Familial Alzheimer’s Gene Alters Children’s Brains
Quick Links
How early does Alzheimer’s disease start? To glean clues to the answer, researchers have been studying young adults who carry genes that destine them to develop the disease as early as their 40s. These investigations revealed that the brains of mutation carriers already function less efficiently in their 20s. Now, scientists with the Alzheimer’s Prevention Initiative (API) have rolled the tapestry back further, providing a glimpse into the brains of preteens and teenagers in Colombia who carry the presenilin 1 “Paisa” mutation. As reported in the June 29 JAMA Neurology, researchers led by Yakeel Quiroz at Massachusetts General Hospital, Boston, found that young, cognitively normal carriers displayed the same functional idiosyncrasies the older carriers did. As expected, these youngsters also overproduced Aβ42. Unexpectedly, however, some abnormalities went in the opposite direction from those in older carriers. The brains of young carriers contained more gray matter in several regions than non-carriers had, and had stronger functional connections.
Overall, the results emphasize that the brains and nervous systems of carriers are already different from non-carriers even at 9 years old, Quiroz told Alzforum. It is unclear if their brains simply develop on a different trajectory, or if these changes represent early neurodegeneration, Quiroz noted. Researchers likewise do not know if the findings will generalize to other mutations or to sporadic disease, though some studies suggest they might. Tantalizingly, infants who inherit the Alzheimer’s risk gene ApoE4 also have structural brain differences from controls, hinting that there may be developmental changes that put people at risk for the disease.
“The authors should be congratulated on evaluating some of the youngest participants for Alzheimer’s biomarker changes. … A better understanding of the earliest changes may be informative for understanding the pathogenesis of Alzheimer’s disease and why these regions appear to be vulnerable for the disease,” Randall Bateman at Washington University in St. Louis, who directs the Dominantly Inherited Alzheimer’s Network (DIAN) Trials Unit, wrote to Alzforum (see full comment below).
Previous studies from both DIAN and API reported that abnormal biomarkers and subtle memory deficits precede AD symptoms by as much as 20 years (see Mar 2011 news; Jul 2012 news). Abnormalities were detected at even earlier ages in the Colombian kindred, who carry the E280A Paisa mutation that leads to excessive production of Aβ42. Among cognitively healthy 18- to 26-year-olds, API researchers found less gray matter in parietal regions and less deactivation of the precuneus and posterior cingulate during a memory task, changes similar to those seen in people with AD dementia (see Nov 2012 news).

Brain Activity Problems Start Early.
In functional MRI scans, the brains of children who carry a presenilin 1 mutation poorly deactivate precuneus and posterior cingulate. [Courtesy of Quiroz et al., © 2015 American Medical Association. All rights reserved.]
The API researchers, led by Francisco Lopera at the University of Antioquia, Medellin, Colombia, and Eric Reiman at Banner Alzheimer’s Institute, Phoenix, wondered how far back these changes might start. In 2011, first author Quiroz began studying biomarkers in children aged 9 to 17 from the Colombian kindred (see Dec 2011 conference news; Aug 2012 conference news). Due to the participants’ young age, the researchers did not perform lumbar punctures, but did collect blood, MRI scans, and cognitive data.
Some findings echoed results from the older group. Among 18 carriers and 19 non-carriers of the Paisa mutation, carriers had about 50 percent more Aβ42 in their blood than non-carriers, along with a similarly elevated ratio of Aβ42/Aβ40, but normal levels of Aβ40. This is in keeping with the mutation increasing production of the more aggregation-prone Aβ42. The children did not undergo PET amyloid scans, but older members of this kindred first develop a measurable brain amyloid burden around age 28. Therefore, abnormal findings in these young brains likely occur in the absence of amyloid plaques.
Children with the mutation performed normally on the Wechsler Intelligence for Children Scale, which includes tests of working memory, verbal IQ, and processing speed. They also performed as well as non-carriers on a memory task that asked them to pair faces with names, but functional MRI revealed that the carriers poorly deactivated posterior parietal brain regions during this task, just as older carriers did (see image above). These regions form part of the default mode network, which is normally suppressed during mental tasks. People with Alzheimer’s have more difficultly turning this network off, as well as switching between brain networks in general (see Jul 2012 news).
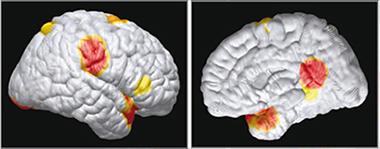
Surprise: Heftier Brains in Young Carriers.
Children with a presenilin 1 mutation had more gray matter in several regions (red indicates the greatest difference from controls). [Courtesy of Quiroz et al., © 2015 American Medical Association. All rights reserved.]
The data also turned up some surprises. Mutation carriers had greater functional connectivity between the posterior cingulate cortex and medial temporal lobe than non-carriers, and greater gray matter in the hippocampus and parietal and temporal lobe regions. This contrasts with the thinning parietal gray matter seen in the 18- to 26-year-olds, and with numerous studies reporting weaker network connections during early disease stages (see e.g., Aug 2013 news; Aug 2014 news).
Quiroz emphasized that these MRI measures should be considered exploratory, as the cohort was small, and not all the differences between carriers and controls remained significant after correction for multiple comparisons. Nonetheless, the findings do dovetail with some previous studies that report thicker brains at preclinical stages of both familial and sporadic disease. A small study in Spain measured thicker precuneus, caudate, and parietotemporal regions compared with controls in six carriers of various PS1 mutations about 10 years before expected symptom onset, while five symptomatic carriers had thinning in these regions (see Fortea et al., 2010). In sporadic disease, infants born with an ApoE4 allele, which roughly triples risk, have thicker frontal, occipital, and posterior parietal cortices than their peers do (see Dec 2013 news; Knickmeyer et al., 2013). In addition, among amyloid-positive adults in the Australian Imaging, Biomarkers, and Lifestyle (AIBL) Study of Ageing, those who were cognitively normal had greater volume in several temporal regions than amyloid-negative or symptomatic patients did (see Chételat et al., 2010).
Tammie Benzinger at Washington University was intrigued by the finding of increased functional connectivity in these young carriers. She noted that a few other studies have reported stronger connections between some brain regions in early stages of AD (see Wang et al., 2007; Zhang et al., 2010; Sheline et al., 2010). She led a DIAN study that saw hypermetabolism in the precuneus and posterior cingulate of mutation carriers more than 10 years before the age of expected symptom onset. The increased glucose use dropped below control levels later in preclinical disease (see Benzinger et al., 2013). “There are a lot of parallels between metabolic and functional imaging, so I think our finding was very similar to the Colombian data,” Benzinger told Alzforum.
Why might the brains of young carriers behave so differently from the older ones? Benzinger noted that there are two popular hypotheses. One holds that pathological processes may already have started in these brains, perhaps causing inflammation that accounts for the increased gray matter. The other maintains that the brains of people with Alzheimer’s mutations have to work harder to accomplish tasks. Hypermetabolism and hyperconnectivity thus may be a form of compensation, which breaks down later in life. To distinguish between these hypotheses will probably require animal studies, Benzinger said. Bill Jagust at the University of California, Berkeley, suggested that there may simply be differences in brain development and wiring in these children. “It’s hard for me to believe that what’s happening in the brain of a cognitively normal 9-year-old is degeneration,” he said. Degeneration would imply that brain functions would drop off over time, he pointed out.
Longitudinal data from mutation carriers will help determine whether their brain functions are already beginning to deteriorate. Quiroz is following the young Colombian cohort to look for any subtle signs of cognitive decline. She is adding an additional cognitive battery, the Spanish-language Evaluación Neuropsicológica Infantil (ENI), and developing shorter, more kid-friendly fMRI tasks (see Rosselli-Cock et al., 2004). In future work, she would like to measure the same imaging and cognitive parameters in young people who carry other familial mutations or ApoE4, to compare age-related changes in these groups.
Does the data suggest that clinicians might need to start treatments at very young ages to prevent familial Alzheimer’s from taking hold? Researchers were cautious on this point. Quiroz noted that since amyloid accumulation begins at age 28 in this kindred, anti-amyloid treatments, at least, would be unlikely to have much effect at earlier ages. The ongoing trial of crenezumab in the Colombian kindred enrolls people from age 30 and up (see Mar 2011 news series; Jul 2014 news). Whether other types of treatment might benefit very young carriers remains a question for the future.—Madolyn Bowman Rogers
References
Mutations Citations
News Citations
- Detecting Familial AD Ever Earlier: Subtle Memory Signs 15 Years Before
- Paper Alert: DIAN Biomarker Data Show Changes Decades Before AD
- API Echoes DIAN: Biomarker Changes Precede Symptoms by 20 Years
- Reeling In Biomarker Data in Young Carriers, API Rocks Staging Boat
- In Big Picture, Familial AD’s Biomarker Data Resemble LOAD
- Communication Breakdown: Multiple Networks Decline in AD Brains
- Brain Connectivity Reveals Preclinical Alzheimer’s Disease
- Neural Circuitry Goes Haywire in Both Sporadic and Familial AD
- Brain Volume, Myelination Different in Infants Carrying ApoE4
- Colombians Come to Fore in Alzheimer’s Research, Mass Media
- Crenezumab Disappoints in Phase 2, Researchers Remain Hopeful
Paper Citations
- Fortea J, Sala-Llonch R, Bartrés-Faz D, Bosch B, Lladó A, Bargalló N, Molinuevo JL, Sánchez-Valle R. Increased cortical thickness and caudate volume precede atrophy in PSEN1 mutation carriers. J Alzheimers Dis. 2010;22(3):909-22. PubMed.
- Knickmeyer RC, Wang J, Zhu H, Geng X, Woolson S, Hamer RM, Konneker T, Lin W, Styner M, Gilmore JH. Common Variants in Psychiatric Risk Genes Predict Brain Structure at Birth. Cereb Cortex. 2013 Jan 2; PubMed.
- Chételat G, Villemagne VL, Pike KE, Baron JC, Bourgeat P, Jones G, Faux NG, Ellis KA, Salvado O, Szoeke C, Martins RN, Ames D, Masters CL, Rowe CC. Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain. 2010 Nov;133(11):3349-58. PubMed.
- Wang K, Liang M, Wang L, Tian L, Zhang X, Li K, Jiang T. Altered functional connectivity in early Alzheimer's disease: a resting-state fMRI study. Hum Brain Mapp. 2007 Oct;28(10):967-78. PubMed.
- Zhang HY, Wang SJ, Liu B, Ma ZL, Yang M, Zhang ZJ, Teng GJ. Resting brain connectivity: changes during the progress of Alzheimer disease. Radiology. 2010 Aug;256(2):598-606. PubMed.
- Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010 Mar 15;67(6):584-7. PubMed.
- Benzinger TL, Blazey T, Jack CR Jr, Koeppe RA, Su Y, Xiong C, Raichle ME, Snyder AZ, Ances BM, Bateman RJ, Cairns NJ, Fagan AM, Goate A, Marcus DS, Aisen PS, Christensen JJ, Ercole L, Hornbeck RC, Farrar AM, Aldea P, Jasielec MS, Owen CJ, Xie X, Mayeux R, Brickman A, McDade E, Klunk W, Mathis CA, Ringman J, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Salloway S, Correia S, Schofield PR, Masters CL, Rowe C, Villemagne VL, Martins R, Ourselin S, Rossor MN, Fox NC, Cash DM, Weiner MW, Holtzman DM, Buckles VD, Moulder K, Morris JC. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):E4502-9. Epub 2013 Nov 5 PubMed.
- Rosselli Cock M, Matute Villaseñor E, Ardila Ardila A, Botero Gómez VE, Tangarife Salazar GA, Echevarría Pulido SE, Arbelaez Giraldo C, Mejía Quintero M, Méndez Losado LC, Villa Hurtado PC, Ocampo Agudelo P. [Neuropsychological Assessment of Children: a test battery for children between 5 and 16 years of age. A Colombian normative study]. Rev Neurol. 2004 Apr 16-30;38(8):720-31. PubMed.
Further Reading
Primary Papers
- Quiroz YT, Schultz AP, Chen K, Protas HD, Brickhouse M, Fleisher AS, Langbaum JB, Thiyyagura P, Fagan AM, Shah AR, Muniz M, Arboleda-Velasquez JF, Munoz C, Garcia G, Acosta-Baena N, Giraldo M, Tirado V, Ramírez DL, Tariot PN, Dickerson BC, Sperling RA, Lopera F, Reiman EM. Brain Imaging and Blood Biomarker Abnormalities in Children With Autosomal Dominant Alzheimer Disease: A Cross-Sectional Study. JAMA Neurol. 2015 Aug;72(8):912-9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University School of Medicine
The authors should be congratulated on evaluating some of the youngest participants for Alzheimer’s biomarker changes. The findings reported on the Colombian kindred are of key interest and support prior findings in the Colombian kindred and in DIAN of increased plasma Aβ42 and Aβ42:40 ratios. Novel findings include a tantalizing report of increased functional connectivity in the posterior cingulate and increased gray matter volumes in the temporal lobe. These findings may also be seen in other dominantly inherited AD populations ( Benzinger et al., 2013 ) and in presymptomatic sporadic AD. Replication of these studies and a better understanding of the earliest changes may be informative to …More
References:
Benzinger TL, Blazey T, Jack CR Jr, Koeppe RA, Su Y, Xiong C, Raichle ME, Snyder AZ, Ances BM, Bateman RJ, Cairns NJ, Fagan AM, Goate A, Marcus DS, Aisen PS, Christensen JJ, Ercole L, Hornbeck RC, Farrar AM, Aldea P, Jasielec MS, Owen CJ, Xie X, Mayeux R, Brickman A, McDade E, Klunk W, Mathis CA, Ringman J, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Salloway S, Correia S, Schofield PR, Masters CL, Rowe C, Villemagne VL, Martins R, Ourselin S, Rossor MN, Fox NC, Cash DM, Weiner MW, Holtzman DM, Buckles VD, Moulder K, Morris JC. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):E4502-9. Epub 2013 Nov 5 PubMed.
University of Arkansas for Medical Sciences
This is a fascinating body of work. It should be noted that it is corroborated also by Sala-Llonch et al., 2015. That study had the additional strengths of being longitudinal and including initially asymptomatic carriers of nine different PSEN1 mutations from 11 different families. Though the subjects were much older (mean age 38.6), they also showed the same sort of trajectory, which the authors characterized as "nonlinear": greater cortical thickness initially, followed by accelerated thinning.
References:
Sala-Llonch R, Lladó A, Fortea J, Bosch B, Antonell A, Balasa M, Bargalló N, Bartrés-Faz D, Molinuevo JL, Sánchez-Valle R . Evolving brain structural changes in PSEN1 mutation carriers . …More
University of Southern California; Imaging Genetics Center
The authors evaluated in 37 children and adolescents how the PSEN1 E280A mutation, which confers autosomal dominantly inherited Alzheimer’s disease (ADAD), was associated with plasma Aβ1-40 and Aβ1-42. They also examined associations of the mutation with structural and functional MRI measures. This is an exciting and important study in a relatively rare population. It serves as a step in identifying which differences seen in preclinical ADAD are progressive and which are developmental, potentially guiding future intervention studies.
Consistent with previous studies in cognitively intact young adult and middle-aged ADAD mutation carriers ( Bateman et al., 2012 ; Fleisher et al., 2015 ), the …More
The authors also found that the volumes of the parahippocampal gyrus and temporal pole were greater in mutation carriers compared with non-carriers. Although some previous studies of ADAD have found thinner regional cortices in preclinical mutation carriers (Mosconi et al., 2006; Knight et al., 2011), another with participants farther from their expected age of disease onset found regional cortical thickness increases in selected temporal and parietal regions in preclinical PSEN1 mutation carriers (Fortea et al., 2010). Together, these studies suggest that early increased regional brain volume, particularly in regions vulnerable to AD pathology, may precede typical AD-patterns of atrophy. Longitudinal study of these participants would help elucidate whether such effects are developmental and how changes in blood and CSF biomarker measures relate to changes in brain structure.
These results provide interesting insights into early AD-related vulnerabilities, and may reflect earliest disease changes. This study sets a strong foundation for future work.
References:
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
Chen M, Inestrosa NC, Ross GS, Fernandez HL. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem Biophys Res Commun. 1995 Aug 4;213(1):96-103. PubMed.
Evin G, Li QX. Platelets and Alzheimer's disease: Potential of APP as a biomarker. World J Psychiatry. 2012 Dec 22;2(6):102-13. PubMed.
Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gutierrez Gomez M, Langois CM, Langbaum JB, Roontiva A, Thiyyagura P, Lee W, Ayutyanont N, Lopez L, Moreno S, Muñoz C, Tirado V, Acosta-Baena N, Fagan AM, Giraldo M, Garcia G, Huentelman MJ, Tariot PN, Lopera F, Reiman EM. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol. 2015 Mar;72(3):316-24. PubMed.
Fortea J, Sala-Llonch R, Bartrés-Faz D, Bosch B, Lladó A, Bargalló N, Molinuevo JL, Sánchez-Valle R. Increased cortical thickness and caudate volume precede atrophy in PSEN1 mutation carriers. J Alzheimers Dis. 2010;22(3):909-22. PubMed.
Knight WD, Kim LG, Douiri A, Frost C, Rossor MN, Fox NC. Acceleration of cortical thinning in familial Alzheimer's disease. Neurobiol Aging. 2011 Oct;32(10):1765-73. PubMed.
Mosconi L, Sorbi S, de Leon MJ, Li Y, Nacmias B, Myoung PS, Tsui W, Ginestroni A, Bessi V, Fayyazz M, Caffarra P, Pupi A. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer's disease. J Nucl Med. 2006 Nov;47(11):1778-86. PubMed.
Toledo JB, Shaw LM, Trojanowski JQ. Plasma amyloid beta measurements - a desired but elusive Alzheimer's disease biomarker. Alzheimers Res Ther. 2013 Mar 8;5(2):8. PubMed.
Make a Comment
To make a comment you must login or register.