Past Webinar
Intraneuronal Aβ: Was It APP All Along?
Quick Links
Introduction
On 16 June 2011, ARF held a Webinar with Eddie Lee, Todd Golde, Gunnar Gouras, Frank LaFerla, Virginia Lee, Lars Nilsson, and Robert Vassar to discuss this study’s implications for intraneuronal Aβ and its role in AD pathogenesis. The Guest Moderator was David Holtzman.
Paper Under Discussion. [Editor's Update: This Paper Has Been Retracted.]
Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, Not Free Abeta Peptides in 3xTg-AD Mice: Implications for Tau versus A{beta}-Mediated Alzheimer Neurodegeneration. J Neurosci. 2011 May 25;31(21):7691-9. Abstract
- Listen to the Webinar
View Comments By:
- Frank LaFerla, Salvatore Oddo — Posted 2 June 2011
- Charles Glabe — Posted 3 June 2011
- Malu G. Tansey — Posted 3 June 2011
- Christian Pike — Posted 6 June 2011
- Thomas Bayer and Oliver Wirths — Posted 7 June 2011
- Nikolaos K. Robakis — Posted 7 June 2011
- Lars Nilsson — Posted 8 June 2011
- Vincent Marchesi, ARF Advisor — Posted 9 June 2011
- Sanjay W. Pimplikar — Posted 10 June 2011
- Estibaliz Capetillo-Zarate, Gunnar K. Gouras, Michael Lin, Davide Tampellini — Posted 13 June 2011
- Dennis Selkoe — Posted 13 June 2011
- Zoia Muresan, Virgil Muresan — Posted 13 June 2011
- A. Claudio Cuello — Posted 14 June 2011
- Gerd Multhaup — Posted 14 June 2011
- P. Hemachandra Reddy — Posted 16 June 2011
- Wataru Araki — Posted 20 June 2011
- Heng Du, Frank Gunn-Moore, Shirley Yan — Posted 20 June 2011
- Jia Yao — Posted 20 June 2011
- Carlos A. Saura — Posted 20 June 2011
Background
Background Text
By Gabrielle Strobel
One of the bigger debates in Alzheimer’s disease research this past decade has been around the hypothesis that Aβ starts accumulating and wreaking havoc inside the neuron before it builds up in the extracellular spaces where amyloid plaques have traditionally been seen. The discovery in 1992 that cells secrete Aβ has stood the test of time and drawn the field’s main focus firmly to the outside of neurons (Seubert et al., 1992; Haass et al., 1992). All the same, a parallel line of inquiry challenging the primacy of this focus has been building steam. It asked whether at least some of the trouble with Aβ originates deep inside nerve cells, with pathogenesis somehow being initiated from within vesicular accumulations of Aβ aggregates.
All along, some scientists had quietly voiced concern that some of the antibody stainings of intraneuronal Aβ might possibly be artifactual in that they represent not true Aβ peptide cleaved out of its parent protein APP, but rather the Aβ sequence within intact APP. A similar technical caveat about exactly what the antibody recognizes has been dogging research on reportedly damaging fragments of other pathogenic proteins. Now, a paper in the May 25 Journal of Neuroscience appears to confirm those concerns for intraneuronal Aβ. First author Matthew Winton and colleagues, led by corresponding author Virginia Lee at the University of Pennsylvania School of Medicine in Philadelphia, publish a two-pronged dataset that shows “unequivocally,” the authors write, that the immunoreactive intraneuronal material is not free Aβ but APP (Winton et al., 2011).
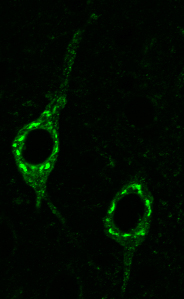
Confocal microscope image of neurons in the 3xTg-AD mice stained for the amyloid-β precursor protein (APP) showing intraneuronal APP (green). Image credit: Edward Lee, Upenn
The UPenn scientists took the 3xTg-AD, aka triple-transgenic, mice made by Frank LaFerla’s group at the University of California, Irvine (Oddo et al., 2003). Combining expression of mutant tau, APP, and presenilin genes, this transgenic strain has drawn wide attention for being a more complete model of AD than most previous mouse strains. It develops both signature pathologies, that is, extracellular amyloid plaques and neurofibrillary inclusions—plus, reportedly, intraneuronal Aβ early on. As per ISI Web of Science, the original paper has been cited 788 times in the scientific literature by May 2011. Triggered in part by the appeal of this model and partly by the mice’s response to an experimental M1 agonist also reported in the original paper, dozens of research groups across the country subsequently used these mice for mechanistic and drug studies. Alzforum featured the paper and some other studies on intraneuronal Aβ, after having previously hosted discussions on the topic (see ARF Live Discussion).
The UPenn group conducted essentially two studies. First, they distinguished staining of free Aβ from staining of Aβ-within-APP using a panel of 12 characterized APP/Aβ antibodies that span the length of APP. Of the dozen, two each bind APPs N-terminal ectodomain or its C-terminus, one binds APP’s BACE cleavage product C99, three bind Aβ epitopes within APP. Two each specifically recognize the ends of Aβ40 or Aβ42, respectively, that exist only on the cleaved peptide. Second, the scientists bred the 3xTg-AD strain with a BACE knockout strain to create a cross that overproduces mutant human APP, presenilin, and tau as before, but cannot process the APP into Aβ.
Immunohistochemistry and biochemistry suggested that there is no free, cleaved intraneuronal Aβ in the triple-transgenic mice. What is there is the Aβ sequence embedded in the APP holoprotein, the authors report. The antibodies that are specific to cleaved Aβ saw nothing inside neurons, but did stain extracellular plaques in these mice, in other transgenic strains, and in human brain. The Aβ-within-APP antibodies did stain inside neurons in the triple-transgenics. The 3xTg/BACE knockouts added genetic evidence. They showed no free Aβ staining or extracellular amyloid plaques, as expected, but they did show similar intraneuronal APP and Aβ-within-APP staining as age-matched 3xTg-AD mice.
What about the intraneuronal accumulation, however? Why would APP do that in the 3xTg-AD strain? By comparison, APP does not accumulate in the Tg2576 mice. Expression or phosphorylation aren’t to blame, the UPenn group reports. The triple-transgenics actually express less APP than do the Tg2576 mice, and APP in both strains becomes similarly phosphorylated as it wends its way through the secretory system. The 3xTg APP does, however, accumulate in vesicles in the area around the nucleus. The authors speculate that this could result from a vesicle trafficking defect brought on by the presenilin-1 and tau mutations the triple-transgenic mice also express. After all, they argue, both presenilin-1 and tau mutations impair vesicle transport and anterograde axonal transport of a variety of cargoes. Hence, APP-bearing vesicles in the 3xTg-AD mice might encounter a holdup at the entrance to the axonal compartment and pile up upstream of it.
And what about tau? One outcome of the 3xTg-AD paper was that intraneuronal Aβ precedes plaques, which in turn precedes tau pathology (Oddo et al., 2003). Is this relationship in timing between intraneuronal Aβ and tau now in question, or indeed also the relationship between amyloid plaques and tau? The latter relationship has since become widely accepted throughout the field as a result not only of this, but also other studies in different model systems. In the 3xTg-AD model, both seem out the window, the authors report. Tau pathology develops similarly in the 3xTg-AD and the 3xTg-AD/BACE knockout mice, meaning that, at least in this model, tau pathology arises independently of Aβ anywhere, inside or outside the neuron.
How far does this study reach? Is this a problem with this mouse model, or with the entire line of investigation into intraneuronal Aβ in the pathogenesis of Alzheimer’s disease? Numerous studies besides LaFerla’s have reported intraneuronal Aβ immunoreactivity. They include studies of normal, AD, or Down’s syndrome brain. They include other mouse models of mutant human APP transgenes, for example, of the Arctic or Austrian mutations, or a 5xTg-AD model. Did those studies truly detect cleaved Aβ peptide? Cell culture experiments modeling intraneuronal Aβ have reported numerous effects including synaptic dysfunction and cell death. How relevant are these to human AD? An Alzforum Papers of the Week search of “intraneuronal amyloid” pulls up 245 citations. To browse a selection, see citations below. Of this body of evidence, what is valid? What needs revisiting? Or is there another explanation altogether for the discrepancies between the UC Irvine and the UPenn group’s findings?
What aspects of intraneuronal Aβ might best be re-evaluated, and which ones stand. What questions are raised in your mind?
Citations on Intraneuronal Aβ:
Achim CL, Adame A, Dumaop W, Everall IP, Masliah E, Neurobehavioral Research Center. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol. 2009 Jun;4(2):190-9. Abstract
Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006 Apr 19;26(16):4277-88. Abstract
Bayer TA, Schäfer S, Breyhan H, Wirths O, Treiber C, Multhaup G. A vicious circle: role of oxidative stress, intraneuronal Abeta and Cu in Alzheimer's disease. Clin Neuropathol. 2006 Jul-Aug;25(4):163-71. Abstract
Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005 Mar 3;45(5):675-88. Abstract
Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004 Oct;165(4):1289-300. Abstract
Christensen DZ, Kraus SL, Flohr A, Cotel MC, Wirths O, Bayer TA. Transient intraneuronal A beta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008 Dec;116(6):647-55. Abstract
Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005;132(1):123-35. Abstract
Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006 Oct 11;26(41):10536-41. Abstract
Dafnis I, Stratikos E, Tzinia A, Tsilibary EC, Zannis VI, Chroni A. An apolipoprotein E4 fragment can promote intracellular accumulation of amyloid peptide beta 42. J Neurochem. 2010 Apr 20. Abstract
Echeverria V, Ducatenzeiler A, Dowd E, Jänne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience. 2004;129(3):583-92. Abstract
España J, Giménez-Llort L, Valero J, Miñano A, Rábano A, Rodriguez-Alvarez J, Laferla FM, Saura CA. Intraneuronal beta-Amyloid Accumulation in the Amygdala Enhances Fear and Anxiety in Alzheimer's Disease Transgenic Mice. Biol Psychiatry. 2009 Aug 6. Abstract
Fujimura RK, Reiner T, Ma F, Phillips V, de Las Pozas A, Dickson DW, Roos BA, Howard GA, Perez-Stable C. Changes in the Expression of Genes Associated with Intraneuronal Amyloid-beta and Tau in Alzheimer's Disease. J Alzheimers Dis. 2009 Sep 11. Abstract
Fujimura RK, Reiner T, Ma F, Phillips V, de las Pozas A, Dickson DW, Roos BA, Howard GA, Perez-Stable C. Changes in the expression of genes associated with intraneuronal amyloid-beta and tau in Alzheimer's disease. J Alzheimers Dis. 2010 Jan;19(1):97-109. Abstract
Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, Xu H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001 Apr 15;21(8):2561-70. Abstract
Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, Fernández-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: A role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31(1):125-47. Abstract
Golde TE, Janus C. Homing in on intracellular Abeta? Neuron. 2005 Mar 3;45(5):639-42. Abstract
Gómez-Ramos P, Asunción Morán M. Ultrastructural localization of intraneuronal Abeta-peptide in Alzheimer disease brains. J Alzheimers Dis. 2007 Mar;11(1):53-9. Abstract
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000 Jan;156(1):15-20. Abstract
Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005 Oct;26(9):1235-44. Abstract
Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001 Apr;125(4):489-92. Abstract
Hasegawa T, Ukai W, Jo DG, Xu X, Mattson MP, Nakagawa M, Araki W, Saito T, Yamada T. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42: implications for the pathogenesis of Alzheimer's disease. J Neurosci Res. 2005 Jun 15;80(6):869-76. Abstract
Hashimoto M, Bogdanovic N, Volkmann I, Aoki M, Winblad B, Tjernberg LO. Analysis of microdissected human neurons by a sensitive ELISA reveals a correlation between elevated intracellular concentrations of Abeta42 and Alzheimer's disease neuropathology. Acta Neuropathol. 2010 May;119(5):543-54. Abstract
Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, Motomura K, Soejima N, Yamasaki R, Hashimoto T. Apomorphine Treatment for Alzheimer’s Mice Promoting Amyloid-β Degradation. Annals of Neurology. 2010 Dec 1Abstract
Iijima-Ando K, Hearn SA, Granger L, Shenton C, Gatt A, Chiang HC, Hakker I, Zhong Y, Iijima K. Overexpression of neprilysin reduces alzheimer amyloid-beta42 (Abeta42)-induced neuron loss and intraneuronal Abeta42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and premature death in Drosophila. J Biol Chem. 2008 Jul 4;283(27):19066-76. Abstract
Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN. Intracellular amyloid-beta 1-42, but not extracellular soluble amyloid-beta peptides, induces neuronal apoptosis. J Biol Chem. 2002 May 3;277(18):15666-70. Abstract
Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, Dewilde A, Vercauteren F, Atifeh R, Ducatenzeiler A, Klein W, Szyf M, Alhonen L, Cuello AC. A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis. 2010 Apr;20(1):113-26. Abstract
Levi O, Dolev I, Belinson H, Michaelson DM. Intraneuronal amyloid-beta plays a role in mediating the synergistic pathological effects of apoE4 and environmental stimulation. J Neurochem. 2007 Nov;103(3):1031-40. Abstract
Lin B, Ginsberg MD, Busto R. Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of beta-amyloid precursor protein. Brain Res. 2001 Jan 5;888(1):107-116. Abstract
Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006 Jan;27(1):67-77. Abstract
Magrané J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci. 2005 Nov 23;25(47):10960-9. Abstract
Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011 Apr 30. Abstract
Moreira PI, Liu Q, Honda K, Smith MA, Santos MS, Oliveira CR. Is intraneuronal amyloid beta-peptide accumulation the trigger of Alzheimer's disease pathophysiology? J Alzheimers Dis. 2004 Aug;6(4):433-4; discussion 443-9. Abstract
Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002 Jan;110(2):199-211. Abstract
Nagele RG, Clifford PM, Siu G, Levin EC, Acharya NK, Han M, Kosciuk MC, Venkataraman V, Zavareh S, Zarrabi S, Kinsler K, Patel N, Nagele EP, Dash J, Wang HY, Levitas A. Brain-Reactive Autoantibodies Prevalent in Human Sera Increase Intraneuronal Amyloid-ß1-42 Deposition. J Alzheimers Dis. 2011 Apr 11. Abstract
Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, Yamaguchi H, Shimohama S, Lee HG, Zhu X, Smith MA, Perry G. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis. 2010 Mar;37(3):731-7. Abstract
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006 Oct 4;26(40):10129-40. Abstract
Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006 Jan;168(1):184-94. Abstract
Ohyagi Y, Tsuruta Y, Motomura K, Miyoshi K, Kikuchi H, Iwaki T, Taniwaki T, Kira JI. Intraneuronal amyloid beta42 enhanced by heating but counteracted by formic acid. J Neurosci Methods. 2006 Jul 20. Abstract
Ohyagi Y. Intracellular amyloid beta-protein as a therapeutic target for treating Alzheimer's disease. Curr Alzheimer Res. 2008 Dec;5(6):555-61. Abstract
Philipson O, Lannfelt L, Nilsson LNG. Genetic and pharmacological evidence of intraneuronal Ab accumulation in APP transgenic mice. FEBS Letters. 2009;583:3021-3026. Abstract
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008 Jun;118(6):2190-9. Abstract
Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem. 2006 Dec 29;281(52):39907-14. Abstract
Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, Ladu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009 Apr 7;106(14):5907-12. Abstract
Rebeck GW, Hoe HS, Moussa CE. Beta-amyloid1-42 gene transfer model exhibits intraneuronal amyloid, gliosis, tau phosphorylation, and neuronal loss. J Biol Chem. 2010 Mar 5;285(10):7440-6. Abstract
Sahlin C, Lord A, Magnusson K, Englund H, Almeida CG, Greengard P, Nyberg F, Gouras GK, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation favors intracellular amyloid-beta production by making amyloid precursor protein less available to alpha-secretase. J Neurochem. 2007 May;101(3):854-62. Abstract
Shie FS, LeBoeur RC, Jin LW. Early intraneuronal Abeta deposition in the hippocampus of APP transgenic mice. Neuroreport. 2003 Jan 20;14(1):123-9. Abstract
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. Abstract
Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci U S A. 2009 Nov 24;106(47):20021-6. Abstract
Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009 Aug 5;29(31):9704-13. Abstract
Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010 Apr 7;30(14):4845-56. Abstract
Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid ß oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011 Jul;89(7):1031-42. Abstract
Van Broeck B, Vanhoutte G, Pirici D, Van Dam D, Wils H, Cuijt I, Vennekens K, Zabielski M, Michalik A, Theuns J, De Deyn PP, Van der Linden A, Van Broeckhoven C, Kumar-Singh S. Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer's disease. Neurobiol Aging. 2006 Nov 15. Abstract
Wegiel J, Kuchna I, Nowicki K, Frackowiak J, Mazur-Kolecka B, Imaki H, Wegiel J, Mehta PD, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Pirttilla T, Frey H, Lehtimäki T, Kivimäki T, Visser FE, Kamphorst W, Potempska A, Bolton D, Currie JR, Miller DL. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol (Berl). 2007 Apr;113(4):389-402. Abstract
Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001 Jun 22;306(1-2):116-20. Abstract
Wirths O, Multhaup G, Bayer TA. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade. J Neurochem. 2004 Nov;91(3):513-20. Abstract
Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006 Nov 24;281(47):36180-6. Abstract
Comment by: Frank LaFerla, ARF Advisor, Salvatore Oddo
Posted 2 June 2011
Winton et al. use histochemistry and a variety of antibodies to conclude that the 3xTg-AD mice lack intracellular Aβ. This study also uses a genetic approach to conclude that Aβ does not modulate tau pathology in 3xTg-AD mice. We find that the data presented do not justify the conclusions. In the following discussion, we point out that the authors’ own data demonstrate the presence of intraneuronal Aβ in the 3xTg-AD mice. We and many others have reported multiple studies over the past decade that argue against these conclusions; however, Winton et al. failed to discuss these studies.
We believe the clear discrepancies between Winton et al. and previous publications likely result from the relative affinities of the various neoepitope antibodies used. To try to detect intracellular Aβ, Winton et al, rely heavily on two neoepitope antibodies, BA27 and BC05. These antibodies are most commonly used for biochemical Aβ ELISA measurements, but it is unclear how effective they are for immunofluorescence analysis, as only low-powered images are provided in the paper. It is well established that antibody sensitivity and specificity can vary in biochemical versus histological uses. Thus, the lack of staining with these two antibodies proves little, especially when no positive control is shown in the fluorescent images. In contrast to these data, we and other groups have used different Aβ42-specific antibodies that clearly demonstrate intraneuronal Aβ in the 3xTg-AD mice (Oddo et al., 2006, Fig. 3), as well as the presence of intracellular oligomers and ADDLs, using multiple antibodies. Importantly, numerous laboratories have independently replicated our findings (to cite a few recent ones, see Khandelwal et al., 2011; Sudol et al., 2009; España et al., 2010). Most recently, one study demonstrated intracellular Aβ by Western blot in 3xTg-AD prior to plaque formation (Himeno et al., 2011, Fig. 3).
Notably, we detect most intraneuronal Aβ within hippocampal CA1 neurons; Winton et al. focus mainly on cortical sections for Aβ, and hippocampal regions for tau analysis. Perhaps the most confusing piece of data presented by Winton et al. is depicted in Figure 5G-I showing 5685 and 4G8 immunofluorescence of 3xTg mice plus a merged image. Only low magnification images are presented, which seem inadequate for making a conclusion about intraneuronal staining. However, after zooming in on these images, one can clearly see cells and portions of cells in Fig. 5I that label with 4G8 only. The double-label presented depicts C-terminal APP labeling in red and Aβ/APP labeling in green. If all of the immunolabeling were the result of APP, one would expect 100 percent colocalization (all yellow). Instead, significant portions of the labeling appear green, indicating Aβ (Fig. 5I). This staining also appears reduced in the 3xTg-AD/BACE-/- mice, as one would expect (Fig. 5J-L); however, no quantification is provided. Surprisingly, the red APP immunolabeling appears decreased in the BACE knockout mice (J vs. G), despite biochemical data suggesting APP is increased in BACE knockouts in line with other groups’ findings.
We have never asserted that there is no neuronal APP expression in a mouse that was specifically designed to overexpress APP in neurons. Rather, we previously showed, and assert here again, that a significant portion, although not all, of the immunolabeling in 3xTg-AD mice represents Aβ. We used similar co-labeling techniques in the past to demonstrate intraneuronal staining in the 3xTg-AD mice (McAlpine et al., 2009).
The lack of immunoreactivity seen in the study by Winton et al. may also be due to the methodology employed, including fixation and pretreatments (we use paraformaldehyde fixation with formic acid pretreatments). Although formic acid and 4 percent PFA fixation are briefly mentioned, the methods section implies that the majority of the mice were fixed with 10 percent formalin, a far stronger fixation approach that is known to reduce antigen sensitivity. It is unclear which, if any, images are derived from formic acid-treated sections.
In summary, it is correct that full-length APP can be detected in the 3xTg-AD mice. However, the data provided in this study does not convince us that no intraneuronal Aβ is present in 3xTg-AD mice.
The second conclusion made by Winton et al. is that tau pathology develops independently of the presence of Aβ. This is based on Fig. 7 displaying that the 3xTg-AD/BACE-/- mice show similar somatodendritic accumulation of tau to 3xTg-AD mice. To our eyes, this figure actually seems to show enhanced AT8 immunoreactivity in 3xTg-AD/BACE-/- mice compared to the 3xTg-AD mice, although no statement is made to this effect. Unfortunately, the paper offers no quantification or time course to fully investigate tau pathology in these mice lacking Aβ. We are puzzled that a lab that established methods for the biochemical analysis of tau provides no biochemical support for this conclusion. As homogenates were prepared for these mice for ELISA measurements in Fig. 3, it would surely have been critical to perform biochemical confirmation of these immunohistochemical images. We asked for samples from the 3xTg-AD/BACE-/-, and were disappointed to find that the mice no longer exist and the lead author has left the lab (J. Trojanowski and V. Lee, personal communication).
We, and many others, have published numerous studies highlighting the influence of Aβ on tau pathology, including in the 3xTg-AD mice. For example, we showed that immunotherapy against Aβ removes somatodendritic human tau (Oddo et al., 2004), a finding that has been replicated in other mouse models (Wilcock et al., 2009) and human Aβ immunotherapy trials (Boche et al., 2010).
We also performed multiple genetic manipulations of the 3xTg-AD mice to alter Aβ production, in the same fashion as the generation of these 3xTg-AD/BACE-/- mice. We have removed the presenilin mutation, and shown that this reduces Aβ42 and leads to a reduction in tau phosphorylation and accumulation (Oddo et al., 2008). Consistent with this, we showed that modulating the ApoE allele in the 3xTg-AD mice alters Aβ accumulation and also provided quantitative biochemical evidence that it alters tau phosphorylation and accumulation (Oddo et al., 2009). Thus, none of these studies support the conclusion that tau pathology develops independently of Aβ in the 3xTg-AD mice.
Perhaps the difference in findings is due to the analysis performed. Detailed biochemical analysis of tau pathology in the 3xTg-AD/BACE-/- as a function of age compared to the 3xTg-AD mice would help to clarify the issue. It is also possible that differences in strain background between 3xTg-AD and the 3xTg-AD/BACE-/- mice were not accounted for. We previously collaborated with one of the authors on this manuscript and detailed the importance of background strain on AD-related pathologies, as have other groups. Notably, we are preparing a manuscript at the moment in which we have produced a novel transgenic mouse that overexpresses both APP and human wild-type tau transgenes. We crossed this to the same BACE knockout mouse used by Winton et al., and detect clear biochemical reductions in hyperphosphorylated tau in even just heterozygous BACE knockout progeny. These findings again support a role for Aβ in facilitating tau pathology, as do many other independent studies.
In summary, we disagree with the conclusions presented in this study.
Comment by: Charles Glabe, ARF Advisor
Posted 3 June 2011
Virginia Lee and coworkers have convincingly demonstrated that the perinuclear, intracellular Aβ immunoreactivity observed in a subset of neurons in the 3xTg-AD mouse is predominantly, if not exclusively, APP and not Aβ. Moreover, they demonstrate that this intracellular immunoreactivity does not require BACE activity, indicating that it does not come from the same pathway that produces secreted Aβ.
Does this mean that intracellular accumulation of APP is insignificant and we can disregard it? As the authors state, these findings “warrant further study as to the role of aberrant APP accumulation.” The accumulation of APP is abnormal because it is age related and it only takes place in a subset of neurons. Similar intraneuronal accumulation of Aβ and APP immunoreactivity is also observed in humans, where it appears to be associated with the earliest signs of AD pathology, while it declines with advancing AD pathology and is largely absent in non-demented individuals that lack AD pathology (1). This suggests that intracellular accumulation of APP may be one of the earliest events of AD pathology. In addition, even if the intracellular Aβ immunoreactivity is APP, it appears to be APP that is misfolded in the same conformation as aggregated Aβ because it reacts with conformation-dependent antibodies that are specific for Aβ aggregates, but not Aβ monomers or APP, such as M16 (2,3) or OC or NU1 (4).
A possible significance of the accumulation of intracellular APP is that it may be the result of seeding by the uptake of extracellular Aβ oligomers. We have previously reported that incubation of APP-expressing cells with Aβ oligomers results in the intracellular accumulation of APP and amyloidogenic fragments of APP in the detergent insoluble fraction of the cell (2). A fraction of this insoluble APP may be slowly converted to insoluble, intracellular Aβ containing “ragged” amino termini by non-specific degradation of the protease-sensitive parts of the insoluble APP (5). If the intracellular accumulation of APP is an early event in AD pathogenesis, it may provide a facile explanation for the recent failure of γ- secretase inhibitors in clinical trials. In those trials, cognitive decline was accelerated in the treated group, possibly because γ-secretase inhibition causes the accumulation of amyloidogenic APP fragments inside the cell. These results can also be explained by the fact that γ-secretase has many other substrates that would also be affected by γ-secretase inhibition; however, a pathogenic role for APP CTF accumulation is parsimonious. 1. Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010 May;119(5):523-41. Abstract
2. Yang AJ, Knauer M, Burdick DA, Glabe C. Intracellular A beta 1-42 aggregates stimulate the accumulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein in transfected cells. J Biol Chem. 1995 Jun 16;270(24):14786-92. Abstract
3. Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004 Apr 7;24(14):3592-9. Abstract
4. Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC. Intracellular Aß-oligomers and early inflammation in a model of Alzheimer's disease. Neurobiol Aging. 2011 Mar 15. Abstract
5. Yang AJ, Chandswangbhuvana D, Shu T, Henschen A, Glabe CG. Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42. J Biol Chem. 1999 Jul 16;274(29):20650-6. Abstract
Comment by: Malu G. Tansey
Posted 3 June 2011
As mentioned in the discussion, we and others had already noted that Aβ peptides were not a major component of the intraneuronal 6E10 immunoreactivity originally reported by the LaFerla lab in the paper by Kitazawa et al., where they showed this was accelerated by chronic systemic inflammation (Kitazawa et al., 2005). We used Dennis Selkoe's C9 anti-APP antibody to distinguish between Aβ and full-length APP/APP-derived fragments, and concluded that the species which accumulates intraneuronally in 3xTg-AD mice exposed to chronic systemic inflammogen (LPS) was likely to be β-CTF. Inhibition of TNF signaling appears to be driving the process (McAlpine et al., 2009). Our biochemical and immunohistological analyses did not reveal robust accumulation of Aβ peptides in 3xTg-AD mice under basal conditions or in response to chronic systemic inflammation. Still, those studies suggested that it may be feasible to selectively target soluble TNF therapeutically to prevent dysregulated APP turnover and/or transport, and perhaps β-CTF accumulation, induced by chronic neuroinflammation. The significance for the AD field is that plenty of evidence indicates that species other than intraneuronal Aβ can exert neurotoxicity, and their formation may be modulated by inflammatory processes.
Comment by: Christian Pike
Posted 6 June 2011
Like Frank LaFerla and Salvo Oddo, I have concerns with the findings and conclusions of Winton and colleagues. Their data clearly demonstrate that several APP and Aβ antibodies share similar patterns of intraneuronal immunoreactivity that persist even in a BACE-/- background that precludes Aβ formation. Although these findings provide compelling evidence that intraneuronal Aβ immunoreactivity in 3xTg-AD mice may represent APP species, there are also a few issues which argue against this conclusion.
In some of my lab’s prior work with 3xTg-AD mice (Rosario et al., 2006; Carroll et al., 2007), we compared the patterns of immunoreactivity for antibodies against Aβ and APP C-terminal fragments to exclude the possibility that the observed intraneuronal Aβ immunostaining largely reflected APP species. We found that Aβ and APP-CTF exhibit qualitatively distinct immunoreactivities that differ both in cellular localization and temporal expression. APP-CTF immunoreactivity in 3xTg-AD mice is localized along the periphery of the soma and is clearly extranuclear, such that large, unstained nuclear regions are visible within neurons. Further, as expected with an APP transgene, the magnitude of APP-CTF expression in 3xTg-AD mice does not exhibit significant change with increasing age and is much higher than that observed in non-Tg mice. In contrast to the APP-CTF pattern, we observed that intraneuronal Aβ immunoreactivity in 3xTg-AD mice has a characteristic punctate appearance that is often present throughout the soma. Further, intraneuronal Aβ immunoreactivity exhibits robust age-related changes, becoming apparent at approximately age three months and strongly increasing at least through age 12 months.
Examination of the figures in the paper by Winton et al. shows that their intraneuronal APP immunolabeling is very similar to our observations, with an even, extranuclear appearance that does not obviously increase over time (five to seven months vs. 10-12 months). Notably, their intraneuronal Aβ immunolabeling is unlike the pattern we observe. It is not punctate in appearance and does not increase with age. Rather, their Aβ immunostaining exhibits either an absence of intraneuronal labeling or intraneuronal labeling that matches the APP-CTF pattern. Thus, it appears that Winton et al. may not be observing the intraneuronal Aβ as reported by LaFerla, my lab, and several other groups. As suggested by LaFerla’s comments, one plausible explanation for the discordant findings of Winton et al. is that their Aβ antibodies and/or immunohistochemical protocol were not optimized for detecting intraneuronal Aβ.
Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006 Dec 20;26(51):13384-9. Abstract
Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007 Nov 28;27(48):13357-65. Abstract
Comment by: Thomas Bayer, Oliver Wirths
Posted 7 June 2011
Winton et al. demonstrated that the 3xTg mouse model suggests that APP accumulates in an age-dependent manner with no signs for intraneuronal Aβ up to the age of 12 months. In previous publications by the LaFerla group (1) and others, the 3xTg-AD mouse has been used as a model with early intraneuronal Aβ pathology. There has been always an issue whether epitopes of diverse Aβ antibodies also cross-react with its parent molecule APP. For example, Aho et al. demonstrated that often it is APP rather than Aβ that is detected intracellularly when using antibodies like 4G8, 6E10, and 82E1 clones that are unable to distinguish Aβ from APP (2). Unfortunately, these antibodies have been widely used in many studies, likely due to their high sensitivity in tissue sections.
This issue has been a major concern also for us, as we have been one of the solicitors for the “intraneuronal Aβ hypothesis” (3). Therefore, we have used different approaches during the last years to analyze whether intraneuronal Aβ is a risk factor for AD.
1. In the APP/PS1KI mouse model, there is early and robust accumulation of diverse intraneuronal Aβ peptides in CA1, frontal cortex, and some cholinergic nuclei. All these brain regions show age-dependent neuron loss between 25 and 50 percent, whereas regions with a comparable plaque load but without intraneuronal Aβ accumulation never develop neuron loss (4-5).
2. In the 5xFAD model, the same is true for cortical layer 5 (6).
3. The TBA42 model exclusively expresses N-truncated Aβ3-42 (without APP), and develops an age-dependent neurological phenotype and severe Purkinje cell loss following intracellular Aβ accumulation (7).
4. Recently, we identified low molecular Aβ pE3 oligomers that can be detected by 9D5, a novel mouse monoclonal antibody against a conformational neoepitope (8). Importantly, we identified 9D5 by screening human AD brain tissue for a specific staining pattern, i.e., strong intraneuronal and only minor plaque staining. The antibody 9D5 did not cross-react with Aβ1-42 monomers, dimers, or other higher molecular structured aggregates, indicating that these oligomers present a unique and novel neoepitope. The therapeutic potential of 9D5 was demonstrated in passively immunized 5xFAD mice as plaque load, and Aβ levels were reduced and behavioral deficits normalized.
We believe that these arguments support the “intraneuronal Aβ hypothesis.” Although most of the data have been obtained in transgenic mouse models, this hypothesis can be principally translated into human pathology, as the example of the 9D5 antibody has demonstrated. 1. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, Laferla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003 Jul 31;39(3):409-21. Abstract
2. Aho L, Pikkarainen M, Hiltunen M, Leinonen V, Alafuzoff I. Immunohistochemical visualization of amyloid-beta protein precursor and amyloid-beta in extra- and intracellular compartments in the human brain. J Alzheimers Dis. 2010;20(4):1015-28. Abstract
3. Wirths O, Multhaup G, Bayer TA. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade. J Neurochem. 2004 Nov;91(3):513-20. Abstract
4. Bayer TA, Wirths O. (2010) Frontiers in Aging Neuroscience 10, 2-8.
5. Wirths O, Bayer TA. Neuron loss in transgenic mouse models of Alzheimer's disease. Int J Alzheimers Dis. 2010;2010. Abstract .
6. Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2010 Jul 8. Abstract
7. Wirths O, Breyhan H, Cynis H, Schilling S, Demuth HU, Bayer TA. Intraneuronal pyroglutamate-Abeta3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009 Oct;118(4):487-96. Abstract
8. Wirths O, Erck C, Martens H, Harmeier A, Geumann C, Jawhar S, Kumar S, Multhaup G, Walter J, Ingelsson M, Degerman-Gunnarsson M, Kalimo H, Huitinga I, Lannfelt L, Bayer TA. Identification of low molecular weight pyroglutamate A{beta} oligomers in Alzheimer disease: a novel tool for therapy and diagnosis. J Biol Chem. 2010 Dec 31;285(53):41517-24. Abstract
Comment by: Nikolaos K. Robakis
Posted 7 June 2011
The current debate catalyzed by the important paper of Winton et al. highlights a serious concern facing the AD field, namely, the relevance of transgenic animal models to the neurodegeneration of AD, the real cause of dementia. Most transgenic models currently used in AD research overexpress high levels of exogenous APP (many times over endogenous APP), an artificial condition not applicable to AD where there is no APP overexpression. Moreover, protein overexpression in brain often causes toxicities due to diverse reasons, including possible interference of the overexpressed protein with normal cellular trafficking, which may not be relevant to disease mechanisms (see also Robakis, 2011; Pimplikar et al., 2010).
The APP-overexpressing transgenic animals may serve as models of amyloidosis. However, it should be kept in mind that although amyloidosis in these animals is driven by overproduction of Aβ peptides (due to overexpression of APP), there is little evidence of Aβ overproduction in sporadic AD, and the mechanism of Aβ fibrillation in human brain (both diseased and normal) remains unknown.
Finally, research in the last quarter of a century has shown that, although both APP and presenilins play critical roles in the etiology of AD, the role of Aβ and AD amyloid, beyond its usefulness for the definition of the disease, remains unclear.
Comment by: Lars Nilsson
Posted 8 June 2011
The study by Winton et al. is a well-designed study that provides important new information on a frequently used animal model of Alzheimer’s disease. However, based on the results presented, I would be cautious to conclude that intraneuronal Aβ accumulation does not occur in 3xTg-AD. We have never had the opportunity to examine the neuropathology of 3xTg-AD, but we do have considerable experience with Tg-ArcSwe and Tg-Swe mice.
Winton et al. used paraffin-embedded material (e.g., Fig. 1), and in our hands, it is difficult to detect intraneuronal Aβ in such tissues. Far greater sensitivity is reached by instead using frozen tissue sections, paraformaldehyde-fixed (4 percent) or unfixed, and immersing them in heated citric acid and formic acid for antigen retrieval. With N-terminal cleavage-specific antibodies, it is quite easy to detect intraneuronal Aβ, but with C-terminal cleavage-specific antibodies, the protocols need to be well optimized (especially with tissues from young mice). Moreover, age and anatomic location are important parameters to consider. We have noticed that intraneuronal Aβ first emerges in neurons with very high APP overexpression and is easiest to detect at the time when extracellular plaques begin to form. Winton et al. also used paraformaldehyde-fixed vibratome sections and 82E1 (an N-terminal neoepitope-specific Aβ-antibody), but, unfortunately, they do not show the results.
Similar to Winton et al., we conducted genetic and pharmacological experiments. We generated double-transgenic Tg-ArcSwe x BACE-knockout mice, administered a γ-secretase inhibitor, and combined that with immunohistochemical detection techniques. Our data strongly suggest that free intraneuronal Aβ peptides do accumulate in Tg-ArcSwe mice (Philipson et al., 2009). The same age-dependent intraneuronal Aβ-phenotype develops in both Tg-ArcSwe and Tg-Swe (Tg-Swe is devoid of the Arctic mutation: Lord et al., 2006). Possibly, intraneuronal Aβ accumulation occurs in some APP transgenic models and not at all in other models, but it is more likely that the levels and thereby ease with which intraneuronal Aβ can be detected differs between models.
Winton et al. highlight the fact that intraneuronal APP is far more abundant than intraneuronal Aβ in transgenic mouse brain. When studying intraneuronal Aβ, it is therefore essential to use neoepitope-specific Aβ antibodies, and to carefully validate and optimize the experimental protocols. Interestingly, 3xTg-AD mice, but not Tg2576, display strong intraneuronal APP-immunostaining, i.e., the phenotype does not simply depend on the extent of APP overexpression. Importantly, Winton et al. observe similar age-dependent tau phenotypes in 3xTg-AD mice with or without murine BACE (and thus human Aβ).
Lord A., Kalimo H., Eckman C., Zhang X-Q., Lannfelt L. and Nilsson LNG. (2006) The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging, 27: 67-77. Abstract
Philipson O., Lannfelt L. and Nilsson LNG. (2009) Genetic and pharmacological evidence of intraneuronal Abeta accumulation in APP transgenic mice. FEBS Lett., 583: 3021-6. Abstract
Comment by: Vincent Marchesi, ARF Advisor
Posted 9 June 2011
How can mice with so many genetic defects possibly reflect the pathophysiology of the earliest, asymptomatic, pre-amyloid accumulation stages of human AD? If this question cannot be answered positively and persuasively, it matters little what form of APP is being analyzed by the different antisera.
Comment by: Sanjay W. Pimplikar
Posted 10 June 2011
It is a healthy sign that the paper by Winton et al. has initiated a debate over intraneuronal Aβ, as more studies are appearing in the literature suggesting that intraneuronal Aβ could play a pathological role in Alzheimer’s disease (AD). This issue can be debated at the technical level in terms of antibody specificity and what a given antibody "sees" or "does not see" in brain sections. The debate can also commence at the biological level to see how the concept of intraneuronal Aβ fits with what we know about the subcellular locations where APP is processed by BACE and γ-secretase.
As pointed out by Thomas Bayer and others, both 6E10 and 4G8 antibodies recognize the Aβ portion in full-length APP and APP-C-terminal fragments. When these antibodies are used in Western blotting, it is easy to distinguish Aβ from other fragments because of its size. Obviously, this is not possible in immunohistochemical analysis. Therefore, scientists rely on double-staining using one of these antibodies in conjunction with a C-terminal antibody (which should see other forms of APP except Aβ) and subtracting one signal from the other (see McAlpine et al., 2009) to get an indirect measure of intracellular Aβ. However, this approach is less than satisfactory, prone to artifacts and subject to interpretation (e.g., see LaFerla’s comments on Fig. 5 of Winton et al.) and might be the root cause of discordance between the findings of Winton et al. and Oddo et al. However, Winton et al. used end-specific antibodies (Ban50 and BC05; Fig. 1), and combined this approach with genetic ablation of BACE (Fig. 6), thereby providing a compelling argument in support of their interpretation. One curious observation, however, is that Winton et al. found no immunoreactivity using BC05 in Fig. 6K. Similarly, Philipson et al. (see comments by Nilsson above) also find no signal using the 40- or 42-specific antibodies that recognize neoepitopes at the C-terminus (Fig. 3 of Philipson et al.). Shouldn’t γ cleavage of α-CTF expose the end of p3 peptide which is recognized by these antibodies? Or is α-CTF poorly or not processed by γ-secretase at all (1)? The lack of this signal is an enigma, since BACE-knockout animals should retain α- and γ-secretase activities and produce large amounts of p3 fragment.
It is also important to consider where in neurons β-CTF peptides are cleaved by γ-secretase, since this is precisely where Aβ peptides are "born." A generally accepted view is that shedding of the extracellular domain of APP is a prerequisite for γ-cleavage (2), and that BACE cleavage takes place in early endosomes (3,4), followed by γ-cleavage in a post-Golgi compartment (5). This makes it difficult to understand how so much intraneuronal Aβ signal is observed in the soma, which is far from the location of early endosomes. On the other hand, full-length APP is known to be accumulated in perinuclear space in the soma, increasing the likelihood that the conclusions of Winton et al. are correct. On a somewhat related note, there are published reports of the presence of Aβ in mitochondria (together with APP and γ-secretase (6,7), which is very difficult to reconcile with the current knowledge of biosynthetic pathways of membrane proteins.
Finally, the sentiments expressed by Nik Robakis (since amyloid-independent mechanisms can also contribute to AD; see, e.g., Alzforum Webinar) or Vince Marchesi above are important. Marchesi’s comments will make some of us cringe uncomfortably, but he raises a valid point. Moreover, if Aβ does accumulate intracellularly, is it present in sufficient amounts to be able to contribute to the disease mechanism? Is it present free in cytosol or bound in membrane vesicles? I believe the intraneuronal Aβ Thomas Bayer is alluding to is the pyroglutamate Aβ, which is different from the one debated here. The Alzforum Webinar organized around this theme (16 June 2011) will be an interesting event where some of these questions might get answered. I hope the panel members will also discuss the "forest" view of intraneuronal Aβ in addition to examining the "trees." Knowing the panel members, there is no doubt that it will be a lively event.
1. Tian Y. et al., (2010) Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J Biol. Chem. vol 285, no 42, 32549-56. Abstract
2. Struhl G and Adachi A. (2000) Requirements for presenilin-dependent cleavage of notch and other membrane proteins. Mol. Cell, vol 6, no. 3, 625-36. Abstract
3. Ehehalt et al., (2003) Amyloidogenic processing of the Alzheimer’s beta-amyloid precursor protein depends on lipid rafts. J Cell Biol., vol 160, no. 1, 113-23. Abstract
4. Rajendran et al., (2008) Efficient inhibition of Alzheimer’s disease beta secretase by membrane targeting. Science, vol 320, no 5875, 520-3. Abstract
5. Cupers et al., (2001) The discrepancy between presenilin subcellular localization and g-secretase processing of amyloid precursor protein. J Cell Biol. vol 154, no. 4, 731-740. Abstract
6. Pavlov P. et al., (2009) Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med, vol 13, no. 10, 4137-45. Abstract
7. Young K and Bennett J (2010) The mitochondrial secret(ase) of Alzheimer’s disease. J Alz. Disease vol. 20, suppl 2, S381-400. Abstract
Comment by: Estibaliz Capetillo-Zarate, Gunnar K. Gouras, Michael Lin, Davide Tampellini
Posted 13 June 2011
We agree with previous comments that Winton and colleagues provide interesting new evidence of APP accumulation and the possible lack of interaction between Aβ and tau in the triple-transgenic mouse. However, this paper is not enough to counter the hundreds of papers now on intraneuronal Aβ in Alzheimer’s disease, and the triple-transgenic mouse remains an important model of both β amyloid and tau pathogenesis. Dystrophic neurites in human AD show APP accumulation; the precise form of this APP isn’t fully clear. It is possible that the development of AD might be characterized by an increase of APP accumulation earlier than Aβ (such as occurs in head injury). Winton et al. also highlight the problem of using Aβ domain antibodies (such as 6E10, 4G8, etc.) to differentiate Aβ from APP.
Our main point in this comment is that intraneuronal Aβ is not easy to see, particularly when you compare it to more easily detectable APP. Aβ is also a changing target. As we well know, it likes to aggregate, but the Aβ42 end-specific antibodies are remarkably specific for the monomer form (Takahashi et al., 2004), so that oligomers become invisible unless you use an oligomer-specific antibody.
How do we know that Aβ42 antibodies are not just seeing APP? Immunofluorescence and immunoelectron microscopy clearly show that antibodies against Aβ42 have different subcellular labeling when compared to APP N- and C-terminal antibodies (Takahashi et al., 2002; Muresan et al., 2009; Gouras et al., 2010: Fig. 2, Aβ42 and APP in human primary neurons). In other words, high-resolution imaging clearly shows that APP and Aβ42 are in different places, which obviously does not fit the idea that they are one and the same. In addition, if Aβ C-terminal end-specific antibodies see APP, this would discount much of AD research on Aβ that has relied on these antibodies for ELISA.
Moreover, the paper by Winton et al. in a way argues against Aβ antibodies seeing APP, since they do not even detect intraneuronal Aβ even when there is a lot of APP. There is another technical point: They see intraneuronal APP only faintly by immunohistochemistry and not at all by immuno-EM in Tg2576 mice. Although APP is normally an abundant protein in neurons, it is overexpressed in these mice, and it has been seen.
Intraneuronal Aβ isn’t easy to see, but it is there. With a good confocal and antibodies, it is quite evident even in wild-type neurons. For recent rigorous technical papers on intraneuronal Aβ, we recommend (as Bayer and Wirths did): Aho et al. (2009) for postmortem human brain; Philipson et al. (2009) on AD transgenic mice; and an important biochemical paper on intraneuronal Aβ42 by Hashimoto et al. (2010).
Aho L, Pikkarainen M, Hiltunen M, Leinonen V, Alafuzoff I. Immunohistochemical visualization of amyloid-beta protein precursor and amyloid-beta in extra- and intracellular compartments in the human brain. J Alzheimers Dis. 2010;20(4):1015-28. Abstract
Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010 May;119(5):523-41. Abstract
Hashimoto M, Bogdanovic N, Volkmann I, Aoki M, Winblad B, Tjernberg LO. Analysis of microdissected human neurons by a sensitive ELISA reveals a correlation between elevated intracellular concentrations of Abeta42 and Alzheimer's disease neuropathology. Acta Neuropathol. 2010 May;119(5):543-54. Abstract
Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci. 2009 Mar 18;29(11):3565-78. Abstract
Philipson O, Lannfelt L, Nilsson LN. Genetic and pharmacological evidence of intraneuronal Abeta accumulation in APP transgenic mice. FEBS Lett. 2009 Sep 17;583(18):3021-6. Epub 2009 Aug 14. Abstract
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. Abstract
Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004 Apr 7;24(14):3592-9. Abstract
Comment by: Dennis Selkoe
Posted 13 June 2011
In my view, the paper by Winton et al. supports the key point that one can prove the existence of intraneuronal Aβ only by using end-specific Aβ antibodies (i.e., to Aβ Ile42 or Val40, and, to a lesser extent, Asp1) that cannot recognize any APP epitopes other than the free Aβ peptide. This would exclude numerous antibodies that are not truly end-specific and recognize epitopes in holoAPP, C99 and C83, and/or APPs-α.
Also, more studies using sensitive biochemical assays to extract free intracellular Aβ from cells and quantify it would be useful, if this has not yet been widely done by multiple labs. One would need to use methods to eliminate any Aβ peptides bound to the outside surface of cells before lysing the cells and quantifying free Aβ inside the cells (or specifically within their membrane vesicles).
Obviously, Aβ is normally generated by β- and γ-secretases within vesicles inside cells (in addition to on the plasma membrane), but these initially intracellular peptides are efficiently secreted under wild-type circumstances. The question is, How much of Aβ at steady state exists inside the cell versus in the extracellular space—in normal brain and then in AD brain. The Winton paper helps shed light on this important question in an APP-overexpressing mouse model.
Comment by: Zoia Muresan, Virgil Muresan
Posted 13 June 2011
We would like to comment on a few issues raised by this paper and previous comments, starting with the now old observations from various groups, Gunnar Gouras’s in particular (1), that in the AD brain Aβ accumulates in a population of neurons in the vulnerable brain regions. Often, some Aβ also accumulates intraneuronally in the non-AD brain. This should not be surprising.
Yet, these data are questioned again and again, mostly based on disbelief that the antibodies used for immunocytochemistry/immunohistochemistry (ICC/IHC) detect genuine Aβ. Indeed, this should be of concern in any ICC/IHC experiment, and this is why adequate controls are required. Antibodies usually recognize epitopes present in immunogens, mostly in the conformation present in the immunogen. Some antibodies work great on immunoblots but are useless in ICC/IHC. Others work well in ICC/IHC but detect the antigen poorly on immunoblots. Numerous other factors may lead to uncertainties as to what the molecular species the antibodies detect in situ really is. The problem is even more complicated with detection of Aβ because the epitopes contained in it are also present in APP and—some of them—in CTFs.
Yet, no matter how one looks at it, Aβ is generated intracellularly. There is no question about this. Unlike α-secretase cleavage, which occurs to a large extent at the cell surface (2), β-secretase cleavage occurs, and Aβ is produced, in intracellular compartments. This happens mainly in endosomes and autophagic vacuoles, but also in early secretory compartments such as the endoplasmic reticulum or the ER-Golgi-intermediate compartment, as Virginia Lee's pioneering papers in the 1990s clearly showed (3,4).
Normally, Aβ is either degraded via the lysosome/autolysosome/proteasome machineries inside cells, or secreted into the extracellular space—some via true secretory vesicles, and most from endosomes, including multivesicular bodies. However, in conditions of AD and in old age, the secretory/endocytic/degradative machineries could fail (see, e.g., the latest study from Randy Nixon’s lab [5]). As a consequence, Aβ is likely to accumulate within cells. A different pool of intracellular Aβ could form by reuptake of Aβ from the extracellular space. The re-internalized Aβ could easily end up in the same endosomes that contain unprocessed and partially processed APP. Thus, it is expected that cells contain a mixture of full-length APP and APP fragments—including Aβ—in the same intracellular compartments.
Much is being discussed about the use of antibodies raised to internal epitopes of Aβ to detect the free peptide in ICC/IHC. In theory, they should detect not only Aβ, but also full-length APP and other APP-derived polypeptides that contain the epitope. Yet this is not exactly true. It boils down to the affinity of the antibody for the Aβ peptide versus APP and APP-derived fragments. As Charlie Glabe noted in his comment, antibodies made to the Aβ peptide preferentially detect the epitope in the conformation that was available in the antigen, i.e., the injected peptide. If so, antibody 6E10 is likely to bind with higher affinity to Aβ proper, compared to APP and APP-derived polypeptides containing the 6E10 epitope. This is particularly true with in-situ detection, such as ICC/IHC, where the native conformation is preserved to a greater extent than in immunoblotting assays, which denature the proteins.
In our experience, the 6E10 antibody in ICC/IHC has a preference for the Aβ peptide compared to full-length APP or CTFβ. We usually rely on co-staining with antibodies to C- and N-terminal epitopes to determine what exactly the antibodies to internal Aβ epitopes stain. Validation with terminal end-specific antibodies should be always done, but these antibodies are usually less well characterized.
The problem with LaFerla's 3xTg-AD mouse is more complicated, since it expresses mutant APP at above physiological levels. We recently showed that, upon overexpression, APP processing becomes abnormal, with much APP remaining uncleaved (6). This might be one explanation for the seemingly increased accumulation of full-length APP detected in the study by Winton et al. Still, we think the 3xTg-AD mouse also accumulates large amounts of genuine Aβ within neurons, as LaFerla and other groups reported over the years. While we did not study this particular mouse, we found intraneuronal accumulations of bona fide Aβ in a subpopulation of neurons from another model of AD—Bruce Lamb’s R1.40 mouse, which expresses a human APPswe mutant at slightly above-normal levels (7). Of course, there is also full-length APP in the neuronal soma in the brain of this mouse, as expected.
We think that the apparent contradiction between the earlier and present studies with the 3xTg-AD mouse is easy to reconcile. Full-length APP and Aβ co-accumulate in certain neurons. We believe this is also true for the human AD brain. In our view, Winton et al. do not argue against the presence of intraneuronal Aβ in AD, and its relevance to the disease process. Virginia Lee was, in fact, an early pioneer of the idea of intracellular Aβ. Instead of being a repudiation of this idea, we think that the paper is a reminder of the difficulties in drawing conclusions when the real culprit may hide in a “sea” of potential culprits. In other words, is the culprit Aβ? Is it full-length APP? Or the CTFs, including AID?
Maybe none of them. We would like to conclude by referring to Nikos Robakis's comment. Like him, we are not convinced that so far the data connecting APP and Aβ to AD support firm conclusions on their roles in the disease process. No one can doubt that they accompany the disease in most cases of bona-fide AD, familial or sporadic. They are part of this disease. Therefore, the focus on APP and Aβ is fully justified, as is the work with animal models of disease (even if they are not perfect), as long as this is not the only focus.
1. Gouras, G.K., et al., Intraneuronal Abeta42 accumulation in human brain. Am J Pathol, 2000. 156(1): p. 15-20. Abstract
2. Thinakaran, G. and E.H. Koo, Amyloid precursor protein trafficking, processing, and function. The Journal of biological chemistry, 2008. 283(44): p. 29615-9. Abstract
3. Cook, D.G., et al., Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med, 1997 3(9): p. 1021-3. Abstract
4. Skovronsky, D.M., R.W. Doms, and V.M. Lee, Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol, 1998 141(4): p. 1031-9. Abstract
5. Lee, S., Y. Sato, and R.A. Nixon, Lysosomal Proteolysis Inhibition Selectively Disrupts Axonal Transport of Degradative Organelles and Causes an Alzheimer's-Like Axonal Dystrophy. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2011. 31(21): p. 7817-30. Abstract
6. Muresan, V., et al., The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci, 2009. 29(11): p. 3565-78. Abstract
7. Muresan, V., B.T. Lamb, and Z. Muresan, DISC1 is required for the formation of intracellular Aβ oligomers, suggesting a link between schizophrenia and Alzheimer’s disease. Annual Meeting of the Society for Neuroscience, San Diego, November 13-17, 2010.
Comment by: A. Claudio Cuello
Posted 14 June 2011
The paper by Winton et al. is a well-written and comprehensive study of the nature of intracellular Aβ-immunoreaction (IR) as observed in very representative transgenic (Tg) animal models. The proposition that only APP material is present intraneuronally in such models is indeed thought provoking. Most of us in the field will accept that the intraneuronal Aβ immunoreactive material might represent a variety of molecular species, notably the APP C-terminal fragment (C-99), APP (as well illustrated in this publication), and a variety of Aβ species—monomeric, soluble oligomeric, and fibrillar forms. The report does not, however, invalidate the comprehensive and well-supported work done by the groups of LaFerla, Gouras, Bayer, our own lab, and others.
In my view, it is risky to assert that a given molecule (Aβ), which exists as N-terminally and C-terminally truncated forms (and is conformationally diverse in its presentation) is not present because it cannot be demonstrated in certain immunohistochemical conditions. In this case, 10 percent formaldehyde fixation, a procedure which will render extensive protein-peptide cross-linking, occluding many antigenic sites and creating steric hindrance to antibody recognition, will make it difficult to label weaker and not abundant epitopes. The bulk of the studies supporting the presence of intracellular Aβ immunoreactive material have been performed in milder formaldehyde (4 percent) fixation procedures.
Another problem with this report is the assertion that Aβ-IR as revealed with 4G8 with confocal microscopy in the triple Tg is fully colocalized with that of antibody 5685. Such colocalization cannot be resolved with the "merge" function with a very low magnification micrograph, as reported in the paper. This can be more convincingly resolved at much higher magnification. Even in Figure 5.I, it is quite clear that some neurons only reveal signal for 4G8 and not for 5685, revealing the holoAPP problem. Even if colocalizations were apparently 100 percent, it should be kept in mind that it will simply mean that both molecules are in the same compartment but not necessarily that two distinct antibodies are recognizing the same molecular species. A single focal plane in confocal microscopy is in the order of 0.7 μm, giving room for thousands of molecules in the same "merged" image. For that reason, proper colocalization analysis must be performed (Manders et al. 1993). Indeed, given that the images provided do not show the same intensity distribution between the two channels (the neurons with the highest 4g8 immunoreactivity are often not the ones with the highest 5685 immunoreactivity), it is likely that colocalization analysis reveals that the antibodies detect different proteins.
Despite these limitations, and in line with LaFerla's and Pike's comments, our lab has been able to differentiate the APP versus Aβ immunoreactions. In our recently described Tg rat (Leon et al., 2010) and mouse (Ferretti et al., 2011) models of AD-like amyloid pathology, we dissociated APP-immunoreactivity with the standard APP N-terminus antibody 22C11 from that of McSA1, which preferentially recognizes Aβ-immunoreactivity independently of state of oligomerization or conformation. With simultaneous immunolabeling with 22C11 and McSA1, and analyzing high magnification and resolution images, we detected a differential pattern of immunoreactivities (as illustrated in Fig. 3 of Ferretti et al., 2011). The 22C11-immunoreactivity appeared diffusely distributed, and that of McSA1-IR (Aβ) was largely associated to granule-like structures in the perinuclear region and in the axon. Little to no subcellular colocalization was observed, suggesting that McSA1 does not recognize the holo-APP protein in 4 percent paraformaldehyde fixed free-floating tissue sections. Furthermore, such intracellular material was also reactive to the Nu1 antibody (Lambert et al., 2007) well characterized for the identification of Aβ oligomers (ADDLs). Again, in our hands, in the McGill-Thy1-APP Tg mice dual immunostaining revealed a differential intracellular staining pattern for Nu1 and APP (Fig. 7 of Ferretti et al., 2011) . Likewise, in the McGill-Thy1-R-APP rat transgenic model of the full amyloid pathology, where we identify a pre-plaque stage of intracellular β amyloid. That immunoreaction with the McSA1 antibody can also be differentiated with confocal microscopy from that of APP, as revealed by the 22C11 antibody. Furthermore, to exclude possible cross-reactivity of the human Aβ-specific antibody used (McSA1) with human holo-AβPP or N-terminal products thereof (e.g., soluble AβPPα or sAβPPα), we pre-absorbed (Fig. S2) McSA1 with synthetic Aβ1-42. This completely abolished intracellular immunostaining, while the same molar concentration of synthetic soluble sAβPPα did not show any significant effect (see supplementary data of Leon et al., 2010.)
The report of Winton et al. postulates that overexpression of holo-APP protein rather than intracellular accumulation of Aβ molecular species is behind the often-reported intracellular Aβ immunoreactivity in transgenic models. It is certainly possible that fairly aggressive transgenesis would facilitate some accumulation of the holo-APP molecule within neurons, but it does not exclude the occurrence of Aβ as recognized by a number of fairly Aβ-specific antibodies. In the McGill-R-Thy1-APP rat transgenic model, that proposition does not apply, as the full amyloid pathology is achieved in the Tg rat with a single APP transgene per allele, making it closer to the progressive, slowly evolving, human pathology.
Winton et al. also postulate that the presence of mutated presenilin protein might provoke perikaryal accumulation of APP due to a vesicular impairment. As our rat and mice transgenic models only carry human mutated forms of APP, that would not apply. Importantly, in both the McGill mice and rat Tg models, we consistently detect Aβ40 and Aβ42 by ELISA in pre-plaque stages, as it has also been reported in other Tg models. This occurrence of Aβ40 and Aβ42 fragments at the stage of intracellular accumulation of Aβ immunoreactivity has been validated in the rat Tg model independently with biochemical analyses by Drs. Lisa Munter and Gerd Multhaup (Freie University of Berlin), colleagues who have a solid record in Aβ biochemistry.
In closing, it is likely that the Aβ-immunoreactivity in transgenic models represents not only Aβ itself, evolving from monomeric to oligomeric to fibrillar as suggested by the report of Ferretti et al. (Ferretti et al., 2011), but APP as well (as illustrated by the work of Winton et al., as well as other APP fragments, notably C-99). The main challenge would be to determine which are the relative proportions of these entities and, ultimately and most importantly, which would be their relative contribution to CNS dysfunction.
Manders et al. Measurement of colocalization of objects in dual colour confocal images. (1993) J. Microsc., V.169 pp. 375-382.
Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, de Wilde A, Vercauteren F, Atifeh R, Ducatenzeiler A, Klein W, Szyf M, Alhonen L and Cuello AC. A Novel Transgenic Rat Model with a Full Alzheimer's-Life Amyloid Pathology Displays Pre-Plaque Intracellular Amyloid-beta-Associated Cognitive Impairment. Journal of Alzheimer's Disease 20 (2010) 113-126. Abstract
Ferretti MT, et al. Transgenic mice as a model of pre-clinical Alzheimer's disease. (Curr Alzheimer Res. 2011). Abstract
Lambert MP et al. Monoclonal antibodies that target pathological assemblies of Abeta.(J Neurochem. 2007). Abstract
Comment by: Gerd Multhaup
Posted 14 June 2011
The intraneuronal pool of Aβ may have a double origin, which should be incorporated in the discussion raised by findings by Winton et al. that question the existence of intraneuronal Aβ in the 3xTg mouse model.
Seminal findings by laboratories headed by Beyreuther and Glabe reported more than 15 years ago that exogenous Aβ42 is taken up from the extracellular space and may result in accumulation of amyloidogenic fragments in different cell lines (Kanuer et al., 1992; Ida et al., 1996). Interestingly, Ida et al. found an Aβ-specific clearance activity inside of neuroblastoma SH-SY5Y cells. Cellular uptake of Aβ was also reported to occur in rat microglia cells when removal of Aβ from the medium and an accumulation in the cells was observed (Ard et al., 1996).
Although the molecular mechanisms for neuronal Aβ internalization have remained unclear, the involvement of numerous receptors in endocytosis of Aβ was described, as well as an accumulation of internalized Aβ in the endosomal/lysosomal system or late endosomes/multivesicular bodies (MVBs) (for a review, see Mohamed and Posee de Chaves, 2011). In glial cells, the purpose of uptake may reflect mainly clearance activity, whereas in neurons Aβ uptake may represent an alternative mechanism in the development of AD. Novel findings from our group strongly support this view.
As presented by Christian Barucker et al. at the last ADPD Conference (Barucker et al., 2011. A novel function of Aβ in gene regulation as an alternative pathway of toxicity. ADPD Conference, Barcelona, Spain, 9-13 March 2011), shorter and longer forms of Aβ are rapidly internalized by SH-SY5Y cells and transported into the nucleus. There, all Aβ peptides accumulated in a time dependent manner. However, we found only specific genes affected by toxic Aβ42 peptides (manuscript in preparation). Also, our findings indicate that this nuclear signaling function is unique for Aβ42 wild-type, and imply that deregulation of Aβ target genes could be an alternative pathway for Aβ-induced neurotoxicity.
Comment by: P. Hemachandra Reddy
Posted 16 June 2011
The current debate on intraneuronal amyloid β (Aβ) in Frank LaFerla’s mice is thought provoking to the entire Alzheimer’s disease (AD) research community because Aβ is a key factor in progression and pathogenesis. In view of that, I read the paper by Winton et al. with great interest. Dr. Lee and colleagues have characterized AD mouse models (3xTg-AD, 3xTg-AD/BACE-/-, BACE-/-, and non-transgenic WT mice) in order to determine the existence of intraneuronal Aβ, and reported that intraneuronal Aβ is absent in 3xTg-AD mice. In our experience, immunostaining analysis using fixed tissues is unreliable and requires extensive optimization. Thus, the conclusion that intraneuronal Aβ is not present in 3xTg-AD mice needs further investigation.
We extensively characterized postmortem brains from AD patients and control subjects, cortical tissues from APP/PS1 mice, and primary neurons from Tg2576 mice using 6E10, 1-40, 1-42, and A11 antibodies. We found excessive amounts 4 kDa Aβ, and also oligomeric Aβ and full-length APP and other APP derivatives in AD patients, APP/PS1 mice, and Tg2576 mice (see Fig. 3 in our recent paper Manczak et al., 2011). Based on immunoprecipitation, Western blotting, and immunohistochemistry data, we concluded that the 6E10 antibody is immunoreactive to full-length APP and its derivatives.
Hyperphosphorylation of tau has been found to be induced by several factors: 1) chronic oxidative stress (Su et al., 2010); 2) Aβ-mediated oxidative stress (Garwood et al., 2011); 3) reduced insulin-like growth factor 1 (Cheng et al., 2005); and 4) mutations in tau gene (Oddo et al., 2003; Lewis et al., 2001; Gotz et al., 2001). The hyperphosphorylation observed in 3xTg-AD mice may likely be caused by a combination of Aβ-mediated oxidative stress and tau mutation.
Several recent studies reported that intraneuronal Aβ is associated with mitochondrial membranes, causing mitochondrial dysfunction and neuronal damage (Caspersen et al., 2005; Crouch et al., 2005; Manczak et al., 2006; Devi et al., 2006; Hansson Petersen et al., 2009; Yao et al., 2009). Several groups have examined the 3xTg-AD for mitochondria abnormalities and oxidative stress (e.g., Yao et al., 2009, Resende et al., 2008). They found that Aβ is associated with mitochondria and increased oxidative stress and decreased respiration (supporting the presence of intraneuronal Aβ in 3xTg-AD mice).
Overall, the data overwhelmingly support intraneuronal Aβ in AD brains and AD mouse models, and support the view that Aβ causes neuronal damage. Therefore, at this point, given the state of Aβ research in the AD field, I believe the conclusions by Winton et al. are questionable.
Cheng CM, Tseng V, Wang J, Wang D, Matyakhina L, Bondy CA. Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology. 2005 Dec;146(12):5086-91. Abstract
Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005 Jan 19;25(3):672-9. Abstract
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006 Aug 30;26(35):9057-68. Abstract
Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008 Oct;14(10):1097-105. Abstract
Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011 Jun 2;2:e167. Abstract
Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001 Aug 24;293(5534):1491-5. Abstract
Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008 Sep 2;105(35):13145-50. Abstract
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001 Aug 24;293(5534):1487-91. Abstract
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004 Apr 16;304(5669):448-52. Abstract
Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006 May 1;15(9):1437-49. Abstract
Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011 Jul 1;20(13):2495-509. Abstract
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003 Jul 31;39(3):409-21. Abstract
Resende R, Moreira PI, Proença T, Deshpande A, Busciglio J, Pereira C, Oliveira CR. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008 Jun 15;44(12):2051-7. Abstract
Su B, Wang X, Lee HG, Tabaton M, Perry G, Smith MA, Zhu X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett. 2010 Jan 14;468(3):267-71. Abstract
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009 Aug 25;106(34):14670-5. Abstract
Comment by: Wataru Araki
Posted 20 June 2011
The paper by Winton et al. has raised a debate on the significance of intraneuronal Aβ. I generally agree with the opinions by Frank LaFerla, Salvo Oddo, Christian Pike, and Gunnar Gouras et al., arguing that the data are insufficient to disprove the presence of intraneuronal free Aβ.
Visualization of intraneuronal Aβ by endo-specific antibodies requires appropriate immunodetection methods, including pretreatment for antigen retrieval (e.g., treatment with formic acid). Therefore, it is possible that the absence of immunopositive signals with endo-specific Aβ antibodies (BC05 and BA27) was due to inefficient antigen retrieval. The authors compared 3xTg-AD mice with 3xTg-AD-BACE knockout mice, but the comparison did not yield information about whether BACE deficiency has any effect on intraneuronal Aβ.
I'd like to emphasize that intraneuronal Aβ, especially Aβ42, is detectable in our mouse model without APP overexpression (Chui et al., 1999). Intracellular Aβ is evident in many cellular models including ours (Takeda et al., 2004). However, it is true that intracellular Aβ is fairly difficult to be detected, as claimed by Gouras et al. Thus, cautious research is necessary in dealing with intraneuronal Aβ.
Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med 5, 560-564, 1999. Abstract
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156, 15-20, 2000. Abstract
Takeda K, Araki W, Tabira T. Enhanced generation of intracellular Abeta42 amyloid peptide by mutation of presenilins PS1 and PS2. Eur J Neurosci 19, 258-264, 2004. Abstract
Comment by: Heng Du, Frank Gunn-Moore, Shirley Yan
Posted 20 June 2011
So APP might also exist inside cells: Didn’t we know this already? The question now appears to be whether there is more APP than shorter forms of Aβ inside cells.
We evaluated accumulation of mitochondrial Aβ in mitochondrial fractions in the presence of digitonin to diminish contamination with other organelles. To investigate whether Aβ was within the organelle (i.e., within a membrane-enclosed compartment) or adsorbed to the outer mitochondrial membrane, we performed protease protection to investigate whether Aβ was within the organelle (i.e., within a membrane-enclosed compartment) or adsorbed to its outer membrane. The data indicated that most of mitochondria-associated Aβ was in a protected, membrane-bound compartment (8). Western blotting, confocal microscopy, and immunoelectron microscopy have shown in human AD patients that Aβ is present intracellularly, including mitochondria (2,8,10,14).
Hansson Petersen et al. demonstrated that Aβ is localized in mitochondria of human aged brain from surgical biopsy tissue by immunogold EM with a specific Aβ antibody. Intracellular proteins that bind to this internal Aβ have been identified in animal models and humans (2,10-11,13), and these proteins were not found to be binding APP. There have been papers showing that these intracellular Aβ interactions or accumulation of intracellular Aβ can be perturbed with reversing of both biochemical and physiological changes (2,9,11-13,15-18).
Thus, it seems that one paper cannot overwhelm a series of findings derived from many pathological and biochemical studies.
Comparing the images presented by Winton et al. with those in other papers using the same mouse model at similar ages (19-20), we notice substantially fewer Aβ plaques in Winton et al. (Fig.1B). Possibilities underlying this discrepancy are, first, Winton used 3xTg-AD mice demonstrating less Aβ production/deposition at the ages up to 12 months, and/or Winton’s immunostaining method was less sensitive because of the methodology. Some appropriate controls would have included pre-immune or non-immune IgG for immunostaining to confirm the specificity of immunoreaction, and qualification of all images. Biochemical approaches to show the size of the Aβ immunoreactive band would have helped to verify the presence of Aβ.
In any event, one cannot conclude that intracellular Aβ is of limited significance in AD-related disturbances because its level is low. For example, Aβ has been shown to directly interact with intracellular proteins ABAD (2), cyclophilin D (10), and Drp1 (18), exacerbating neuronal and mitochondrial stress. These data suggest intracellular Aβ, though its level might be low, plays a role in Aβ-induced intracellular perturbations in AD pathogenesis.
1. Oddo S, et al. (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3):409-421. Abstract
2. Lustbader JW, et al. (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304(5669):448-452. Abstract
3. Wilson CA, Doms RW, & Lee VM (2003) Distinct presenilin-dependent and presenilin-independent gamma-secretases are responsible for total cellular Abeta production. J Neurosci Res 74(3):361-369. Abstract
4. Cataldo AM, et al. (2004) Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging 25(10):1263-1272. Abstract
5. Almeida CG, Takahashi RH, & Gouras GK (2006) Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci 26(16):4277-4288. Abstract
6. Yang AJ, Chandswangbhuvana D, Margol L, & Glabe CG (1998) Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res 52(6):691-698. Abstract
7. Takuma K, et al. (2009) RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci U S A 106(47):20021-20026. Abstract
8. Caspersen C, et al. (2005) Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. Faseb J 19(14):2040-2041. Abstract
9. Manczak M, et al. (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15(9):1437-1449. Abstract
10. Du H, et al. (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 14(10):1097-1105. Abstract
11. Yao J, et al. (2009) Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 106(34):14670-14675. Abstract
12. Dragicevic N, et al. (2010) Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis 20 Suppl 2:S535-550. Abstract
13. Yao J, et al. (2011) Inhibition of Amyloid-{beta} (A{beta}) Peptide-Binding Alcohol Dehydrogenase-A{beta} Interaction Reduces A{beta} Accumulation and Improves Mitochondrial Function in a Mouse Model of Alzheimer's Disease. J Neurosci 31(6):2313-2320. Abstract
14. Hansson Petersen CA, et al. (2008) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105(35):13145-13150. Abstract
15. Takuma K, et al. (2005) ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. Faseb J 19(6):597-598. Abstract
16. Yao J, et al. (2007) Interaction of amyloid binding alcohol dehydrogenase/Abeta mediates up-regulation of peroxiredoxin II in the brains of Alzheimer's disease patients and a transgenic Alzheimer's disease mouse model. Mol Cell Neurosci 35(2):377-382. Abstract
17. Ren Y, et al. (2008) Endophilin I expression is increased in the brains of Alzheimer disease patients. J Biol Chem 283(9):5685-5691. Abstract
18. Manczak M, Calkins MJ, & Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Human molecular genetics 20(13):2495-2509. Abstract
19. Rohn TT, et al. (2008) Lack of pathology in a triple transgenic mouse model of Alzheimer's disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci 28(12):3051-3059. Abstract
20. Hirata-Fukae C, et al. (2008) Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res 1216:92-103. Abstract
Comment by: Jia Yao
Posted 20 June 2011
I agree with Dr. Reddy's comments. The mitochondrial localization of various Aβ species constitutes direct evidence of intraneuronal Aβ accumulation rather than APP. We have used the 6E10 antibody to detect multiple Aβ species in purified mitochondrial fractions by Western blot. In the 3xTg-AD mouse model, we observed at least three distinct bands with molecular weight at full APP size (~130 kD, ~56 kD, and a 16 kD size band). Although we did not observe the 4 kD amyloid monomer in our mitochondrial prep, we believe the 16 kD band is likely to represent the Aβ tetramer and cannot be explained by abnormally folded full-length APP.
Similarly, in the Tg2576 model, Reddy's group demonstrated a 4 kD Western band in a purified brain mitochondrial prep. In mutant APP-expressing N2A cell lines, this group identified oligomers of Aβ at various sizes (mainly 16 kD, 27 kD, and ~40 KD).
The small immuno-bands (4 kD, 16 kD, 27 kD, 56 kD) observed in isolated mitochondrial fraction from the 3xTg-AD mice, Tg2576 mice, and other APP transgenic models more or less provide evidence of intraneuronal, or, more specifically, mitochondrial accumulation of Aβ oligomers.
Noteworthy: APP is also observed in the same mitochondrial prep in our study in the 3xTg-AD mouse model and in other models.
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A, 2009; 106: 14670-5. Abstract
Yao J, Irwin R, Chen S, Hamilton R, Cadenas E, Brinton RD. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiology of Aging, 2011. Abstract
Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Human molecular genetics, 2006; 15: 1437-49. Abstract
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science, 2004; 304: 448-52. Abstract
Comment by: Carlos A. Saura
Posted 20 June 2011
There is compelling evidence that Aβ generated from the sequential cleavage of APP by β-secretase and PS/γ-secretase is both secreted at the extracellular space and accumulated intracellularly in cultured cells and brains from transgenic mouse models and AD. The general picture raised by a large number of studies from independent labs, including our group, is that intracellular Aβ is commonly detected in the brains of several AD transgenic mouse models including 3xTg-AD, APP/PS1KI, 5xFAD, R1.40, APPSw,Ind, and APPInd (Tampellini et al. 2010; Oddo et al., 2003; Casas et al., 2004; Oakley et al., 2006; Gimenez-Llort et al., 2007; España et al., 2010; Manczak et al., 2011).
Winton and colleagues add a new perspective and raise doubt about previous studies in 3xTg-AD mice. Our recent studies of Aβ staining with 2G3 antibody, specific for Aβ40, and an Aβ42 antibody, revealed robust intraneuronal Aβ in the basolateral amygdala (BLA), and to a lesser extent in CA1/CA3 hippocampus (20-40 percent of cells) and layer V of the neocortex (20 percent of neurons of this layer) in six-month-old 3xTg-AD mice (España et al., 2010). Staining was not detected in non-transgenic controls. This indicates that intracellular Aβ staining is almost absent in the majority of cortical layers in 3xTg-AD mice at six months (Fig. 1A, C of Winton et al.), but is detected within neurons of the hippocampus and amygdala. Winton et al. do not show pictures of BLA and hippocampus at six months.
We observed an age-dependent increase of intracellular Aβ/APP staining with 2G3 (Aβ40) and 6E10 antibodies, but unchanged APP/APPs labeling with 8E5 antibody. Since full-length APP expression detected by Western blotting and 8E5 staining is unchanged in 3xTg-AD mice during aging, 2G3 and 6E10 stainings seem to represent intracellular Aβ species. In agreement with Christian Pike´s comment, our studies indicate that APP staining was found to be widespread in the soma, whereas Aβ staining was punctuate and perinuclear. The differential staining pattern for Aβ/APP in 3xTg-AD mice of the present report clearly differs from the previous reports, raising doubt in my mind whether the lack of BA27 (Aβ40) and BC05 (Aβ42) staining truly represents absence of intracellular Aβ. It is possible that BA27 and BC05 (regretfully JFR/c40 and c42 staining is not shown) do not recognize the conformational state of intracellular Aβ but are able to detect extracellular aggregated Aβ in plaques (Fig. 1A-D). This may also explain why BA27 and BC05 labeling disappears in 10- to 12-month-old 3xTg-AD;BACE-/- mice (Fig. 4A-D).
From the studies with 3xTg-AD; BACE-/- mice, it seems that the experimental staining conditions with 6E10/4G8 antibodies used here (dilution of 6E10 and 4G8 antibodies may be also important) allow detection of human APP but not soluble Aβ. Still, the lack of staining with BA27 and BC05 antibodies in the 3xTg-AD neocortex, a brain region not abundant in Aβ-containing neurons, does not necessarily prove the absence of intraneuronal Aβ accumulation in 3xTg-AD mice.
We performed many control experiments to rule out the possibility of APP detection with Aβ-specific antibodies. Using these approaches, we also detected APP expression (8E5) and intraneuronal Aβ staining with 6E10 (APP/Aβ), Aβ40 (2G3), and Aβ42 antibodies, but not amyloid plaques in the hippocampus, specific neocortical layers, and BLA in two other transgenic lines (APPInd and APPSw,Ind mice from Lennart Mucke´s lab). The consequences of abnormal intracellular accumulation of Aβ in these models and their relevance for AD pathology need further investigation.
Casas C, Sergeant N, Itier J, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer T, Benoit P, Pradier L (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165:1289-1300. Abstract
España J, Gimenez-Llort L, Valero J, Miñano A, Rabano A, Rodriguez-Alvarez J, Laferla FM, Saura CA (2010) Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer's disease transgenic mice. Biol Psychiatry 67 513-521. Abstract
Gimenez-Llort L, Blazquez G, Canete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, Fernandez-Teruel A (2007) Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev 31:125-147. Abstract
Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20:2495-2509. Abstract
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129-10140. Abstract
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409-421. Abstract
Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer's disease transgenic mice. J Neurosci. 2010;30:14299-14304. Abstract
References
Webinar Citations
- Intraneuronal Aβ Accumulation—More Evidence, Less Controversy?
- Reimagining Alzheimer's Disease—Time for Bright New Ideas?
Paper Citations
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992 Sep 24;359(6393):325-7. PubMed.
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992 Sep 24;359(6393):322-5. PubMed.
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011 May 25;31(21):7691-9. PubMed. RETRACTED
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003 Jul 31;39(3):409-21. PubMed.
- Achim CL, Adame A, Dumaop W, Everall IP, Masliah E, . Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol. 2009 Jun;4(2):190-9. PubMed.
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006 Apr 19;26(16):4277-88. PubMed.
- Bayer TA, Schäfer S, Breyhan H, Wirths O, Treiber C, Multhaup G. A vicious circle: role of oxidative stress, intraneuronal Abeta and Cu in Alzheimer's disease. Clin Neuropathol. 2006 Jul-Aug;25(4):163-71. PubMed.
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005 Mar 3;45(5):675-88. PubMed.
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004 Oct;165(4):1289-300. PubMed.
- Christensen DZ, Kraus SL, Flohr A, Cotel MC, Wirths O, Bayer TA. Transient intraneuronal A beta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008 Dec;116(6):647-55. PubMed.
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005;132(1):123-35. PubMed.
- Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006 Oct 11;26(41):10536-41. PubMed.
- Dafnis I, Stratikos E, Tzinia A, Tsilibary EC, Zannis VI, Chroni A. An apolipoprotein E4 fragment can promote intracellular accumulation of amyloid peptide beta 42. J Neurochem. 2010 Nov;115(4):873-84. PubMed.
- Echeverria V, Ducatenzeiler A, Dowd E, Jänne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC. Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the beta-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience. 2004;129(3):583-92. PubMed.
- España J, Giménez-Llort L, Valero J, Miñano A, Rábano A, Rodriguez-Alvarez J, Laferla FM, Saura CA. Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer's disease transgenic mice. Biol Psychiatry. 2010 Mar 15;67(6):513-21. PubMed.
- Fujimura RK, Reiner T, Ma F, Phillips V, de Las Pozas A, Dickson DW, Roos BA, Howard GA, Perez-Stable C. Changes in the Expression of Genes Associated with Intraneuronal Amyloid-beta and Tau in Alzheimer's Disease. J Alzheimers Dis. 2009 Sep 11; PubMed.
- Fujimura RK, Reiner T, Ma F, Phillips V, de Las Pozas A, Dickson DW, Roos BA, Howard GA, Perez-Stable C. Changes in the expression of genes associated with intraneuronal amyloid-beta and tau in Alzheimer's disease. J Alzheimers Dis. 2010;19(1):97-109. PubMed.
- Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, Xu H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001 Apr 15;21(8):2561-70. PubMed.
- Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, Laferla FM, Fernández-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31(1):125-47. PubMed.
- Golde TE, Janus C. Homing in on intracellular Abeta?. Neuron. 2005 Mar 3;45(5):639-42. PubMed.
- Gómez-Ramos P, Asunción Morán M. Ultrastructural localization of intraneuronal Abeta-peptide in Alzheimer disease brains. J Alzheimers Dis. 2007 Mar;11(1):53-9. PubMed.
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000 Jan;156(1):15-20. PubMed.
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005 Oct;26(9):1235-44. PubMed.
- Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001 Apr;125(4):489-92. PubMed.
- Hasegawa T, Ukai W, Jo DG, Xu X, Mattson MP, Nakagawa M, Araki W, Saito T, Yamada T. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42: implications for the pathogenesis of Alzheimer's disease. J Neurosci Res. 2005 Jun 15;80(6):869-76. PubMed.
- Hashimoto M, Bogdanovic N, Volkmann I, Aoki M, Winblad B, Tjernberg LO. Analysis of microdissected human neurons by a sensitive ELISA reveals a correlation between elevated intracellular concentrations of Abeta42 and Alzheimer's disease neuropathology. Acta Neuropathol. 2010 May;119(5):543-54. PubMed.
- Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, Motomura K, Soejima N, Yamasaki R, Hashimoto T. Apomorphine Treatment for Alzheimer’s Mice Promoting Amyloid-β Degradation. Annals of Neurology. 2010 Dec 1;
- Iijima-Ando K, Hearn SA, Granger L, Shenton C, Gatt A, Chiang HC, Hakker I, Zhong Y, Iijima K. Overexpression of neprilysin reduces alzheimer amyloid-beta42 (Abeta42)-induced neuron loss and intraneuronal Abeta42 deposits but causes a reduction in cAMP-responsive element-binding protein-mediated transcription, age-dependent axon pathology, and prem. J Biol Chem. 2008 Jul 4;283(27):19066-76. PubMed.
- Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN. Intracellular amyloid-beta 1-42, but not extracellular soluble amyloid-beta peptides, induces neuronal apoptosis. J Biol Chem. 2002 May 3;277(18):15666-70. PubMed.
- Leon WC, Canneva F, Partridge V, Allard S, Ferretti MT, DeWilde A, Vercauteren F, Atifeh R, Ducatenzeiler A, Klein W, Szyf M, Alhonen L, Cuello AC. A novel transgenic rat model with a full Alzheimer's-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment. J Alzheimers Dis. 2010;20(1):113-26. PubMed.
- Levi O, Dolev I, Belinson H, Michaelson DM. Intraneuronal amyloid-beta plays a role in mediating the synergistic pathological effects of apoE4 and environmental stimulation. J Neurochem. 2007 Nov;103(3):1031-40. PubMed.
- Lin B, Ginsberg MD, Busto R. Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of beta-amyloid precursor protein. Brain Res. 2001 Jan 5;888(1):107-116. PubMed.
- Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006 Jan;27(1):67-77. PubMed.
- Magrané J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci. 2005 Nov 23;25(47):10960-9. PubMed.
- Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011 Jul 1;20(13):2495-509. PubMed.
- Moreira PI, Liu Q, Honda K, Smith MA, Santos MS, Oliveira CR. Is intraneuronal amyloid beta-peptide accumulation the trigger of Alzheimer's disease pathophysiology?. J Alzheimers Dis. 2004 Aug;6(4):433-4; discussion 443-9. PubMed.
- Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002 Jan;110(2):199-211.
- Nagele RG, Clifford PM, Siu G, Levin EC, Acharya NK, Han M, Kosciuk MC, Venkataraman V, Zavareh S, Zarrabi S, Kinsler K, Thaker NG, Nagele EP, Dash J, Wang HY, Levitas A. Brain-reactive autoantibodies prevalent in human sera increase intraneuronal amyloid-β(1-42) deposition. J Alzheimers Dis. 2011;25(4):605-22. PubMed.
- Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, Yamaguchi H, Shimohama S, Lee HG, Zhu X, Smith MA, Perry G. Intraneuronal amyloid beta accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis. 2010 Mar;37(3):731-7. PubMed.
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006 Oct 4;26(40):10129-40. PubMed.
- Oddo S, Caccamo A, Smith IF, Green KN, Laferla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006 Jan;168(1):184-94. PubMed.
- Ohyagi Y, Tsuruta Y, Motomura K, Miyoshi K, Kikuchi H, Iwaki T, Taniwaki T, Kira J. Intraneuronal amyloid beta42 enhanced by heating but counteracted by formic acid. J Neurosci Methods. 2007 Jan 15;159(1):134-8. PubMed.
- Ohyagi Y. Intracellular amyloid beta-protein as a therapeutic target for treating Alzheimer's disease. Curr Alzheimer Res. 2008 Dec;5(6):555-61. PubMed.
- Philipson O, Lannfelt L, Nilsson LN. Genetic and pharmacological evidence of intraneuronal Abeta accumulation in APP transgenic mice. FEBS Lett. 2009 Sep 17;583(18):3021-6. PubMed.
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008 Jun;118(6):2190-9. PubMed.
- Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem. 2006 Dec 29;281(52):39907-14. PubMed.
- Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, Ladu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009 Apr 7;106(14):5907-12. PubMed.
- Rebeck GW, Hoe HS, Moussa CE. Beta-amyloid1-42 gene transfer model exhibits intraneuronal amyloid, gliosis, tau phosphorylation, and neuronal loss. J Biol Chem. 2010 Mar 5;285(10):7440-6. PubMed.
- Sahlin C, Lord A, Magnusson K, Englund H, Almeida CG, Greengard P, Nyberg F, Gouras GK, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation favors intracellular amyloid-beta production by making amyloid precursor protein less available to alpha-secretase. J Neurochem. 2007 May;101(3):854-62. PubMed.
- Shie FS, Leboeuf RC, Jin LW, LeBoeur RC. Early intraneuronal Abeta deposition in the hippocampus of APP transgenic mice. Neuroreport. 2003 Jan 20;14(1):123-9. PubMed.
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
- Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci U S A. 2009 Nov 24;106(47):20021-6. PubMed.
- Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009 Aug 5;29(31):9704-13. PubMed.
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010 Apr 7;30(14):4845-56. PubMed.
- Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011 Jul;89(7):1031-42. Epub 2011 Apr 12 PubMed.
- Van Broeck B, Vanhoutte G, Pirici D, Van Dam D, Wils H, Cuijt I, Vennekens K, Zabielski M, Michalik A, Theuns J, De Deyn PP, Van der Linden A, Van Broeckhoven C, Kumar-Singh S. Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer's disease. Neurobiol Aging. 2008 Feb;29(2):241-52. PubMed.
- Wegiel J, Kuchna I, Nowicki K, Frackowiak J, Mazur-Kolecka B, Imaki H, Mehta PD, Silverman WP, Reisberg B, Deleon M, Wisniewski T, Pirttilla T, Frey H, Lehtimäki T, Kivimäki T, Visser FE, Kamphorst W, Potempska A, Bolton D, Currie JR, Miller DL. Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration. Acta Neuropathol. 2007 Apr;113(4):389-402. PubMed.
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001 Jun 22;306(1-2):116-20. PubMed.
- Wirths O, Multhaup G, Bayer TA. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade. J Neurochem. 2004 Nov;91(3):513-20. PubMed.
- Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006 Nov 24;281(47):36180-6. PubMed.
- Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet. 2011 Jun 1;20(11):2091-102. PubMed.
- Sudol KL, Mastrangelo MA, Narrow WC, Frazer ME, Levites YR, Golde TE, Federoff HJ, Bowers WJ. Generating differentially targeted amyloid-beta specific intrabodies as a passive vaccination strategy for Alzheimer's disease. Mol Ther. 2009 Dec;17(12):2031-40. PubMed.
- Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, Motomura K, Soejima N, Yamasaki R, Hashimoto T, Tabira T, Laferla FM, Kira J. Apomorphine treatment in Alzheimer mice promoting amyloid-β degradation. Ann Neurol. 2011 Feb;69(2):248-56. PubMed.
- McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, Das P, Golde TE, Laferla FM, Oddo S, Blesch A, Tansey MG. Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009 Apr;34(1):163-77. PubMed.
- Oddo S, Billings L, Kesslak JP, Cribbs DH, Laferla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PubMed.
- Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer's disease. Neuroscience. 2009 Mar 31;159(3):1055-69. Epub 2009 Jan 19 PubMed.
- Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, Nicoll JA. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropathol. 2010 Jul;120(1):13-20. PubMed.
- Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, Cribbs DH, Laferla FM. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J Neurosci. 2008 Nov 19;28(47):12163-75. PubMed.
- Oddo S, Caccamo A, Cheng D, Laferla FM. Genetically altering Abeta distribution from the brain to the vasculature ameliorates tau pathology. Brain Pathol. 2009 Jul;19(3):421-30. PubMed.
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010 May;119(5):523-41. PubMed.
- Yang AJ, Knauer M, Burdick DA, Glabe C. Intracellular A beta 1-42 aggregates stimulate the accumulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein in transfected cells. J Biol Chem. 1995 Jun 16;270(24):14786-92. PubMed.
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004 Apr 7;24(14):3592-9. PubMed.
- Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC. Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer's disease. Neurobiol Aging. 2011 Mar 15; PubMed.
- Yang AJ, Chandswangbhuvana D, Shu T, Henschen A, Glabe CG. Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42. J Biol Chem. 1999 Jul 16;274(29):20650-6. PubMed.
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, Laferla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005 Sep 28;25(39):8843-53. PubMed.
- McAlpine FE, Lee JK, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, Das P, Golde TE, Laferla FM, Oddo S, Blesch A, Tansey MG. Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009 Jan 22; PubMed.
- Rosario ER, Carroll JC, Oddo S, Laferla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006 Dec 20;26(51):13384-9. PubMed.
- Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, Laferla FM, Pike CJ. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007 Nov 28;27(48):13357-65. PubMed.
- Aho L, Pikkarainen M, Hiltunen M, Leinonen V, Alafuzoff I. Immunohistochemical visualization of amyloid-beta protein precursor and amyloid-beta in extra- and intracellular compartments in the human brain. J Alzheimers Dis. 2010;20(4):1015-28. PubMed.
- Wirths O, Bayer TA. Neuron loss in transgenic mouse models of Alzheimer's disease. Int J Alzheimers Dis. 2010;2010 PubMed.
- Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2012 Jan;33(1):196.e29-40. PubMed.
- Wirths O, Breyhan H, Cynis H, Schilling S, Demuth HU, Bayer TA. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009 Oct;118(4):487-96. PubMed.
- Wirths O, Erck C, Martens H, Harmeier A, Geumann C, Jawhar S, Kumar S, Multhaup G, Walter J, Ingelsson M, Degerman-Gunnarsson M, Kalimo H, Huitinga I, Lannfelt L, Bayer TA. Identification of low molecular weight pyroglutamate A{beta} oligomers in Alzheimer disease: a novel tool for therapy and diagnosis. J Biol Chem. 2010 Dec 31;285(53):41517-24. PubMed.
- Robakis NK. Mechanisms of AD neurodegeneration may be independent of Aβ and its derivatives. Neurobiol Aging. 2011 Mar;32(3):372-9. PubMed.
- Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci. 2010 Nov 10;30(45):14946-54. PubMed.
- Tian Y, Crump CJ, Li YM. Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J Biol Chem. 2010 Oct 15;285(42):32549-56. PubMed.
- Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000 Sep;6(3):625-36. PubMed.
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003 Jan 6;160(1):113-23. PubMed.
- Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knölker HJ, Simons K. Efficient inhibition of the Alzheimer's disease beta-secretase by membrane targeting. Science. 2008 Apr 25;320(5875):520-3. PubMed.
- Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W. The discrepancy between presenilin subcellular localization and gamma-secretase processing of amyloid precursor protein. J Cell Biol. 2001 Aug 20;154(4):731-40. PubMed.
- Pavlov PF, Hansson Petersen C, Glaser E, Ankarcrona M. Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med. 2009 Oct;13(10):4137-45. PubMed.
- Young KJ, Bennett JP. The mitochondrial secret(ase) of Alzheimer's disease. J Alzheimers Dis. 2010;20 Suppl 2:S381-400. PubMed.
- Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid-beta precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. J Neurosci. 2009 Mar 18;29(11):3565-78. PubMed.
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008 Oct 31;283(44):29615-9. PubMed.
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997 Sep;3(9):1021-3. PubMed.
- Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol. 1998 May 18;141(4):1031-9. PubMed.
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011 May 25;31(21):7817-30. PubMed.
- Ferretti MT, Partridge V, Leon WC, Canneva F, Allard S, Arvanitis DN, Vercauteren F, Houle D, Ducatenzeiler A, Klein WL, Glabe CG, Szyf M, Cuello AC. Transgenic mice as a model of pre-clinical Alzheimer's disease. Curr Alzheimer Res. 2011 Feb 1;8(1):4-23. PubMed.
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007 Jan;100(1):23-35. PubMed.
- Knauer MF, Soreghan B, Burdick D, Kosmoski J, Glabe CG. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc Natl Acad Sci U S A. 1992 Aug 15;89(16):7437-41. PubMed.
- Ida N, Masters CL, Beyreuther K. Rapid cellular uptake of Alzheimer amyloid betaA4 peptide by cultured human neuroblastoma cells. FEBS Lett. 1996 Sep 30;394(2):174-8. PubMed.
- Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer's amyloid beta-protein by microglia in culture. J Neurosci Res. 1996 Jan 15;43(2):190-202. PubMed.
- Cheng CM, Tseng V, Wang J, Wang D, Matyakhina L, Bondy CA. Tau is hyperphosphorylated in the insulin-like growth factor-I null brain. Endocrinology. 2005 Dec;146(12):5086-91. PubMed.
- Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005 Jan 19;25(3):672-9. PubMed.
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006 Aug 30;26(35):9057-68. PubMed.
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008 Oct;14(10):1097-105. PubMed.
- Garwood CJ, Pooler AM, Atherton J, Hanger DP, Noble W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011;2:e167. PubMed.
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001 Aug 24;293(5534):1491-5. PubMed.
- Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008 Sep 2;105(35):13145-50. PubMed.
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001 Aug 24;293(5534):1487-91. PubMed.
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004 Apr 16;304(5669):448-52. PubMed.
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006 May 1;15(9):1437-49. PubMed.
- Resende R, Moreira PI, Proença T, Deshpande A, Busciglio J, Pereira C, Oliveira CR. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med. 2008 Jun 15;44(12):2051-7. PubMed.
- Su B, Wang X, Lee HG, Tabaton M, Perry G, Smith MA, Zhu X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett. 2010 Jan 14;468(3):267-71. PubMed.
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009 Aug 25;106(34):14670-5. PubMed.
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999 May;5(5):560-4. PubMed.
- Takeda K, Araki W, Tabira T. Enhanced generation of intracellular Abeta42 amyloid peptide by mutation of presenilins PS1 and PS2. Eur J Neurosci. 2004 Jan;19(2):258-264. PubMed.
- Wilson CA, Doms RW, Lee VM. Distinct presenilin-dependent and presenilin-independent gamma-secretases are responsible for total cellular Abeta production. J Neurosci Res. 2003 Nov 1;74(3):361-9. PubMed.
- Cataldo AM, Petanceska S, Terio NB, Peterhoff CM, Durham R, Mercken M, Mehta PD, Buxbaum J, Haroutunian V, Nixon RA. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004 Nov-Dec;25(10):1263-72. PubMed.
- Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res. 1998 Jun 15;52(6):691-8. PubMed.
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005 Dec;19(14):2040-1. PubMed.
- Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis. 2010;20 Suppl 2:S535-50. PubMed.
- Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, Guo L, Chen D, Stern DM, Gunn Moore FJ, Xi Chen J, Arancio O, Yan SS. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J Neurosci. 2011 Feb 9;31(6):2313-20. PubMed.
- Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, Trillat AC, Stern DM, Arancio O, Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005 Apr;19(6):597-8. PubMed.
- Yao J, Taylor M, Davey F, Ren Y, Aiton J, Coote P, Fang F, Chen JX, Yan SD, Gunn-Moore FJ. Interaction of amyloid binding alcohol dehydrogenase/Abeta mediates up-regulation of peroxiredoxin II in the brains of Alzheimer's disease patients and a transgenic Alzheimer's disease mouse model. Mol Cell Neurosci. 2007 Jun;35(2):377-82. PubMed.
- Ren Y, Xu HW, Davey F, Taylor M, Aiton J, Coote P, Fang F, Yao J, Chen D, Chen JX, Yan SD, Gunn-Moore FJ. Endophilin I expression is increased in the brains of Alzheimer disease patients. J Biol Chem. 2008 Feb 29;283(9):5685-91. PubMed.
- Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA, Head E. Lack of pathology in a triple transgenic mouse model of Alzheimer's disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci. 2008 Mar 19;28(12):3051-9. PubMed.
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, Tsukagoshi-Nagai H, Horikoshi-Sakuraba Y, Mughal M, Rebeck GW, Laferla FM, Mattson MP, Iwata N, Saido TC, Klein WL, Duff KE, Aisen PS, Matsuoka Y. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008 Jun 24;1216:92-103. PubMed.
- Yao J, Irwin R, Chen S, Hamilton R, Cadenas E, Brinton RD. Ovarian hormone loss induces bioenergetic deficits and mitochondrial β-amyloid. Neurobiol Aging. 2011 Apr 22; PubMed.
- Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer's disease transgenic mice. J Neurosci. 2010 Oct 27;30(43):14299-304. PubMed.
Other Citations
Further Reading
Papers
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000 Mar 31;101(1):57-66. PubMed.
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011 May 25;31(21):7691-9. PubMed. RETRACTED
Panelists
-
 Edward B. Lee, M.D., Ph.D.
University of Pennsylvania
Edward B. Lee, M.D., Ph.D.
University of Pennsylvania
-
 Todd E. Golde, M.D., Ph.D.
Goizueta Institute @ Emory Brain Health
Todd E. Golde, M.D., Ph.D.
Goizueta Institute @ Emory Brain Health
-
 Gunnar Gouras, Dr.
Lund University
Gunnar Gouras, Dr.
Lund University
-
 Frank LaFerla, Ph.D.
University of California, Irvine
Frank LaFerla, Ph.D.
University of California, Irvine
Current Scientific Advisory Board Member -
 Virginia Lee, PhD
University of Pennsylvania
Virginia Lee, PhD
University of Pennsylvania
-
 Lars Nilsson, Ph.D.
University of Oslo
Lars Nilsson, Ph.D.
University of Oslo
-
 Robert Vassar, PhD
Northwestern University Feinberg School of Medicine
Robert Vassar, PhD
Northwestern University Feinberg School of Medicine
-
 David Holtzman, B.S., M.D.
Washington University
David Holtzman, B.S., M.D.
Washington University
Current Scientific Advisory Board Member
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.