From Stem Cell Exosomes to Restoring ZZZs: New Ideas to Protect the Brain
Quick Links
The dearth of new drugs to treat neurodegenerative disease is making researchers cast their nets wide as they explore new ideas for prevention and treatment. Some creativity was on display at the annual Society for Neuroscience conference, held November 12-16 in San Diego. There, speakers proposed restoring sleep patterns to prevent amyloid plaque accumulation, and regulating an inflammatory astrocyte pathway to ward off vascular dementia. Studying the resilience of some elders’ brains to Alzheimer’s pathology, other scientists unearthed a new explanation: These brains generate high numbers of neural stem cells that release exosomes packed with a suite of regulatory microRNAs that preserve synapses. Most of these ideas have far to go before they could become usable therapies, but the approaches sparked excitement at SfN.
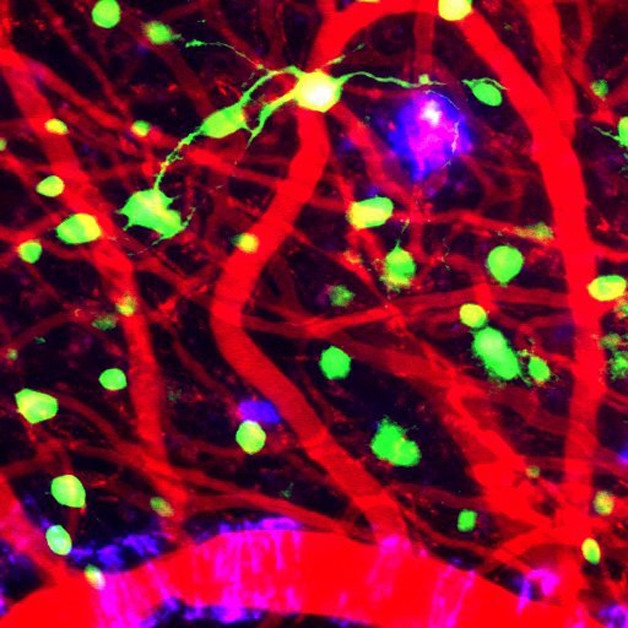
Tracking Amyloid Accumulation. A cranial window allows researchers to follow the development of plaques (purple), and their relationship to neurons (green) and blood vessels (red), in the cortices of live AD model mice after sleep therapy. [Courtesy of Brian Bacskai and Ksenia Kastanenka.]
New Clues to Cognitive Reserve
Researchers have long been tantalized by the observation that some people resist the deleterious effects of Alzheimer’s pathology into very old age (see Oct 2016 news). For example, Changiz Geula of Northwestern University, Chicago, previously reported that the brains of some old people with superior cognitive ability harbored extensive amyloid plaques (see Dec 2008 conference news). At an SfN press conference on AD, Geula extended this with a detailed look at eight people who had died in their late 90s, having maintained the recall prowess of a 50- or 60-year-old. Postmortem, their brains revealed a wide range of pathology, he said. Two had limited plaques and tangles equivalent to Braak stage I or II, four were classified as Braak stage III or IV, and the remaining two brains contained enough plaques and tangles to meet a pathological diagnosis of Alzheimer’s disease. However, all these brains contained healthy-looking neurons, not the extensive cell death seen in people with clinical symptoms of AD. In addition, brains of similar cases sported more postsynaptic markers than the brains of age-matched controls with average cognitive abilities. The data suggest that these people possess resilience factors that protect their brains, Geula concluded, and he is trying to identify those from among a range of genetic and environmental parameters.
In a separate talk, Maria Micci, who works in collaboration with Giulio Taglialatela at the University of Texas Medical Branch, Galveston, proposed an explanation for why some older people maintain their synapses so well. The researchers had noticed that the brains of people who died with preserved cognition but extensive Alzheimer’s pathology contained more neural stem cells (NSCs) than did AD or MCI brains. The higher the number of these cells, the better the person’s score had been on their last MMSE (see Briley et al., 2016). When they examined the brains more closely to see why cognition might be spared, Taglialatela and colleagues found that postsynapses remained devoid of Aβ oligomers. This contrasted with the high levels of synaptic Aβ in AD brains, as seen by antibody staining of isolated synapses (see Bjorklund et al., 2012).
How might the NSCs protect synapses? Perhaps, Micci thought, via exosomes. After all, these vesicles are packed with cargo and can be taken up by bystander cells. To test this, she treated mouse hippocampal slice cultures with exosomes secreted by cultured rat NSCs, then later added preformed oligomers of recombinant Aβ42. Pretreatment with NSC exosomes spared these hippocampal synapses from accumulating Aβ, she reported. The treatment also preserved long-term potentiation and prevented synaptic excitability.
Micci saw similar results when she injected rat NSC exosomes into the hippocampi of wild-type mice, waited four hours, then isolated synaptosomes and treated them with Aβ in vitro. Again, synapses stayed clean. Next, the researchers examined nestin-δ-HSV TK-eGFP mice, in which neurogenesis can be switched off. After disabling neurogenesis and isolating synapses, Aβ treatment resulted in excessive oligomers crowding these structures. As in wild-type mice, this experimental inundation of synapses was prevented by injecting NSC exosomes into the brain before isolating synapses. In other experiments, the researchers have found that NSC exosomes shield synapses from tau oligomers as well, Taglialatela told Alzforum.
Notably, exosomes from mature neurons conferred no protection in any of these scenarios. To find out what makes NSC exosomes special, the researchers broke them open and compared their contents to those of neuronal exosomes. That revealed a unique signature of eight miRNAs in the NSC exosomes. Now, Micci and Taglialatela are exploring whether this set of miRNAs affects how synapses function, and whether it can protect them from damaging oligomers. Micci will also isolate exosomes from postmortem AD and control brains to see if they contain the same set of miRNAs.
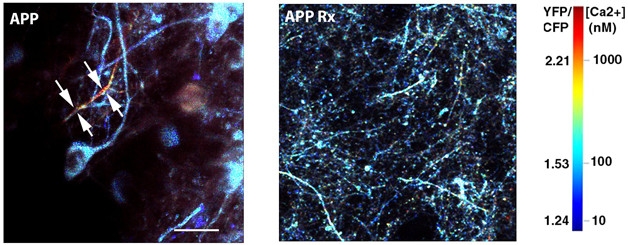
Calming Calcium Spikes. In AD model mice (left), high calcium levels (yellow and red) flood neurons (blue), but disappear when slow-wave sleep is restored (right). [Courtesy of Brian Bacskai and Ksenia Kastanenka.]
Can Refreshing Circadian Rhythms and Sleep Block Plaque Accumulation?
Researchers have long known that AD patients sleep poorly, but recent studies have switched things around to suggest disrupted sleep may precede and contribute to the development of dementia (see Aug 2012 conference news; Oct 2013 news). In part, this may be because the brain clears waste proteins such as amyloid during slumber (see Jun 2014 news; May 2014 conference news). Speakers at SfN suggested two different approaches to normalizing sleep, with an eye to preventing amyloid buildup.
Trongha Xuan Phan, who works with Li-Huei Tsai at the Picower Institute at Massachusetts Institute of Technology, Cambridge, and Robert Vassar at Northwestern University, Chicago, focused on the circadian clock and the genes it regulates. Some of those might be therapeutic targets for restoring Aβ clearance, he reasoned. Because histone deacetylase 1 (HDAC1) knockout mice have abnormal circadian rhythms, he investigated which genes this enzyme controls. In knockout mice, preliminary results suggest that only 1,421 genes oscillated with the sleep-wake cycle, compared to 2,822 genes in wild-type mice, he reported. Thus, HDAC1 appears to control about half of normally cycling genes. Loss of cycling correlated with worse contextual memory in these mice; this dovetails with other studies tying circadian disruptions to poor hippocampal function (see Wardlaw et al., 2014).
Among the genes with disrupted cycling, aquaporin 4 stood out to the researchers. This astrocytic water channel has been associated with clearance of Aβ from the brain, and has been found to be in short supply in AD brains (see Aug 2012 news). Surprisingly, Phan found that the HDAC1 knockouts expressed twice as much aquaporin 4 as controls, rather than less. Looking more closely at previous research for an explanation, he found that the site of aquaporin 4 expression matters. Global brain expression has been reported to be elevated in AD patients, even while aquaporin 4 near blood vessels drops (see Hoshi et al., 2012; Dec 2016 news). Since Phan measured global aquaporin 4 expression in the hippocampus, the findings are consistent with the literature, he told Alzforum.
The researchers have now crossed HDAC1 knockout mice with an AD model to find out if loss of circadian rhythms affects pathology. In the future, Phan plans to examine how aquaporin 4 expression in the perivasculature changes during the day. Global aquaporin 4 expression peaks during sleep in people, but these oscillations dampen with age, Phan noted. He also wants to study how light therapy, which helps improve sleep in older adults and people with AD, affects HDAC1, circadian gene cycling, and Aβ clearance.
Ksenia Kastanenka, working with Brian Bacskai of Massachusetts General Hospital, Charlestown, took a different tack. Recent studies have suggested that amyloid accumulation in the brain can disrupt slow-wave sleep, which is essential for memory consolidation (see Jun 2015 news; Varga et al., 2016). Kastanenka used mice to take a closer look at this issue. To visualize slow-wave oscillations, she applied a voltage-sensitive dye to the cortices of APPswe/PS1DE9 mice through a cranial window. She found that the power, but not the frequency, of the oscillations dropped starting at three months of age, before plaques formed.
What might explain this? One clue was that these mice have low levels of the inhibitory neurotransmitter GABA. When the researchers injected exogenous GABA into their brains, it restored the power of slow-wave sleep, suggesting that overactive neurons were responsible for the disruptions. Other studies agree that neuronal hyperactivity contributes to memory problems and degeneration in AD (see Dec 2011 news; Mar 2015 news).
To see if Aβ by itself could disrupt slow waves, Kastanenka and colleagues collected Aβ oligomers secreted by cultured neurons isolated from Tg2576 mice. They added these oligomers to the cortices of wild-type mice through a cranial window. Again, they saw a drop in the power of slow-wave oscillations.
Could restoring slow-wave sleep help neurons? The researchers expressed light-activated channelrhodopsin 2 in frontal cortical neurons of three-month-old APPswe/PS1DE9 mice, waited one month, then drove neuronal activity with blue light delivered to the cortex. When light flickered at the frequency of slow waves, the power of these oscillations returned to normal. The treatment boosted GABA to wild-type levels and dampened neuronal calcium, another marker of hyperactivity, to normal levels (see image above).
Moreover, during two months of light treatment, no new amyloid plaques formed. These mice normally deposit numerous plaques at that age. Altogether, the data suggest that restoring slow-wave sleep in AD patients might prevent neurodegeneration, Kastanenka said. An enthusiastic audience peppered her with questions, and wanted to know how these findings might be applied. Kastanenka suggested that the key might be to enhance the activity of inhibitory interneurons to restore balance to the circuitry in AD brains.
Targeting Astrocytes to Ameliorate Vascular Dementia
Could astrocytes be the ticket to new therapies beyond AD? Christopher Norris of the University of Kentucky, Lexington, thinks as much. He focuses on vascular dementia, the second leading cause of dementia that co-occurs in almost half of AD cases. Despite numerous hypotheses, researchers have reached no consensus for how vascular problems cause cognitive decline. Because astrocytes act as liaisons between blood vessels and neurons, Norris wondered if they might play a role. To investigate, he used a mouse model of vascular dementia developed by colleague Donna Wilcock at Kentucky. Wild-type mice are fed a diet rich in methionine and low in folate, boosting levels of homocysteine in the blood, damaging blood vessels, and causing cognitive decline (see Sudduth et al., 2013; Sudduth et al., 2014). In these mice, the astrocyte end-feet that wrap around blood vessels become disrupted, losing crucial ion channels, Wilcock found (see Sudduth et al., 2016).
What might explain these changes? In AD models, Norris and others previously reported that Aβ stimulates calcium release in astrocytes, thus activating the phosphatase calcineurin, which in turn triggers the transcription factor NFAT. This pathway turns on inflammation and triggers loss of synapses and cognitive decline (see Oct 2009 news; Dec 2009 news; Feb 2010 news). Norris thought this pathway might be activated by vascular pathology, as well. After all, astrocytes near microinfarcts in postmortem brain sections express high levels of activated calcineurin (see Pleiss et al., 2016).
If so, could turning down NFAT signaling ameliorate vascular dementia? Norris and colleagues injected a viral vector carrying the NFAT inhibitor VIVIT into the brains of two-month-old wild-type mice. An astrocyte-specific promoter limited expression to these cells. Two months later, the researchers placed the mice on the vascular dementia diet for three months. Mice whose NFAT was blocked preserved normal long-term potentiation and synapse number around the injection area, whereas controls had deficits on these readouts. In addition, the treated brain region maintained normal cerebral blood flow and healthy neurons. Norris is now testing behavior in treated mice. He suggested that NFAT inhibition might be a viable strategy for preventing vascular dementia. In previous work, he reported that the same approach can rescue synapses and cognition in animals models of AD and traumatic brain injury (see Furman et al., 2012; Furman et al., 2016).—Madolyn Bowman Rogers
References
News Citations
- The Making of a “Superager”: New Research Examines Neuroanatomy that Keeps Older Adults Sharp
- DC: Amyloid-Laden Brains—What Do They Mean for Healthy Seniors?
- Night Owl? Early Bird? Good Night’s Sleep May Protect the Brain
- From ApoE to Zzz’s—Does Sleep Quality Affect Dementia Risk?
- While You Were Sleeping—Synapses Forged, Amyloid Purged
- Glymphatic Flow, Sleep, microRNA Are Frontiers in Alzheimer’s Research
- Brain Drain—“Glymphatic” Pathway Clears Aβ, Requires Water Channel
- Dearth of Water Channels a Sign of ‘Glymphatic’ Breakdown in Alzheimer’s?
- Does Amyloid Disturb the Slow Waves of Slumber—and Memory?
- Research Brief: Hippocampal Hyperactivity Tied to Early MCI Atrophy
- More Evidence That Epilepsy Drug Calms Neurons and Boosts Memory
- The Skinny on NFATs—Mediators of Aβ Toxicity?
- Chicago: NFATs, Calcineurin—Mediators of AD, PD Pathogenesis?
- Calcium Hypothesis—Studies Beef Up NFAT, CaN, Astrocyte Connections
Research Models Citations
Paper Citations
- Briley D, Ghirardi V, Woltjer R, Renck A, Zolochevska O, Taglialatela G, Micci MA. Preserved neurogenesis in non-demented individuals with AD neuropathology. Sci Rep. 2016 Jun 14;6:27812. PubMed.
- Bjorklund NL, Reese LC, Sadagoparamanujam VM, Ghirardi V, Woltjer RL, Taglialatela G. Absence of amyloid β oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer's disease neuropathology. Mol Neurodegener. 2012;7:23. PubMed.
- Wardlaw SM, Phan TX, Saraf A, Chen X, Storm DR. Genetic disruption of the core circadian clock impairs hippocampus-dependent memory. Learn Mem. 2014 Aug;21(8):417-23. Print 2014 Aug PubMed.
- Hoshi A, Yamamoto T, Shimizu K, Ugawa Y, Nishizawa M, Takahashi H, Kakita A. Characteristics of aquaporin expression surrounding senile plaques and cerebral amyloid angiopathy in Alzheimer disease. J Neuropathol Exp Neurol. 2012 Aug;71(8):750-9. PubMed.
- Varga AW, Wohlleber ME, Giménez S, Romero S, Alonso JF, Ducca EL, Kam K, Lewis C, Tanzi EB, Tweardy S, Kishi A, Parekh A, Fischer E, Gumb T, Alcolea D, Fortea J, Lleó A, Blennow K, Zetterberg H, Mosconi L, Glodzik L, Pirraglia E, Burschtin OE, de Leon MJ, Rapoport DM, Lu SE, Ayappa I, Osorio RS. Reduced Slow-Wave Sleep Is Associated with High Cerebrospinal Fluid Aβ42 Levels in Cognitively Normal Elderly. Sleep. 2016 Nov 1;39(11):2041-2048. PubMed.
- Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab. 2013 May;33(5):708-15. Epub 2013 Jan 30 PubMed.
- Sudduth TL, Weekman EM, Brothers HM, Braun K, Wilcock DM. β-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res Ther. 2014;6(3):32. Epub 2014 Jun 9 PubMed.
- Sudduth TL, Weekman EM, Price BR, Gooch JL, Woolums A, Norris CM, Wilcock DM. Time-course of glial changes in the hyperhomocysteinemia model of vascular cognitive impairment and dementia (VCID). Neuroscience. 2017 Jan 26;341:42-51. Epub 2016 Nov 25 PubMed.
- Pleiss MM, Sompol P, Kraner SD, Abdul HM, Furman JL, Guttmann RP, Wilcock DM, Nelson PT, Norris CM. Calcineurin proteolysis in astrocytes: Implications for impaired synaptic function. Biochim Biophys Acta. 2016 Sep;1862(9):1521-32. Epub 2016 May 20 PubMed.
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease. J Neurosci. 2012 Nov 14;32(46):16129-40. PubMed.
- Furman JL, Sompol P, Kraner SD, Pleiss MM, Putman EJ, Dunkerson J, Mohmmad Abdul H, Roberts KN, Scheff SW, Norris CM. Blockade of Astrocytic Calcineurin/NFAT Signaling Helps to Normalize Hippocampal Synaptic Function and Plasticity in a Rat Model of Traumatic Brain Injury. J Neurosci. 2016 Feb 3;36(5):1502-15. PubMed.
Further Reading
News
- Cognitive Reserve—More Evidence It Prevents Neurodegeneration
- A Life of Cognitive Enrichment May Fend Off Dementia. But How?
- Sleep and Brain Cleansing—Fresh Insights into Regulation and Disruption
- Wake Up and Smell the … Orexin? Peptide Percolates in Alzheimer’s Brain
- Sleep Patterns, Circadian Clock Linked to Aβ Oxidative Stress
- Paper Alert: Does Plaque Steal Shuteye?
- Could Toning Down Calcineurin Neutralize α-Synuclein?
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.