
The trial enrolled 856 participants, who were randomized into one of five dose groups or placebo using a Bayesian adaptive randomization algorithm. As the trial proceeded, frequent analyses of blinded study data steered new participants into dose arms most likely to be effective. Early on, that looked to be the top two doses, of 10 mg/kg monthly and 10 mg/kg every two weeks, hence nearly half the participants ended up in these groups.
But partway through, the European Medicines Agency requested that APOE4 carriers not be put on the highest dose, and also that all APOE4 carriers who had been on that dose for less than six months discontinue. The regulators’ concern was ARIA-E, a temporary inflammatory reaction seen in the first few months of Aβ immunotherapy, which appears more commonly in APOE4 carriers receiving higher doses of antibody. The investigators complied, but the EMA request left APOE4 carriers underrepresented in the highest group. By the end of the trial, they made up only 30 percent of that arm, compared with 71 percent of the placebo group and 89 percent of the next-lower-dose group, who received 10 mg/kg monthly.
As reported at AAIC, the highest dose performed the best, registering a statistically significant 30 percent less decline on the ADCOMS compared with the decline in the placebo group. They also declined 47 percent less on the ADAS-COG, and 26 percent less on the CDR-SB. In the 10 mg/kg monthly group—the one with the highest percentage of APOE4 carriers—there was a trend toward slower cognitive decline on all three measures, but none was statistically significant.
According to new data presented at CTAD, APOE4 status did not affect the rate of cognitive decline in the placebo group. In untreated participants, the downward trajectories in ADCOMS, ADAS-COG, and CDR-SB scores, measured every three months, were virtually the same for carriers and noncarriers. The factors that did contribute to placebo participants’ progression included their clinical stage, whether they were taking medication for AD, and their baseline ADCOMS score—not their APOE4 status.
Did APOE4 affect response to treatment? The subgroup analysis said yes, Swanson reported. Carriers on the highest dose had less cognitive decline at 18 months than the noncarriers or the group overall. On the ADCOMS, for example, where the highest dose group declined 30 percent less than placebo, APOE4 carriers declined 63 percent less and noncarriers only 7 percent less. This result indicates that the treatment effect on the high dose was likely not due to the lack of APOE4 carriers, Swanson said. In fact, he thinks the imbalance may have led the trial to underestimate BAN2401’s effect. However, the trial was not powered to show statistical significance in the subgroup analysis, and the groups were small. The high-dose cohort was left with only 10 APOE4 carriers among the 79 people who completed the 18-month regimen—and those 10 had been on that dose for six months or longer before the EMA intervened, and did not drop out due to ARIA-E.
To boost those numbers, the investigators combined the 10 mg/kg biweekly and 10 mg/kg monthly groups. The merged cohort comprised 273 APOE4 carriers and 141 noncarriers, a similar makeup to the placebo group. This combined group declined 21 percent less than placebo. Once again, the APOE4 carriers had a better response, declining 25 percent less than placebo, whereas noncarriers declined just 6 percent less.
In other subgroup analyses, BAN2401 appeared to reduce progression in both the MCI and mild AD groups, and regardless of whether participants took AD medications. BAN2401 treatment substantially reduced brain amyloid, by as much as 93 percent in the high-dose group, and was similar across all subgroups.
Analysis of CSF biomarkers in a substudy of participants hinted at treatment effects on pathology. Previously, the investigators had shown that Aβ concentrations in cerebrospinal fluid increased 300-fold after treatment, while total tau levels trended downward over the course of the study. New data on neurogranin, a marker of synaptic damage, and phosphoTau181, a marker of tau pathology downstream of amyloid, revealed that their median concentrations dipped by 11 and 13 percent, respectively, after 18 months of treatment. The placebo group showed less or no change. Neurofilament, a marker of axonal damage, rose in all groups, but its rise was halved in the treatment group. These results are exploratory because they included only 23 people from the two high-dose arms and 16 from the placebo group.
Swanson showed additional analyses supporting the idea that the antibody slows disease progression. Calculating change on ADCOMS over time, investigators found that treatment reduced the slope of decline from 0.0083 to 0.0060 points per month, a statistically significant difference. This indicates that the changes at 18 months might produce larger clinical benefits when treatment continues longer, Swanson said. According to a joint press release, Biogen and Eisai are planning an open-label extension for participants in this study, with enrollment expected to begin this year.
Reisa Sperling, Brigham and Women's Hospital, Boston, called it “very encouraging,” that APOE status did not affect decline in the placebo group, and appeared to increase a person’s benefit from treatment. However, she noted that anyone who developed ARIA-E was removed from the study. APOE4 carriers have higher rates of ARIA-E, which could have led to more of them discontinuing the study, further confounding the results. Swanson said the team had not specifically looked at this, but would.
Robert Vassar, Northwestern University in Chicago, said he was struck that, despite the dramatic reduction in amyloid on the high dose, clinical progression continued, albeit at a lower rate. Was the decline independent of Aβ? Could this be related to the tau reduction that was observed in CSF? Swanson reiterated that they did see a correlation between clinical measures and amyloid PET across the dose range, but that other factors could be involved, too.
Swanson said Eisai and Biogen are discussing next steps with regulators. Meanwhile, slides of the CTAD presentation are posted on Eisai’s website.
Samantha Budd Haeberlein of Biogen presented data on the company’s other anti-amyloid antibody, aducanumab. By now, aducanumab has reached 48 months of continuous dosing in its open-label extension of the Phase 1b PRIME trial in prodromal or mild Alzheimer’s disease. Continuing on from the 24- and 36-month data (May 2018 conference news), aducanumab still clears amyloid at 48 months. Removal seems to bottom out at a reduction of 75 centiloid units, which brings people below the 1.1 cutoff on the SUVR scale for brain-wide amyloid positivity. The highest aducanumab dose group of 10 mg/kg reached that cutoff by 24 months of treatment, and stayed there. By 48 months, the next-highest dose of 6 mg/kg group joined them.
At 48 months, the 10 mg/kg group continued to show slower decline in MMSE and CDR-SB compared with lower doses, including the 6 mg/kg group. ApoE4 status does not seem to affect disease progression or treatment effects in this study, Budd said. The safety profile of aducanumab remains unchanged, according to 48-month data presented by Philipp von Rosenstiel, also of Biogen. Biogen is testing aducanumab in two large Phase 3 studies. Both are now fully enrolled, with a total of just over 3,200 participants, said Budd.—Pat McCaffrey
It seems blood tests for Aβ have moved off the wish list and are becoming reality—almost. Building a knowledge base for plasma measures as a proxy for brain amyloid, multiple methods applied to different research cohorts are now consistently linking low plasma Aβ42/40 to clinical, cognitive, and biomarker evidence of Alzheimer’s disease. At the 11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, in presentation after presentation, researchers made the case that currently available blood tests are suitable pretests to reduce or replace amyloid PET scans for screening participants in clinical trials. This would save time and money, lighten the burden on volunteers, and expand access to therapeutic trials beyond the catchment areas of centers with PET capability. Current work to standardize the tests focuses on pre-analytic sample handling to minimize error. While several candidate assay platforms are vying for clinical samples to help them validate their product, at least one recruitment effort plans to incorporate multiple assays to compare and contrast their performances in real life.
In a keynote that opened the meeting, Randy Bateman of Washington University, St. Louis, recapped Aβ blood assay research over the last 18 months. It happened fast. Until 2017, plasma Aβ assays had low accuracy and yielded mixed results. A meta-analysis of more than two dozen studies found high variability and no significant difference between people with AD and healthy controls (Apr 2016 news on Olsson et al., 2016; Alzbiomarker).
From his lab’s kinetics work measuring production and clearance of Aβ in people, Bateman knew half the peptide produced in the brain ended up in blood, and that brain amyloid deposition throttled this transfer. To find a blood signature of this change, his lab developed an ultrasensitive assay for blood Aβ based on immunoprecipitating and quantitating the peptides with mass spectrometry. The technique uncovered a subtle but reproducible signal, whereby the ratio of Aβ42/40 in blood nudged downward by 14.3 percent in people with brain amyloid (Ovod et al., 2017). Compared with what’s found in cerebrospinal fluid, blood Aβ concentrations were much lower, and the drop in amyloid-positive people less dramatic, but nonetheless, blood Aβ42/40 ratios were able to distinguish amyloid-positive and -negative people with 89 percent accuracy.
Since then, Bateman has validated the assay in two additional cohorts, including one with longitudinal data. Over time, blood Aβ follows a similar pattern as CSF Aβ, whereby the Aβ42/40 ratio drops early in the disease, both preceding and predicting brain amyloidosis. Among amyloid PET-negative, cognitively normal people, a lower Aβ42/40 ratio signaled who would become PET-positive during two to seven years of follow-up.
It’s Complicated. Rather than relying on a single cutoff value, schemes that combine plasma Aβ concentration, age, and APOE4 status could produce fine-grained predictions of amyloid PET positivity. [Courtesy of S. Schindler, Y. Li, and R. Bateman.]
The relationship between plasma Aβ42/40 and brain amyloid holds regardless of a person’s age or ApoE4 status, and when these three factors are combined, they predict amyloid status with 95 percent accuracy. This suggests blood measures could be as good as CSF or PET at detecting brain amyloid in the general population, Bateman claimed. For secondary prevention trials, a blood amyloid prescreen would halve the number of PET scans needed to identify amyloid-positive, asymptomatic participants, he said. For primary prevention studies seeking participants who are unlikely to be amyloid-positive, such as younger or ApoE4-negative people, prescreening with a blood test might reduce PET scans as much as 85 percent, he calculated.
Slicing the data another way, Bateman suggested that instead of instituting a single cutoff value for positivity or negativity, researchers might instead consider a range of probabilities based on a person’s Aβ42/40 concentration, age, and ApoE4 status combined. Inge Verberk of VU University Medical Center, Amsterdam, suggested this same approach at AAIC last summer (Verberk et al., 2018).
Since Bateman’s ultrasensitive blood Aβ assay development, the field is becoming crowded with options. They include an independent mass spec technique (Feb 2018 news), a fully automated ELISA system, and several other antibody-based detection systems (Verberk et al., 2018). Consistently, each assay points to lower Aβ42/40 ratios in blood as a specific and sensitive indicator of brain amyloid.
The newest entry is an enhanced immunoassay based on single molecule optimized array (Simoa) detection. It is already moving toward commercialization. Developed in Charlotte Teunissen’s lab at Amsterdam University Medical Center, the assay better distinguishes amyloid-positive from -negative than the commercially available Simoa assay from Quanterix (Aug 2018 conference news). Jeroen Vanbrabant, ADx Neurosciences, Gent, Belgium, told Alzforum that ADx has begun scale-up and commercial production of Teunissen’s assay. ADx scientists have replicated her lab’s results and have scaled up production to 100-kit batches. Going forward, Vanbrabant said they’ll continue to test and validate the assay, generate standard operating procedures for sample collection, storage, and analysis, and expand clinical data to compare the blood test with amyloid PET. Hugo Vanderstichele, also of ADx, said researchers can already obtain prototype kits.
The existence of multiple competing assays presents a challenge for standardization and replication between labs and cohorts, issues the field has begun to address. One effort, spearheaded by Kaj Blennow, University of Gothenburg, Sweden, will generate a panel of 50 plasma samples and ship them to different labs by the end of the year. For CSF assays, pre-analytical sample handling was a nettlesome source of variability, and the situation is shaping up to be similar for blood. At CTAD, Tobias Bittner and colleagues at Roche presented an analysis of common aspects of sample handling—all the boring little details—that might muddle results in Roche’s automated Elecsys blood Aβ and tau immunoassays. The goal was to issue a recommended sample-handling protocol. Some details did not matter: The time of day blood was drawn, what plastic the tube was made of, up to five transfers from tube to tube, and up to three freeze-thaw cycles all did not affect results. But other little things did: Choice of anticoagulant, storage temperature, and how long samples sat around before being tested or frozen all affected detection of Aβ40, Aβ42, and tau. Filling tubes less than halfway led to loss of tau signal, the scientists found.
Vanderstichele agrees controlling pre-analysis is important. In agreement with Roche’s data, the ADx protocol calls for collection of EDTA plasma, and freezing samples within an hour. “The assays need strictly controlled collection and storage. Time and temperature are critical,” he said. Another assay, offered by Araclon Biotech-Grifols, Zaragoza, Spain, strictly requires people to fast overnight before giving a blood sample, said Ian Sherriff of Araclon.
Araclon’s assay had its own, company-sponsored, session at CTAD, rolling out data from academic collaborators and multiple cohorts. Araclon’s Pedro Pesini reviewed its automated Aβ40 and Aβ42 assay, which the company sells as an in-house test service (Pérez-Grijalba et al., 2016). The assay distinguishes between free and total Aβ peptides, which are measured in undiluted or diluted plasma, respectively. Both correlate with other biomarkers of brain amyloid deposition, though the ratio of total Aβ42 to 40 (TP42/40) appears to correlate best.
Lower TP42/40 was associated with amyloid positivity and with accumulation of amyloid over time in cognitively healthy normal people in the Australian Imaging, Biomarker, and Lifestyle Flagship Study (Fandos et al., 2017). At CTAD, Victor Villemagne, University of Melbourne, Australia, showed data for the entire AIBL cohort spanning from healthy control to mild cognitive impairment to AD. He confirmed that the association with amyloid status held across the clinical spectrum. In AIBL, the relationship of blood Aβ to PET was similar to that of CSF Aβ42 to PET, with TP42/40 dropping logarithmically with increasing amyloid PET signal. The decrease in plasma ratio was linear over time, and faster in people who were accumulating amyloid.
Early warning? Modeling indicates that, in a healthy person who accumulates amyloid, TP42/40 drops below mean values a decade before he or she reaches the brain-wide cutoff for amyloid positivity (bottom panel). For amyloid-PET, the first signs of deposition come seven years before the threshold is reached. [Courtesy of V. Villemagne.]
Modeling the changes over time in healthy elderly controls, Villemagne estimated that blood Aβ values start to drop 10 to 11 years before a person crosses the threshold to PET positivity (see image above). By comparison, PET starts showing deposits in isolated brain regions about seven years before a person crosses the threshold for brain-wide amyloid positivity.
To illustrate the value of the blood test as a prescreen, Villemagne offered a hypothetical trial enrolling 150 amyloid PET-positive, cognitively normal people. For that, researchers expect to have to perform an average of 3.3 PET scans per recruitment. The blood test could reduce that to 1.4 scans per recruited person. At a cost of $5,000 per PET scan versus $140 for the blood test, that reduction would add up to a savings of $1.4 million dollars, he calculated.
At CTAD, Anne Fagan of Washington University, St. Louis, presented more recent data published this month by the Spanish group (Pérez-Grijalba et al., 2018). It indicates that TP42/40 is associated with FDG PET, amyloid PET, and risk of progression to AD dementia in a group of people with amnestic MCI. The AB255 study, sponsored by Araclon, draws volunteers from memory clinics in Spain, Italy, Sweden, and France. The investigators enrolled 228 people age 65–85, either cognitively normal or with amnestic cognitive impairment (aMCI), and followed them for two years.
At baseline, the average TP42/40 was lower in amnestic MCI than cognitively normal participants, and came with cortical hypometabolism suggestive of a high risk of AD. Indeed, a lower TP42/40 increased a person’s risk of progressing from aMCI to Alzheimer’s dementia over the next two years by 70 percent. In a subgroup, TP42/40 correlated with Aβ42 in CSF and inversely with PET-PIB. Finally, Fagan showed that TP42/40 separated amyloid PET-positive and -negative groups with a sensitivity and specificity similar to Bateman’s mass spec assay. Were there limitations to the Araclon assay? Sure. The values in the two groups overlap a fair amount, the effect size is modest, and there is not yet a universal cutoff for amyloid positivity, Fagan said. “Although I do not endorse one method over another, as they each have their strengths and weaknesses, the similarities of the results in both studies is exciting since it gives further support to the potential viability of the ratio as a useful biomarker,” she told Alzforum.
Does the relationship between TP42/40 and brain amyloid hold for subjective cognitive decline? Yes, sort of, said Agustin Ruiz of the Fundació ACE in Barcelona. His data came from the Fundació’s Healthy Brain Initiative (FACEHBI) (Rodriquez-Gomez et al., 2017), a study that recruits people who complain of memory issues yet score normal on cognitive tests. In those participants who also received florbetaben PET scans, the investigators once again found an inverse association between TP42/40 and PET positivity. However, the blood ratio discriminated less well here than in some other clinical cohorts, probably because FACEHBI included fewer people with brain amyloid, Ruiz said. Even so, an empirical cutoff that pinned amyloid positivity with 83 percent sensitivity and 59 percent specificity cut by half the number of people who would need PET scans for a trial targeting that early stage of AD, and still capture only PET-positive people.
Sure enough, blood tests are already wending their way into prescreening efforts. The Trial-Ready Cohort for Preclinical/Prodromal Alzheimer’s Disease (TRC PAD) project, run jointly by Reisa Sperling at Boston’s Brigham and Women’s Hospital, Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas, and Paul Aisen at the University of Southern California Alzheimer’s Therapeutic Research Institute (ATRI), aims to include measures of plasma Aβ when it begins in-clinic screening to build its cohort early in 2019, said Gustavo Jimenez-Maggiora of USC.
Aisen confirmed that he is seeking funds to evaluate three plasma assays: the two immunoprecipitation/mass spectrometry methods reported by WashU and by Akinori Nakamura and Katsuhiko Yanagisawa at the National Center for Geriatrics and Gerontology in Aichi, Japan, and the Elecsys immunoassay. “The first stage would be comparative validation of the methods, as well as assessment of pre-processing approaches. The second stage would be incorporation of plasma Aβ ratio testing into our risk algorithm, to yield a highly accurate predictor of brain amyloid elevation,” Aisen wrote to Alzforum, adding “Ultimately, we hope a plasma assay may obviate the need for amyloid PET or lumbar puncture in the selection process for the trial-ready cohort.”—Pat McCaffrey
It’s Official: Tau PET Sees Tangles, and Staging Tangles Predicts Decline
Do tau PET tracers truly detect tangles? How will we use them? The cutting edge on these evolving questions was on display at the 11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, Spain.
Mark Mintun of the Eli Lilly subsidiary Avid Radiopharmaceuticals, Philadelphia, presented postmortem validation data that confirmed the flortaucipir signal indeed picked up tau tangles in the brain, at least at advanced Braak stages. Other scientists advanced newer tau tracers from Merck, Roche, and Life Molecular Imaging (formerly Piramal). Their data showed that these second-generation tracers are more sensitive and have less noise than flortaucipir, or other early tracers that have since faded, such as THK5351. Practical applications of tau imaging are starting up, from screening people for clinical trials to picking up subtypes of Alzheimer’s disease. Tau PET talks generated intense interest from an engaged audience.
“These are incredible studies,” said Eric Siemers, who retired from Lilly and now consults at Siemers Integration LLC in Indianapolis.
More Signal, Less Noise. New tau tracers, such as PI-2620 shown here, have little background. [Courtesy of Andrew Stephens.]
It’s Official: Flortaucipir Measures Tangles in Late-Stage Tauopathy
Lilly first announced findings from its Phase 3 postmortem study in a press release (Sep 2018 news), and in Barcelona, Mintun showed detailed data. The trial recruited people who had less than six months to live and were willing to have a flortaucipir scan and donate their brains. Out of 64 participants who came to autopsy, 49 had been clinically diagnosed with dementia and one with mild cognitive impairment. Their average age was 82. On postmortem examination, 39 of them had tangle pathology consistent with Braak stage 5 or 6; the other 25 were at earlier stages.
For one primary study endpoint, Lilly measured flortaucipir’s ability to predict Braak stage 5/6 pathology. Tracer signal in AD brain regions had to be 65 percent higher than in cerebellum in order to count as a positive scan. Five independent, blinded readers evaluated the scans. They correctly identified 36 of the 39 advanced-stage brains as positive, for a diagnostic sensitivity of 92 percent. However, they also called positive the scans from five people who were at earlier Braak stages, leading to a specificity of only 80 percent.
In answer to a question from Gil Rabinovici of the University of California, San Francisco, Mintun said these false positives were mostly Braak stage 4. Rabinovici challenged the decision to select such an advanced stage of disease as the criteria for a positive scan. Mintun said Lilly deemed this a useful clinical threshold and is examining the use of lower thresholds as an exploratory outcome. Others noted both publicly and to Alzforum that identifying people at earlier Braak stages would be important for selecting trial participants. It is unclear how well flortaucipir would perform at this stage of disease, when the tracer signal is lower and likely to be more muddied by background off-target binding.
As a co-primary endpoint, Lilly researchers evaluated flortaucipir’s ability to discern high levels of AD neuropathology as per NIA/AA criteria (Hyman et al., 2012; Jan 2012 news). Results were similar to the Braak analysis, with scan reads reaching a sensitivity of 95 percent and specificity of 81 percent. The area under the curve was 0.94, which Minton noted was good for a diagnostic test.
Brains with AD neuropathology had tracer signal in the posterior lateral temporal and occipital lobes. At the most advanced stages of disease, parietal and frontal regions lit up as well. For both endpoints, the readers agreed on how to interpret a scan 90 percent of the time. False-positive calls varied the most between readers, and Mintun said Lilly will look into the reasons behind this variability.
Researchers in Barcelona greeted the data with enthusiasm. “This is a major advance. I’m very excited,” said Stephen Salloway of Brown University in Providence, Rhode Island. At the same time, Salloway expressed concern that using a negative/positive dichotomy for tau scans could limit research. After all, the new NIA/AA research framework describes tau pathology as a continuum. Mintun agreed that quantifying tau might be more useful than assigning a cutoff for positivity. In a separate, higher-resolution study of three additional postmortem brains, researchers measured the amount of tangles in several brain regions and compared it to the SUVR signal. Mintun said the signal strength correlated well with absolute levels of pathology. “We have good hope of treating tau pathology as a continuum,” he said.
See Tau Early. Roche’s tau tracer RO-948 better discriminates AD patients from controls based on tangles in Braak regions 1/2 than in 3/4. People with cognitive decline (SCD/MCI) took up the same amount of tracer as age-matched controls (OC) in all regions. [Courtesy of Gregory Klein.]
Sharper Image with New Tracers?
One limitation of flortaucipir is its high background signal in the basal ganglia and choroid plexus, which sits right above the hippocampus. New tracers appear more specific. In Barcelona, Gregory Klein at Roche reported findings for that company’s tau tracer RO-948 (May 2018 news). Oskar Hansson and colleagues at Lund University, Sweden, used RO-948 to scan 223 participants in BioFinder-2, who included 30 young controls, 21 cognitively healthy older controls, 84 people with some cognitive impairment, 50 people diagnosed with AD, and 30 with other dementias, as well as a few whose diagnosis was uncertain.
The RO-948 signal distinguished people with an AD diagnosis from age-matched controls with high significance. P values stayed in the 0.0005 range whether the researchers examined tracer binding in transentorhinal regions corresponding to Braak stage 1/2, limbic regions corresponding to Braak 3/4, or cortical regions corresponding to Braak 5/6. In fact, the difference was most pronounced in regions 1/2 (see image above).
In people with mild or subjective cognitive impairment, on the other hand, tracer binding was only elevated in the transentorhinal cortex, and the average was not much different from binding in age-matched cognitively healthy controls. This suggests some transentorhinal cortex binding may represent normal aging-related changes, though the uptake range in this group was wide. Does this suggest that tau PET clearly flags people only once their clinical symptoms advance beyond MCI? Supporting this, the authors found that more tangles in limbic regions correlated with lower MMSEs, with SUVRs of 1.5 or higher corresponding to an average MMSE below 25.
The SUVR signal in this cohort ran from zero to 4, providing a wide dynamic range. RO-948 had little off-target binding, with 9 percent of participants having a small signal in the basal ganglia, and 14 percent in the choroid plexus. Almost a third of the cohort had a signal in their meninges, however; in the seven with the strongest binding here, the authors speculated that it could interfere with measuring tau in cortical regions of interest. In addition, off-target binding in the substantia nigra and retina was common.
Andrew Stephens of Life Molecular Imaging presented similar data for the tracer PI-2620, on 12 people diagnosed with AD and 10 healthy controls. (For previous PI-2620 data, see Apr 2017 conference news; Dec 2017 conference news). In AD patients, the researchers saw increased tracer binding in a mesial temporal cortical composite region, as well as in a temporoparietal composite. In both cases, the Cohen’s d effect size was larger than 2, i.e. the difference between AD and controls exceeded two standard deviations. This tracer generated no signal in basal ganglia or choroid plexus (see image above). Like RO-948, PI-2620 had peak SUVRs of 4 or more. It performed best when scans were done 30 to 90 minutes after injection. In this time window, test-retest variability remained below 5 percent, but was higher at later time points.
This contrasts with Merck’s MK-6240. Previous work had suggested an optimal time window of 70 to 90 minutes for scanning with this tracer (May 2018 conference news), but in Barcelona, Tharick Pascoal of McGill University, Montreal, reported that MK-6240 gives more reliable results when measured between 90 and 110 minutes after injection. His studies indicate the tracer is slow, reaching equilibrium in regions with low tauopathy after 60 minutes but taking 90 minutes in regions with high pathology (Pascoal et al., 2018). In a study of 16 participants, Pascoal and colleagues found that scanning sooner than 90 minutes after injection led to underestimates of tangle burden in regions with SUVRs above 2. This gets more pronounced the higher the tau burden. In a therapeutic trial using tau PET as an outcome measure, this could underrate a drug effect, Pascoal said, because the pretreatment signal would underestimate tau burden more than the post-treatment signal would.
Putting Tau PET to Use
As tau PET imaging becomes more reliable, researchers are investigating how it might work in practice. In Barcelona, Adam Fleisher of Avid said tau scans could help sites screen participants for therapy trials, picking out those who are most likely to decline cognitively in the near future but are not too advanced to benefit from therapy. Fleisher analyzed data from selected participants in the Phase 3 EXPEDITION3 and a Phase 2 flortaucipir trial. This ad hoc cohort comprised 65 people with prodromal AD and 181 with AD dementia, with an average age of 73. Both trials ran for 18 months, with repeat cognitive testing, and the selected participants had baseline flortaucipir and florbetapir scans. Twenty percent of the cohort were amyloid-negative, and all of that group were also tau-negative. Among the amyloid-positive group, 77 percent were tau-positive, too.
The researchers evaluated tau burden in two different ways. In the first method, a purely quantitative one, they divided the group into quartiles based on their levels of tau tracer signal. Curiously, participants with AD dementia were equally distributed across the tau quartiles. In the second method, a regional one, researchers determined by visual read whether tau scans had an “AD pattern.” As in the autopsy study, this was defined as uptake in the posterior lateral temporal and occipital lobes, with the most advanced cases also having parietal and frontal uptake.
By either method, people with more tau pathology declined faster on cognitive tests. For the quartile method, those in the first quartile stayed about stable, while those in the second or third quartile slid by an average of 5 points from their baseline performance on the ADAS-Cog11, and those in the fourth quartile dropped a whopping 11 points. For visual reads, participants broke into two categories. Those without parietal tau barely declined, while those with tau there lost six points on the ADAS-Cog11. The pattern for MMSE scores and tau burden was similar.
Overall, the distribution of tau better predicted cognitive decline than the quantity, Fleisher noted. People whose tangles had spread beyond the posterior lateral temporal (PLT) lobe declined equally fast regardless of whether they fell into the first, second, or third quartiles. Tau extending beyond this region seems to trigger cognitive decline, Fleisher concluded. Frontal tau uptake marked another key event, as everyone in this category fell into the fourth quartile, with the fastest decline.
Combining quantitative and qualitative data best captured disease progression, Fleisher said. People whose florbetapir SUVR exceeds 1.10, with tangles in the PLT, are at risk for imminent cognitive decline, while those with an SUVR of 1.46 or higher and uptake in the frontal cortex have global tauopathy and will decline steeply. These two thresholds define an optimal window for therapeutic intervention, Fleisher proposed.
A different application of tau imaging came from Rik Ossenkoppele of VU University Medical Center, Amsterdam. Ossenkoppele wondered whether the uptake pattern could reveal subtypes of Alzheimer’s. Some people have a limbic-predominant form of the disease, with atrophy mostly in the hippocampus, while others have a hippocampal-sparing form, with atrophy mostly in the neocortex. In typical AD, both regions shrink (Murray et al., 2011; Whitwell et al., 2012; Ferreira et al., 2017). Can tau PET tell them apart? Ossenkoppele and colleagues took a look.
They analyzed data from 260 amyloid-positive people with symptomatic AD, who were in the Swedish BioFinder study or were seen at the University of California San Francisco AD Research Center or the Memory Disorder Clinic of Gangnam Severance Hospital in Seoul, South Korea. The researchers used MRI volumetry to divide this cohort into the three AD subtypes based on their atrophy in these respective regions. This categorized 70 participants as typical, 77 as limbic-predominant, and 76 as hippocampal-sparing AD. The remaining 37 participants had little evidence of brain atrophy. Several differences emerged. People with typical AD had more white-matter hyperintensities than the other groups. Participants with the hippocampal-sparing form tended to be younger and were less likely to carry ApoE4. They declined the fastest on the MMSE over four years, but were less likely than the other subtypes to have memory problems.
What about tau, though? As expected, flortaucipir scans unmasked a distinct pattern of tracer uptake for each subtype. Limbic-predominant AD had most tangles in the entorhinal cortex, hippocampal-sparing AD in parietal and frontal cortex, while typical AD was marked by tangles in lateral temporal as well as frontal and occipital cortices. In short, tau PET can be used as a proxy for atrophy, Ossenkoppele concluded.
As researchers train their sights on tau pathology as a therapeutic target, being able to measure and track tangles will be crucial, noted Lennart Mucke of the Gladstone Institute of Neurological Disease in San Francisco, in a plenary. “I’m excited that the pipeline is filling up with tau biomarkers,” he said.—Madolyn Bowman Rogers
Pascoal TA, Shin M, Kang MS, Chamoun M, Chartrand D, Mathotaarachchi S, Bennacef I, Therriault J, Ng KP, Hopewell R, Bouhachi R, Hsiao HH, Benedet AL, Soucy JP, Massarweh G, Gauthier S, Rosa-Neto P.
In vivo quantification of neurofibrillary tangles with [18 F]MK-6240.
Alzheimers Res Ther. 2018 Jul 31;10(1):74.
PubMed.
Amyloid PET Aids Diagnosis. But Could CSF Do Just as Well?
Clinicians have been eagerly awaiting results of the enormous Imaging Dementia–Evidence for Amyloid Scanning (IDEAS) study, which may determine whether amyloid scans will be covered by insurance. Preliminary findings previously indicated that such scans do sway diagnoses and treatment plans when used for patients with uncertain etiology. At the 11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, Spain, Gil Rabinovici of the University of California in San Francisco presented final results for this aspect of the study. In a cohort of 11,409 people, clinicians changed treatment plans for two-thirds of them after seeing amyloid PET scans, he reported. This is well over the study’s endpoint of 30 percent. Yet the positive finding does not guarantee insurers will cover the scans. For payers, a crucial question is whether those treatment changes improved the health of participants, and IDEAS researchers are still collecting data on that. They will get that answer in 2019.
Even so, Rabinovici believes amyloid scans have shown their clinical worth. “This is the strongest Phase 4 data to date supporting the use of amyloid PET,” he told the audience in Barcelona.
Another issue for payers is cost-effectiveness. An amyloid PET scan runs thousands of dollars, far exceeding the cost of lumbar punctures or blood draws. In the past, cerebrospinal fluid tests have had too much variance to make good diagnostics, but with the advent of automated assays and certified reference materials, measurements are becoming more reliable. In Barcelona, several groups reported that automated CSF measures agree closely with amyloid scan results, suggesting that these markers could reduce the need for amyloid scanning in clinical diagnostics.
The Centers for Medicare & Medicaid Services (CMS) supports the IDEAS study under its coverage with evidence development mechanism (Apr 2015 news). IDEAS was planned as a four-year, $100 million study of 18,200 Medicare recipients seen at doctor’ offices around the U.S. It finished enrolling in January 2018, ahead of schedule and under budget. IDEAS admitted people who met the appropriate-use criteria for amyloid scanning by having measurable cognitive impairment but an uncertain diagnosis (Jan 2013 conference news). At the 2017 AAIC in London, Rabinovici presented findings from the first third of this cohort (Aug 2017 conference news).
At CTAD, Rabinovici showed data for the whole cohort. IDEAS’ first aim was to see if amyloid scanning affected treatment. Clinicians first developed a treatment plan without knowing the patient’s amyloid status, then revisited it once they knew. Researchers registered 16,008 people for this part of the study. (The remaining participants are registered for the second part of the study, which looks at health outcomes after a year.) Rabinovici noted a large number of dropouts. About 2,000 people did not show up for their amyloid scans, another 1,800 skipped their post-PET visits, and about 800 more were disqualified for various violations of study protocol. This left 11,409 participants for analysis. Dropouts did not differ significantly from people who remained in the study, Rabinovici said.
Among the 11,409 participants, 60 percent were clinically diagnosed with mild cognitive impairment, the others with dementia. Their average age was 75. Scans determined that 55 percent of the MCI group were amyloid-positive, as were 70 percent of those with dementia.
Physicians adjusted their diagnoses after seeing scans. A quarter of the cohort were initially deemed to have AD but changed to a non-AD diagnosis, and another 11 percent started with a different diagnosis but later told they had AD. Overall, among all those with a positive amyloid scan, AD diagnoses jumped from 80 to 96 percent, while in the amyloid-negative group, they fell from 72 percent to 10 percent. As in the preliminary findings, scans had their largest effect in ruling out AD.
Treatment plans changed accordingly for about 60 percent of people with MCI, and 64 percent of those with AD. Most often, the changes involved prescription of the approved Alzheimer’s drugs, acetylcholinesterase inhibitors and memantine. Clinicians typically added these drugs after seeing a positive scan, with their use climbing from 40 to 82 percent in the MCI group and from 63 to 91 percent in the dementia group. Clinicians were much less likely to cancel prescriptions, with only a small drop in AD drug use after PET imaging.
Doctors changed other medications in about a quarter of the cohort, and recommended counseling for safety and long-term care planning in a quarter of participants as well. Clinicians reported that the PET result drove these decisions 87 percent of the time.
In addition, doctors cut plans for additional testing and imaging by half after seeing scans. The scan data also refined referrals for AD clinical trials. Although physicians referred slightly fewer people, the percentage of those who were amyloid-positive jumped from 66 to 93. This may result in fewer screen failures in those trials, Rabinovici said in his talk.
Rabinovici noted that this study represented real-world specialty care, a middle ground between primary care and select tertiary academic centers. Participants were seen at 595 dementia clinics nationwide; about 80 percent of these were private practices. That said, the study was limited by recruiting a fairly homogenous population that was 88 percent white and highly educated, with 44 percent having a college degree. By design, the study only analyzed the benefit of PET scanning for people with measurable cognitive impairment and atypical presentations. To address these shortcomings, Rabinovici said that IDEAS researchers were discussing a proposed follow-on study with the CMS. IDEAS2 would enroll people with typical AD and earlier-stage disease, and would strive to recruit more under-represented participants (Oct 2018 conference news). The new study will also collect DNA and plasma samples and store them in a biorepository, Rabinovici told the audience.
Cheaper Option?
In the meantime, PET scans remain uncovered by most insurance companies, and thus unaffordable for many patients. Could CSF biomarkers fill the gap? In Barcelona, Samantha Burnham of Commonwealth Scientific and Industrial Research Organisation in Canberra, Australia, suggested they eventually will. She presented data from 202 participants in the Australian Imaging, Biomarkers, and Lifestyle (AIBL) study, 70 percent of whom were cognitively healthy. All underwent lumbar punctures and PET scans with either PiB, florbetapir, or flutemetamol. CSF was analyzed by Elecsys automated assays in Gothenburg, Sweden. Amyloid scans flagged 42 percent of the cohort as amyloid-positive, and CSF biomarker ratios—whether of total tau/Aβ42, p-tau/Aβ42, or Aβ42/40—agreed with the scans 90 percent of the time. Each of the three ratios gave an AUC of 0.94. Single biomarkers performed less well, with an Aβ42 cutoff agreeing with scans only 81 percent of the time.
Timo Grimmer of the University of Munich showed similar data coming from the Phase 3 CREAD studies of the therapeutic antibody crenezumab. In these trials, 109 people, whose average age was 70 and CDR 0.5 to 1, participated in a sub-study directly comparing CSF and PET. They underwent florbetapir scanning and donated CSF for analysis by Elecsys. Similar to the AIBL data, Aβ42 alone agreed with PET scans 85 percent of the time. There were 16 people with discordant findings; nine were CSF-positive and PET-negative, while seven were CSF-negative and PET-positive. As in the AIBL study, tau/Aβ42 ratios performed better, giving 90 percent concordance with PET.
Another presentation, from Alberto Lleó of the Hospital de Sant Pau in Barcelona, compared florbetapir PET to a different automated platform, the LUMIPULSE system from Fujirebio Inc. The researchers analyzed 94 people from the Sant Pau Initiative on Neurodegeneration (SPIN) cohort, whose average age was 73 and whose diagnoses ranged from cognitively normal to dementia. Amyloid scans were positive in 63 percent. As in the other studies, CSF ratios gave high agreement with scans, from 86 to 88 percent, while single biomarkers performed less well.
Other new studies add to these data. Researchers at Washington University in St. Louis recently reported that CSF ratios determined by Elecsys can discriminate PiB-positive and PiB-negative people (Schindler et al., 2018). Likewise, researchers led by Oskar Hansson at Lund University, Sweden, found that CSF cutoffs determined in the BioFinder cohort agreed with PET imaging in the ADNI cohort 90 percent of the time, and predicted cognitive decline over the next two years (Hansson et al., 2018). Furthermore, Hansson and other PET experts agree that semiquantitative analyses of PET scans agree with visual reads only 90 percent of the time, suggesting that CSF biomarkers are already pushing against the ceiling of current amyloid PET scan accuracy.—Madolyn Bowman Rogers
How Does The NIA-AA Framework Measure Up Against Real Data?
This past spring, the National Institute on Aging and Alzheimer’s Association proposed a new research framework in an effort to nudge the field toward a biological definition of Alzheimer’s disease (Apr 2018 news). The framework defined AD as the presence of plaque and tangle pathology, regardless of symptoms, and offered a systematic definition of pathological changes based on biomarkers for brain amyloid, tau, and neurodegeneration. In parallel, it outlined a six-stage symptomatic scheme that covers the spectrum from cognitively normal to severe dementia. The package came aligned with recent FDA guidance on clinical trials for pre-dementia (Mar 2018 news).
How does this framework hold up in real life? Does it even jibe with natural history data? At the Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, Spain, the field got some initial answers to these questions. From early data testing the framework in large research cohorts assembled to date, it appears the biomarkers do a good job of predicting impending decline in people who are already cognitively impaired. On the other hand, an initial attempt at defining tests that enable staging of symptoms highlighted the critical need for measures that can tell apart the earliest phases of disease, and pick out the first, subtle signs of decline.
Leslie Shaw, University of Pennsylvania, Philadelphia, showed that a combination of brain amyloid, tau tangles, and neurodegeneration predicted prognosis in participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Shaw analyzed data from all 505 participants with a clinical diagnosis of mild cognitive impairment (MCI) at baseline, who underwent a spinal tap and an FDG-PET scan. He classified them according to the A/T/N scheme, using CSF Aβ1-42 for brain amyloid (A), CSF pTau181 for aggregated tau (T), and FDG-PET for neurodegeneration (N). Each participant’s values were deemed normal or abnormal based on cutoffs determined in ADNI. The binary classification of each of these three biomarker types bins people into eight possible profiles, ranging from completely negative (A-/T-/N-) to triply positive (A+/T+/N+). Shaw asked whether a person’s specific combination predicted his or her progression to AD dementia, and rate of decline in memory, cognition, and function.
Among these 505 people with MCI, 58 percent were above the cutoff for amyloid positivity based on CSF Aβ1-42. Their rate of progression to dementia increased with each additional positive marker: Only 11.6 percent of people with A+/T-/N- developed dementia over four years, but 40 percent did when T was also positive, and 80.9 percent when all three were positive. Shaw called this consistent with the idea that amyloid positivity on its own represents an early disease stage with a long horizon to dementia, while T and N reflect a later stage with additional downstream tau pathology and neurodegeneration.
People with brain amyloid and low FDG PET, but negative tau (A+/T-/N+), also showed substantial rates of decline, with 56.4 percent reaching dementia in four years. Under the NIA-AA criteria, this group is considered to have Alzheimer’s disease, which requires abnormal amyloid and tau for the diagnosis. They were on a trajectory of decline from a different disease, said Shaw. To understand which one, researchers urgently need α-synuclein, TDP-43, and vascular dementia markers.
In contrast, all amyloid-negative groups progress more slowly, ranging from 8.1 percent progression to dementia in triple-negative participants, to 16.5 for the A-/T+/N-, and 28.4 or 31 percent for A-/T+/N+ and A-/T-/N+, respectively.
How about annual rates of decline for MMSE, CDR-SB, and functional scores? They largely paralleled the rates of progression across the groups, Shaw said.
What’s the take-home? “If a person is amyloid-positive, it’s important to know other things, too,” Shaw said. “If we only know amyloid, we have a huge range of progression rates. But we can refine these progression rate predictions when we have baseline data for other markers, too.”
In other words, a person’s biomarker evaluation should include amyloid and tau status, and markers for neurodegeneration. How the “N” is measured will depend on local circumstances, Shaw said. The A/T/N approach makes a patient’s staging granular and improves his or her individual risk assessment. At this point, the field needs to do this work with other cohorts and other biomarkers for the A/T/N categories, Shaw concluded.
Enter Samantha Burnham, Commonwealth Scientific and Industrial Research Organization, Highett, Australia. At CTAD, she reported a similar analysis of A/T/N biomarkers in the Australian Imaging, Biomarker & Lifestyle Study of Aging. Her results in cognitively normal participants and those with MCI echoed Shaw’s, though with her smaller sample size, the differences were less dramatic.
Burnham analyzed data from 200 AIBL participants who had undergone lumbar puncture and were followed for a mean of 4.5 years, including 27 with AD, 33 with MCI, and 140 who were cognitively normal. Like Shaw, her group measured CSF Aβ1-42 for A and pTau181 for T, but for N they assayed CSF total tau and based their cutoff on a comparison of the AD and normal controls in their study.
For her analysis, Burnham collapsed the eight categories into four. They were: all normal markers, Alzheimer's pathological change (A+ only), AD (A+/T+), and non-AD pathological change (A-). Among the cognitively normal group, 67 percent were amyloid-negative, and nearly half of those had three normal biomarkers. In the MCI group, 75 percent were amyloid-positive, and half the MCIs were positive for all three biomarkers. In the AD group, 66 percent were triply positive.
When Burnham looked at baseline cognitive performance for people in the normal cognition group, those with biomarker evidence of AD (A+/T+) performed at the lower end of the normal range on the AIBL-PACC cognitive composite and the MMSE. Burnham’s group sizes were small and the differences not statistically significant. Even so, this result was consistent with Shaw’s result of more positive biomarkers predicting worse cognitive performance, and with Reisa Sperling’s previous finding in A4 that cognitively normal people with both amyloid and tau pathology performed worse at baseline than people who only had brain amyloid.
Burnham had too few people progress to MCI or AD to draw conclusions about progression, but will do this analysis in larger cohorts, including in the new CONCORDE-AD network. Short for Connecting Cohorts to Diminish Alzheimer’s Disease, this public-private collaborative will combine data from AIBL, the Swedish BioFinder study, and five other cohorts covering more than 20,000 people across the disease spectrum, in Australia, North America, and Europe (Burnham et al., 2018).
Burnham told the CTAD audience that CSF concentrations of pTau181 and total tau strongly correlate, indicating that total tau is not an appropriate measure of neurodegeneration. She is looking for other measures of N, including neurofilament light protein or structural MRI (Nov 2018 news).
What About Symptoms?
Besides biomarker-based classification, the NIA-AA framework also includes a six-stage ladder of symptomatic disease severity. The scheme postulates three stages of early disease, whereby people in stage 1 are completely cognitively normal with no complaints of memory problems; people in stage 2 report subjective memory concerns or display subtle abnormalities on sensitive cognitive tests, and people in stage 3 have obvious abnormalities on cognitive tests and mild functional impairment. Stages 4 to 6 encompass mild, moderate, and severe dementia.
How can scientists know which tests best identify people in each stage? Which tests best track a person over time as he or she moves from one stage to the next? That’s especially important in stages 1 and 2, as these people are increasingly becoming prime targets for enrollment in clinical trials.
Roos Jutten, VU University Medical Center, Amsterdam, pulled together test data on 1,213 people from four cohorts: the Harvard Aging Brain Study (HABS), ADNI, the National Alzheimer’s coordinating center (NACC), and the Amsterdam Dementia Cohort (ADC). All have brain amyloid as per PET or CSF, and had at least two cognitive assessments. For judging cognition, Jutten drew on the MMSE or Montreal Cognitive Assessment (MoCA) and devised a memory retention score based on items recalled from a story or list. She generated scores for subjective memory decline based on a visit to memory clinic or self-reported concerns, and for functional impairment based on the CDR-SB or global CDR score. Jutten created stage-specific cutoffs for each test and tested different ways of combining all the measures that would place the most people into one of the NIA-AA stages.
In the end, Jutten’s scheme placed 87 percent of participants clearly into one category or another. Most of the cognitively normal people, or those with subjective memory decline, fell into stage 1 or 2, while those formerly described as MCI mainly clustered in group 3. Thirteen percent of people could not be classified in this way, mostly because measures were not congruent. For example, a person with a high MMSE score but clear impairment on the CDR would not fit into a single category.
The different cohorts did not all shake out the same way. All HABS participants fell into stages 1 and 2, while in ADNI, most of the 258 subjects Jutten used were in stage 3. The NACC skewed toward stages 3 and 4, while the ADC contained no stage 1, and mostly stages 3 and 4.
How did the different tests change over time? Jutten compared baseline, one-, and three-year scores. For MMSE, the one-year decline was fastest for people in stage 4, and significant in stage 3. The MMSE clocked no change at one year in stages 1 or 2. After three years, Jutten did detect a drop in MMSE scores in stages 2, 3 and 4, but still no change in stage 1. Delayed recall was similar, with a measurable decline in test scores only in stages 3 and 4 after one or three years. Jutten also saw a ceiling effect on the MMSE in stages 1 and 2, and a floor effect on the delayed recall in stage 4.
Only the category fluency test registered change in stage 1 participants over time, and that took three years to detect. Jutten detected changes within one year on that test in stages 2, 3, and 4. Letter fluency only declined in stage 3 and 4 subjects after three years.
In essence, no single test picked up one-year change at each of the clinical stages. This means the field needs stage-specific cognitive measures, especially for stages 1 and 2, Jutten said. Her group will continue to investigate other available tests with the goal of combining them to create a sensitive, stage-specific battery that can then be compared to later outcomes. Researchers also need to investigate which changes are specific to AD, or change over time with aging alone, Jutten said.—Pat McCaffrey
Fits and Starts: Trial Results from the CTAD Conference
This is Part 1 of a two-part story. Click here for Part 2.
As in years past, trial results offered up a mixed bag of goods and duds at the Clinical Trials on Alzheimer’s Disease Conference, held October 24–27 in Barcelona, Spain. In the first Phase 3 AD trial ever conducted in China, a seaweed oligosaccharide was claimed to have halted or even reversed the slide into dementia of people with mild to moderate Alzheimer’s. In a more familiar story, another offbeat approach—a plasma exchange therapy—was reported to slow decline in people with moderate, but not mild, AD. Not so for the fyn kinase inhibitor saracatinib, which hit the end of the road. New Aβ vaccines advanced, and the first clinical trial of an antiviral drug potentially offers a new angle on AD. Finally, the neurosteroid allopregnanolone inched ahead, though estrogen receptor-modulating PhytoSERMs did not.
First, news from the home team. Antonio Páez of the Barcelona-based blood products company Grifols, presented topline data from the Phase 2b/3 AMBAR (Alzheimer’s Management by Albumin Replacement) trial. The14-month regimen of repeated plasma exchange and albumin replacement (PE-A) slowed cognitive decline in people with AD, Páez said.
Most Aβ in the blood is bound up with the protein albumin. AMBAR is a form of plasmapheresis designed to exchange Aβ-laden, oxidized albumin with clean protein. Siphoning off peripheral Aβ should draw Aβ peptides from the brain, the thinking goes, while fresh albumin boosts the blood’s antioxidant, immune-modulatory, and anti-inflammatory capacities (Boada et al., 2014).
Last year at CTAD, Grifols presented data from a pilot study suggesting trends toward improvement in cognition (Dec 2017 conference news). The new trial enrolled 350 volunteers aged 55–85 with mild to moderate AD, and ran at 41 sites in Spain and the U.S. Participants were randomized either to exchange and replacement, or to an elaborate sham procedure that kept patient, caregiver, and clinical raters blinded. After six weekly treatments, the trial switched to monthly sessions for one year. It compared three treatment conditions: two doses of albumin in combination with intravenous immunoglobulin (IVIG), and one dose of albumin alone. IVIG contains immunomodulators and human antibodies including anti-Aβ; some preparations had shown promise in AD but failed in later-stage trials (e.g., Gammagard®, Octagam®10%) (May 2013 news). The investigators hoped they might see an added effect of the albumin-IVIG combination on the primary outcomes of change from baseline on the ADAS -Cog and ADCS-ADL after 14 months.
Exchange for the Better? Plasma exchange and albumin replacement slowed worsening in ADAS-Cog (top) and ADCS-ADL (bottom) in people with moderate AD. Participants received six weekly total plasma exchanges (TPE), followed by 12 monthly low-volume plasma exchanges (LVPE) and albumin replacement. [Courtesy of Grifols.]
Alas, it was not to be. While each of the three treatment groups showed between 50 and 75 percent less decline on the ADAS-Cog than placebo, the differences were not statistically significant. When the investigators lumped the three treatment arms into one group, the combined cohort declined 66 percent less than placebo, which just skirted significance (p=0.06). On the ADCS-ADL, the pooled group declined 52 percent less than placebo, which did reach significance, with a p value of 0.03.
A prespecified analysis split the trial cohort into two subgroups of high versus low MMSE scores, each having 39 people on placebo and 122 on treatment. This unmasked a treatment benefit in the more-affected people, Páez said. The mild dementia group, whose scores ranged from 22 to 26, did not decline over the study period, with or without treatment. In the moderate group, with scores between 18 and 21, the placebo group dropped from baseline both on ADAS-Cog and ADCS-ADL, and treatment reduced that decline by 61 percent on both measures.
With 4,709 procedures completed, the study was the largest series of plasma exchanges ever dedicated to a single disease, Páez said. Seventy-two percent of volunteers completed the study, and the most frequent complication was local reactions to the catheter. Jeffrey Cummings, Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas, praised the investigators for achieving this many plasmapheresis sessions in people with AD with virtually no serious adverse events. “It’s an achievement to bring this fairly intense procedure to bear on patients substantially compromised by AD,” he said.
Researchers debated the lack of decline in the mild group, which was unexpected. The investigators suspect placebo effects, which can be powerful in intervention trials, especially of intensive procedures. Lon Schneider, University of Southern California, Los Angeles, offered another explanation. “This is a potentially compelling set of outcomes, but you’ve a priori split the sample into these mild and moderate groups, and then we start to speculate,” he said. A number of factors could be masking an effect in the mild group, Schneider suggested, including their ADAS-Cog baseline scores, cholinesterase use, and age. “If you had shown these coefficients and baseline scores, we might be having a different discussion,” he said.
What now? Páez said Grifols plans to run at least one new clinical trial, perhaps trying different frequencies of plasma exchange in moderate AD. The company also has other plasma protein fractions to test, he said.
From Blood Products to Gifts From the Sea Meiyu Geng, Chinese Academy of Sciences, Shanghai, presented Phase 3 results of GV-971, a seaweed-derived oligosaccharide. In July, its developer, Shanghai Green Valley Pharmaceuticals, claimed that taking this oral drug for 36 weeks improved ADAS-Cog scores in people with mild to moderate AD (press release). Back then, the company announced it would seek approval to market the drug in China this year.
Green Valley bills GV-971, aka oligo-mannurarate, as a jack of all trades. Unpublished preclinical data suggests the compound inhibits Aβ fibril formation, is anti-inflammatory, and even normalizes gut microbiome changes connected to Alzheimer’s disease, Geng told Alzforum. In the trial, 818 participants with clinically diagnosed mild to moderate AD took 900 mg in two capsules, or placebo, twice a day for 36 weeks. ADAS-Cog scores were higher in the treatment versus placebo group at four, 12, 24 and 36 weeks, with a mean difference at 36 weeks of 2.54 points. There was a trend toward improvement on the Clinicians Interview-Based Impression of Change, a secondary outcome, Geng said at CTAD.
Other secondary outcomes did not budge, including ADCS-ADL, the neuropsychiatric inventory, or FDG-PET in a subset of participants. A subgroup analysis revealed the drug had its largest effect on the more severely affected people with MMSE scores between 11 and 15.
The treatment was well-tolerated, with 80 percent of people completing the trial and similar adverse event rates between treatment and placebo groups. The company is planning a global trial of GV-971 to confirm the findings in China, and will add biomarkers to probe the potential mechanism of action, Geng said. According to ClinicalTrials.gov, the investigators are recruiting for a small study in China that will assess safety and pharmacokinetics of higher doses.
Some audience members said that the results reminded them of the trials for first-generation cholinesterase inhibitors or memantine. People in this study were not taking those medications, Geng said. In China, she noted, AD is rarely diagnosed, and thus often untreated. Most people have no access to those drugs.
Vaccines Inch Forward
Two amyloid vaccination strategies are slowly moving along. Ajay Verma, of United Neurosciences in Hauppauge, New York, reported on an Aβ1-14 immunogen. The peptide elicits antibodies that bind Aβ oligomers, fibrils and plaques, but not monomers. The vaccine’s claim to fame is safety. It caused no amyloid-related imaging abnormalities in a Phase 1 trial, and so far hasn’t caused any ARIA through the first half of a Phase 2a trial in people with mild AD (Dec 2017 conference news). That trial, which administered 12 doses over 60 weeks, followed by 18 weeks of observation with periodic PET, MRI, and cognitive tests, is now complete. The data is still blinded, but safety signs continue to be encouraging: 41 of 43 participants completed the study, with no sign of ARIA-E. Verma promised top line results by the end of the year and full data at the AD/PD conference in March in Lisbon, Portugal.
Safety is also looking good for another active, N-terminal Aβ immunotherapy. Bjørn Sperling, H. Lundbeck A/S Valby, Denmark, presented interim data on its ongoing first-in-human Phase 1b study of LuAF25013. Taking place in Sweden, Finland, and Austria, the trial has enrolled 40 of a planned 50 people with mild Alzheimer’s and CSF biomarker-confirmed brain amyloid, who will receive vaccine at four different doses for up to two years. LuAF25013 elicits antibodies to aggregated forms of Aβ, which recognize plaques in brain, Sperling showed at CTAD. As of September, safety looked good, Sperling said, without ARIA-E or other serious adverse events related to treatment. Most events were mild injection-site reactions; none prompted dropouts. Six participants developed at least one new ARIA-H while on the trial; all were asymptomatic. Sperling said Lundbeck will enter Phase 2 in the first half of 2019, vaccinating 200 people with CSF biomarker-confirmed AD. The primary outcome will be change in brain amyloid measured by PET, with secondary measures of cognitive and behavioral impairment plus antibody titers and bio markers of neurodegeneration.
Going Viral?
With recently renewed rumblings about a role for infection in the etiology of some cases of AD (Jun 2018 news; Jun 2018 news), a Swedish company has jumped in with a treatment idea. Nina Lindblom of Apodemus AB in Solna, Sweden, and colleagues at the Karolinska Institute presented results from their trial of an antiviral treatment. They target enterovirus, the most common human pathogenic virus and the cause of 1 billion infections annually just in the U.S.
Thinking that repeated infections might stoke neuroinflammation in adults with AD, the researchers asked if they could slow the rate of AD progression by preventing new infections or treating persistent, low-grade infections. To do that, they use Apovir, a combination of the experimental anti-enterovirus agent pleconaril, originally developed to treat the common cold, and the hepatitis C treatment ribavirin, which was added to prevent the emergence of resistance. In a Phase 2a study, they treated 69 people with mild AD with Apovir or placebo for nine months, then followed them for another 12 months.
During the treatment phase the dropout was high, Lindblom reported. Side effects of ribavirin caused half the treatment group to leave. By nine months, there were but 18 people left in the treated group, and 31 taking placebo. Nonetheless, there were some hopeful hints. The ADAS-Cog worsened steadily in the placebo group over time, but the treated group improved on this measure by three points over baseline at nine months. CDR-SB scores worsened in the placebo but not the treated group. After treatment stopped, both groups progressed at the same rate.
A post-hoc analysis suggested these results were not due to the dropouts, Lindblom claimed. Moreover, change in ADAS-Cog scores correlated with plasma concentrations of pleconaril, suggesting that this drug is more important for the treatment effect than ribavirin.
Lindblom told Alzforum that Apodemus is continuing development with pleconaril alone. A new, Phase 2 trial started in Poland and the Czech Republic in July 2018. Lindblom said it is similar to the previous trial but will evaluate 600 mg/day pleconaril alone versus placebo as an add-on to acetylcholinesterase inhibitors or memantine. The trial will initially enroll 120 patients, and use change on ADAS-Cog 11 at one year as primary endpoint. Apodemus expects to perform an interim analysis in the third quarter of 2019.
Going Hormonal? Roberta Diaz Brinton, Arizona University, Tucson, reported on a completed Phase 1 trial of allopregnanolone, a naturally occurring neurosteroid that stimulates neurogenesis and dampens inflammation and Aβ production in mice. Brinton had previously shown partial results of this small trial (May 2017 conference news). It tested 12 weekly infusions of 2, 4 and 6 mg of allopregnanolone or placebo in 12 women and 12 men with mild cognitive impairment or early AD. The treatment continues to be safe across these doses; Brinton reported no adverse events, no ARIA, and clinical results in the normal range. When they ramped up dosing to find a maximum tolerated dose, women became sleepy at 10 mg, men at 6 mg.
Brinton also spotted some MRI and functional MRI signals associated with allopregnanolone. Compared with placebo, treatment slowed left hippocampal atrophy, and this effect was more pronounced in APOE4 carriers. High-dose treatment improved functional connectivity between the right parietal lobe and posterior cingulate cortex. The 4 mg dose evinced stronger connectivity between the default mode network and the left thalamus or the left putamen compared with placebo. Do these changes translate into better cognition? Brinton is not sure, but called the results encouraging evidence for changes in synaptic function. Exploratory cognitive measures showed high variability and no significant differences between groups, though CogState battery scores did move in the direction of improvement with treatment.
To predict a person’s response to allopregnanolone, Brinton’s team generated induced pluripotent stem cells (IPSCs) from blood cells of patients in the trial, which they then differentiated into neuronal progenitors and treated with allopregnanolone. At CTAD, Brinton drew a correlation between allopregnanolone induction of mitochondrial respiration in a person’s IPSC-derived neuronal stem cells and his or her change in hippocampal volume. In people whose mitochondria responded to the hormone, the hippocampus atrophied less. With these tools in hand, Brinton has submitted a proposal to the NIA for a Phase 2 study of 200 participants, treated for up to 18 months.
On a less-cheery note, USC’s Schneider presented final results from a Phase 1/2 trial of PhytoSERMs, a plant-based estrogen receptor-beta targeting formulation he and Brinton developed. It zeroes in on brain estrogen receptors while minimizing effects at peripheral alpha receptors (for background, see Dec 2014 conference news).
Schneider and Brinton evaluated whether the supplement would improve symptoms in peri- and postmenopausal women with cognitive complaints and hot flashes. They gathered safety, pharmacokinetic, and efficacy data for up to 12 weeks of daily dosing in 71 healthy women age 45–60 who reported at least one cognitive complaint and one vasomotor-related symptom per day, i.e., hot flashes or night sweats.
The PhytoSERMs were safe but failed to improve cognitive symptoms. The vasomotor endpoints suggested a potential for efficacy. Schneider said that if Brinton and he were to take PhytoSERMs forward, it would be as a food supplement focusing on those, not on cognitive symptoms.
The end also came for the fyn kinase inhibitor saracatanib, aka AZD0530. Christopher van Dyck, Yale University, presented data on a negative Phase 2a trial of the compound, originally developed by AstraZeneca to treat cancer. The Yale group worked with the Alzheimer’s Therapy Research Institute at the University of Southern California to repurpose saracatanib for AD, because fyn mediates Aβ synaptotoxicity and tau dysfunction in model systems (Um et al., 2012; Ittner et al., 2010).
The investigators knew from Phase 1 that the drug had a narrow therapeutic index, meaning that pushing the dose to achieve effective brain levels would bump up against toxicity (Nygaard et al., 2015).
Called CONNECT, the current trial tested whether 12 months of saracatinib would slow disease in 159 people with AD. Its design was innovative, using FDG PET as a primary outcome. Alas, the drug did not slow the disintegration of the FDG-Pet signal, or change any of the other outcomes. At the same time, adverse events and the number of people who quit the study showed that dosing was maxed out. “I don’t think this can be pursued at any higher doses than we did,” said van Dyck. While the drug failed, FDG acquitted itself well as an outcome measure, he said. It correlated with clinical measures, and boasted tight confidence intervals, implying it might allow detection of smaller drug effects than the cognitive tests, he believes.—Pat McCaffrey
Trials of Diabetes-Related Therapies: Mainly a Bust
This is Part 2 of a two-part story. Click here for Part 1.
The highs and lows of diabetes-related therapies were on display at the Clinical Trials on Alzheimer’s Disease Conference, held October 24–27 in Barcelona, Spain. This time around, it was mostly the lows. Intranasal insulin presented a puzzle, the RAGE inhibitor azeliragon attempted another artful save to keep more study going, and MCI prevention with pioglitazone got a lessons-learned epilogue.
Special Delivery?
CTAD afforded a first look at results from a Phase 2/3 trial of intranasal insulin, which attempted to reverse the waning brain concentration and function of this hormone in people with AD. Interest in this approach was sparked by a single-site pilot study of 104 people with amnestic MCI or AD, which reported that four months of insulin treatment appeared to stave off cognitive decline and improve brain glucose uptake (Craft et al., 2012). In the SNIFF trial, Suzanne Craft, Wake Forest School of Medicine, Winston Salem, North Carolina, and colleagues expanded to 25 sites with a 12-month, blinded, placebo-controlled treatment phase plus six months of open-label extension. The blinded portion wrapped up in June of 2018, and Craft presented primary data. The upshot: Insulin had no effect on cognition. Craft believes a change in the delivery device partway through the trial may have influenced the results.
The study enrolled 289 volunteers with MCI or mild AD, who took 20 units twice a day of insulin or placebo. The primary outcome was their ADAS12-Cog score, measured quarterly. They started out using Kurve Technology’s ViaNase spray device, the same as in the pilot trial. However, the device had been redesigned and frequently malfunctioned, so the investigators switched to NeuroPharma’s Precision Olfactory Delivery™ device for the bulk of the participants. At the end of the trial, they decided to conduct the primary analysis with the 240 people in that cohort. They found the treatment had no effect on change from baseline on ADAS12-Cog, or secondary cognitive outcomes, or CSF Aβ and tau biomarkers. Treatment did result in more hippocampal shrinkage, and a trend toward more entorhinal cortex thinning.
When Craft looked separately at the 49 people who used the ViaNase device in secondary analyses, she saw different results. In them, insulin appeared to slow the worsening on ADAS12-Cog, as in the pilot study. The difference was significant only at six months; at nine or 12 months there was a trend in the same direction (p=0.09). The ViaNase group was similar in size to the pilot cohort, but was recruited from multiple sites. Safety and compliance were good with both devices, Craft said.
Still to come are results of a prespecified responder analysis by APOE genotype, disease severity and AD biomarker status, and analysis of the open-label extension, which will wrap up this year, Craft said.
What about drug delivery and exposure? The investigators did not fully investigate these aspects before conducting the SNIFF trial, but will now, Craft said. They collected CSF samples at baseline and one year for more than 150 participants, and can analyze those to estimate insulin exposure. They did test acute delivery by dosing and then collecting CSF to analyze for insulin with the Kurve device. This generated a dose-response curve, Craft said. This was not done with the new device prior to its use in this trial, but is now underway.
Pioglitazone Epilogue
The TOMMORROW secondary AD prevention trial is over, and at CTAD researchers got to see its data, presented by Robert Alexander, Takeda, Cambridge, Massachusetts. TOMMORROW tested whether the diabetes drug pioglitazone would prevent mild cognitive impairment in asymptomatic people at genetic risk for AD. It stopped earlier this year after a futility analysis gave it only a 15 percent chance of success (Jan 2018 news).
The trial enrolled 3,494 cognitively normal participants between 65 and 83, who were judged to be at risk of developing cognitive impairment in the next five years by an algorithm that weighed their APOE and TOMM40 genotypes and ages. The treatment plan called for low-dose pioglitazone or placebo to continue until 202 people reached the primary endpoint, which was progression to MCI. As a group, participants skewed slightly female and were overwhelmingly Caucasian. Most took the drug or placebo for less than three years.
Pioglitazone did not slow progression. In the treatment group, 2.8 percent developed MCI, the same as placebo, and time to progression was also the same in both groups. Pioglitazone had no effect on secondary endpoints assessed out to 36 months. The cognitive composite battery score increased over time in both groups, while ADCS-ADL scores remained constant.
A second objective of the study was to test the algorithm’s ability to identify high-risk people. To do that, investigators included 433 low-risks participants, who were treated with placebo. When the study was halted, 73 people had mild cognitive impairment, including 3.3 percent of the high-risk group risk and 1 percent of the low risk group. That gave an adjusted risk ratio of 3.26 for the high-risk participants, which was statistically significant, Alexander said. The time from enrollment to MCI was faster in the high-risk group, too, with a median time of 383 days compared with 634 days in the low-risk group.
More than 60 percent of people in the high-risk group had APOE4. Attendees questioned whether the investigators detected any contribution of the TOMM40 polymorphism above and beyond age and APOE4. Alexander said they haven’t had a chance to look at the relative contribution of each component, but are working on that.
Subgroup to the Rescue?
In contrast to pioglitazone’s postmortem, a different drug was presented as still offering a faint glimmer of hope. Azeliragon has been in clinical trials since before 2005. It is an inhibitor of the receptor for advanced glycation end products (RAGE). Advanced glycation end products are overproduced in chronic hyperglycemia; the receptor also counts Aβ fibrils among its ligands. Azeliragon was pursued in hopes it would block brain inflammation and amyloid toxicity. Earlier this year, its current owner, vTv Therapeutics of High Point, North Carolina, terminated a Phase 3 trial after it failed to show any benefit over placebo on cognitive endpoints in patients with probable Alzheimer’s disease (Apr 2018 news).
At CTAD, Marwan Sabbagh, Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, presented—no surprise to veteran observers of Alzheimer’s trials—a subgroup analysis that he said may offer a lifeline for this compound. Asking what kind of patient might have high levels of RAGE, or its ligands, led the researchers to look at people with elevated concentrations of glycated hemoglobin HbA1c, a RAGE ligand linked to tissue damage and vascular complications of diabetes. Lo and behold, in the subgroup of trial participants who had diabetes and high HbA1c, the researchers reportedly observed a significant benefit of azeliragon on both the ADAS-Cog and CDR-SB.
The group sizes are tiny, with but 22 placebo and 33 treated participants. Still, to Sabbagh’s mind, the result suggests that azeliragon could help patients who have both Type 2 diabetes and AD. Carmen Valcarce of vTv said her company will try to raise money to pursue this subgroup signal in a bigger study.—Pat McCaffrey
To speed up recruitment into Alzheimer’s trials, researchers have created innovative new platforms that establish trial-ready cohorts of people at early disease stages. The farthest along is the European Prevention of Alzheimer’s Dementia Trial-Ready Cohort (EPAD-TRC), followed by the Trial-Ready Cohort for Preclinical/Prodromal Alzheimer’s Disease (TRC-PAD) in the U.S. Both launched in 2014. At the 11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, Spain, leaders of both initiatives gave updates. EPAD is about to release baseline data from its first 500 participants, and will begin its first therapeutic trial in a year, while TRC-PAD is filling up its online registry and will begin in-person screening next year. Researchers hope that TRCs will be game-changers for trial enrollment. “The goal is to have no screen failures in trials,” Ritchie said.
Both initiatives take similar approaches, recruiting from existing registries and trials and screening those participants for amyloid positivity. Those who pass become part of a deeply phenotyped longitudinal cohort that provides a pool of participants to be tapped on an ongoing basis for therapy trials (Aug 2016 conference news; Aug 2016 conference news).
In Barcelona, Lisa Vermunt of VU University Medical Center, Amsterdam, and Pieter Jelle Visser of Maastricht University in the Netherlands showed an analysis of how efficiently EPAD had recruited. They examined a subset of participants drawn from four feeder cohorts: the population-based Generation Scotland, the ALFA study in Barcelona, the Dutch Brain Health Registry, and the French Trial Registry. Together, these cohorts comprised 16,877 non-demented participants. Using a prescreening algorithm that took their age, APOE status, memories, and family histories into account, researchers invited 3,009 of those 16,877 people for clinic visits. Alas, only 414 people took them up on the offer and met all inclusion/exclusion criteria. Of those, 332 so far have undergone lumber punctures. 110 people were under the cutoff for amyloid positivity, with CSF Aβ42 below 1,098 pg/ml.
What predicted successful enrollment? The researchers found that older or less-educated people were less likely to show up for a scheduled in-person screening. Since both those factors increase the risk of AD, this results in losing many high-risk people from the cohort, Visser suggested. In addition, women were less likely than men to come in, again depleting a potentially higher-risk group. On the other hand, people with family histories of dementia were more likely to follow through with screening.
The large Generation Scotland cohort, 13,681 strong, had the lowest response rate to the study invitation, perhaps because researchers notified them by regular mail, Visser said. On the other hand, people recruited from memory clinics and smaller registries were more likely to come in and to stick with the process. Almost half of those from the French Trial Registry, which recruits from memory clinics throughout France, ended up undergoing CSF testing, while only about one in 200 of those from Generation Scotland did. Selecting from higher-risk populations boosts the screen success rate, Visser noted.
Among those who donated CSF, people who were older or carried an APOE4 allele were more likely to be amyloid-positive, as expected. Adding a plasma Aβ test before drawing CSF could reduce screen failures at this last step, Visser suggested (see Part 3 of this series).
Early Decline in EPAD. The RBANS cognitive composite picks up subtle deficits in people at CDR 0.5; these are most pronounced in those with amyloid plaques. [Courtesy of Craig Ritchie.]
So where does the EPAD trial-ready cohort stand now? Craig Ritchie of the University of Edinburgh, Scotland, who co-leads EPAD, told researchers in Barcelona that as of October 2018, 1,111 people had been enrolled. Initially EPAD aimed for a cohort size of 6,000 but it has since dropped this target, trying now to recruit only as many people as are needed for trials. After a slow start in 2016, enrollment accelerated, and sites are now adding 100 participants per month. Since mid-2017, study investigators have been prioritizing recruiting from clinics over population cohorts, Ritchie said. He added that almost all participants have stayed in the study so far.
Because recruitment will continue throughout EPAD’s lifetime, investigators periodically lock data sets to facilitate comparisons of research findings from this cohort over time. They have just locked the first set, comprising baseline data from the first 500 participants. Ritchie noted that EPAD collects a vast amount of data, following good clinical practice (GCP) standards to support its use later on as people enter therapy trials. Participants are followed over time, providing run-in data for these trials.
The first 500 participants have a mean age of 66, and 52 percent are female. A majority, 59 percent, have a family history of dementia, but only 37 percent carry an APOE4 allele. Ritchie noted this is close to the population average of around 25 percent APOE4 carriers, and well below the proportion seen in most clinical and trial cohorts. “This is almost a community-based cohort,” he explained. A large majority are cognitively healthy, with a cohort mean MMSE of 28.6; 83 percent of them have a CDR of zero, though 17 percent are mildly impaired with a CDR of 0.5.
On biomarker analysis, 35 percent met the CSF Aβ42 cutoff for positivity. EPAD does not disclose amyloid status to participants. Amyloid-positive participants were more likely to be older, carry an APOE4 allele, and have a CDR of 0.5. Other characteristics, including sex, education, and family history, did not affect amyloid status. Based on these data, researchers developed an algorithm for predicting amyloid positivity using age, genotype, and CDR. The algorithm achieved a positive predictive value of 80 percent, and negative predictive value of 65 percent. Ritchie hopes its use may boost the success rate of screening as EPAD continues to enroll.
Comparing several different cognitive tests, researchers found the RBANS to be the most sensitive measure of decline. It related closely to the CDR, with people at 0.5 testing slightly lower on several RBANS subtests, particularly those related to memory. People who were CDR 0.5 and amyloid-positive had a more marked drop.
Michael Ropacki of Strategic Global Research & Development, Half Moon Bay, California, elaborated on this in a poster. At baseline, amyloid-positive and –negative participants differed on the RBANS, but did not vary much on the MMSE or on neuropsychological tests, he reported. Moreover, 37 percent of the cohort hit the ceiling on the MMSE, while 71 percent scored at floor on the Amsterdam Instrumental Activities of Daily Living Questionnaire, indicating no functional impairment. Ritchie said the RBANS will be the primary outcome measure in the EPAD trial (Ritchie et al., 2017). “It is looking good as a tool to measure change over time,” Ritchie said.
This first EPAD dataset, V500.0, will be released to members of the EPAD consortium in December. Consortium members have six months of privileged access, and in summer 2019 the data will become publicly available worldwide. Ritchie expects to release the first year of follow-up data on these 500 participants, data lock V500.1, to the consortium in summer 2019; if recruitment stays on pace, baseline data on the first 1,500 participants (V1500.0) will come out at that time, too. The first proof-of-concept trial is expected to start in 2019 or 2020, and will include three interventions in different arms.
The TRC-PAD cohort is not as far along. This initiative is funded by the National Institute on Aging, and led by Paul Aisen of the University of Southern California, San Diego, Reisa Sperling of Brigham and Women’s Hospital, Boston, and Jeffrey Cummings of the Cleveland Clinic Lou Ruvo Center for Brain Health in Las Vegas. Initially, the Global Alzheimer’s Platform (GAP) planned to support the project as well, but was not able to provide any funding, Aisen noted.
The APT Webstudy is an internet-based platform that launched in December 2017. Upon joining, participants fill out forms on their demographic data, family and medical history, lifestyle, and medications. They take Cogstate card tests and the Cognitive Function Index (CFI) at baseline and every three months thereafter. Tests take less than 20 minutes to complete. In addition, participants are referred for APOE genotyping.
Currently, 7,400 people are in the APT Webstudy, out of a planned 50,000. Seventy percent are women, and 93 percent are white. They tend to be highly educated, with 85 percent having at least some college. Their mean age is 64. They come from every state, but participants cluster in the southwest U.S., the mid-Atlantic states, and Florida. About 40 percent of them access the website via mobile devices.
To select people for the trial-ready cohort, the researchers will use an algorithm that takes test scores and APOE genotype into account, as well as prior biomarker or clinical findings available from participants. The selected participants will be invited to come to a clinic for in-person screening, and if eligible, will be enrolled in TRC-PAD. This in-person screening will begin in 2019, Jimenez-Maggiora said. Screening will start at eight sites and ramp up to 30 sites across the U.S., Aisen told Alzforum. Researchers will screen for plasma Aβ and follow up with CSF or PET to confirm amyloid status. Participants will take the PACC cognitive composite. Those enrolled into TRC-PAD will continue to take quarterly online cognitive tests, and return to the clinic for semi-annual visits. In this way, there will be run-in data for each participant before he or she enrolls in trials. Jimenez-Maggiora noted that TRC-PAD will expand to other countries as well. Japan’s effort will be led by Takeshi Iwatsubo at the University of Tokyo, and France’s by Bruno Vellas of Gerontopole in Toulouse.—Madolyn Bowman Rogers
Which Are the Right Tests to Satisfy New FDA Guidance?
As researchers push toward testing Alzheimer’s drugs at presymptomatic stages, the hurdle before them—how to show efficacy and get a drug approved—looks different than before. In its February 2018 draft guidance for industry, the U.S. Food and Drug Administration signaled a willingness to consider a slowing of cognitive decline alone as the basis for approval. At the 11th Clinical Trials on Alzheimer’s Disease conference, held October 24–27 in Barcelona, Spain, leaders in the field wrestled with how this new guideline might be put into play, and what cognitive tests might fit the agency’s bill.
Suzanne Hendrix of Pentara Corp., Salt Lake City, made a case for analyzing longitudinal data to select the measures that change the most as disease gets worse. Such measures should be tailored to the specific disease stage and population under investigation. A slowing of decline on those scales would reflect slowing in the underlying disease process, Hendrix argued. “If you can measure progression with a test, then you can measure a treatment effect that is disease-related,” she said. Researchers broadly agreed on the need for such customized measures.
New Guidance Engenders Debate
The FDA guidance defines four stages of sporadic Alzheimer’s disease based solely on clinical and cognitive criteria, without reference to biomarkers. In this scheme, stage 1 has no detectable impairment, stage 2 is marked by subtle cognitive decline, stage 3 by cognitive decline and functional impairment, and stage 4 is dementia. Stages 1 and 2 would be considered preclinical disease, and stage 3 prodromal, researchers noted in Barcelona. Maria Carrillo of the Alzheimer’s Association said the guidance represents two years of effort from a working group that convened in 2015 and used the best data available at that time. As new research refines scientists’ understanding of AD, however, the picture may change again. “The guidance is meant to encourage the field to test hypotheses,” Carrillo said. “Some of the revisions in the guidance are already paying off, and some already need revising.”
Case in point: It is not so clear, after all, whether stage 1 disease actually exists, noted Paul Aisen of the University of Southern California, San Diego. Recent studies have found measurable cognitive decline even in people with sub-threshold levels of amyloid accumulation (Aug 2018 conference news). In people with amyloid encroaching on the brain, performance slides slightly faster over time than it does in age-matched controls, even while cognitive scores remain in the normal range. Thus, all preclinical disease may effectively be stage 2.
However, Aisen noted that the difference in decline is slight in preclinical disease, detectable over three years on a sensitive cognitive battery such as the PACC, and over six years on the CDR-SB. In other words, detecting this modest decline would require either long trials or even more sensitive cognitive measures.
Samantha Budd Haeberlein of Biogen in Cambridge, Massachusetts, would rather see the latter than the former. She celebrated the FDA’s willingness to consider new neuropsychological tools. “Industry welcomes this openness, but we’re not as enthused about long trials,” she noted drily. Others agreed. “The guidance is encouraging,” said Gary Romano of Janssen Pharmaceuticals in Philadelphia, adding, “The catch is to demonstrate a relationship between the early and late manifestations of disease.”
Researchers are committed to going early. As Ron Petersen of the Mayo Clinic in Rochester, Minnesota, put it, “Stage 2 is where the money is." These are people whose cognitive decline can be measured, but who are still early enough in disease that interventions could stem the worst of AD. In the draft guidance, the FDA says it will accept cognitive measures alone as the basis for approval in stage 2. It also says a drug’s benefit must be robust and consistent across multiple domains, and that the application will be stronger if supported by biomarker evidence. Petersen suggested mining longitudinal data to identify other early impairments, such as mild neurobehavioral symptoms, that could strengthen the case for a clinical benefit. “Perhaps we could marry subtle behavioral features with tau pathology,” Petersen said.
Industry faces another hurdle in demonstrating that even a slight slowing of cognitive decline is meaningful to people’s daily lives. Chris Edgar of the U.S./Australian testing company Cogstate noted that the previous FDA guidance used the term “meaningful” only twice, but the new draft guidance mentions it 25 times. “This indicates a shift in focus by the FDA,” he said in Barcelona. Alas, the FDA does not define the term. “We’re some ways from having a consensus on what it means,” Edgar said.
He noted that the FDA gives great weight to patient and caregiver perspectives, implying that these should be incorporated into outcome measures. Jason Hassenstab of Washington University in St. Louis agreed. “We’re taking that approach in DIAN,” he said. DIAN investigators are attempting to measure qualities that might be considered intangible, such as a person’s contentment, confidence, and sense of identity, to evaluate whether the benefit of a therapy was meaningful to them, Hassenstab said.
Aisen suggested that the Cognitive Function Index (CFI), a brief survey that asks about small functional deficits in people at preclinical stages, may help get at the question of meaningfulness (Mar 2015 news). The CFI distinguishes between amyloid-positive and –negative people at baseline. “However, I’m not confident it will perform well longitudinally,” Aisen said. Currently, many companies are moving toward single measures that incorporate cognitive and functional tests, such as the CDR-SB, ADCOMS, and the integrated Alzheimer’s Disease Rating Scale (iADRS, Wessels et al., 2015). Overall, Aisen believes more work is needed to translate the FDA guidance into concrete outcome measures. “The framework is not that easy to operationalize,” he said.
Which Test for Progression? Average scores for normal people (blue dots) cluster together, while MCI scores (pink dots in middle) worsen over two years, and mild AD scores (pink dots on left) worsen from baseline to 12 and 24 months. These values define disease progression (red axis). Scores for individual tests (yellow dots) often diverge from this axis, with ADAS-Cog scores (yellow line from upper right to lower left) farthest from it, CDR-SB scores (yellow line from bottom right to upper left) closer, and ADCOMS scores (middle yellow line) fitting best. Vertical axis (green line) shows cognition-related scores on top, functional tests on bottom. [Courtesy of Suzanne Hendrix.]
The Hunt for Best Cognitive Tests Is On
So how to find that most sensitive, meaningful test? In Barcelona, Hendrix proposed one way. She visualized neuropsychological test scores in three-dimensional space to find those measures that correlate most closely with disease progression. Using the ADNI data set as an example, she graphed baseline, 12-, and 24-month data from cognitively normal controls, people with mild cognitive impairment, and with mild AD. Scores for controls clustered at one side of the space, while those for MCI ended up in the middle, and the mild AD group landed on the far side. For the controls, all three time points bunched up, but for the MCI group they separated, and for the mild AD group they were spaced far apart, in keeping with the rate of decline at each disease stage.
Hendrix showed a line drawn from the heart of the clustered baseline control scores through the MCI group to end in the center of the 24-month mild AD scores. This line defines the axis of disease progression from normal to mild AD, Hendrix said. With the axis established, researchers can look at lines drawn through specific patient populations longitudinally and see how well performance on each individual test over time parallels the axis of disease progression—in other words, how well that test reflects an actual worsening of disease.
A complication arises because the axis of disease progression is not perfectly straight. Scores on some tests change more during the MCI stage and others during the AD stage, resulting in different slopes or directions for progression at each stage. This means that most tests work best at a specific stage (Nov 2018 conference news).
For example, in the ADNI data, the ADAS-Cog aligns with the axis of progression at the mild AD stage, but not with progression in MCI. The CDR-SB matches up a little better with the slope of progression in MCI, but still not well. To find a better measure for MCI, Hendrix collaborated with researchers at Eisai to develop ADCOMS. They fit a partial least squares (PLS) model using neuropsychological data from four different cohorts, then looked for those individual test items that correlated best with disease progression in people with MCI over one year. The graphical representation in three-dimensional space corresponds to the mathematical model using a PLS regression, and makes it easier to understand the math. The model selected four items from the ADAS-Cog, two from the MMSE, and all six from the CDR-SB, weighted them according to their relative contributions to disease progression, and combined them into ADCOMS (Wang et al., 2016). The resulting composite includes both cognitive and functional measures. In Barcelona, Hendrix showed that ADCOMS performed in the ADNI data set as it was designed to do, lining up well with the axis of disease progression in MCI.
However, ADCOMS is not a panacea either. It performs poorly in preclinical disease, because this relatively healthy population is outside the dynamic range of its functional measures. For preclinical disease, Hendrix teamed up with colleagues at the Alzheimer’s Prevention Initiative to use the same PLS model to develop the API Preclinical Cognitive Composite (APCC). This battery was optimized to detect cognitive change that occurs over the decade before people got their MCI diagnosis; it includes sensitive cognitive measures but no functional ones (Ayutyanont et al., 2014).
The researchers adjusted APCC scoring to account for the effects of normal aging-related cognitive decline. At the MCI stage, aging-related decline has little relative effect, but at preclinical stages, it can be a significant confounder, Hendrix told Alzforum. An earlier version of the APCC, called the API-LOAD, did not include this correction for normal aging. She noted that this aging correction, and the longitudinal derivation, are the main differences between the APCC and the Preclinical Alzheimer Cognitive Composite (PACC) developed by A4 researchers (Jun 2014 news). The PACC includes a broader range of cognitive performance and does not correct for age-related cognitive decline.
So which test is best for preclinical disease? Hendrix said it depends what researchers want to measure. Some treatments, such as cognitive enhancers, might improve progressive and non-progressive aspects of cognition. In that case, the PACC might be good for evaluating efficacy. Interventions such as nutritional or lifestyle interventions might be expected to slow age-related decline as well as Alzheimer’s pathology, and the API-LOAD might be the best test to evaluate efficacy (Langbaum et al., 2014). For a therapy targeting a specific AD pathology, such as amyloid or tau, the APCC is likely to paint a clearer picture of its effects. “It comes down to figuring out what kind of change you have in an untreated population, and what aspects of that change you’re targeting with your treatment,” Hendrix told Alzforum.
How Might This Work?
In Barcelona, Hendrix showed an example of how this method could help a clinical trial. Danone Nutricia Research recently reported that the LipiDiDiet trial of its nutriceutical drink Souvenaid missed its primary endpoint but met two secondary ones, hinting at some activity (Mar 2016 conference news; Nov 2017 news). Nutricia contracted with Hendrix to perform a post hoc analysis assessing Souvenaid’s performance on the ADCOMS.
Souvenaid showed statistical significance on the ADCOMS. Graphing the data in three-dimensional space, Hendrix found that LipiDiDiet’s primary outcome measure, the neuropsychological test battery (NTB), bore almost no relationship to disease progression. The axis of NTB scores angled 75 degrees away from the axis of disease progression. The NTB measures several aspects of cognition, such as processing speed and attention, that vary greatly from day to day based on a person’s physical and mental state, i.e., how rested or focused he or she is. These factors are not expected to change in a consistent way in this stage of disease, Hendrix noted. Some of the NTB’s executive function tasks may measure inherent abilities that, again, change little with disease progression in this stage.
By contrast, ADCOMS scores lined up more closely with disease progression in the LipiDiDiet data set, as they had in the ADNI dataset. When Hendrix reanalyzed LipiDiDiet data using ADCOMS, she saw a 36 percent slowing of cognitive decline, with a p value of 0.023. Cohen’s d was 0.31, considered a relatively small effect size. Breaking ADCOMS down by its component tests, Hendrix found that the CDR-SB items provided most of the discrimination in this study. Had the LipiDiDiet trial used ADCOMS, or another measure optimized for disease progression in MCI, it would have posted a positive result, Hendrix concluded.
Hendrix claims that other trials, too, would have performed better with a more tailored outcome measure. She performed post hoc analysis on the public results from the EXPEDITION3 solanezumab trial, in which solanezumab slowed decline on the ADAS-Cog, but not by enough to achieve statistical significance (Jan 2018 news). A global test statistic that is similar to ADCOMS does show statistical significance in this data set, although solanezumab’s benefit remains small, Hendrix said.
Hendrix ran a similar analysis for seven other recent trials (see table above). Each trial that had a significant effect or a trend on the ADAS-Cog, MMSE, or CDR-SB turned out to be clearly significant on ADCOMS, while trials without a significant benefit were more clearly negative on ADCOMS. Using the right measure gives more accurate results for both positive and negative trials, Hendrix argues. “Some studies we think have failed may not have failed, and studies we think succeeded may not have succeeded. We’re in muddy waters a lot of the time,” she told Alzforum.
The key is to use longitudinal data to evaluate neuropsychological tests and find those that actually reflect disease progression, Hendrix stressed. Measures should combine a large change over time with a small standard deviation, giving them the best chance to detect a slowing of cognitive decline. In studies where different outcome measures conflict, a three-dimensional analysis can show which measure more closely reflects disease progression and deserves more weight, she added.
This strategy may help identify tests that satisfy the FDA, Hendrix believes. “We need to find the most sensitive measures of progression in a given population, and use those as outcomes,” she said. She believes measures that closely reflect disease progression are more likely to be clinically meaningful. “We need to make sure our outcome measures reflect the symptoms that progress,” Hendrix said.—Madolyn Bowman Rogers





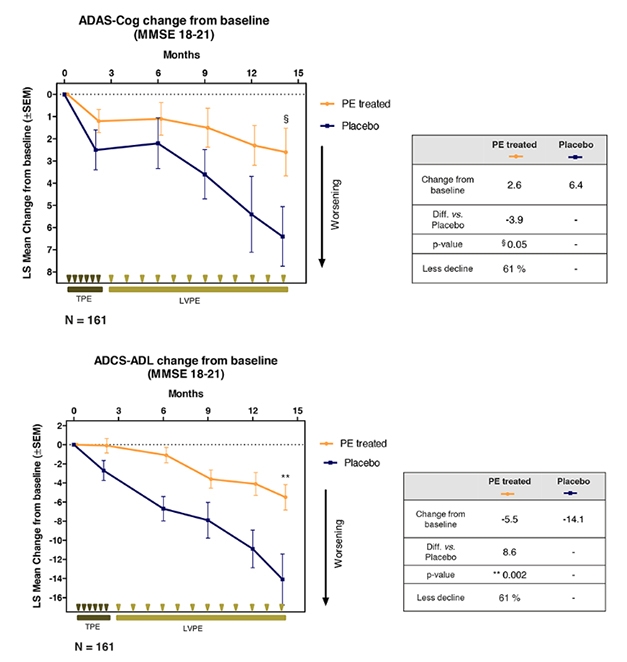


Comments
No Available Comments
Make a Comment
To make a comment you must login or register.