CONFERENCE COVERAGE SERIES
AAT-AD/PD Focus Meeting 2018 - Advances in Alzheimer’s and Parkinson’s Therapies
Torino, Italy
15 – 18 March 2018

CONFERENCE COVERAGE SERIES
Torino, Italy
15 – 18 March 2018
Elusive as it has been for years, the goal of a blood test to detect Alzheimer’s disease appears suddenly achievable. First came the debut of two mass spectrometry methods and an ELISA that quantified Aβ in the blood reliably enough to pick out people with brain amyloidosis (Feb 2018 news; Dec 2017 conference news; Jul 2017 conference news). And now, researchers at the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting, held March 15–18 in Turin, Italy, have brought plasma p-tau181 into play, as well.
Dubbed AAT-AD/PD, the new conference merges the International Geneva/Springfield Symposium on Advances in Alzheimer Therapy with the International Conference on Alzheimer’s and Parkinson’s Diseases. It will be held every two years, alternating with AD/PD. The inaugural meeting convened 1,336 attendees from 64 countries to the former Fiat main factory building, which was reconfigured into a congress center in the early 1990s and still has a racetrack on its roof.
In Turin, researchers led by Charles S.Y. Yang at the biotech company MagQu Co., based in New Taipei City, Taiwan, presented a method that uses magnetic spin to detect minute quantities in blood of a given target protein, in this case tau phosphorylated at position 181. This enabled detection of consistent differences between healthy controls, people with prodromal AD, and those with dementia, Yang reported. Meanwhile, Michelle Mielke at the Mayo Clinic in Rochester, Minnesota, in collaboration with Jeffrey Dage and colleagues at Eli Lilly and Co., Indianapolis, described an ELISA-based method for quantifying plasma p-tau181 that also distinguished these diagnostic groups.

The rooftop racetrack on former FIAT manufacturing building, site of the AAT-AD/PD conference. [Photo: André P. Meyer-Vitali/Creative Commons.]
“Plasma p-tau181 appears promising, with an impressive area under the curve,” noted Kaj Blennow of the University of Gothenburg, Sweden, in an overview lecture on fluid biomarkers. Blennow is collaborating with Yang to validate his assay. Others at the meeting reacted to the data with enthusiasm. “This is what we’ve been waiting for,” said Hilkka Soininen of Kuopio University Hospital, Finland.
Few previous studies had examined plasma p-tau, although last year researchers reported that the marker distinguished people who had sustained head injuries from healthy controls better than did total tau in plasma. Likewise, Mielke and Dage found plasma total tau to be a dubious marker, reporting that its levels in healthy controls only weakly correlated with later cognitive decline and progression to AD (Sep 2017 news). Total tau is believed to reflect neuronal injury and neurodegeneration, whereas phosphorylated tau appears to be more specific for the pathology present in Alzheimer’s (Aug 2016 conference news).
Because neurofibrillary tangles contain hyperphosphorylated tau, p-tau levels probably rise early in the pathogenesis of AD, before tangles form, Yang suggested at AAT-AD/PD. The question is, can it be detected specifically in the blood? This presents a challenge because, at a few pg/ml, its levels are extremely low, about 10 times lower than plasma t-tau. To overcome this, Yang used a technique developed by MagQu called immunomagnetic reduction (IMR). The researchers coat magnetic particles with antibodies against the protein of interest, then mix these particles with a blood sample. When p-tau181 binds to the particles, its mass slows down their rotation in a magnetic field (see image below and company webpage). This change in rotation can be detected with a powerful magnetic sensor, the superconducting quantum interference device (SQUID). Yang said that spinning the particles so fast that non-specific antibody binding is removed allows the researchers to quantify only the protein of interest.

Antibodies bound to magnetic particles capture p-tau181 in plasma. The bound tau slows the particles’ spin, allowing detection of tiny quantities. [Courtesy of Charles Yang.]
Yang and colleagues used IMR to compare p-tau181 levels in 23 healthy controls, 29 people with mild cognitive impairment due to AD, and 21 people with mild AD. The method measured rising levels, from an average of 2.46 pg/ml in controls, to 4.41 in MCI and 6.15 in AD. The difference was significant, with a p value of 0.001, Yang said.
The method has potential as a diagnostic test, Yang asserted. A cutoff of 3.1 pg/ml separated controls from MCI with an area under the curve of 86 percent, while a cutoff of 5.7 pg/ml separated MCI and AD with an AUC of 78 percent. Moreover, p-tau181 better detected AD progression than other plasma biomarkers did. Using the same method, t-tau and the Aβ42/Aβ40 ratio each distinguished healthy controls from MCI, but not MCI from AD, Yang reported. These data appeared January 23 in Alzheimer’s and Dementia (Yang et al., 2018).
In Turin, the audience peppered Yang with questions. Some asked what forms of tau IMR detects; Yang replied that it cannot distinguish between tau monomers or oligomers. Others asked how plasma p-tau181 relates to other AD biomarkers, and Yang said his company is preparing studies in the U.S. and Japan to correlate p-tau181 with tau PET and serum neurofilament light (Nov 2017 news). He is currently repeating the findings in a Swedish cohort in collaboration with Blennow and colleagues.
For their part, Mielke and Dage took a different approach to measuring plasma p-tau. They used a sandwich immunoassay in which both antibodies against p-tau181 were monoclonals. This improved specificity over traditional ELISAs, Dage told Alzforum. He noted that the assay was able to detect p-tau181 in 97 percent of the tested plasma samples, with measurements varying about 8 percent between runs.
Following up on their earlier t-tau study, the researchers used this assay to compare plasma p-tau181 and t-tau levels in 172 healthy controls, 57 people with MCI, and 40 with AD participating in the Mayo Clinic Study on Aging. As did Yang and colleagues, they, too, found a statistically significant rise in plasma p-tau181 with advancing disease, from 5.1 pg/ml in controls to 5.7 in MCI and 11.8 in AD. Moreover, p-tau181 correlated with both amyloid and tau PET, Mielke reported in Turin. Plasma p-tau181 predicted the presence of brain amyloid with an accuracy of 75 percent. This is as good a predictor as using age and ApoE status, she noted. However, it is lower than current plasma Aβ tests, which detect amyloidosis with an accuracy of up to 90 percent. Mielke suggested p-tau181 could make a good screening measure for trials.
Plasma t-tau, on the other hand, correlated strongly with cortical atrophy but only weakly with tau PET. T-tau levels did not relate to a person’s diagnosis, and correlated with p-tau181 only among those who were amyloid-positive, Mielke said. Total tau in plasma predicted brain amyloid only 55 percent of the time, in line with previous findings that this marker flags degeneration and injury more generally, not AD pathology specifically.
Lilly is now replicating its p-tau and t-tau findings using samples from its clinical trials. In addition, the researchers plan to test the p-tau181 ELISA in non-AD tauopathies.—Madolyn Bowman Rogers
Remember the epitope debate around therapeutic Aβ antibodies? N-terminal versus mid-region versus C-terminal—which would be most effective? At the Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held March 15–18 in Turin, Italy, a similar conversation erupted for tau antibodies. Alzheimer’s researchers are increasingly targeting tau. Neurofibrillary tangle pathology correlates with cognitive decline, but tau also is thought to occur as small, misfolded aggregates that researchers want to catch to interrupt templated seeding and spreading through the brain. Several tau therapeutic antibodies are in development, from preclinical stages to Phase 2, and many groups are trying to figure out which antibody characteristics may work best. In Turin, multiple groups reported that antibodies directed against the middle region of tau stopped seeding of tau extracted from AD brain in cellular assays and mouse models. By contrast, antibodies against the N terminus—which are known to clear neurofibrillary deposits—poorly inhibited seeding and propagation of AD-derived tau in these models. Most tau antibodies currently in the clinic target the N terminus, causing hallway debates about their chance of success.
Other researchers cautioned that it remains unclear whether the finding will hold up in AD brain, which contains different forms of tau than do the cellular and mouse models used. “Ongoing clinical trials will start to give answers to this,” Marc Mercken at Janssen told Alzforum. Meanwhile, Luc Buee at the University of Lille, France, urged that more preclinical work be done in multiple experimental models of propagation before advancing antibodies to human trials. “We don’t want to go too fast and make mistakes,” Buee said.
Researchers believe that neurons pass misfolded tau to nearby cells, allowing the pathology to invade anatomically connected brain regions (Mar 2013 conference news; Aug 2013 news; Nov 2016 news). If so, antibodies might be able to intercept these toxic forms in the extracellular space, halting the advance of the disease. Several groups have generated antibodies that target extracellular, aggregated forms of tau, with those from Biogen, AbbVie, and Roche currently the farthest along in development (Apr 2017 conference news; Aug 2017 conference news). Complicating these efforts, however, is the fact that researchers still do not know exactly which species of tau transmit pathology.

Location, Location. Experimental antibodies “D” and “C,” which bind to the mid-region of tau (red and purple bands), are highly active in seeding assays, while N-terminal antibodies “A” and “B” (binding at blue and navy bands) act weakly. [Courtesy of Jean-Philippe Courade.]
To start to answer this question, researchers at UCB Biopharma in Brussels tested the efficacy of a broad spectrum of tau antibodies. In Turin, Jean-Philippe Courade described the strategy. Led by Martin Citron, the UCB team generated dozens of antibodies by immunizing animals with recombinant tau fibrils bearing different post-translational modifications, as well as paired helical filaments (PHFs) from AD brain. They purified 94 of these antibodies and used surface plasmon resonance to determine what region of tau each one bound. The researchers selected 51 for screening in a functional assay. For the assay, they expressed human P301S tau in HEK 293 cells and added pathological tau isolated from pooled human AD samples to trigger aggregation. Antibodies were added at the same time as the toxic tau. Two days later, the researchers measured the amount of fibrillar tau in the cells by fluorescence resonance energy transfer (FRET).
Only one of the antibodies, UCB0107, strongly suppressed aggregation, Courade reported. UCB0107 targets amino acids 235–246, which lie at the end of tau’s second proline-rich region, right before its microtubule-binding domain. UCB0107 dose-dependently inhibited fibrillization, achieving nearly 100 percent inhibition at an antibody concentration of 300 nM, Courade said. It performed equally well against pathologic tau isolated from progressive supranuclear palsy and frontotemporal dementia tissue samples, he added. UCB is advancing this antibody to the clinic and has started a Phase 1 trial.
Other antibodies only partially suppressed seeding. For example, one that targeted phosphorylated S202/T205 in the mid-region of tau potently lowered seeding, but reached a plateau at around two-thirds inhibition. This suggests that not all seeds are phosphorylated at this site, and points to heterogeneity in the phosphorylation of seeds, Courade said.
Notably, antibodies against the N-terminal region of tau had little effect on seeding, Courade said. The first wave of tau antibodies currently in trials target this region because they were chosen based on affinity, and antibodies tend to bind most strongly here. For a side-by-side comparison, UCB researchers synthesized antibodies with the same complementarity-determining region (CDR) as existing antibodies based on the patent literature and tested them in their seeding assay. They found partial activity for AbbVie’s C2N 8E12, but no activity for Biogen’s BIIB092, which recognizes N-terminal fragments of tau. “The highest-affinity antibodies were not the most efficacious in this assay,” Courade said. He believes the binding epitope is a more important factor for clinical efficacy than affinity.
Researchers at Janssen Pharmaceuticals in Beerse, Belgium, reported similar findings using a slightly different assay. They also generated a panel of antibodies from a variety of antigens, including recombinant aggregated tau and PHFs from AD brains. Unlike UCB, they used these antibodies to immunodeplete pathogenic tau from AD and transgenic mouse brain extracts. Then they added the immunodepleted extract to cells expressing P301S tau, and measured fibrillization.
At AAT-AD/PD, Janssen’s Kristof Van Kolen reported that antibodies against tau’s mid-region best removed pathogenic seeds, whereas antibodies to the N-terminal region of tau only weakly suppressed seeding. The difference was particularly pronounced for AD brain extract, less so for mouse brain extract.
Why don’t N-terminal antibodies stop seeding? “We believe this is due to N-terminal truncation of aggregated tau,” Mercken told Alzforum. The N-terminal tails stick out from the clumped core of tau fibrils and are known to be clipped off by proteases. This means the proteopathic tau seeds may not have the tau N-terminus anymore. Human tau fibrils may be particularly vulnerable to proteolysis because tangles linger for decades in the brain, giving the process more time to occur than in mouse brain, Mercken suggested. Still, he believes some percentage of aggregated tau retains its tails, explaining the partial activity of N-terminal antibodies.
The Janssen team did not report side-by-side comparisons in Turin, but told Alzforum that they saw some efficacy for both C2N 8E12 and BIIB092 in their assays, in contrast to UCB’s results. Janssen also took an antibody active against seeding into Phase 1.
The same pattern occurred in vivo, Mercken told Alzforum. The researchers injected AD brain material enriched for PHFs into the brain of a transgenic tau mouse to spur fibrillization. When they co-injected various antibodies, they again found that those that recognized the mid-region of tau were most effective, while N-terminal antibodies could not fully block seeding.

Preventing Seeding. AD brain lysate triggers accumulation of toxic tau (brown, stained with AT8) in the hippocampi of tau transgenic mice (right). This is reduced by intraperitoneal treatment with mid-region tau antibody UCB0107 (left). [Courtesy of Luc Buee and UCB.]
Buee and colleagues reported similar findings from mouse models. The French researchers injected pathological tau from AD brain into the hippocampi of young tau transgenic mice to trigger pathology. This was followed by six weekly intraperitoneal tau antibody injections. In Turin, Morvane Colin in Buee’s group said that a mouse version of UCB0107 prevented tau aggregates from forming.
While the results suggested that UCB0107 inhibits seeding in vivo, it was unclear if the antibody could also stop the spread of misfolded tau once present, Colin noted. To tease this out, the researchers used a different mouse model. In this paradigm, they injected an aggregated tau fragment, K18, which consists of only the microtubule-binding domain. Because UCB0107 does not recognize this fragment, it could not prevent these seeds from triggering local tau aggregation. But injected UCB0107 did prevent tau fibrils from appearing on the opposite side of the brain. “This suggests the antibody stops the spread of pathogenic species,” Colin told Alzforum.
“The message from these studies is that you have to characterize where on tau therapeutic antibodies bind,” Rakez Kayed of the University of Texas Medical Branch, Galveston, told Alzforum. While the first-generation therapeutic tau antibodies were mostly N-terminal, newer ones typically bind the mid-region, he added. One old mid-region antibody is MC1, generated as an experimental tool by Peter Davies of the Litwin Zucker Center for Alzheimer's Research, Long Island, New York (Jicha et al., 1997); it is now being developed as a therapeutic in collaboration with Eli Lilly.
However, speakers were careful to emphasize that it is unclear whether these findings will translate to human brain. The brain lysates used in these assays contain both intracellular and extracellular tau, and so may not resemble the seeds that spread Alzheimer’s pathology, Mercken said. Figuring out which forms of toxic tau are extracellular will be crucial for therapy. For example, Mercken speculated that toxic S422 p-tau may remain intracellular, perhaps explaining the failure of the Roche RG7345 antibody that targeted this form. Buee raised a different concern, suggesting that some of the N-terminal truncation of tau seen in the postmortem human and mouse brain lysates might occur after death. If so, N-terminal antibodies could be more effective in patients than in these assays, he suggested.
Tau mouse models have another limitation, Randall Bateman of Washington University in St. Louis pointed out. They express tau mutations, such as P301S, that predispose people to frontotemporal dementia, not AD. Thus, they may not model the tau pathology present in Alzheimer’s disease, Bateman said. He noted that a robust AD tauopathy model is a needed tool that has yet to be developed. In the meantime, he suggested using models that express wild-type mouse tau.
John Trojanowski and Virginia Lee of the University of Pennsylvania, Philadelphia, have found that, just as was seen in transgenic animals, injection of toxic tau aggregates into wild-type mice causes misfolding and propagation of endogenous tau along the anatomical connectome of the injection site (Nov 2017 news). Trojanowski and Lee reported further details of these studies at AAT-AD/PD.
In addition, factors other than epitope affect the therapeutic efficacy of a given antibody. Mercken said that, in his team’s hands, antibodies against specific tau phosphorylation sites had different activity for human and mouse brain lysate. “That has to be taken into account if you want to progress a phosphorylation-specific antibody to the clinic,” he told Alzforum.
Finally, some antibodies recognize monomeric tau as well as aggregates. In Turin, Trojanowski cautioned that antibodies that bind tau monomers, as does UCB0107, might have to be administered at high doses to prevent physiological tau from soaking up all of the therapeutic. Mercken agrees this is a concern, noting that Janssen has selected an antibody that recognizes phosphorylated PHFs, but not physiological tau, to take into Phase 1. However, Courade suggested that monomeric tau exists at low concentration in the extracellular space, making this less of an issue.—Madolyn Bowman Rogers
Some ongoing prevention trials enroll people carrying familial Alzheimer’s disease mutations because they are destined to develop the disease on a predictable timescale. But will findings from these trials generalize to the vastly larger sporadic AD population? In other words, is dominantly inherited AD the same or different from sporadic AD? And what role does aging play? These questions spark debate whenever they come up, and researchers with the Dominantly Inherited Alzheimer Network set out to answer them once and for all. They are comparing the DIAN dataset to that from observational studies of late-onset disease. At the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held March 15–18 in Turin, Italy, John Morris and Randall Bateman at Washington University in St. Louis reported preliminary data from the study. The findings to date indicate that despite having different causes, familial and sporadic AD on the whole share the same pathophysiology and progression. The few distinctions between the diseases are mostly due to the older ages of people with sporadic AD, and the quixotic effects of particular AD mutations, the scientists noted.
“We are cautiously optimistic that lessons learned from DIAN will translate to LOAD,” Morris said in Turin. This has big implications, because it suggests that a mechanism-based drug that proves its mettle in a DIAN study could be approved for sporadic disease.
One original aim of the DIAN project, which started enrolling in 2008, was to compare the features of symptomatic AD in dominantly inherited and the common late-onset sporadic populations, Morris noted (Nov 2008 news series). Both diseases are marked by altered Aβ42 homeostasis, but autosomal-dominant disease is caused by both an overproduction and impaired clearance of the peptide, while sporadic is believed to be due to impaired clearance (Jul 2010 conference news; Dec 2010 news; Jun 2013 news).

Autosomal-dominant and sporadic AD have different triggers, but converge on the same pathophysiologic process. [Courtesy of Randall Bateman.]
An obvious comparison for the DIAN data set is ADNI, the longitudinal observational study of late-onset disease that started in 2004 (Mar 2005 conference news; Oct 2008 news series). DIAN’s initial protocols were based on ADNI’s to facilitate comparisons. However, over time, protocols in both studies diverged a bit. For one, ADNI replaced PiB amyloid imaging with florbetapir, while DIAN stuck with PiB. For another, the field has evolved, as in the move toward automated, more reliable cerebrospinal fluid assays (Aug 2015 conference news; Dec 2017 conference news). As a result, the data cannot be directly compared as originally gathered.
For this reason, researchers at DIAN and ADNI recently launched a joint effort to harmonize data from the two studies. They reprocessed all imaging scans using the centiloid scale, which renders a simple unit value for each scan and enables side-by-side comparisons between different tracers (Feb 2013 conference news; Nov 2014 news). They reanalyzed all CSF samples on the Roche Elecsys automated platform, and also compared Aβ isoforms by mass spectrometry, Morris said.
The researchers faced another challenge in comparing cohorts, namely the mean 30-year age difference between DIAN and ADNI participants. Because participants could not be matched by age, the researchers decided to match them instead by dementia severity. Progression to a CDR sum of boxes score of 1 indicated a person’s year of symptom onset, and served as the anchor point for the comparison. For some participants, these data were directly available, whereas for others who were already symptomatic when they enrolled, the researchers extrapolated back to the likely year of onset. For asymptomatic DIAN participants, researchers used the estimated year of onset based on their affected parent’s age at onset.

Faster Accumulation. People with a familial AD mutation (red lines) experience more aggressive amyloidosis than do people with sporadic AD (blue lines). [Courtesy of John Morris.]
The analysis comprised 559 ADNI participants and 302 mutation carriers in DIAN. Besides age, researchers noted a few demographic discrepancies between the cohorts. ADNI participants had more years of education than DIAN participants on average, and they were all from North America whereas DIAN is worldwide. In addition, ADNI, having been funded earlier, has more years of follow-up.
Despite these differences, most clinical and biomarker data were indistinguishable between the groups, Morris said. This is the main finding of the comparison study. Clinically, both diseases primarily involved defects in memory and executive function. Brain atrophy and hypometabolism occurred with similar patterns in each group. CSF biomarkers moved in the same ways, except the drop in CSF Aβ42 prior to symptom onset was steeper in DIAN than LOAD.
Ron Thomas of the University of California, San Diego, who heard the talks in Turin, agreed that the curves of biomarker change in autosomal dominant AD and LOAD resemble each other. “There is substantial similarity between the cascade of processes,” he wrote to Alzforum.
One difference cropped up in amyloid PET imaging, namely more deposition in the basal ganglia in people with AD mutations compared to sporadic AD. This has been reported previously, and occurs with specific presenilin 1 mutations (Jun 2007 news; Aug 2017 conference news). In addition, people with familial AD tended to accumulate a heavier amyloid burden than those with sporadic disease (see image above). Tau PET was only recently added to these studies and is still being analyzed, Morris noted.
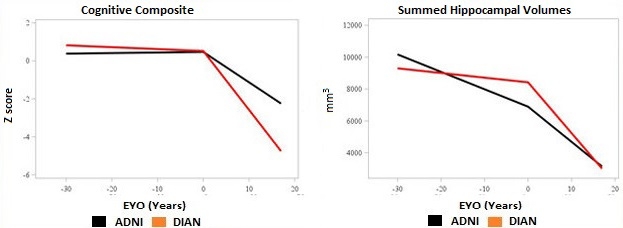
Earlier, Slower Decline. People with sporadic AD (blue lines) have some age-related decline before symptoms appear, but then progress more slowly than those with an AD mutation (red lines). [Courtesy of John Morris.]
The other discrepancies between the cohorts were attributable to age, Morris said. ADNI participants tended to have more brain shrinkage and cognitive decline prior to symptom onset than did DIAN participants, as might be expected in older adults. However, after symptom onset the pattern reversed, with DIAN participants experiencing steeper declines in brain volume and memory, Morris noted (see image above). “Symptomatic AD progresses more aggressively in dominantly inherited cases than in LOAD,” Morris said. This resembles the pattern seen in people with high cognitive reserve, who maintain cognitive function to a point of heavy amyloid load but then decline more quickly than people with low reserve.
Neuropathology data from participants who came to autopsy also exposed the aging component of LOAD. The younger DIAN participants developed pure AD pathology, whereas ADNI participants had comorbidities such as TDP-43 accumulation, argyrophilic grain disease, age-related tau astrogliopathy, hippocampal sclerosis, or vascular infarcts. Notably, in addition to amyloid plaques and neurofibrillary tangles, both DIAN and ADNI participants frequently developed α-synuclein deposits. Lewy bodies were present in 44 percent of ADNI cases and 54 percent of DIAN. This suggests that α-synuclein deposition is linked mechanistically to AD pathophysiology, Bateman noted. “There is important biology going on here that we do not understand,” he said in Turin.
Overall, dominantly inherited and sporadic AD are not identical, but they progress in the same way and have all the same core features, such that the diseases are difficult to distinguish clinically, Morris concluded. One indication of this is that a small fraction of people with late-onset AD turn out to have a familial mutation that went unrecognized. In fact, Bateman estimated that there might be 10 times as many mutation carriers with late-onset than early onset disease. In clinical practice, a 75-year-old person with symptomatic AD is much less likely to get tested for APP or presenilin mutations than a 50-year-old, but when scientists do look for mutations in aged cohorts, they tend to find some.
How will trials of LOAD deal with this? Samantha Budd Haeberlein of Biogen told Alzforum the company does not test for familial AD mutations when enrolling participants in aducanumab Phase 3 trials. Participants may be as young as 50, hence these trials may well include some carriers. When the trial data are analyzed, Biogen researchers will examine whether carriers responded to the treatment in the same way noncarriers did, Budd said.
DIAN researchers are comparing their dataset to other sporadic disease data, as well, including the National Alzheimer’s Coordinating Center (NACC) data set and the Adult Children Study at WashU, Morris noted. Participants in the latter are of a similar age to DIAN participants, removing one source of variation. Those data will be published soon. Virginia Buckles at WashU presented some preliminary findings at the 2016 Alzheimer’s Association International Conference in Toronto (Aug 2016 conference news).

Slow Clearance in ADAD/LOAD. Once amyloidosis sets in, mutation carriers (red line, left graph) clear Aβ42 as slowly as do people with sporadic late-onset AD (red line, right graph). [Courtesy of Randall Bateman.]
Also in Turin, Bateman presented data from stable isotope labeling kinetics (SILK) studies, which reveal the dynamics of Aβ production and clearance. SILK shows that Aβ isoform clearance slows by 2.5-fold from young adulthood to old age for everybody, regardless of mutation status, Bateman said. On the production side, mutation carriers make about 25 percent more Aβ42 than noncarriers throughout life. However, once amyloidosis starts, clearance slows identically in both carriers and noncarriers, Bateman stressed (see image above). Though the diseases start differently, their pathophysiology merges very early in the disease course, 15 or 20 years before symptoms emerge, he said. After symptoms appear, the disease lasts for about 10 years in either case.
The data suggest that treatments for dominantly inherited AD are likely to translate to LOAD. By way of an analogy, Bateman noted that statins work extremely well to prevent atherosclerosis and heart attacks in the rare cases of dominantly inherited familial hypercholesterolemia, and statins also work reasonably well in the general population in which this disease evolves more heterogeneously with an added aging component.
Thomas called the findings from these analyses important. “It would be a major step forward for AD research if autosomal-dominant AD can be considered as the same disease process, albeit accelerated, as LOAD,” he wrote.
Now if only the AD field had an effective drug. Several, such as Lilly’s solanezumab, Janssen’s BACE inhibitor, and Roche/Genentech’s crenezumab are currently being tested in both populations, allowing direct comparison of their effects.—Madolyn Bowman Rogers
No Available Comments
The advent of PET to image Alzheimer’s pathology in people has handed researchers new tools for staging disease during life, but PET also presents mysteries. At the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD) March 15–18 in Turin, Italy, Swedish researchers introduced a new method for analyzing amyloid accumulation in brain networks that could become an extremely early biomarker in people who have not yet reached the brain-wide cutoff for amyloid positivity. Other speakers debated an unexpected conundrum in tau PET, where older AD patients have less tau signal than younger ones. Do other pathologies fuel dementia in older people, or does the finding reflect a difference in how the brain handles tau with age? Meanwhile, other Swedish researchers showed the latest on an antibody-based PET ligand that detects oligomeric Aβ. The technique remains preclinical, but new data demonstrates that the ligand can detect a drop in Aβ after mice are treated with a BACE inhibitor. Beyond these three news flashes, AAT-AD/PD featured no major developments on PET, although longitudinal and multi-biomarker studies are ongoing.
Can the Pattern of Amyloid Deposition Predict Progression?
Previous work from researchers at Lund University, Sweden, indicated that Aβ42 levels in cerebrospinal fluid drop a few years before amyloid PET scans reach the brain-wide cutoff for positivity (Aug 2016 conference news). But PET studies have also shown that the brain accumulates patches of amyloid here and there before that brain-wide threshold is reached. Joana Pereira at Karolinska Institute in Stockholm wondered if this regional PET amyloid accumulation could become a biomarker in amyloid CSF–positive but PET-negative people. In collaboration with the Lund group, Pereira examined data from ADNI2 participants. In this cohort, 291 were CSF–/PET–, 81 CSF+/PET–, and 272 CSF+/PET+. She compared their amyloid PET to MRI scans to determine where in their brains amyloid accumulated, and used graph theory to analyze network relationships between 72 different regions.
In Turin, Pereira reported that amyloid burdens in particular brain regions correlated more closely with each other in CSF+ people than in controls. This correlation suggests a connection between these regions, Pereira noted. She saw this increased connectivity primarily for regions within the default mode network (DMN), while regions outside the DMN had less correlated amyloid loads than in controls.

Amyloid Network. Red dots indicate regions with early amyloid accumulation, blue lines the connections between them. [Courtesy of Joana Pereira.]
Pereira also examined whether the correlations between regions were direct, or whether two given regions were connected through a third region. This measure is called network path length. Network paths within the DMN were shorter in CSF+/PET– people than in controls, she found, indicating a more direct connection between regions with high amyloid burden. This suggests amyloid is propagating through connected regions, Pereira said. In CSF+/PET+ people, network paths were shorter both within and outside the DMN.
Then Pereira analyzed a third measure. Clustering quantifies whether regions with correlated amyloid loads are close to each other in the brain. Here she saw no difference in CSF+ people. This, Pereira noted, implies that amyloid tends to accumulate in distant rather than neighboring brain regions; in other words, that the propagation happens via functional connections.
Of the three measures—connectivity, path length, and clustering—changes in network path length best discriminated between AD stages, Pereira said. She suggested this network measure could help pick out people on the path to AD and assess disease progression.
In addition, Pereira analyzed whole brain networks, and identified a subnetwork made up of regions that showed changes in connectivity and path length (see image above). This subnetwork expanded to include more regions in people with more amyloid pathology, again suggesting propagation along brain connections. It starts in medial regions, and spreads to lateral and frontal regions later in the disease. “This subnetwork could be more vulnerable to the effects of amyloid,” Pereira told Alzforum. The data were published in the January Cerebral Cortex (Pereira et al., 2018).
“This is very impressive data,” commented session co-chair Flavio Nobili of the University of Genoa, Italy. Many researchers asked technical questions. Some wanted to know how the amyloid network correlated with structural and functional changes. Pereira said she sees little change in brain volume at this early stage of the disease, when people are still 15 to 20 years away from developing symptoms. She is analyzing functional MRI data now. Previously, she found that the loss of structural connectivity correlated with neurodegeneration, but not amyloid, in the Swedish BioFinder cohort (Pereira et al., 2017). Other work by the Swedish group has linked nascent amyloid accumulation in regions of the default mode network (DMN) with a loss of functional connectivity within that network, and between the DMN and other networks (Nov 2017 news).
Tau Pathology Looks Different in Middle-Aged and Elderly
Researchers have had less time to image tau than amyloid, and emerging findings still hold surprises. In Turin, Michael Weiner of the University of California, San Francisco, presented the latest tau PET data from ADNI, which by now has 240 baseline and 76 follow-up scans with AV1451. Tauopathy is believed to accelerate during symptomatic disease and for many years hopes were high that tau would be a straightforward progression marker. But surprisingly, in ADNI the tau PET signal was not higher in people with AD than in those with mild cognitive impairment. This finding appears to be age-dependent, Weiner noted. The average age of these ADNI participants was 79, whereas in a cohort of UCSF patients with an average age of 63, the tau signal did shoot up at the dementia stage (Maass et al., 2017). What is going on here? The answer is unknown, but will be important, Weiner said. He suggested the older group may have pathologies other than AD that contribute to their dementia.
Simon Lovestone at Oxford University added to this discussion when he presented preliminary findings from a pilot study of 24 people with mild AD, who agreed to undergo frequent extensive testing on a wide range of clinical and biomarker measures. The participants took clinical and cognitive batteries, gave samples of cerebrospinal fluid, blood, urine, and saliva, and underwent multiple brain-imaging techniques, including MRI, amyloid and tau PET, and EEG. They wore devices that recorded their gaits and navigation, and completed telephone assessments of verbal fluency, episodic memory, mood, and sleep patterns. Why gather so much data? There has long been dissatisfaction with the spotty, even misleading, results coming out of studies where measures that can be quite variable from day to day are taken but a few times, many months or a year apart (see Dec 2017 conference story). New approaches are trying to measure outcomes much more frequently or even continuously. The ultimate goal is to find biomarkers that could serve as outcome measures in secondary prevention trials, Lovestone said, and his current pilot study addressed whether it is feasible to collect this much information. The answer was a resounding yes, as the participants completed nearly all of the roughly 7,000 data points requested, Lovestone reported.
In his study, too, Lovestone was surprised to see that a participant’s tau burden was lower the older he or she was. He agreed with Weiner that dementia in older people could be due to factors other than AD pathogenesis; however, he also noted that older people appeared more susceptible to the toxic effects of tau, showing more cognitive impairment than a younger person with the same tau burden (Koychev et al., 2017). Thus, older people with high tau pathology simply may not show up in a group selected for mild dementia, he suggested.
Keith Johnson of Massachusetts General Hospital, Boston, agrees the most parsimonious explanation for the data is a survival effect, where older people with advanced tau pathology are more likely to die or progress to later disease stages, and thus are underrepresented in studies. He noted that amyloid accumulation follows a similar pattern as tau, with a robust age dependence that flattens out at old age and even dips slightly in the oldest old, supporting this idea. The age dependence of tau PET could complicate the interpretation of clinical trial data, and might require careful age balancing of treatment groups, Johnson wrote to Alzforum. Many current AD treatment trials include people ranging from age 50 to 80 in a given group.
For his part, William Jagust at the University of California, Berkeley, believes the age difference in tau PET scans may be saying something fundamental about the biology of tau. Jagust noted that several groups have found an inverse relationship between age and tau pathology. Research groups at UC Berkeley, UCSF, and in Sweden recently reported that younger AD patients have widespread neocortical tau, while older patients accumulate more focal tau in the hippocampus and medial temporal regions (Ossenkoppele et al., 2016; Schöll et al., 2017). Moreover, tau PET correlates more strongly with cognitive decline in younger than in older people (Aug 2017 conference news).
“[These findings] are part of a larger story about age and AD that has been around for decades,” Jagust wrote to Alzforum. He notes that AD tends to present slightly differently in younger and older people. People with early onset AD develop more cortical hypometabolism than do late-onset cases (Mielke et al., 1992; Kim et al., 2005). Because tau pathology correlates with metabolic decline, it is not surprising for PET to see more cortical tau in younger patients as well, Jagust said. He believes this reflects a distinction between typical late-onset AD and a hippocampal-sparing form of the disease that affects mostly younger people (Murray et al., 2011). “The phenomenology fits together and is quite consistent, but the biology is unexplained,” Jagust noted.

See Oligomers in Vivo? A new PET tracer detects a drop in this form of Aβ in mouse brain after treatment with BACE inhibitor NB-360. [Courtesy of Silvio Meier and Dag Sehlin.]
PET Ligand Detects Treatment Effect on Soluble Aβ
While tracers for fibrillary amyloid and tau paired helical filaments are being used more widely, scientists are working to develop new ligands for other players in AD pathogenesis. Researchers at Uppsala University, Sweden, previously introduced an antibody directed against oligomeric forms of Aβ that could detect soluble protofibrils in transgenic mice (Feb 2016 news; Sehlin et al., 2017). The tracer is based on the murine version of BioArctic’s BAN2401 antibody.
In Turin, Dag Sehlin at Uppsala showed new data. Silvio Meier in his group treated Tg-ArcSwe mice, which generate high levels of oligomeric peptide, with Novartis’ NB-360 BACE inhibitor from 10 to 13 months of age. At this age, the PET signal rises rapidly in untreated controls. In response to treatment, however, the PET signal dropped by about half, bringing it down to near baseline (see image above). The researchers confirmed this visual drop in soluble Aβ by also directly measuring the peptide in brain tissue of treated and untreated mice.
The findings suggest that the oligomeric tracer can detect a treatment effect, Sehlin noted. However, he said that the mice had few plaques at the tested age, leaving unclear whether the tracer could still detect a drop in soluble Aβ at more advanced stages of plaque pathology. In addition, technical hurdles remain before this tracer can be tested in people.—Madolyn Bowman Rogers
No Available Comments
In the last few years, numerous studies have reported that dementia incidence is falling in the developed world. The drop in new cases has been attributed to better public health, in particular improved blood pressure control, but this conclusion may not tell the whole story. At the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held March 15–18 in Turin, Italy, Walter Rocca of the Mayo Clinic in Rochester, Minnesota, summarized recent research from his group and others that suggests the incidence of neurodegenerative diseases such as Parkinson’s, amyotrophic lateral sclerosis, and early onset Alzheimer’s is going up, not down. The reasons are unclear but could be environmental factors. Rocca argued this trend may extend to late-onset AD as well. He attributed the drop in all-cause dementia in recent decades to fewer cases of vascular dementia and stroke, which he thinks could be masking an increase in pure AD pathology.
Other data at AAT-AD/PD supported the related idea that vascular factors drive much of the risk of dementia in the very old. Claudia Kawas of the University of California, Irvine, reported findings from her 90+ study that found vascular disease conferred twice as much dementia risk in the oldest old as did having Alzheimer’s pathology. Together, the data highlight the role of vascular pathology in brain health, but also the need to distinguish between underlying biologic mechanisms of neurodegeneration, and to reduce risk factors behind each mechanism in order to improve public health.
Much has been made of studies in the U.S. and Europe that have found lower rates of dementia from about the year 1990 onward. The average drops about 25 percent over a decade in most studies (May 2013 news; Feb 2016 news; Nov 2016 news). Much of the improvement appears to stem from better cardiovascular health, although more education, less smoking, and other lifestyle changes have also been credited (Jul 2014 news; Apr 2016 news; Sep 2017 news). Importantly, these epidemiological studies did not distinguish between AD and other causes of dementia.
In Turin, Rocca warned that at the same time researchers are seeing these encouraging dementia trends, they are watching the incidence of parkinsonism rise. His group found that from 1976–2005 in Minnesota, the incidence in men increased by 30 percent (Jul 2016 news). Studies in Finland and Taiwan report similar numbers, with increases of 10 percent per decade (Isotalo et al., 2017; Liu et al., 2016). At the same time, new cases of ALS have climbed in Denmark (Seals et al., 2013).
Why is this happening? Scientists do not know. Epidemiological studies have found an inverse correlation between PD and cigarette smoking, smokeless tobacco use, and exposure to secondhand smoke, hinting that something in tobacco could be protective (Li et al., 2015; O’Reilly et al., 2005; Searles et al., 2012). If this is the case, falling smoking rates could be fueling a rise in PD, some researchers propose. For his part, Rocca mentioned head trauma, infectious agents, or alcohol consumption as possible culprits for the PD surge (Jul 2016 news; Dec 2016 news).
In particular, exposure to pollutants and pesticides has been implicated in Parkinson’s and ALS risk, and Rocca believes this may be the major reason for the increase he sees (Apr 2011 news; Lee et al., 2016; May 2016 news). Agricultural pesticide use surged after World War II, meaning more people now consume these substances in their diet, Rocca told Alzforum. Urban crowding and exposure to pollutants have also increased.
Could pollutants and pesticides bump up AD incidence as well? AD has been linked to pesticide exposure (Jan 2014 news), but evidence is scarce. One bit comes from a Canadian study, which reported a 24 percent rise in dementia in people between the ages of 50 and 64 from 2002–2013 (Cerasuolo et al., 2017). Since younger people with dementia typically develop a pure neurodegenerative pathology such as AD or frontotemporal dementia, without other age-related disorders or vascular pathology, this hints that AD could be climbing, Rocca said.
The drop in all-cause dementia seen epidemiologically could be explained simply by better cardiovascular health, Rocca suggested. Stroke and heart disease have been on the wane for up to 50 years, and Canadian researchers report drops of up to 40 percent in new cases since 2002 (Jones and Greene, 2013; Sposato et al., 2015). Stroke and vascular disease may account for a significant portion of dementia, Rocca said. One large neuropathological study of 5,715 brains in the National Alzheimer’s Coordinating Center database found signs of vascular disease in 60 percent to 80 percent of all dementia cases (Toledo et al., 2013).
In Turin, Kawas’ presentation bolstered Rocca’s view. Her 90+ study has 1,603 participants, 76 percent of whom are women. Their mean age is 96; one-third each are cognitively healthy, mildly impaired, or have dementia. So far, 309 have come to autopsy. Kawas reported that only 60 percent of participants with dementia had plaques and tangles, not much higher than the 42 percent of non-demented people who did. These non-demented people with high amyloid burden tended to have protective factors, such as an ApoE2 allele or high education, Kawas noted.
Overall, having AD pathology about doubled a very old person’s risk of having dementia, but other conditions conferred far more dementia risk. Vascular disease quadrupled it, Lewy body disease jacked up the odds sixfold, and hippocampal sclerosis 10-fold. In fact, hippocampal sclerosis accounted for one-third of dementia cases in this cohort. This condition correlated with having thyroid disease and thyroid antibodies in the blood, Kawas said. In the oldest old, pathologies other than AD account for much of the dementia risk, she concluded, estimating that eliminating AD pathology entirely would only halve the number of dementia cases in the oldest old.
“The dementia developing de novo in this group is very different in causes and manifestations from dementias with earlier onset,” Rocca noted. “This important work supports the hypothesis that improvements in cardiovascular health may be a major hope for the prevention of dementia.”
Will population dementia rates continue to fall? It’s hard to predict, Rocca said. Worsening obesity and diabetes may offset gains from better cardiovascular health, while factors such as pesticide use may drive an increase in pure neurodegeneration, he argues (Rocca, 2017). Even if incidence falls, prevalence will continue to rise worldwide due to the aging population and less access to healthcare and to higher education in the developing world (Aug 2015 news; Oct 2016 news; May 2017 news).
Rocca compared epidemiologic trends to a mobile blowing in the wind—a relatively small perturbation can cause it to flip around. “Disease risks change over historical epochs, and the trends reflect complex interactions. It’s a delicate balance,” he said.—Madolyn Bowman Rogers
No Available Comments
Current treatments for Parkinson’s disease target motor symptoms, and do nothing to halt progression or address other aspects of the disease. To get beyond this state of affairs, researchers are targeting the underlying disease pathology. They take guidance from genetic studies that have highlighted the major molecular players, such as α-synuclein, GBA, LRRK2, and tau. At the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held March 15–18 in Turin, Italy, speakers debated therapies aimed at all of these molecules. Several treatments are in or headed toward the clinic, and most could potentially work for dementia with Lewy bodies (DLB) as well. Researchers agreed the therapies—if they work—mark a sea change for synucleinopathy treatment.
“We are at a watershed moment for PD trials,” said Jesse Cedarbaum of Biogen. “If the drugs we’re developing work as we hope, they should address non-motor as well as motor symptoms.” Showing they do, however, will require new outcome measures to track change. “We have to rethink the clinical trial playbook,” Cedarbaum said.
Johan Luthman of Eisai stressed that PD is a multisystem disorder. “We need to get away from thinking of PD as a disease of dopamine cell loss, and instead think of it as a synucleinopathy,” he said. This shift in the PD field parallels the evolution in Alzheimer’s research, where scientists moved their focus from cholinergic deficits to the molecular pathogenesis of Aβ and tau.
Pharma Crosshairs Locked on α-Synuclein
In this conception of PD, preventing α-synuclein pathology is a primary approach. Several groups have designed antibodies they hope will arrest the spread of misfolded protein from cell to cell, with Biogen and Roche’s furthest along. Biogen did not present in Turin, but is recruiting for a Phase 2 trial of its antibody BIIB054, which targets aggregated α-synuclein. Biogen researchers will present Phase 1 data at the American Academy of Neurology annual meeting in Los Angeles later this month (May 2017 conference news). Meanwhile, Roche’s RG7935/PRX002, developed in partnership with Prothena, has also completed Phase 1 (May 2017 conference news) and did present in Turin. Based on pharmacokinetic data from this trial, the company is confident it is reaching the brain concentration necessary for target engagement, said Frank Boess of Roche.
Boess described the plan for the Phase 2 trial, dubbed PASADENA. A multicenter trial in the U.S., Austria, France, Germany, and Spain, it will enroll 300 people who have been diagnosed with PD within the last two years but are not yet taking dopaminergic medication. They will be between 40 and 80 years of age, and can be on a monoamine oxidase-B inhibitor such as rasagiline or selegiline.
Roche chose this population based on clinical data from the Parkinson’s Progression Markers Initiative and other studies that measured rates of change on the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS). “We think at this point in disease there is the highest dynamic change in PD symptoms measured by the MDS-UPDRS, so we have a high chance of detecting a reduction in disease progression,” Boess said.
For the first year, one-third of the cohort will receive placebo and one-third a low dose of antibody, 1,500 mg. The remainder will take a high dose, which will vary by body weight—3,500 mg for people under 65 kg; 4,500 for those 65 kg or heavier. The high dose is roughly equivalent to the highest dose tested in Phase 1, 60 mg/kg, but transformed into flat doses to simplify administration, Boess said.
The primary outcome measure will be change on the MDS-UPDRS after one year; secondary measures include decline in DaT-SPECT imaging signal, which detects loss of dopaminergic synapses, the time before a participant starts symptomatic dopaminergic treatment, as well as other clinical measures, and drug pharmacokinetics. Researchers will monitor clinical features such as tremor, bradykinesia, gait, and dexterity using smartphones as an exploratory measure. Phase 1 data suggest this technology outperforms paper-and-pencil tests or clinician assessments (May 2017 conference news).
After one year, placebo patients will cross over to one of the two active doses; patients and researchers will remain blinded to dose and the trial will run for an additional year. The FDA recommends this type of crossover design as a way to detect disease modification. The idea is that if the placebo group fails to catch up to the benefit seen with active treatment, then that suggests the treatment slows disease progression (Mar 2018 news).
Vienna-based AFFiRiS AG completed Phase 1 testing of its Affitope PD03A vaccine, a peptide designed to generate antibodies against α-synuclein. In Turin, Werner Poewe of Innsbruck Medical University, Austria, who ran a clinical site, presented data showing that this one-year trial enrolled 36 early stage Parkinson’s patients who were on dopamine medication. They received four subcutaneous shots of either 15 mg or 75 mg antigen or placebo, at monthly intervals, and a booster at nine months. The trial met its safety and immunogenicity goals, Poewe said. Participants tolerated the injections, and no safety concerns or signs of encephalitis on MRI scans cropped up. The antigen provoked an immune response, with antibodies peaking one month after the third injection, and jumping again after the booster. The antibodies bound to α-synuclein in vitro, Poewe added. As shown also in Turin, a second vaccine, Affitope PD01A, performed similarly in a Phase 1 trial of 24 PD patients.
GBA Bears Blame for Some of the Worst Parkinson’s Cases
Other researchers are taking a different tack against PD pathology. Mutations in the glucocerebrosidase (GBA) gene occur in about 10 percent of Parkinson’s patients, and generally come with earlier onset, faster decline, and higher odds for psychosis or dementia. GBA accounts for a similar proportion of DLB cases. The mutations lower the activity of GBA’s protein product, the enzyme Gcase, by about two-thirds. This leads to a buildup of sphingolipids, in turn causing α-synuclein levels to rise. In a vicious cycle, aggregated α-synuclein may further depress Gcase activity, though the mechanism behind these relationships is unclear, Pablo Sardi of Sanofi Genzyme said.
Intriguingly, people with idiopathic PD and DLB also have weak Gcase activity, suggesting therapies that target this enzyme might help them, too (Gegg et al., 2012; Chiasserini et al., 2015). Low levels of Gcase activity correlate with earlier disease onset and worse non-motor symptoms. Sanofi has a GBA therapy in Phase 2, but did not present data in Turin (Dec 2016 conference news).
Other researchers are developing the cough medicine Ambroxol as a GBA therapy. In Turin, Anthony Schapira of University College London explained that Ambroxol seems to act as a chaperone, binding mutant GBA in the endoplasmic reticulum and escorting it through the Golgi apparatus to the lysosome for degradation. In cellular and animal models of PD, the drug lowered α-synuclein levels, he said. Ambroxol is in Phase 2 PD trials in Canada and London, with a Phase 1 trial starting in Florida.
Schapira led the London trial, AIM-PD, which enrolled 12 GBA carriers and 10 people with idiopathic PD to look at safety and establish proof of principle. Participants took 1,200 mg/day of Ambroxol for six months. About 10 percent of the drug entered the central nervous system, based on the serum:CSF ratio. Blood levels of Gcase rose 30 percent in GBA carriers and 15 percent in those with idiopathic PD, suggesting the drug was hitting its target, Schapira said. Participants tolerated the treatment well, with no serious safety issues. In answer to an audience question, Schapira said that Ambroxol seems to help all GBA mutations tested so far. This pilot study collected efficacy data as an exploratory outcome measure, but Schapira did not discuss it in Turin. A larger Phase 2 study is enrolling. Nir Giladi of Tel Aviv University praised the target engagement signal in the AIM-PD study. “We are in a really exciting time. Our ultimate aim is to detect people at risk, treat early, and prevent symptoms altogether,” Giladi said in Turin.
Other Genetic Targets Moving Into Trials
LRRK2 variants, while less prevalent in PD than GBA mutations, drive up a carrier’s risk as much as 16-fold (Feb 2012 news). Because pathogenic variants increase LRRK2 activity, several groups are developing inhibitors. Denali Therapeutics in South San Francisco has two molecules in Phase 1, but did not present at AAT-AD/PD (press release). Other groups, including Pfizer, Merck, Glaxo-SmithKline, and Genentech, are beginning to test their own compounds.
No research groups are known to specifically target tau in PD, though mutations in the MAPT tau gene boost risk for PD (Charlesworth et al., 2012), and the gene takes second place in the Top Results list on PDGene.

No tau, no α-synclein? Antibodies to oligomeric tau (right) prevented Lewy bodies (red) in mice that overexpress α-synuclein (left). Nuclei are stained blue. [Courtesy of Gerson et al., Molecular Neurodegeneration.]

Oligomers Interact? Antibodies to oligomeric tau (right) abolished oligomeric α-synuclein (green) in PD model mice (left). [Courtesy of Gerson et al., Molecular Neurodegeneration.]
In Turin, Rakez Kayed of the University of Texas Medical Branch, Galveston, said that all α-synuclein mouse models have tau pathology, and the two proteins have been found to interact whereby one triggers the other to form fibrils (Apr 2003 news; Jul 2013 news). Kayed treated seven-month-old A53T mice, which overexpress mutant human α-synuclein, with a single injection of tau oligomer-specific antibody (TOMA), and tested them two weeks later. The treatment lowered tau oligomers to almost wild-type levels, while rescuing nest-building behavior, novel object recognition, and gait (Gerson et al., 2018). “Tau immunotherapy helps β-synuclein mice,” Kayed concluded.
Diabetes drug for PD?
Some approaches lack a genetic basis. In fact, GWAS for PD, and also for Alzheimer’s disease, show no significant hits in diabetes genes. Still, the diabetes drug exenatide has been reported to halt motor decline in a small Phase 2 trial of 62 patients (Aug 2017 news). Exenatide is an analogue of glucagon-like peptide-1 (GLP-1), which stimulates the pancreas to release insulin. In a plenary session, Fabrizio Stocchi of IRCCS San Raffaele Pisana, Rome, noted that neurons express the GLP-1 receptor, particularly in the basal ganglia, and Christian Hölscher of Lancaster University, U.K., said that insulin-signaling falters in the PD brain. GLP-1 receptor agonists resensitize cells to insulin signaling. In animal models, exenatide protects dopaminergic neurons.
Hölscher wondered if other diabetes drugs might improve on GLP-1 receptor agonists. Some diabetes drugs target both GLP-1 and its cousin, glucose-dependent insulinotropic polypeptide (GIP). The scientists modified dual GLP-1/GIP receptor agonists to better enter the brain, and tested several in the MPTP mouse model of toxin-induced parkinsonism, as well as the OHDA rat model. The researchers gave the drug once daily for six days. Compared with the GLP-1 receptor agonist liraglutide, the dual agonists better protected synapses and dopaminergic neurons and reduced inflammation, Hölscher reported in Turin. The drugs restored levels of dopamine and growth factors such as insulin, BDNF, and GDNF, while reducing proinflammatory cytokines like TNF-a and IL-1b. The treatment improved the rodents’ gait, performance on the rotarod, and grip strength. Longer-lasting dual agonists worked best (Jalewa et al., 2017; Feng et al., 2018). The data suggest that dual agonists could be more effective than GLP-1 analogues alone, Hölscher said.
The most potent molecule, DA5, will go into the clinic for PD and indeed AD, said Hölscher, who holds patents on the compounds (Cao et al., 2018). He noted that these drugs do not directly affect glucose levels, and so can be taken by non-diabetics. In answer to audience questions, he said he believes the drugs act by restoring the apparatus that produces and releases dopamine.—Madolyn Bowman Rogers
No Available Comments
Researchers keep turning up unexpected links between Parkinson’s disease and the immune system. At the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), March 15–18 in Turin, Italy, Michael Schlossmacher of the Ottawa Hospital Research Institute, Canada, reported that the PD-linked gene LRRK2 helps mice fight bacterial and viral infections. Together with previous findings that animals release α-synuclein in response to infection, the data support the idea that microbial exposure could play a role in neurodegenerative disease. And if bad bacteria can hasten PD, could good bacteria delay it? A hint in this direction came from Roberto Grau of Rosario National University, Argentina, who reported that feeding probiotic bacteria to worms prevented α-synuclein accumulation and neurodegeneration. The bacteria turned on longevity genes, which may control α-synuclein folding and degradation, Grau suggested. He is about to test his idea in an upcoming PD trial.
If confirmed, the studies imply that microorganisms, and the immune response to them, may modify PD risk, for good or ill. “We think that environmental triggers such as microorganisms interact with the genetic makeup of the host organism to initiate inflammatory responses, and those responses determine the outcome of the illness,” Schlossmacher told Alzforum. His team recently drew attention to this complex disease model of PD pathogenesis and is testing this idea in animal and human studies (Schlossmacher et al., 2017).
The idea that systemic infections might be at the root of some cases of PD has been gaining some ground in the last decade (July 2011 news series). Researchers recently reported that gut infections stimulate the release and aggregation of α-synuclein (Oct 2016 news; Jul 2017 news). The protein is expressed in the peripheral and central nervous system neurons, as well as in red blood cells and platelets (Barbour et al., 2008; Scherzer et al., 2008). In mice, the normal gut microbiome accelerates α-synuclein pathology, while in people, the composition of gut microflora changes early in the disease (Dec 2016 news; Apr 2017 news).
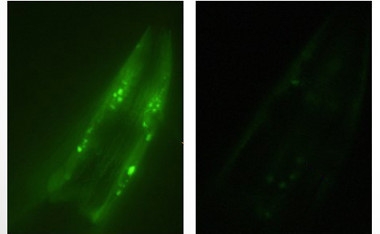
PD model worms (left) accumulate α-synuclein deposits (green), but not when fed probiotic bacteria (right). [Courtesy of Roberto Grau.]
Why does the body pump out α-synuclein in response to an infection? Last year, Schlossmacher and colleagues reported that the protein helped mice survive a virulent immune challenge, with α-synuclein knockouts succumbing sooner to a deadly virus (May 2017 news). Others have similar findings (Beatman et al., 2015). In Turin, Schlossmacher added new data, proposing a comparable immune modulatory role for LRRK2. This large multifunctional protein is highly expressed in microglia, monocytes, and adaptive immune cells, in addition to its expression in neurons and glia.
To plumb its role in infection, the researchers infected newborn LRRK2 knockout mice with reovirus T3D (Gauvin et al., 2013), and adult knockouts with salmonella bacteria. In both paradigms, the knockouts had evidence of more disease than wild-type, amassing about double the viral or bacterial burden. Female mice lacking LRRK2 were more susceptible to infection than males. Intriguingly, the pathological outcomes of female LRRK2 heterozygotes mirrored those of male knockouts, while male heterozygotes were more likely to be unaffected, responding to infection like wild-type mice. This work is currently in revision (Shutinoski B, Hakimi M, et al.).
PD-linked LRRK2 risk alleles are not knockouts, however. They are mostly gain-of-function variants that increase the protein’s kinase activity. What do those do in response to infection? Schlossmacher and colleagues tested mice that expressed the main PD risk variant, G2019S. In contrast to knockouts, they dealt with infections handily, keeping their viral or bacterial load low. Female mice survived bacterial sepsis better than males but succumbed sooner to a viral brain infection, though the number of mice examined thus far was small.
The G2019S variant is known to boost myeloid cell recruitment, and a different LRRK2 variant is a known risk factor for an excessive inflammatory response in people with leprosy caused by Mycobacterium leprae (Fava et al., 2016). Schlossmacher speculated that G2019S may augment inflammation, which helps the body fight off a virulent infection but could possibly harm the brain. If so, G2019S’s efficacy against infections may explain why this variant has survived in humans during evolution.
If independently confirmed, LRRK2 would be the first PD gene shown to have a strong gender effect in animal studies. The reason why is unclear, but the sex difference mirrors findings in people. In the Ashkenazi Jewish population, women carrying a G2019S variant are more likely than male carriers to develop PD, and their symptoms appear five years earlier on average (Cilia et al., 2014; Marder et al., 2015). This flips the normal gender effect in PD, where men are typically more susceptible.
In future work, Schlossmacher is investigating whether his LRRK2 findings relate to PD. He believes the protein may influence how the body mobilizes α-synuclein in response to an infection. “We think LRRK2 is the horse, and α-synuclein the buggy; where the buggy goes depends on which way the horse pulls,” he told Alzforum. This raises a question for ongoing trials of α-synuclein antibodies, Schlossmacher noted. If the protein helps defend the body against infection, lowering it too much could leave people more vulnerable. “Trialists should monitor patients for signs of infection,” he suggested.
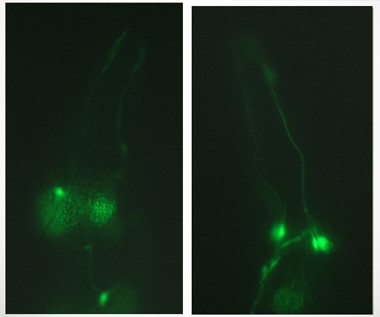
In PD model worms fed the standard diet of E. coli (left), dopaminergic neurons (green) degenerate; neurons stay healthy when the worms eat probiotics (right). [Courtesy of Roberto Grau.]
Working in Rosario, Grau focused on good bacteria instead of bad. He previously reported that the probiotic Bacillus subtilis delayed aging in the roundworm C. elegans by reducing insulin signaling (Donato et al., 2017). Because aging is a risk factor for PD, he wondered if the probiotic could protect against it. He used two worm models, one that overexpressed human α-synuclein, and another that expressed a risk variant in the parkin gene. These worms develop several phenotypes mimicking PD, including clumsy movement, constipation, and impairment in dopamine-dependent behaviors such as reproduction and avoiding danger.
When the mutant worms were raised on media containing Bacillus subtilis, however, nearly all these behaviors reverted to wild-type levels. The probiotic suppressed accumulation of α-synuclein into Lewy bodies by 75 percent, and prevented the degeneration of dopaminergic neurons (see images above). The worms lived as long as wild-types. The data suggest that probiotics could possibly prevent the development of PD pathology, Grau said.
The protective effect of Bacillus subtilis depended on its ability to form a biofilm, a slimy sheet of cells surrounded by an extracellular matrix. When the researchers used a mutant strain without that ability, few benefits accrued to the worms. Bacillus lives as a biofilm in the gut, and this organization allows the cells to communicate more efficiently among themselves and with the host, Grau explained.
In his previous study, he found that Bacillus biofilms dialed down insulin-signaling genes such as DAF-2 in the host. This suppression leads to the induction of protective genes, including FOXO and heat shock factor 1. HSF1 in turn mediates the response to cellular stress, and Grau believes it may turn on molecular chaperones that assist in the degradation of α-synuclein.
Grau plans to treat PD patients with the Bacillus subtilis strain DG101, along with their normal medications, to see if the probiotic might improve health, lower symptoms, or extend lifespan. This strain robustly induces FOXO and HSF-1, he noted. The trial is expected to start later this year. He plans to enroll more than 100 people in Argentina who are being treated for PD at private hospitals and follow them for six to 12 months.
In the Alzheimer’s field, treatments that target the aggregation of pathogenic proteins have not worked in symptomatic populations, possibly due to the long prodrome of the disease. This has fueled a push toward preventative trials.

This Japanese breakfast food of fermented soybeans is made with B. subtilis. [Courtesy of Wikimedia.]
Some people around the world already consume B. subtilis. For example, a Japanese breakfast food called natto is made by fermenting soybeans using the probiotic. The Japanese credit consumption of this food as one reason for their longevity. Grau told Alzforum that natto helped inspire his studies of B. subtilis’ health benefits.
The strain that makes natto produces the protease nattokinase, which degrades amyloid in mice and promotes non-amyloidogenic processing of Aβ (Hsu et al., 2009; Fadl et al., 2013). It is unclear if the food affects PD or Alzheimer’s risk. The incidence of Parkinson’s disease in Japan is somewhat lower than in most Western countries (Muangpaisan et al., 2009).
Schlossmacher called Grau’s work elegant, with fascinating implications. He noted that probiotics might trigger shifts in the relative abundance of other species in the gut microbiome, leading to changes throughout the intestine. In theory, such microbiome changes could also help prevent PD by keeping the α-synuclein response to infection in check, he said. Signs of ongoing α-synuclein aggregation occur in healthy people in the GI tract, notably in the appendix (Gray et al., 2013), and chronic constipation is associated with PD risk. Gut motility, which is influenced in part by the GI tract’s microbiome constituents, could thus be a modifiable risk factor, Schlossmacher argues.—Madolyn Bowman Rogers
No Available Comments
As Parkinson’s researchers push beyond symptom management to disease modification, they need far better tools to gain a deeper understanding of how α-synuclein misbehaves in the brain. Topping their wish list is a PET tracer to visualize Lewy bodies in living people, and at the first Advances in Alzheimer’s and Parkinson’s Therapies Focus Meeting (AAT-AD/PD), held March 15–18 in Turin, Italy, there finally was some anticipatory buzz based on a candidate that will head to human trials this year. Researchers also need better animal models, and here, too, new data showed promise when a Dutch group presented a marmoset model that appears to mimic PD better than rodents. Other researchers explored how pathology, risk factors, and symptoms interact, with one reporting that chronic stress accelerates the spread of injected α-synuclein through the rodent brain, while another tied cognitive decline in PD to a loss of dopamine signaling in the precuneus.
“The field will be completely transformed by new technology,” Walter Koroshetz of the National Institute of Neurological Disorders and Stroke (NINDS) predicted at the conference.
Such is the need for an α-synuclein PET tracer that the Michael J. Fox Foundation, after funding research into it for years, has offered a $2 million prize to the first group to produce one. So far, AC Immune in Lausanne, Switzerland, seems to have a leg up on the contest. Last year, company researchers described a compound that selectively bound α-synuclein fibrils, but not Aβ amyloid, and had good brain uptake in animals (May 2017 conference news). Tracer selectivity for α-synuclein has been a big challenge, because the human brain contains far less α-synuclein than Aβ, hence many candidate tracers also tend to bind Aβ.
In Turin, Jan Stöhr of AC Immune reported that the company has anointed a new lead compound, dubbed AA, which reportedly has improved properties over the earlier candidate. AA has sub-nanomolar affinity for Lewy bodies in brain sections of people who died with PD, and 500-fold selectivity for α-synuclein over Aβ, Stöhr said. In radio-binding assays, the compound bound to recombinant α-synuclein fibrils and more importantly bound to PD brain-derived α-synuclein aggregates with high affinity, again demonstrating target engagement on patient-derived protein. The researchers have now radiolabeled AA with fluorine 18 and tested uptake in animal brains. In rodents, 6.5 percent of the tracer reached the brain, with uptake in one minute and washout in 45, Stöhr reported. In primates, 4 percent reached the brain, with uptake in five minutes and washout in 20. The company, together with its partner Biogen, is planning the first human study for later this year.
“These data are the most impressive I’ve seen [for α-synuclein tracers],” said John Trojanowski of the University of Pennsylvania, Philadelphia. Jamie Eberling of MJFF agreed. “Selectivity has been a big stumbling block for α-synuclein tracer development. The AC Immune compounds are the first I’ve seen that looked to be extremely selective, along with other favorable characteristics,” she wrote to Alzforum. Another researcher at the AAT-AD/PD talk wondered whether the compound would enter cells to bind α-synuclein deposits. Stöhr said the fact that it passes through the blood-brain barrier so quickly suggests that it will also cross the cell membrane.
PET tracers will identify α-synuclein pathology directly. In the meantime, researchers gauge this pathology indirectly by imaging dopaminergic degeneration with 123I-FP-CIT SPECT, which detects dopamine transporters (e.g., Jung et al., 2018). Can this type of scan relate degeneration to symptoms? At AAT-AD/PD, Andrea Pilotto of the University of Brescia, Italy, described how he used CIT SPECT to investigate the relationship between dopaminergic transmission and cognition. One in two Parkinson’s patients decline cognitively, but researchers cannot predict who has the more severe prognosis.
Working in the lab of Alessandro Padovani, Pilotto scanned 67 PD patients, 34 of whom had mild cognitive impairment. Overall, he saw no link between striatal binding and whether the person was impaired. Pilotto replicated previous work correlating low uptake in the right caudate and right putamen with low MMSE and verbal fluency, respectively, but saw no difference between MCI and controls in this regard. Where he did find a difference was in the precuneus, a piece of parietal cortex hit early on by amyloid plaques in preclinical Alzheimer’s disease. Low CIT binding there did correlate with MCI. Dopamine helps activate the precuneus to perform executive and attention tasks, Pilotto noted, saying his data suggest that a lack of dopamine in the precuneus could underlie cognitive deficits.
In another study, Pilotto used FDG PET to examine brain metabolism in 54 people with PD. One in three had an atypical scan that resembled the FDG PET pattern of AD and dementia with Lewy bodies (DLB); only these patients developed dementia (Pilotto et al., 2018). Once again, the atypical PD and the AD patterns shared waning metabolism in the precuneus. Precuneus dysfunction could be the key to cognitive impairment in PD, Pilotto suggested in Turin. Intriguingly, other research has correlated tau tangle pathology in the precuneus with cognitive impairment in PD and DLB (Sep 2016 news). Pilotto is currently validating his findings with PPMI data, and is following his original cohort longitudinally.

These small monkeys have behaviors similar to people, perhaps making them a better model for Parkinson’s disease. [Courtesy of Wikimedia.]
Besides tracers for imaging PD pathology, researchers also need animal models that more fully recapitulate the human disease. Marmosets are sometimes used to make primate models because these New World monkeys weigh less than 2 pounds, breed quickly, yet have some behaviors that resemble those of humans. Over the last three decades, some groups have used marmosets for PD research by injecting the toxin MPTP to kill dopaminergic neurons, but recent refinements to this approach may bring it closer to human disease.
Ingrid Philippens of VU University, Amsterdam, injects a low, 0.5 mg/kg dose of MPTP weekly. This dosage schedule leads to a slowly progressing form of parkinsonism that shares features with the human disease. In the brain, dopaminergic neuron damage activates compensatory mechanisms and triggers α-synuclein pathology, Philippens told Alzforum. At the level of behavior, this approach models prodromal stages of PD, with subtle motor deficits plus other premotor symptoms. For example, the monkeys develop REM sleep behavior disorder. Just like people with prodromal Parkinson’s, affected males kick their female partners during sleep (yes, marmosets do sleep with their partners, see Verhave et al., 2011). The researchers also see genetic differences in the monkeys’ susceptibility to the disease that correlate with neurotransmitter levels (Franke et al., 2016).
In Turin, Philippens described how her team monitored the behavior of freely moving monkeys in their home cages through telemetry. The marmosets wear necklaces that contain an actimeter, similar to the wrist actimeters people wear. As the animals bustle about their cages, the actimeters send signals to a receiver using Actiwatch software. In addition, the marmosets are implanted with receivers that measure EEG activity along with other physiological parameters such as body temperature. The combination of these two devices detects sleep disturbances. The approach provides continuous data, similar to what some PD trials are now collecting to better track small, day-to-day changes (May 2017 conference news). In addition, the researchers place a touchscreen in the marmoset’s home cage to evaluate changes in its learning and memory. Such tests typically require the animal learn which of two images on the screen will provide a food reward, and then unlearn this association when the reward is paired with the other object (e.g., Kangas et al., 2016). The use of tests more comparable to those used with people may facilitate translation of therapies to the clinic, the scientists noted.
Other researchers are investigating ways to induce parkinsonism in marmosets without a toxin. Fifteen years ago, a human α-synuclein transgene introduced into marmosets was found to cause pathology and dopaminergic neuron loss (Feb 2003 news) and more recently, Japanese scientists triggered Lewy body pathology, spread, and some dopaminergic neuron loss by injecting synthetic α-synuclein fibrils into the basal ganglia of non-transgenic marmosets (Shimozawa et al., 2017).
Koroshetz noted that the value of a given model depends on its purpose. MPTP models were essential in deciphering circuit disruption due to loss of dopaminergic neurons and led to the development of deep-brain stimulation as a therapy. However, they do not address the spread of α-synuclein pathology believed to underlie PD. “The marmoset model from Japan may be very helpful if they have pathology that resembles what we see in PD patients,” he wrote to Alzforum. In Turin, Koroshetz noted that the behavioral features of this model look identical to PD.
To better understand how α-synuclein spreads and affects function, other scientists are incorporating additional drivers of PD into the models they already have in an effort to build more “complete” representation of the human disease. Consider, for example, an attempt to build stress and depression into a mouse model by Johannes Burtscher in the lab of Hilal Lashuel of École Polytechnique Fédérale de Lausanne, Switzerland.
Burtscher noted in Turin that anxiety and depression increase a person’s risk for PD. These emotions are processed in the amygdala, where Lewy bodies are common; what’s more, aggregated α-synuclein injected into mouse striatum spreads to many connected regions, including the amygdala (Popescu et al., 2004; Luk et al., 2012). Burtscher found that aggregated α-synuclein levels in the amygdala peaked 30–90 days after injecting preformed fibrils into the dorsal striata of wild-type mice.
To see what the arrival of this pathology might do to the amygdala, Burtscher injected 5–10 μg of preformed α-synuclein fibrils into the striatum and checked behavior one month later. The mice were less afraid of open spaces than controls, but seemed passive, showing less interest in new objects and moving less during a forced swim test. Because an overactive amygdala is linked to anxious behavior, these data suggest the opposite process, namely an underactive amygdala, Burtscher said. He noted that apathy and emotional numbing are also seen in the prodromal stages of PD.
Because chronic stress also increases risk for PD, Burtscher wondered if stress could affect pathology. He slipped corticosterone into the mice’s drinking water for a month to stress them. In response, the mice gained weight and showed signs of depression on classic rodent tests, giving up quickly in forced swim tests and showing no interest in sugar water. Burtscher then injected the aggregated α-synuclein into their striata while continuing with corticosterone for another month. In the stressed mice, α-synuclein pathology spread farther than in controls, reaching the entorhinal cortex, whereas in controls it stopped at the substantia nigra and amygdala. Burtscher also saw more degeneration of dopaminergic neurons in the stressed group. Chronic stress predisposes α-synuclein to spreading, Burtscher concluded.—Madolyn Bowman Rogers
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.