This year’s annual meeting drew more than 23,000 scientists and featured 1,000 presentations on Alzheimer’s disease. Researchers soaked up new data on topics from the basic biology of tau and Aβ to the physiology of sleep and the microbiome. Alzforum covers the highlights.
Tau Snapshots from Neuroscience 2017
This year’s Society for Neuroscience meeting, held November 11–15 in Washington, D.C., drew 23,526 attendees from around the globe. For researchers in the Alzheimer’s disease field, the meeting has lost some of its vibrancy of late, and this year seemed no exception. Only one of 10 press briefings featured AD, a nanosymposium on “tauopathies” focused on brain effects of anesthetics and only two of its nine abstracts mentioned tau. Many leading AD researchers nowadays devote their travel to other meetings, but SfN remains a must for the basic science relevant to AD, and for students with broad interests across neuroscience.
This year, our SfN news coverage balances two stories on emerging areas—sleep and the microbiome in brain health and AD (see Parts 2 and 3 of this series)—with two stories on preclinical and mechanistic advances on the old but insufficiently understood topic of tau. Some researchers tried to curtail toxic forms of tau using intrabodies and zinc-finger suppressors (see below). Other labs revealed how tau compromises mitochondria, the nucleus, and the vasculature, and how microglia and synapses may contribute to the prion-like spread of tau in the brain (see Part 4 of this series).
Zinc-Finger Vectors. AAV vector injected into the hippocampus (left) limits zinc finger to that locale, whereas a single retro-orbital injection of a PHP.B vector achieves brain-wide expression (right). [Courtesy of the Hyman lab.]
Cool Ways to Temper Tau Sarah DeVos and Susanne Wegmann from Bradley Hyman’s lab at Massachusetts General Hospital, Charlestown, described a way to cut tau production in vivo using a zinc-finger-based gene-suppression system. DeVos and Wegmann collaborated with Bryan Zeitler and colleagues at Sangamo Therapeutics, a Richmond, California-based company that develops this technology for a variety of conditions. The researchers fused zinc-finger (ZF) proteins that specifically recognize the tau gene onto a transcriptional repressor. The Kruppel-associated box, aka KRAB domain, of the human ZF protein KOX1 served as the business end of these tau repressors. KRAB domains enlist co-repressors in the nucleus to shut down transcription.
Wegmann and DeVos tested different suppressor and Adeno-associated virus (AAV) vector combinations. Out of three different repressors, one dubbed ZFP-TF89 worked best to specifically knock down tau. Wegmann made a construct to drive expression of ZFP-TF89 from a cytomegalovirus promotor, and packaged it into an AAV9 virus. A nuclear localization signal ensured this engineered suppressor would be delivered next to DNA, while a Venus yellow fluorescent protein allowed her to track infected cells. In vitro, Wegmann saw increasing knockdown of tau with increasing dose of the vector.
To try ZFP-TF89 in wild-type mice, Wegmann injected the AAV vector into the hippocampus on one side of the brain and saline into the other. Venus fluorescence confirmed widespread ipsilateral expression of the vector in the hippocampus. Immunohistochemistry, western blot, and mRNA analysis indicated about 88 percent suppression of endogenous tau expression six weeks after injection. Remarkably, even 11 months later, tau mRNA levels were still down by 75 percent, while the neuronal marker NeuN indicated no major change in neurons. However, Wegmann saw a transient uptick in glial fibrillary acidic protein and in microglial activation, suggesting the AAV vector may cause a mild inflammatory reaction. A control vector expressing only GFP evoked a similar reaction.
To avoid untoward glia responses, the scientists swapped out the CMV promoter, which turns on ZFP-TF89 in most cell types, for promoters specifically active in neurons, including those for synapsin-1, CaMKIIa, and MeCP2. These barely expressed ZFP-TF89 in glia while efficiently suppressing tau in N2a neuroblastoma cells as well as in neurons in vitro and in vivo.
Would the vectors protect mice from AD pathology? Wegmann tested them in 4.5-month-old APPPS1 animals, which overproduce Aβ42. Previously, several groups had reported that Aβ was much less toxic when tau was knocked down or knocked out (May 2007 news). Wegmann injected AAV containing synapsin-1 ZFP-TF89 into one side of the brain, and a control vector expressing red fluorescent protein into the other. Ten weeks later, she recorded 35 percent fewer dystrophic neurites on the treated side of the brain than on the control side. Both the amount and size of Aβ plaques were the same on both sides of the brain. Wegmann said this fits with the idea that knocking down tau protects against various manifestations of Aβ toxicity, including seizure, hippocampal atrophy, and learning and memory deficits. Zeitler claimed this ZFP-TF based approach is the first to target all tau forms within neurons using a single AAV administration.
Could such a system someday treat tauopathies? It’s possible—the FDA just approved an AAV-based gene therapy for treating retinal dystrophy, and an AAV9-based therapy for spinomuscular atrophy 1 made runner-up for breakthrough of the year in Science magazine (see press release; Science, 22 Dec 2017).
Still, the viruses generally don’t express that broadly in the human brain, and researchers are seeking alternatives. Last year, Benjamin Deverman and colleagues at the California Institute of Technology, Pasadena, reported that a viral capsid variant called AAV-PHP.B transfers genes throughout the CNS about 40 times more efficiently than the commonly used AAV9 (Deverman et al., 2016). When used with the neuron-specific synapsin-1 promoter, these researchers were able to widely express a TDP43 transgene in neurons in a mouse brain after a single intravenous injection of the virus (Jackson et al., 2016).
Taking advantage of this, DeVos tested a synapsin1 ZFP-TF AAV-PHP.B construct in mice. She reported that one week after a single retro-orbital injection to place the virus into the capillary bed behind the eye, tau mRNA and protein levels fell by up to 70 percent across the brain and spinal cord. CSF tau plummeted by 80 percent after 10 weeks and this persisted for six months until the end of the study.
DeVos injected 4.5-month-old APPPS1 mice once with AAV-PHP.B capsids carrying the ZF suppressor. Much like Wegmann, she found a 50 percent reduction in neuritic dystrophies around Aβ plaques 2.5 months later.
Researchers at SfN asked about off-target effects or changes to DNA. DeVos emphasized that the ZF strategy does not alter the DNA; it only prevents transcription, and that the transcription factor only binds the tau gene. The researchers tested for changes in cellular gene expression with Affymetrix microarrays, said DeVos, and found the tau suppression to be highly specific, with extremely low-off target gene modulation in the two regions they have examined so far, the frontal cortex and the hippocampus.
Others wondered if expression of the zinc finger in the periphery might be a problem, and DeVos noted that when using the synapsin-1 promoter, very little repressor is made outside the brain. Zeitler emphasized that they use human proteins for the transcription factor component, minimizing any chance for an immune response. How about microtubule function? Might it be compromised? The lab is working on this question, but DeVos noted that knocking down tau, or even completely knocking it out, has little effect (Aug 2013 news).
Another hybrid approach for targeting tau came from Marshall Goodwin, from Todd Golde’s lab at the University of Florida, Gainesville. He uses intrabodies, single-chain antibodies that can be expressed inside cells. The rationale is that because tau aggregates intracellularly, intrabodies could be better at binding and removing it than regular antibodies, which don’t easily penetrate cells, or even the brain.
Goodwin and colleagues first used tau antibodies generated by Peter Davies, including CP13, PHF1, and Tau5. They spliced DNA for the variable motifs from the heavy and light chains of the antibodies, and packaged them into viral vectors. When injected into the brains of newborn P301L mouse pups, these tau-transgenic mice expressed single-chain (scFv) intrabodies widely in the CNS, said Goodwin. However, while the CP13 intrabody reduced the amount of tau aggregates by six months, it did not make the mice live much longer, extending their survival from 10.2 months only to about 12. “These intrabodies would need to be optimized for therapeutics,” said Goodwin.
Goodwin has since fused the intrabodies to other domains to promote degradation of tau. He coupled the CP13 intrabody to the catalytic domain of several E3 ubiquitin ligases. He predicted that once these hybrid intrabodies bound tau, the ligases would tag it with ubiquitin, signaling the proteasome to degrade it. Similarly, in another hybrid, he spliced the scFV to a motif that targets proteins for chaperone-mediated autophagy (CMA).
Golde’s group has set up a three-step system to expedite screening of these antibodies. They test them first in HEK models of tau aggregation, then in wild-type brain slices infected with tau genes that created neurofibrillary tangles, and then finally in a mouse AAV model that accumulated tangles within three months. So far, Goodwin has tested the modified intrabodies only in the cell model. While the CP13 intrabody decreased insoluble tau in the cells, adding the CMA motif almost totally ablated tau aggregates. Several of the ligase versions also outperformed the normal intrabody.
Researchers at the meeting considered the approach exciting, but wondered about safety. Goodwin said that the parent intrabodies have been tested in normal mice and do not appear to be toxic and are being tested for any effects on behavior. He plans to test the modified versions in neurons and in vivo.—Tom Fagan
Disturbed Sleep Exerts Toll on Memory and Neurodegeneration
The indispensable role of a good night’s sleep in memory and brain health was the subject of several talks at the Society for Neuroscience annual meeting, held November 11–15 in Washington, D.C. How might sleep-driven memory processes falter with age and Alzheimer’s pathology? It appears that as the brain gets older, accumulating Aβ and tau desynchronize the coordinated network oscillations that convert short-term memories to long-term ones during sleep. Sleep loss, in turn, ramps up production of Aβ and phosphorylated tau and kills off neurons in deep brain structures, never to be replaced.
While You Were Sleeping
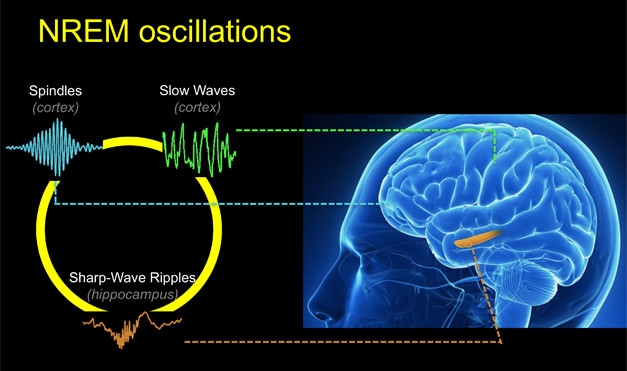
Non-rapid eye movement sleep makes up about 80 percent of our sleeping hours. During NREM sleep, a trio of neuronal oscillations coordinates to consolidate memories formed during the day (Diekelmann and Born, 2010). Two of these oscillations, called the sleep spindle and the slow cortical wave, occur near the surface of the brain and can be measured by electroencephalography (EEG). Sleep spindles, which originate in the thalamus then propagate through the cortex, oscillate at 10 to 15 Hz, and get their name from the characteristic spiky trace they leave on EEG charts (see image below). The slow waves of the cortex hum along at about 0.8 Hz. Scientists believe that when these both align with the hippocampal sharp wave, then the brain is poised to convert short-term memories to long-term ones.
Synchronous Sleep. Coordination between three oscillations—hippocampal sharp-wave ripples, spindles, and slow waves—is crucial for consolidating memories while we sleep. [Courtesy of the Walker lab.]
Randolph Helfrich, working with Robert Knight and Matthew Walker at the University of California, Berkeley, wondered if the synchrony of the slow cortical wave and sleep spindle changes with age, and how that might affect memory. They recruited 20 participants around 20 years old and 32 older adults who averaged 74 years old. Each spent a night at the sleep lab hooked to an EEG monitor. Before they retired, Helfrich trained them on a word-pairing task. They learned to associate 120 real words with 120 nonsense ones. Helfrich tested subjects before they fell asleep and again after they woke up about eight hours later.
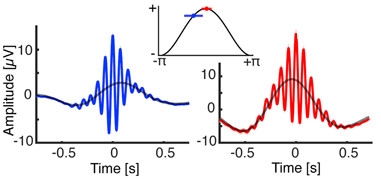
Helfrich’s findings were published in the December 14 Neuron (Helfrich et al., 2017). He found that the older people were less able to recall word pairs after a night’s rest. Their sleep oscillations were slightly off, too. While the spindle peaked just after the zenith of the slow cortical wave in the 20-year-olds, it crested about 125 ms earlier in the older adults (see image below). The more out of synch the two oscillations were, the worse the person performed on the memory task. Some of the younger volunteers did have premature spindles as well, but these seemed to affect memory performance only weakly. “Older adults express both waves, but they don’t come together as well in time,” said Helfrich. “If they don’t talk to one another, then there’s no reactivation of a short-term memory to put it in long-term storage.”
Twin Peaks.
In young adults (right) the sleep spindle (red) peaks slightly after the slow cortical wave (black curve). In older adults (left) the spindle (blue) peaks just before. [Neuron, Helfrich et al.]
Knowing that the brain shrinks with age, Helfrich wanted to test whether atrophy in any particular brain region predicted perturbed coordination of the two oscillations. The researchers analyzed magnetic resonance imaging (MRI) scans of each participant. They found that a smaller medial prefrontal cortex correlated with poor synchronization between spindles and slow waves. The medial prefrontal cortex plays an important role in the generation of slow cortical waves (Saletin et al., 2013).
In his paper, Helfrich and co-authors proposed that noninvasive stimulation of the brain during sleep might be able to restore the precise coordination between spindles and slow cortical waves to match that of young adults. Perhaps that would reduce memory decline in aging, they suggested.
Could Alzheimer’s pathology further skew this delicate synchrony? Walker’s group previously found that Aβ deposition in the cortex correlated with fewer slow cortical waves (Jun 2015 news). Joseph Winer, in the lab of Bill Jagust at UC Berkeley, wondered if tau accumulation in the medial temporal cortex might disrupt the sleep spindles that originate close by in the thalamus.
To test this hypothesis, Winer recruited 23 cognitively normal subjects from the Berkeley Aging Cohort Study who had had an amyloid and a tau PET scan. Their average age was 76. Each had an overnight sleep study that measured sleep spindles and slow cortical waves by EEG.
As previously reported by Walker, Aβ deposition in the cortex correlated with disrupted slow cortical waves. Wave amplitudes were smaller in many areas where plaques accumulated. The strongest association was in the prefrontal cortex, where slow waves originate; the more plaques there, the lower the wave amplitude. On the other hand, tau accumulation in the hippocampus decreased the frequency of sleep spindles. This correlation was strongest in parietal areas, where spindle frequencies tend to hover around the top of the 10–15 Hz range; the more tau there, the lower the spindle frequency.
Further, tau deposition correlated with poor synchrony between slow cortical waves and sleep spindles. “These findings give us a window into how [AD] pathology in healthy people disrupts important brain processes during sleep,” Winer told Alzforum.
Jagust agreed. “We need to study more subjects to be certain, but I think our data points to the idea that at least some of the changes in coupling might be related to the deposition of tau,” he wrote to Alzforum.
Researchers at SfN asked if people with Aβ or tau pathology report sleep problems. Winer replied that only amyloid deposition correlates with complaints on sleep questionnaires. The relationship is likely bi-directional, he said, because sleep helps clear waste from the brain, and Aβ pathology in turn impairs sleep (May 2014 conference news). However, it remains to be seen whether better sleep will improve memory once Aβ and tau have already deposited, Winer added. The team next plans to examine whether Aβ/tau-related sleep disturbances correlate with worse memory.
While You Were Not Sleeping
Could pulling a few all-nighters per week to binge-watch the latest hit show permanently damage the brain? Possibly, according to preliminary research by Sigrid Veasey at the Perelman School of Medicine, University of Pennsylvania, Philadelphia. Her work suggests that temporarily depriving mice of their normal sleep accelerates tau phosphorylation in the locus coeruleus, which leads to rampant neurodegeneration there. Nestled in the brainstem, the locus coeruleus is active when people are awake, and it projects all over the brain. It has been proposed as a start site for AD pathology (Elobeid et al., 2012).
Veasey previously found that chronic sleep loss in wild-type mice led LC neurons to produce more Aβ, which in turn prompted tau hyperphosphorylation (unpublished data). She reported that sleep loss leads to a drastic loss of neurons in the LC (Zhang et al., 2014). In the current study, she tested whether tau contributed to LC neuron loss, and whether those neurons were replaced.
The troubling answers appear to be yes and no. Veasey and colleagues subjected eight-week-old mice to one month of chronic sleep loss. For three consecutive days each week, tempting new climbing toys encouraged mice to play instead of rest during eight of their normal 12 daytime sleep hours. While they were allowed to rest and catch up on sleep for the remainder of the 24-hour period, they only made up about half of what they had lost.
Four weeks after intervention ended, Veasey found about 30 percent fewer neurons in the LC of sleep-deprived mice than in the LC of mice that slept soundly. Meanwhile, there were four times more activated astrocytes and twice as many activated microglia in the LC of sleep-deprived than control mice. In addition, p-tau, as detected with the AT8 antibody, turned up in neurons and in microglia.
Nine months after this one month of sleep loss, the mice had trouble remembering as judged by an object placement task. This test measures how long mice spend exploring each of two familiar objects, one having been moved to a new location. In addition, 30 percent of neurons in the amygdala had died and p-tau had not gone away, but had instead spread to the hippocampus. Repeating the experiment with tau knockouts, or in mice injected with tau siRNA just before sleep deprivation, completely protected against neuron loss.
While normal tau mediated injury from sleep loss, mutant tau was even worse. P301S tau mice that were sleep-deprived lost twice as many LC neurons as did wild-type animals put through the same wringer. Again, these neurons were not replaced nine months later. “Sleep loss can irreversibly injure the brain,” Veasey said. “This is the first work to show that conclusively.” This neuronal loss came with massive increases in p-tau and tau aggregates labeled by AT8 and MC-1, respectively, in both the LC and entorhinal cortex.
To rule out overstimulation of mice as the cause of this damage, as opposed to lack of sleep, Veasey’s group is looking at a second type of deprivation, namely fragmented sleep, seen in mice housed in cages that gently nudge the animals awake every minute. In those mice, too, the researchers are seeing more p-tau and neuron loss in the LC.
Audience members clamored to know if there was a way to reverse the impact of temporary sleep loss. Veasey emphasized that the p-tau remained nine months after the treatment and the LC neurons never came back. She believes the changes are permanent, but there may be some compensation for the lost neurons. Eckhard Mandelkow of the DZNE in Bonn, Germany, thought that likely. While he was shocked by the amount of neuron loss, he cautioned about overinterpreting the tau data, pointing out that p-tau is not the same as tangles and that the MCI antibody is not specific for tangles either.
Still, in keeping with Winer’s data, Veasey thinks there may be a vicious cycle where sleep loss causes tau abnormalities, which in turn cause more sleep problems. One person in the audience noted that people with Down’s syndrome, who are at extremely high risk for early onset AD, accumulate a lot of tau in their LC in their teenage years, and they also suffer from disturbed sleep.—Gwyneth Dickey Zakaib
At the Society for Neuroscience annual meeting, held November 11–15 in Washington, D.C., scientists presented further hints that bacteria in the gut may influence the symptoms and pathology of Alzheimer’s and other neurodegenerative diseases. Tweaking the bacterial profile in the digestive tracts of transgenic mice—either by feeding them probiotics or supplementing wild-type microbiota—reduced amyloid plaques, soothed inflammation, and improved memory. ApoE genotype correlated with different species of bacteria in the mouse gut, and certain bacterial metabolites prevented misfolded proteins from aggregating. Together, these studies showed how researchers are tackling the nexus between the microbiome and neurodegeneration.
“This area of research is set to explode in the next few years,” said Dave Morgan, now at Michigan State University College of Human Medicine in Grand Rapids.
Suzana Petanceska, National Institute on Aging, agreed. “Microbiome research is not just an emerging, but an expanding area of research into a variety of disease areas—from obesity and cancer to neurodevelopmental and neuropsychiatric disorders," she said.
Over the past two years, scientists have reported that some species in the microbiome can promote protein aggregation in certain neurodegenerative diseases, including Parkinson’s and Alzheimer’s (Dec 2016 news). Gut microbes seem to exacerbate Aβ pathology in mouse models of amyloidosis, and obliterating them ameliorates pathology (Feb 2017 news; May 2016 conference news). Could shifting the gut microbiota in mice help ameliorate AD-like symptoms?
To find out, some scientists, including Hyunjung Choi, who works in the lab of Inhee Mook-Jung at Seoul National University in South Korea, manipulated the gut microbiome of the ADLPAPT mouse. The researchers created this new model of AD by crossing the 5XFAD strain with JNPL3 tau animals. By about seven months, ADLPAPT mice accumulate both plaques and neurofibrillary tangles, and they lose hippocampal neurons. The latter distinguishes them from most other AD mouse models, which poorly recapitulate the neurodegeneration of AD.
When the ADLPAPT mice were two months old, Choi started transferring fecal microbiota from other mice into their guts. Six ADLPAPT mice received bacteria from age-matched wild-type animals five days a week for 16 weeks, while control ADLPAPT mice got bacteria from other ADLPAPT mice. Four months later, microbiota in the treated ADLPAPT mice resembled that of the wild-type donor. Choi then examined them for changes in Aβ and tau pathology.
Mice receiving wild-type microbiome had half as many Aβ plaques in their frontal cortices and hippocampi as did control ADLPAPT mice. Their hippocampi contained 60 percent less total tau, and their brains had half as much inflammation as measured by staining for Iba1 and GFAP, markers of activated microglia and astrocytes, respectively. These results suggest that the gut microbiome can modulate AD pathology, though the number of animals treated thus far was very small, Choi said.
Cortical boost: Wild-type mice on a control diet (left) have twice as many cortical neurons (red with blue nuclei) as AD mice (middle) at nine months. Give those AD mice a probiotic (right), and their cortical neurons stay at control numbers. [Courtesy of Krista McMurry.]
What about using a probiotic to deliver live “good” bacteria in the gut? Krista McMurry, from the lab of Paola Sacchetti, University of Hartford, Connecticut, supplemented the diets of either wild-type or APPSweTau301 transgenic mice with a probiotic or control inoculum for 12 weeks. The researchers used mice that were five to six months old, the age at which pathology starts. Delivered on a vanilla wafer cookie, the probiotic mixture contained two Lactobacillus strains. The control wafer had saline. Lactobacillus strains commonly appear in probiotic supplements and clinical data suggests they improve symptoms in people with irritable bowel syndrome and reduce inflammation of joints in a mouse model of arthritis (Tiequn et al., 2015; Liu et al., 2016). At the end of the experiment, she examined whether Lactobacillus treatment affected neuron loss, neuroinflammation, or memory.
By the end of the supplementation, APPSweTau301 mice on the control diet had half the number of neurons in their entorhinal cortices as did wild-type mice; however, APPSweTau301 mice on the probiotic maintained neurons at, or even above, control levels (see image above). And while the number of activated astrocytes in the cortices of the control transgenic mice was double that in wild-type mice, probiotic-treated mice had normal astrocyte numbers.
This neuroprotection seemed to carry no functional benefit on the Barnes maze, an illuminated circular platform with 20 holes around the edge, only one of which leads to safety. Mice that ate probiotics made more exploratory head pokes than did wild-type mice, indicating poorer navigational memory. While the probiotic mice explored half as many outlets as did AppSw/Tau controls, they took as long to escape the maze, suggesting they learned no faster.
McMurry said she next will compare activated microglia between samples, analyze neuron and glia levels in the hippocampus, and examine differences in pathological Aβ and tau species.
Similarly, Harpreet Kaur, from the lab of Colin Combs at the University of North Dakota, Grand Forks, found only a hint that a probiotic mix of eight strains of bacteria, including Bifidobacteria and Lactobacilli, improved memory in another mouse model of AD, the NL-G-F mutant APP knock-in. While seven-month-old NL-G-F mice given the probiotic for eight weeks tended to explore novel rather than familiar arms of a cross maze, the difference was not significant. Fewer activated microglia in the temporal cortices of the treated mice suggested the probiotic had some effect on the brain, but levels of soluble and insoluble Aβ40/42 in the temporal cortex were no different than in controls. Steve Estus, University of Kentucky, Lexington, who was not involved in the study, commented that this probiotic might reduce anxiety and related behavior, rather than cognition.
Are there genetic influences on the microbiome? ApoE is a strong risk factor for AD and other diseases, so Estus was curious if there was any cross-talk between its isoforms and the microbiome. Ishita Parikh, then a graduate student in the Estus lab, now a postdoctoral fellow in the lab of Ai-Ling Lin, also at the University of Kentucky, used ApoE targeted replacement (TR) mice, in which one of the human ApoE isoforms occupies the spot in the genome reserved for mouse ApoE in the wild-type. Parikh analyzed fecal DNA from 223 animals, a large number for an academic mouse study. Poop came from four- and six-month-old ApoE2, ApoE3, and ApoE4 TR mice, as well as offspring from TRx5XFAD mouse crosses. The researchers identified different bacteria by DNA sequencing.
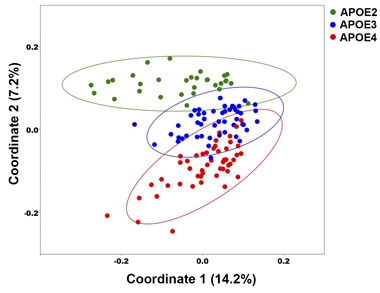
ApoE Shifts Microbiome?
Principal coordinate analysis, based on 14.2 percent and 7.2 percent of the total genetic variation found, displays how similar or dissimilar two microbiomes are. The microbiome of each mouse is represented by a single data point. Similar bacterial profiles appear close together. Bacteria of ApoE2, ApoE3, and ApoE4 mice separate into distinct clusters. [Courtesy of Ishita Parikh.]
Next the researchers used a method called principal coordinate analysis (PCoA) to quantify the similarities and dissimilarities among the different microbiomes. ApoE2, ApoE3, and ApoE4 mice had different bacterial profiles, regardless of whether the mice carried the 5XFAD mutations. Relative to the other genotypes, ApoE2 mice hosted more members of the Ruminococceae family, bacteria that digest starches to short-chain fatty acids. ApoE4 mice had more Lactobacilleae, which have been linked with good gut health. The profile of the ApoE3 mice lay in between. Estus plans to test for ApoE-related microbiome differences in other models of disease.
The upshot of these studies is that the gut microbiome might somehow protect the brain, though the mechanisms are not understood. Lap Ho, from the lab of Giulio Pasinetti at the Icahn School of Medicine at Mount Sinai, New York, examined whether certain bacteria metabolites affected dimerization of synthetic α-synuclein in vitro. Of six compounds Ho tested, one—valeric acid, a short-chain fatty acid produced when bacteria digest dietary fiber—prevented α-synuclein monomers from pairing up and aggregating into fibrils. These scientists previously reported that short-chain fatty acids can inhibit Aβ40 and Aβ42 aggregation (Ho et al., 2018). “This is a side of short-chain fatty acids that I hadn’t considered,” said Estus. “I usually think of them acting in an epigenetic fashion.” Estus wondered what concentration of valeric acid was needed to block α-synuclein dimerization, and how that compared to the concentrations found naturally in the human brain.
These aren’t the only ways intestinal flora could affect the brain. “The human microbiome provides a wealth of highly pro-inflammatory neurotoxins such as lipopolysaccharide, endotoxins, and the enzyme fragilysin,” Walter Lukiw, Louisiana State University, New Orleans, told the audience at SfN. These toxins are normally confined to the human GI tract, he said, but with aging or disease-related breakdown of the GI tract or blood-brain barrier, these neurotoxins may gain access to CNS compartments.
These conference presentations were preliminary and warrant more exploration, researchers agreed. “These early signals are piquing people’s interest,” Petanceska told Alzforum. “We need to be scientifically rigorous, put these early findings to the test, and expand the repertoire of microbiome manipulations to gain more confidence that there is something to them. Equally important is to complement studies in animal models with robust human studies to understand the role of the microbiome in brain aging and AD neurodegeneration.”—Gwyneth Dickey Zakaib
Besides the protein aggregates that mark tauopathies, in turns out that mutant forms of tau damage neurons in myriad other ways. Many were scrutinized at the Society for Neuroscience conference, held November 11–15 in Washington, D.C. Scientists reported how tau slows down mitochondria, disrupts the nuclear membrane and transcription, and even tangles up whole vascular networks. The meeting showed how after decades of research into the microtubule binding protein, it continues to surprise.
Tau and Organelles
Although tau is predominantly a cytoplasmic protein, several groups have begun to home in on what mutant tau does to the nucleus. Bahareh Eftekharzadeh, from Bradley Hyman’s lab at Massachusetts General Hospital, Charlestown, wondered if tau might disrupt nucleocytoplasmic transport. After all, transport falters in cells containing aggregates of other cytoplasmic proteins, such as TDP-43 and mutant huntingtin. Eftekharzadeh tested this idea using a dextran exclusion assay. It exploits the fact that if large dextran molecules of more than 500 kDa can access the nuclei, then these organelles must be compromised.
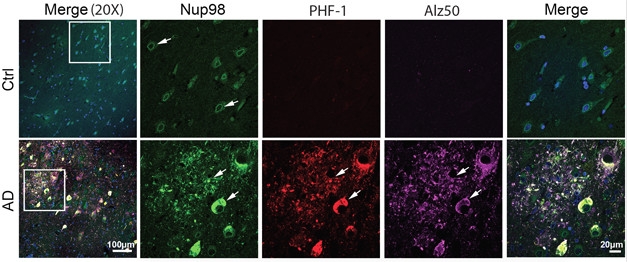
Nuclear Tau. Immunohistochemistry shows tau (PHF-1, Alz50) co-localizes with Nup98 in AD cases but not controls. [Courtesy of Bahareh Eftekharzadeh.]
To estimate the integrity of nuclei from people with AD, Eftekharzadeh tested tissue samples from the hippocampi and cerebella of patients who had died at Braak stages I, III, or VI. In all cases, nuclei from the cerebellum appeared to be intact, allowing only small dextrans of around 25 kDa to pass. By comparison, nuclei from hippocampi at Braak stage I appeared intact, but 30 percent of nuclei isolated from stage III and 50 percent of those isolated from stage VI tissue were leaky.
Why was that? Eftekharzadeh found that considerably more tau bound to nuclear pore complex proteins in the hippocampi of AD than control brains. In fact, in AD tissue about 60 percent of phospho-tau co-localized with nucleoporin 98 (Nup98). Turning to the Tg4510 mouse model of tauopathy to investigate further, she found tight co-localization of Nup98 and phospho-tau in neurofibrillary tangles. These animals express human tau with the P301L mutation and accumulate tangles in the cortex and hippocampus by four months. Almost all tangles in sections from human brain tested positive for Nup98 as well.
Eftekharzadeh thinks Nup98 might promote the formation of tangles and compromise the nuclear pore complex in the process. In support of this, she showed that, in vitro, the nucleoporin promoted aggregation of P301L tau. She needed half as much Nup98 to aggregate mutant tau as she did for wild-type tau. Surface plasmon resonance confirmed that the two proteins directly bound. Intriguingly, Eftekharzadeh said her findings support the idea that liquid-liquid phase separation might drive formation of tau tangles, because both Nup98 and tau, as Susanne Wegmann of Hyman’s lab and others reported earlier this year, form liquid droplets (May 2017 conference news; Jul 2017 news; Aug 2017 news). Fusion of the two types of droplets might set the stage for aggregation, she thinks.
In a related study, collaborator Gavin Daigle from Jeff Rothstein’s group at Johns Hopkins University, Baltimore, reported that tau binds to nucleoporins in postmortem tissue from frontotemporal dementia patients as well, and that this interferes with nucleocytoplasmic transport. Along the same theme, Garrett Lee Cornelison from Bess Frost’s lab at the University of Texas Health, San Antonio, reported that nuclear envelope invaginations in tauopathy models are enriched for polyadenylated RNA.
Invaginations and other aberrations in the nuclear envelope and nucleoskeleton have been studied before. They are a hallmark of laminopathies, where the lamin proteins that make up the nucleoskeleton fail. Since the lamin nucleoskeleton provides a scaffold for packaging DNA into highly condensed heterochromatin, transcription is altered in laminopathies.
Nuclear Invaginations. In nuclei of fruit flies that express R406W tau, lamin staining (red) outlines nuclear invaginations (left) that accumulate polyA RNA (green, center). DAPI (blue) outlines nuclei (right). [Courtesy of Garrett Cornelison.]
What does this have to do with tau? Last year, Frost reported that lamin nucleoskeleton breaks down in fly models of tauopathy, causing invagination of the nuclear envelope and neurodegeneration. She also found lamin network disruptions in tissue samples from AD patients (Frost et al., 2016). In D.C., Cornelison showed that nuclear membrane invaginations disrupt RNA processing in a tau model. He noted that these invaginations always contain nuclear pores, altering cellular homeostasis. “Diffusion from sites of transcription to nuclear pores is the rate-limiting step for RNA export,” he emphasized. Having pores in close proximity to sites of RNA synthesis, such as the nucleolus, might accelerate release of RNAs into the cytoplasm, and that could have dire consequences for the cell.
Cornelison tested this idea by blocking RNA translocation into the cytosol. The upshot: Either genetically or chemically suppressing RNA export from the nucleus protected the flies against tau toxicity. Cornelison crossed fruit flies expressing mutant tau with flies lacking Nxt1 and Nxf1, two components of the RNA export machinery. He also treated the tau flies with Leptomycin B or KPT-350, two molecules that banjax exportin 1, which ushers RNA and protein out of the nucleus. All treatments protected against tau-mediated neurodegeneration.
Further, Cornelison thinks that the nucleoplasmic reticulum actively promotes RNA export. In the R406W tau flies, he found that 43 percent of nuclei that had invaginations had more polyadenylated RNA. In fact, 32 percent of nuclear invaginations had puncta indicative of RNA accumulation in the invagination itself. Cornelison said that polyA RNA associated with nuclear invaginations in AD patients as well. He plans to investigate if particular RNAs may be involved.
Others thought these findings interesting. Eckhard Mandelkow, DZNE, Bonn, Germany, asked whether invaginations might be stabilized by tau binding to actin filaments, and what happens to invaginations if there is no tau. Cornelison said he did not know, but agreed this was an important question. He noted that nuclear invaginations happen in non-neuronal cells, particularly in cancers, which would suggest tau is not required. Mandelkow also wondered if the concentration of tau and RNA in the tiny space of the invagination might create an environment for tau to aggregate and form tangles. That would be in keeping with Eftekharzadeh’s data that Nup98 and tau co-aggregate. Frost told Alzforum that it might be the direct interaction between tau and pores that brings tau into the invaginations. Cornelison noted that flies do not seem to form the type of tangles found in the human brain, but agreed it would be interesting to test for aggregation near these invaginations.
“Collectively our studies point to the convergence of multiple tau toxicity mechanisms occurring at arguably one of the most important cellular organelles,” Eftekharzadeh told Alzforum. She noted growing evidence of pathological tau interfering with the nuclear membrane, including work on human induced pluripotent stem cell-derived neurons being studied at Rick Livesey’s lab at the University of Cambridge, England. Frost added that the leaky nuclei seen in the Hyman lab fit with pore containing invaginations, which may be less able to restrict the flow of diffusible molecules.
Another major organelle, the mitochondria, also suffers at the hands of tau. At SfN, James Johnson, who works at Michael Ashby’s lab at the University of Bristol, England, reported that mitochondria in Tg4510 mice behave erratically from an early age. Johnson infected young mice with a virus encoding a green fluorescent mitochondrial and a red fluorescent neuronal marker and repeatedly tracked both with a two-photon microscope over a cranial window. He first took measurements when the mice were 15 weeks old, and then every two weeks until week 37.
Johnson showed that while about 15 percent of neuronal mitochondria moved along axons in control mice, only 10 percent did in tau mice. As the tau mice aged, more of their mitochondria paused. They paused for longer than in normal mice, and even the mitochondria that did move were only half as fast as those from wild-type.
Whether these sluggish mitochondria explain behavioral deficits in Tg4510 mice remains to be seen. However, last March Ashby reported that dendritic spines in these animals turned over faster than those in control mice, while presynaptic spines were more stable (Jackson et al., 2017). In keeping with this, Johnson found that mitochondria in Tg4510 mice were more attracted to terminal boutons at the ends of axons and less attracted to “en passant” boutons along the axon shaft. This penchant might explain the stability of the presynapses.
Beyond Organelles
Mutant tau seems to cause problems at the tissue level, as well. Rachel Bennett, also from Hyman’s lab, reported disruptions to the vasculature in Tg4510 mice. She used two-photon microscopy to examine capillaries, showing how at 15 months, these mice have about 22,000 capillaries per cubic millimeter to 16,000 in wild-type mice. The narrowest vessels accounted for most of the difference, with the transgenic animals having many more 3 mm diameter and smaller capillaries than the wild-type. Further, Bennett reported that the small vessels twist strangely, and have nearly four times as many static white blood cells than regular vessels and very little blood flow.
Vascular Dystrophy.
Two-photon microscopy of 15-month-old mouse brain shows blood vessel abnormalities in Tg4510 mice (right) compared with wild-type controls (left). [Courtesy of Rachel Bennett.]
What causes these morphological changes? Bennett surmised that either old vessels were collapsing or that new, aberrant ones were forming. To test for the latter, she looked for expression of hypoxia-induced angiogenesis genes in endothelial cells and microglia. She found many that were more highly expressed in 15-month-old transgenics, including Pgf, Plau, Met, Serpine1, VegfA, and MMP9. Looking in mice of different ages, Bennett found that in capillaries, plasminogen activation inhibitor 1 (PAI-1), which is encoded by the Serpine1 gene, began to tick up when the animals were nine months old. It reached maximum levels when they were a year old, just before their cerebrovasculature began to morph. PAI-1 expression co-localized with cells expressing Iba1 and binding the tau aggregate antibody, MC1, hinting that microglial tau might play a role in the vasculature changes in these mice, and maybe even in some human tauopathies.
Support for this came from labmate Sarah Hopp, who outlined how microglia process tau from other cells and then release tau seeds. Hopp isolated microglia from Tg4510 mice and from APP/PS1/rTg21221 animals, which express wild-type human tau. She used quantitative PCR to measure gene expression, a Simoa assay to measure minute amounts of tau, and a cell-based assay to test for the presence of tau seeds.
Hopp found that while microglia from the tau transgenics do not express the tau gene, they do contain seeds that aggregate tau in the in vitro cell assay, indicating that the cells must have phagocytosed the protein. Even the conditioned medium from cultured microglia bore seeds, indicating that the cells then release some of the tau they consumed. Seeding activity in the medium decreased over time, suggesting a continual process of absorption and release of tau; however, it did not completely disappear, leading Hopp to speculate that the microglia cannot totally degrade the protein.
What about human microglia? Hopp isolated these cells from tissue taken from people who had just died at Braak stage VI AD or FTLD. She digested the tissue with enzymes to free the microglia, then cultured them for three days. They also harbored tau seeds and released them into the culture medium. Finally, she showed that microglia isolated from wild-type mice reduce but do not completely eliminate seeding activity in extracts from Tg4510 brain.
Hopp concluded that microglia are able to take up and break down fibrillogenic tau, but not efficiently enough to fully abolish it. She acknowledged that many questions remain open, including how the cells degrade tau, how they release it, and to what extent they contribute to the transmission of pathological tau in vivo.
Others wondered whether astrocytes might also take up and release the protein. Hopp agreed that astrocytic tau is important to understand, especially for some tauopathies, such as corticobasal dementia, where tau pathology has been documented in astrocytes. She said she has not seen tau co-localize with astrocytes, but noted that they are more difficult to isolate than microglia. On whether microglia release tau in exosomes, Hopp said it was hard to separate free from exosomal tau, though she might address the question using an ELISA with and without detergent. In response to concerns about contamination from macrophages, Hopp said she will address this by identifying cells with Tmem119, recently reported to be a microglial-specific marker (Satoh et al., 2016).
Research from many labs has indicated that while tau predominantly resides inside the cell, it can also spread trans-synaptically from neuron to neuron (Feb 2012 news). In D.C., Emily Miyoshi from Karen Gylys’ lab at the University of California, Los Angeles, proposed that this process might be spurred by Aβ. Evidence for this came from the study of human synaptosomes. Like intact neurons, these cell-free preparations respond when depolarized with potassium chloride. Previous work from Gylys’ lab showed that this caused the synaptosomes to release a C-terminal truncated form of tau (Sokolow et al., 2015).
In her poster, Miyoshi showed that the synaptosomes release tau and small vesicles, and the synaptosome medium tests positive for exosomal markers including CD9, CD63, and CD81. She then tested synaptosome extracts in the same cell-based seeding assay Hopp used. Miyoshi reported that synaptosomes from AD tissue carried almost tenfold the tau seeds as those from control samples. Furthermore, synaptosomes from two other tauopathy cases, who were at the same Braak stage as the AD patients, had no more tau seeds than control synaptosomes. Miyoshi thinks that the Aβ in the AD brain somehow makes the synaptic transmission of toxic tau more likely.
Miyoshi also claimed to have molecules that block propagation of tau. She tested this by collecting exosomes from cells that had been seeded with tau, and incubating them with naïve HEK293T cells. Two inhibitors, dubbed DDL111 and DDL112, blocked this propagation. Miyoshi declined to reveal much about these molecules beyond saying that they block the release of exosomes.—Tom Fagan





Comments
No Available Comments
Make a Comment
To make a comment you must login or register.